 Andrea Bonnin-Marquez
Andrea Bonnin-Marquez Sanne L. Maas
Sanne L. Maas Melissa Corcini-Berndt1
Melissa Corcini-Berndt1 Emiel P. C. Van der Vorst
Emiel P. C. Van der Vorst- 1Department of Internal Medicine I, Aachen-Maastricht Institute for Cardio-Renal Disease (AMICARE), and Institute for Molecular Cardiovascular Research (IMCAR), University Hospital Aachen, RWTH Aachen University, Aachen, Germany
- 2Institute for Cardiovascular Prevention (IPEK), Ludwig-Maximilians-Universität, Munich, Germany
Introduction: Chronic kidney disease (CKD) is a progressive condition associated with increased mortality and morbidity, placing a substantial burden on healthcare systems globally. CKD often coexists with cardiovascular disease (CVD), further complicating patient outcomes. This study investigates the kinomic profile of hyperlipidemic mice to understand the signaling mechanisms underlying CKD progression and its cardiovascular consequences.
Methods: Apoe−/− mice were subjected to a Western-type diet, with or without adenine supplementation to induce CKD. Kinase activity was profiled using PamGene® assays on renal cortex samples collected at early (4 weeks) and late (12 weeks) stages of CKD.
Results: It could be demonstrated that CKD led to significant increases in peptide phosphorylation related to both tyrosine and serine-threonine kinases, which were particularly pronounced in the late-stage model. Therefore, the kinase activity in the kidney increased upon CKD development in a CKD-stage-dependent manner. Notably, the activity of cyclin-dependent kinases (CDKs) was reduced at early disease stages but remained unaffected in late stages. Pathway analysis revealed stage-specific alterations in cell cycle regulation, inflammation, oxidative stress, lipid metabolism, and fibrosis pathways associated with kinase activity changes throughout disease progression.
Discussion: These findings highlight critical kinases involved in CKD development and suggest their potential roles in mediating pathological processes such as inflammation and fibrosis. Targeting specific kinases may offer novel therapeutic strategies for mitigating CKD progression and its cardiovascular complications. Future research should explore the causal relationships between newly identified kinases and CKD development.
1 Introduction
Chronic kidney disease (CKD) is a complex, progressive disease with substantial consequences globally. CKD patients suffer from increased mortality and morbidity, and treatment, particularly in advanced stages, poses a significant economic burden (Foreman et al., 2018; Jha et al., 2023; Levin et al., 2017; Stewart et al., 2024). According to current international guidelines, CKD is defined as abnormalities in kidney function or structure persisting for at least 3 months, with consequences for patient health (Kidney Disease: Improving Global Outcomes CKDWG, 2024; Stevens et al., 2013). Disease severity is classified based on glomerular filtration rate (GFR), albuminuria, and underlying cause (Kidney Disease: Improving Global Outcomes CKDWG, 2024; Stevens et al., 2013). These alterations to the function and structure of the kidney are irreversible and, in the early stages, often asymptomatic, leading to late diagnoses (Webster et al., 2017). Moreover, there is no effective cure, and patients who progress to end-stage kidney disease (ESKD) require kidney replacement therapy, either via dialysis or kidney transplant (Webster et al., 2017). Despite advancements in diagnostic guidelines and treatments, CKD remains underdiagnosed and undertreated worldwide (Stewart et al., 2024). In addition, CKD has not been included in the World Health Organization’s list of major drivers of non-communicable disease-related early mortality, limiting both resource allocation and recognition of the disease’s impact (Francis et al., 2024). Consequently, CKD-related mortality has continued to rise over the last decades, and the disease is projected to become the fifth leading cause of years of life lost globally by 2040 (Foreman et al., 2018; Stewart et al., 2024; Collaboration, 2020).
In addition to its direct health burden, CKD patients are at an increased risk for cardiovascular disease (CVD), with the majority of CKD patients succumbing to cardiovascular events rather than from ESKD (Foreman et al., 2018; Kidney Disease: Improving Global Outcomes CKDWG, 2024; Webster et al., 2017; Vaduganathan et al., 2022). Furthermore, CKD can also occur as a comorbidity of various pathologies, including diabetes, hypertension, heart failure, and atherosclerosis (Jankowski et al., 2021; Marx-Schutt et al., 2025; Noels et al., 2025; Rangaswam et al., 2019). Patients with such comorbidities face particularly poor prognoses, with reduced survival and diminished quality of life (Fraser et al., 2015). Notably, CKD patients exhibit a markedly increased risk for atherosclerosis, which is the main underlying cause of CVD (Jankowski et al., 2021; Marx-Schutt et al., 2025). CKD and CVD share various pathological mechanisms, including inflammation, oxidative stress, fibrosis (Kumar et al., 2019), and hyperlipidemia (Visconti et al., 2016). The most common dyslipidemia pattern in CKD patients consists of hypertriglyceridemia and reduced high-density lipoprotein levels (Reiss et al., 2015). In recent years, triglycerides and triglyceride-rich lipoproteins, which are not the typical target of lipid-lowering therapies, have gained more recognition as a causal factor in atherosclerosis and as an independent risk factor for cardiovascular complications (Xu et al., 2024). Despite these well-established mechanistic links between CVD and CKD, the field of cardiorenal research only began to expand from the early 2000s (Lv et al., 2023). As a result, a deeper understanding is still needed to uncover additional contributors and critical regulatory pathways involved in CKD and CKD-induced atherosclerosis.
Major regulators of cellular processes relevant to CKD are protein kinases, which mediate signal transduction via phosphorylation (Ubersax and Ferrell, 2007), resulting in diverse functional effects on cellular activity (Manning et al., 2002). Kinases are commonly classified into two main groups, tyrosine-specific kinases and serine/threonine-specific kinases (Ubersax and Ferrell, 2007). Kinases are one of the largest gene families, making up approximately 2% of the genome, and are estimated to phosphorylate about 30% of all cellular proteins on at least one residue (Ubersax and Ferrell, 2007), underscoring their critical biological importance.
Given this central role, we decided to employ a kinomics approach to explore the signaling mechanisms at play in CKD. More specifically, we studied the kinomic profile of the kidneys of hyperlipidemic mice with or without CKD, as hyperlipidemia is a major characteristic of both CKD and atherosclerosis, and particularly a driver of CKD-induced atherosclerosis. In addition, we evaluate kinase activity at both early and advanced stages of disease to observe changes associated with disease progression.
2 Materials and methods
2.1 Mice
All mice were apolipoprotein E (Apoe)−/− and bred on a C57/Bl6 background for more than 10 generations (Jackson Laboratory; Strain#: 002052). Starting at 8 weeks of age, mice were fed a Western-type diet (WTD) containing 21% fat and 0.15%–0.20% cholesterol (Altromin 132010, Sniff TD88137), supplemented with or without 0.3% adenine for 2 weeks to induce CKD. Subsequently, the mice were fed a WTD with or without 0.15% adenine for another 2 or 10 weeks to maintain CKD and stimulate CKD-induced hyperlipidemia.
To verify CKD induction, mice were placed in individual metabolic cages overnight to collect urine at baseline and at the end of the 4 or 12-week feeding period. Blood samples were collected at the indicated time points by retro-orbital bleeding after inhalational anesthesia using isoflurane and subsequently processed to obtain either serum or plasma. For functional renal analyses, urea, creatinine, and total protein levels were measured in urine and serum using a clinical renal test analyzer according to the manufacturer’s instructions. Plasma cholesterol and triglyceride concentrations were measured by using enzymatic assays (Roche diagnostics) following the manufacturer’s protocol.
Euthanasia was performed under deep, non-antagonisable anaesthesia (10 mg/kg xylazine and 100 mg/kg ketamine) with subsequent blood collection via retro-orbital puncture using anticoagulant-coated capillaries. All animal studies performed were approved by the local ethical committee (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Germany, approval number 81-02.04.2019.A364). All procedures are in line with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
2.2 Kinase activity profiling - Analysis
Snap-frozen renal cortex pieces (10 mg) were thawed on ice and lysed in M-PER™ Mammalian Protein Extraction Reagent (ThermoFisher Scientific) containing 1% Halt™ Protease Inhibitor Cocktail (ThermoFisher Scientific) and 1% Halt™ Phosphatase Inhibitor Cocktail (ThermoFisher Scientific) for 10 min. The tissue was homogenized with a metal bead in the TissueLyser LT (Qiagen) for 7 min at 50 Hz, after which the lysates were centrifuged at 15.000 x g for 15 min at 4°C. Protein amounts were measured using the Nanodrop One Microvolume UV-Vis Spectrophotometer (ThermoFisher Scientific).
The PamChip® peptide tyrosine kinase and Ser/Thr Kinase assay microarray systems were used on the PamStation®12 (PamGene International) to define the profiles of Tyrosine kinase (PTK) and Serine-Threonine kinase (STK), respectively. The PTK-PamChip® includes 196, and the STK-PamChip® array contains 144 individual phospho-sites. For PTK and STK activity profiling, all reagents were used by PamGene International B.V.
In the PTK assay, 10.0 μg of protein (n = 4 per condition) was used following the standard protocol provided by PamGene. Lysates were prepared by adding PTK Basic Mix, consisting of 4 μL of 10x protein PTK reaction buffer (PK), 0.4 μL of 100x BSA, 0.4 μL of 1 M dithiothreitol (DTT) solution, 4 μL of 10x PTK additive, 4 μL of 4 mM ATP, and 0.6 μL of monoclonal anti-phosphotyrosine FITC-conjugated detection antibody (clone PY20). Distilled water was added to bring the total volume to 40 μL. Before loading the samples, a blocking step was performed by applying 30 μL of a 2% BSA solution to the center of each array, followed by a wash with PTK solution for PamChip® preprocessing. Subsequently, 40 μL of the PTK Basic Mix was added to each array of the PamChips®. The microarray assays were then run for a total of 94 cycles on the PamStation®12. An image was recorded using a CCD camera at kinetic read cycles 32–93, with exposure times of 10, 50, and 200 ms, and an end-level read cycle at 10, 20, 50, 100, and 200 ms. Spot intensities at each time point were quantified and corrected for local background using BioNavigator software version 6.3 (PamGene International, ‘s-Hertogenbosch, The Netherlands).
In the STK assay, phosphorylated Ser/Thr residues were detected using 2.0 μg of protein per sample (n = 4 per condition) and 400 μM ATP, combined with the STK Basic Mix. The Basic mix consisted of 1× protein PTK reaction buffer (PK), 1x bovine serum albumin (BSA), and 1x STK antibody mix. Before loading the samples, a blocking step was performed using 30 µL of 2% BSA, followed by washing with PTK solution for preprocessing. Subsequently, 40 µL of STK Basic Mix was applied to each array. Samples were incubated for one hour at 30°C, during which the samples were actively pumped back and forth through the porous material to maximize binding kinetics and minimize assay time. Phosphorylation events were detected using a FITC-conjugated antibody. Imaging was done using an LED-based imaging system, and the spot intensities at each time point were quantified and corrected for local background using the BioNavigator software version 6.3 (PamGene International, ‘s-Hertogenbosch, The Netherlands).
2.3 Kinase activity profiling - Visualization
The distribution of the phosphorylated peptides is shown in the volcano plots created with the R package EnhancedVolcano v1.26.0 (Blighe et al., 2025). Blue dots depict peptides with an adjusted P value < 0.05.
A principal component analysis (PCA) was performed with the help of the R package stats V4.3.1 (Team, 2013), depicting the singular peptide decomposition, examining the covariances/correlations between the samples. Results were visualized as two-dimensional PCA plots generated with ggplot2 (Wickham, 2016), enabling the identification of sample clustering patterns, variability, and potential outliers.
To classify and visualize kinases identified in the assay, a kinase coral tree was generated using the Coral web tool (http://phanstiel-lab.med.unc.edu/CORAL). This approach organizes kinases based on their evolutionary and functional relationships into families and subfamilies, as defined by the Human Kinome Project classification system. Input data included the list of kinases detected as active in the PTK or STK PamChip® assays. Visualization parameters were set to reflect relative kinases; node/branch colors represent mean kinase statistics (<0.05), and node sizes represent the median final scores (>1.2). The hierarchical representation in the coral tree allowed the identification of family-wide trends, highlighting condition-specific kinase activation patterns, and supporting the functional interpretation of the kinase profiles.
Over-representation analysis (ORA) was performed for kinases with significant differences compared to control samples (median final scores >1.2 and adjusted p-value <0.05) using the Gene Ontology, Kyoto Encyclopedia of Genes and Genomes (KEGG), and WikiPathway databases. ORA was conducted using the R packages clusterProfiler v4.8.1 (Wu et al., 2021), and the data was visualized with the R package ggplot2 v3.4.2 (Wickham, 2016).
2.4 Statistical analysis
Statistical analyses were performed with GraphPad Prism v.10 (GraphPad Software, LLC). Gaussian distribution was tested using the D’Agostino & Pearson test, after which the Mann-Whitney U test was used to analyze the data. *P < 0.05; ****P < 0.0001.
3 Results
3.1 CKD increases peptide phosphorylation in the kidneys of hyperlipidemic mice.
Apoe−/− mice were fed with a pro-atherogenic Western-type diet (WTD) to induce hyperlipidemia, or a WTD enriched with adenine (WTD-A) to induce CKD in hyperlipidemic mice, for either 4 weeks (early-stage) or 12 weeks (late-stage). Subsequently, the renal cortex was collected to compare the kinomic profiles of hyperlipidemic mice with or without CKD (Figure 1A). Serum urea and creatinine levels significantly increased in WTD-A-fed mice, while urine urea and creatinine decreased, coinciding with an increased amount of urine protein, suggesting a successful induction of kidney damage (Supplementary Figures S1A-F, S2A-F). Circulating cholesterol and triglyceride levels did not change, while the body weight was slightly reduced in WTD-A-fed mice, again aligning with an induction of CKD in these mice (Supplementary Figures S1G-I, 2G-I). Kinase activity was measured with the PamGene® platform, revealing specific peptide phosphorylation as a proxy for kinase activity. Apparent differences in peptide phosphorylation patterns are observed between WTD-A and WTD-fed mice at both early- and late-stages of CKD (Figures 1B–E; Supplementary Tables 1-4). Particularly for the PTK, the magnitude of these differences increases upon disease progression, reaching its peak in the late-stage CKD model (Figures 1B,C). Further analysis reveals a significant increase in phosphorylation of peptides related to tyrosine kinases, with changes being more pronounced at 12 weeks than at 4 weeks (Figures 1F,G). Notably, 38 out of the total 75 differentially phosphorylated peptides are upregulated in both early and late stages, indicating substantial overlap (Figure 1H). Nevertheless, disease progression contributes to the phosphorylation of 35 additional peptides exclusively in the late-stage CKD group (Figure 1H).
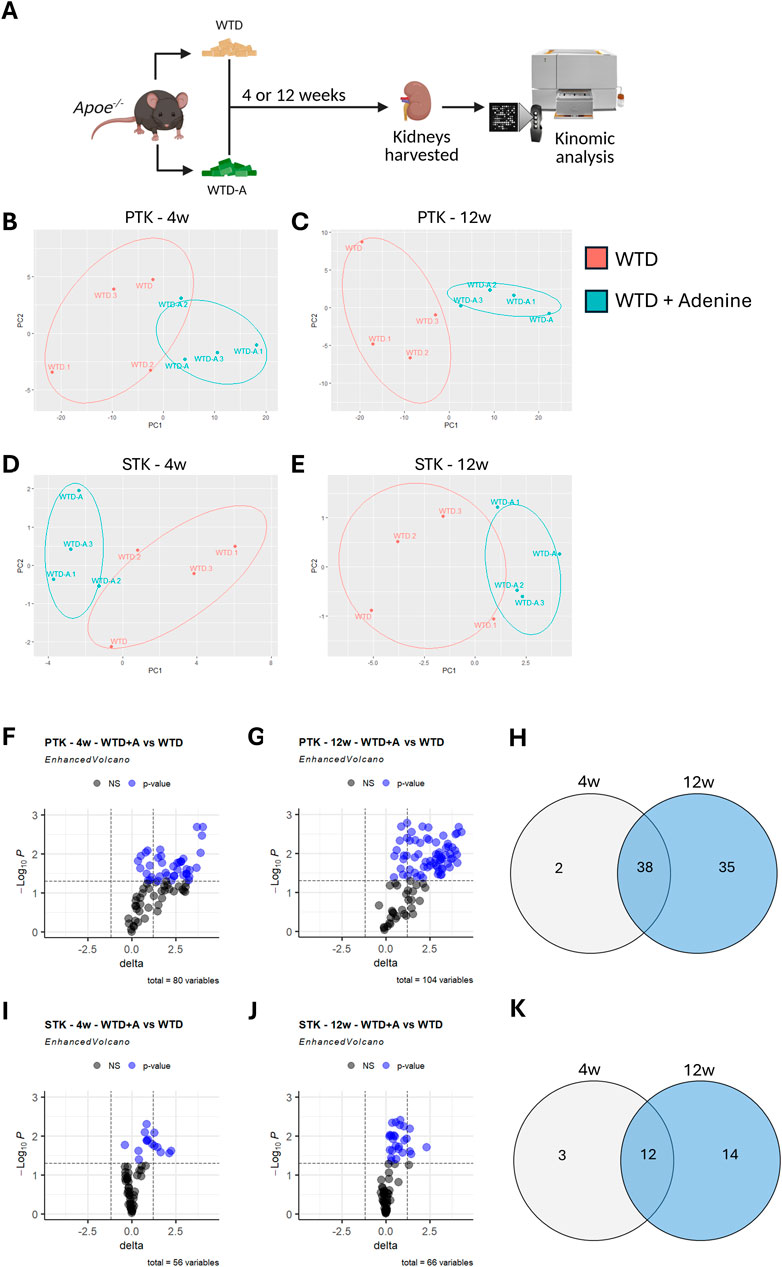
Figure 1. Peptide phosphorylation is increased in hyperlipidemic mice with CKD and upon disease progression. (A) Schematic diagram of the study design. (B–E) Principal component analysis (PCA) depicting the variance in peptide phosphorylation profiles in kidney lysates from control (WTD) and CKD mice (WTD + Adenine) for (B) PTK-related peptides after 4 weeks of diet, (C) PTK-related peptides after 12 weeks of diet, (D) STK-related peptides after 4 weeks of diet, or (E) STK-related peptides after 12 weeks of diet. (F,G) Volcano plots displaying differentially phosphorylated PTK-related peptides. Blue dots represent significant differential phosphorylation of peptides between the two groups (p < 0.05). Total variables reflect the number of peptides with measurable amounts of phosphorylation. (H) Venn diagram showing the overlap and unique sets of differentially phosphorylated PTK-related peptides in both early (4 weeks) and late (12 weeks) stages of CKD. (I,J) Volcano plots displaying differentially phosphorylated STK-related peptides. Blue dots represent significant differential phosphorylation of peptides between the two groups (p < 0.05). Total variables reflect the number of peptides with measurable amounts of phosphorylation. (K) Venn diagram summarizing differentially phosphorylated STK-related peptides shared or unique between early- and late-stage CKD. All data represent four biological replicates per condition.
A similar, although less pronounced, pattern is observed for peptides related to serine-threonine kinases (Figure 1I-K). For these peptides, 12 out of the total 29 differentially phosphorylated peptides overlap between the early- and late-stages of CKD, while 14 additional peptides are phosphorylated only in the late-stage (12 weeks), compared to the early-stage (4 weeks) CKD model (Figure 1K).
Overall, CKD in hyperlipidemic mice results in an increased phosphorylation of peptides related to both tyrosine and serine-threonine kinases in the kidney. Furthermore, the amount of phosphorylation increases with disease progression.
3.2 Differential kinase activity in kidneys of hyperlipidemic mice upon CKD development.
We deployed an Upstream Kinase Analysis to determine which kinases are responsible for the differentially phosphorylated peptides observed in CKD. In line with the observed changes in peptide phosphorylation patterns, tyrosine kinase (TK) activity is strongly increased in mice fed with WTD-A, compared to WTD-fed mice (Figures 2A,B). Furthermore, while overall the activity of the same tyrosine kinases is significantly increased upon CKD development, kinases are slightly more active in the late-stage CKD model (12w) compared to the early-stage model (4w) (Figure 2C). A similar trend is observed for serine-threonine kinases, as the kinase activity is increased more in the late-stage CKD model (12w) compared to the early-stage model (4w) (Figure 2D).
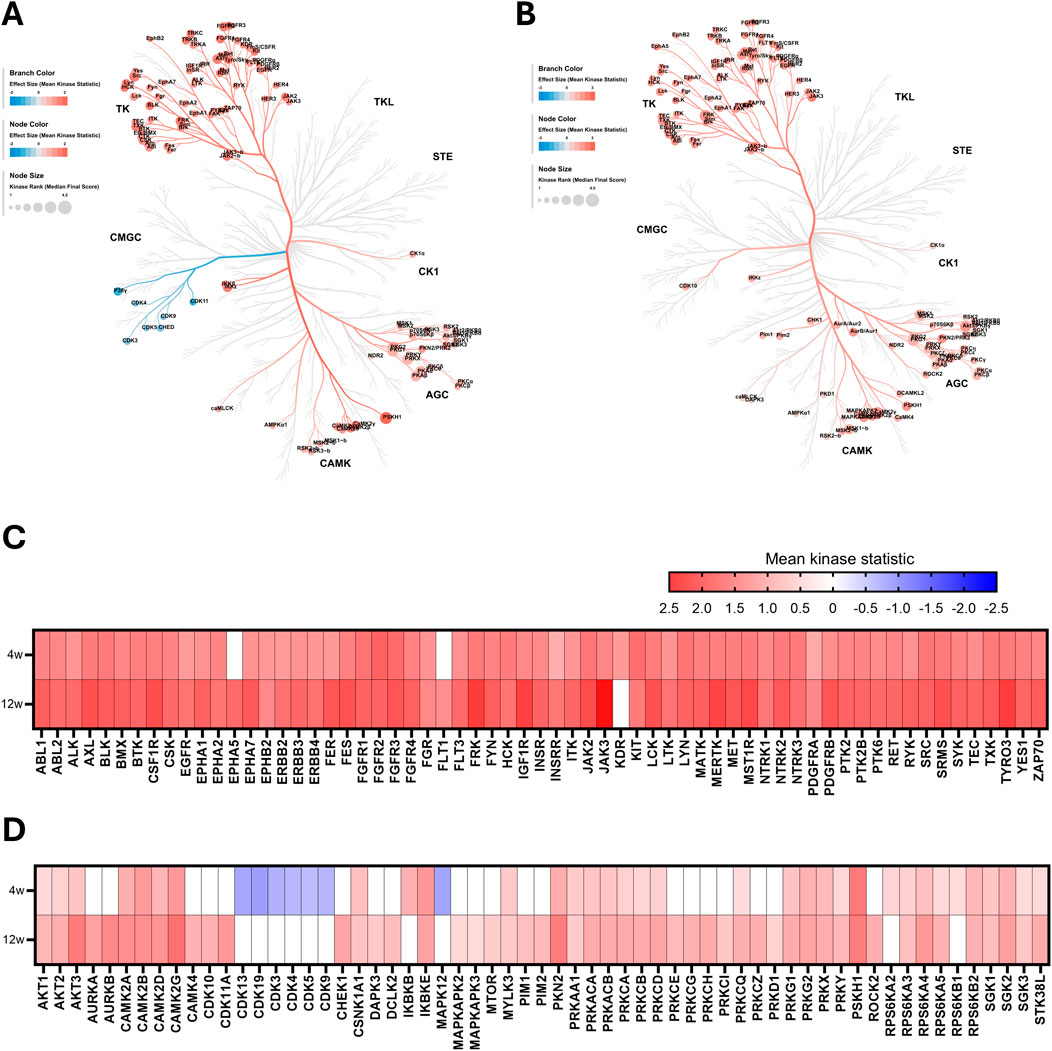
Figure 2. Overall kinase activity is elevated in hyperlipidemic mice with CKD. (A,B) Kinome coral trees depicting kinase rank (node size) and the effect size (node and branch color) of significantly differentially regulated kinases in kidney lysates after (A) 4 weeks and (B) 12 weeks of diet. Both STK and PTK are represented. (C,D) Heatmaps showing effect size (mean kinase statistic) for (C) PTK and (D) STK after 4 and 12 weeks of diet, sorted alphabetically. All data represent four biological replicates per condition.
Among all predicted kinases, Janus kinase 3 (JAK3) emerges as the most highly upregulated tyrosine kinase, particularly in late-stage CKD (Figure 2C). JAK3 is well known for its key role in lymphocyte homeostasis, proliferation, and activation (Borie et al., 2003). The most upregulated serine-threonine kinase is calcium/calmodulin-dependent protein kinase II gamma (CAMK2G) (Figure 2D). This kinase is known to modulate various processes, including calcium homeostasis, cell cycle, cytoskeletal organization, and a regulatory role in the inflammatory signaling through its function as an intracellular calcium sensor (Junho et al., 2020).
Interestingly, despite the general trend of increased kinase activity in mice with CKD, compared to mice without CKD, there is a distinct cluster of kinases that is strongly downregulated in the early-stage model (4w). This group includes several cyclin-dependent kinases (CDKs), best known for their critical role in cell cycle and gene transcription regulation (Malumbres, 2014), as well as mitogen-activated protein kinase 12 (MAPK12, also known as p38γ), which is known to play a role in inflammatory and metabolic pathways (Qi and Chen, 2023) (Figure 2D).
3.3 Cell cycle-related pathways in the kidney are affected by CKD development.
Pathway analysis was performed using ORA to understand the complex interplay of significantly differentially activated kinases. Given that CDKs showed a clear and distinct downregulation compared to other kinases, we first examined pathways in which these kinases are involved. As expected, these kinases are primarily associated with cell cycle regulation, a process of particular interest in CKD due to its role in maladaptive cell repair and promoting a pro-fibrotic phenotype.
Although CDK downregulation is observed only in early-stage CKD (4 weeks), most of the same cell cycle-related pathways remain active in kidneys with more advanced disease (Table 1). Notably, with disease progression, there is a significant shift towards increased activity of kinases involved in these selected pathways (Figure 3A).

Table 1. Cell cycle-related pathways.
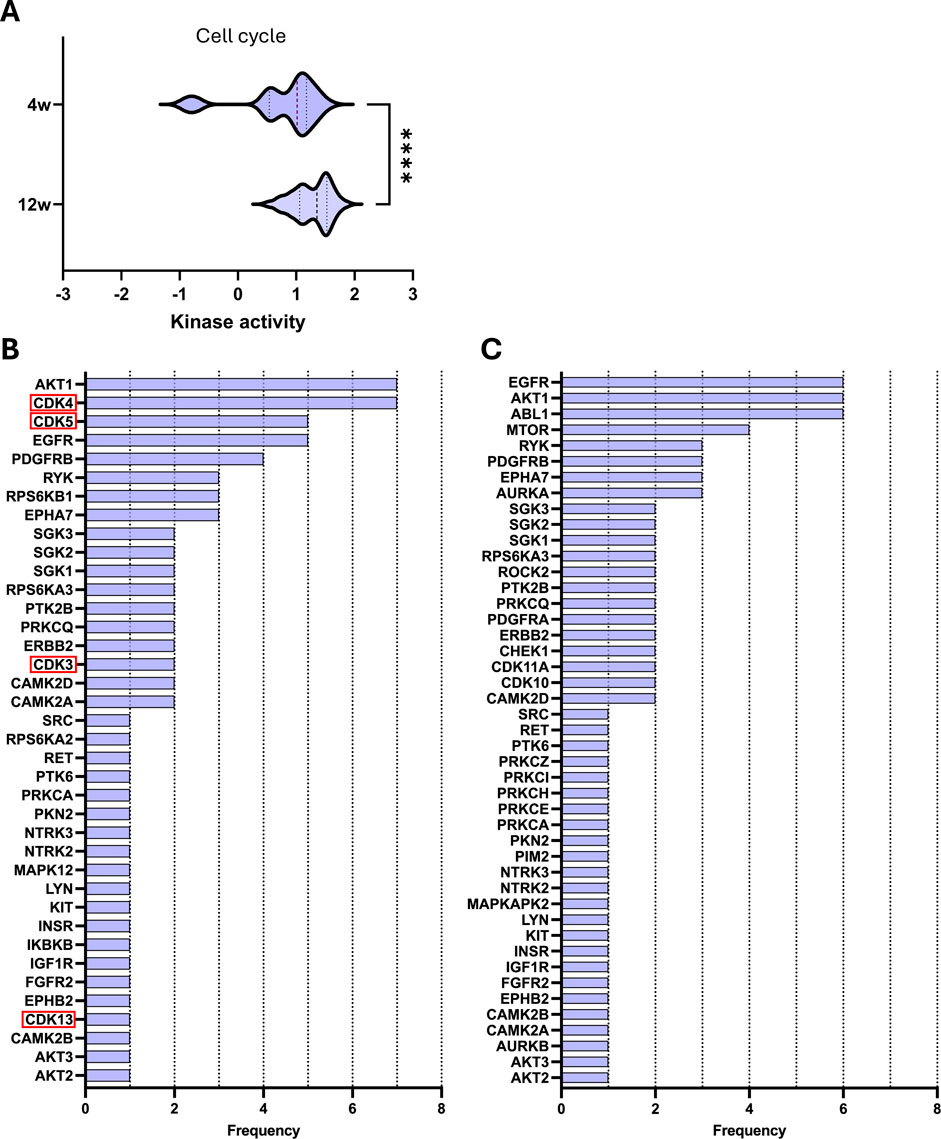
Figure 3. Key kinases play a significant role in selected cell cycle pathways during CKD development. (A) Violin plot of kinase activity in cell cycle-related pathways. Kinase activity is reflected by the mean kinase statistic of each kinase involved in these pathways. (B,C) Bar graphs of kinase frequency of kinase involvement across cell cycle pathways after (B) 4 weeks and (C) 12 weeks of diet. Downregulated kinases are highlighted with a red box. See Table 1 for all pathways (e.g., a frequency of 1 denotes that the kinase is involved in one pathway, while a frequency of 5 denotes that the kinase is involved in five distinctive pathways). Statistical significance was assessed by the Mann-Whitney U test. ****P < 0.0001.
Next, we examined the kinases that were significantly differentially activated within these selected pathways to identify those with the highest recurrence across pathways (Figures 3B,C). In the early-stage CKD, the most frequently repeated kinases are AKT1 and CDK4, which are both involved in 7 out of 11 listed pathways (Table 1; Figure 3B). Meanwhile, in late-stage CKD, CDKs are no longer differentially regulated; however, AKT1 remains highly represented, now accompanied by epidermal growth factor receptor (EGFR) and Abelson murine leukemia viral oncogene homolog 1 (ABL1). All three kinases are involved in 7 out of 7 of the listed pathways (Figure 3C). Overall, the same pathways are significantly different independent of the stage of CKD; however, the composition of active kinases shifted with disease progression. These changes highlight potential stage-specific kinase regulators that may contribute to CKD advancement.
3.4 CKD development affects kinases involved in key pathological processes
Expanding on these apparent changes in cell cycle, we further focused on four key pathological processes involved in the progression of CKD (Yuan et al., 2022), namely, inflammation, oxidative stress, lipid metabolism, and fibrosis. For each process, the top 15 pathways ranked according to adjusted p-value were selected, excluding broad pathways that substantially overlapped across the selected processes as well as repeated pathways (Tables 2–5). Comparing kinase activity within these processes reveals a significant difference in kinase activity between the early-stage (4 weeks) and late-stage (12 weeks) CKD, with a general increase in activity at the more progressed CKD stage (Figures 4A–D). To identify the most influential kinases, we examined their presence across multiple pathways and ranked them by frequency of occurrence (Figures 4E,F). Similarly to the cell cycle analysis, AKT1 emerges as the most prominently involved kinase across the selected pathways. While some highly recurrent kinases, such as SRC, have already been studied in the field of CKD, others remain unexplored. A consistent trend was observed as certain kinases maintain high activity across both disease stages, whereas others show marked changes in both their activity and relevance as CKD progressed. These effects can also be directly appreciated by examining the various pathways (Table 2–5), which show considerable overlap between early and late stages, yet also stage-dependent shifts in the significance of the pathways. In contrast, some key pathways retain strong relevance throughout disease progression as evidenced by their significance (Table 2–5).

Table 2. Top 15 selected pathways related to inflammation.

Table 3. Top 15 selected pathways related to oxidative stress.

Table 4. Top 15 selected pathways related to lipid metabolism.

Table 5. Top 15 selected pathways related to fibrosis.
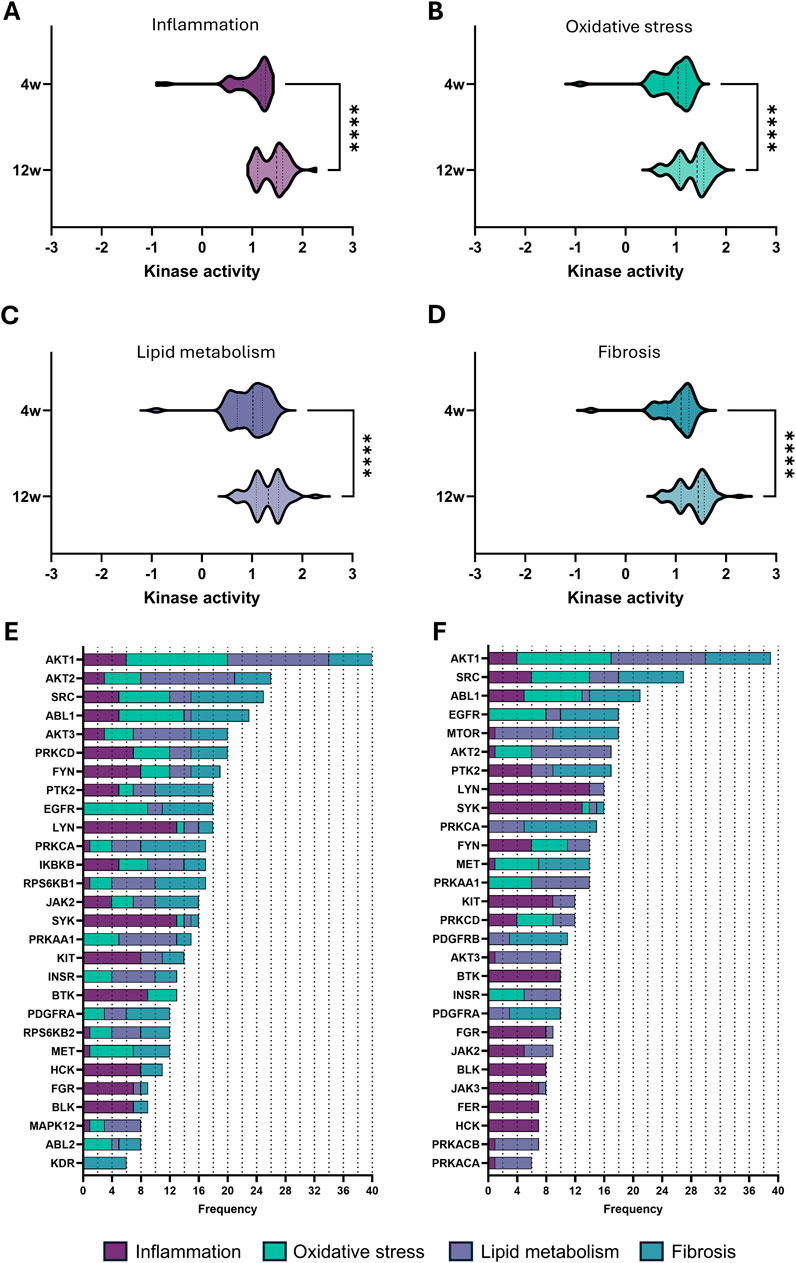
Figure 4. Kinase activity in the top 15 pathways related to major pathological processes in CKD. (A–D) Violin plot of kinase activity in pathways related to (A) inflammation, (B) oxidative stress, (C) lipid metabolism, and (D) fibrosis, based on the top 15 pathways identified (see Tables 2–5). Kinase activity is shown as the mean kinase statistic of each kinase involved in these pathways. (E,F) Bar graphs of kinase frequency among the top 15 pathways in each of the four pathological processes (See Tables 2–5 for all pathways; e.g., a frequency of 1 denotes that the kinase is involved in one pathway, while a frequency of 5 denotes that the kinase is involved in five distinctive pathways) after (E) 4 weeks and (F) 12 weeks of diet. Statistical significance was assessed by the Mann-Whitney U test. ****P < 0.0001.
4 Discussion
CKD, particularly when accompanied by cardiovascular complications, remains a major global health burden, yet the underlying mechanisms are still not fully understood. To gain more insights into these mechanisms, we deployed an unbiased kinomics approach on the renal cortex of hyperlipidemic mice with or without CKD. Thereby, we could identify numerous kinases that are differentially regulated in early- or late-stage CKD development. These kinases were related to several key pathological processes of CKD, including inflammation, oxidative stress, and fibrosis, as well as lipid metabolism pathways.
Interestingly, one of the highly upregulated kinases that was also most prominent in the selected pathological processes is Akt1, while its family members, Akt2 and Akt3, were also prominently represented in these processes. This observation also validates our study, as it has already been well described that Akt signaling plays an essential role in CKD (Lan and Du, 2015; Wang H. et al., 2024). For instance, genetic deletion of Akt1 in a mouse model that mimics the transition of acute kidney injury to CKD resulted in an attenuation of tubulointerstitial fibrosis and tubular apoptosis (Kim et al., 2023). Another kinase group strongly associated with CKD is the Src family kinases, consisting of eight family members: Src, Fyn, Yes1, Lyn, Fgr, Hck, Blk, and Lck (Wang and Zhuang, 2017). Particularly, Src, Fyn, and Hck have already been widely studied in CKD and have been identified as critical mediators of renal fibrosis. For example, inactivation of Src in a mouse model of renal interstitial fibrosis resulted in reduced renal fibroblast activation and attenuated extracellular matrix protein deposition (Yan et al., 2016). Furthermore, Fyn-deficient mice have also been shown to exhibit attenuated renal fibrosis upon ureteral obstruction (Seo et al., 2016), further demonstrating the importance of Src family kinases in renal diseases. Strikingly, all eight family members were significantly upregulated in both the early- and late-stage CKD in our study, again validating our results. Interestingly, the other Src family kinases (Blk, Fgr, Hck, Lck and Lyn) are still relatively unexplored in CKD. However, we observed similar upregulation of these kinases upon CKD development, compared to, for example, Src or Fyn. Therefore, these may also be interesting kinases to further study in this context, especially since many Scr family kinases have redundant roles (Wang and Zhuang, 2017).
Examining additional kinases involved in the selected pathological pathways revealed several that have already been linked to CKD in the literature, although less widely studied so far. For example, ABL has been demonstrated to drive fibroblast activation and differentiation during renal fibrosis, through the regulation of RACK1 stabilization (Bao et al., 2024). Furthermore, PRKCD (PKCδ), is upregulated in fibrotic kidneys, while the PKCδ inhibitor, rottlerin, alleviated both renal fibrosis and inflammation (Wang D. et al., 2024). PTK2, also known as FAK, is another kinase that has already been shown to be upregulated during the transition from acute kidney injury to CKD. In line with this, mice that have a renal collecting duct-specific Ptk2 deficiency demonstrated reduced oxidative stress and attenuated renal fibrosis (Gao et al., 2025).
Due to the unbiased approach of our study, we were also able to identify kinases that have not yet been described in the context of CKD. For example, BTK, which is considered an essential regulator of immunoregulation (Weber et al., 2017), was also identified as a kinase that is increased upon CKD development in our mice and involved in the selected key pathways. Although some studies are showing the involvement of BTK in diabetic nephropathy (Zhao et al., 2021), hemolytic-uremic syndrome (Kroller et al., 2023), and lupus nephritis (Chalmers et al., 2018), the role of BTK in CKD has not yet been explored. Another notable kinase, which was not involved in the selected pathways, yet was still highly upregulated upon CKD development, is PSKH1. Although PSKH1 has recently been identified as a crucial factor in kidney development, no studies have yet elucidated its potential pathological role in kidney disease. Based on our results, this would be an interesting kinase to study in the future. Therefore, our unbiased kinome profiling provides the opportunity to identify novel mediators involved in CKD development and is thus a valuable source for future lead identification.
Another interesting observation of our unbiased kinomics analysis is that CDKs are significantly less active in the early phase of CKD development, while this inhibition is abolished upon disease progression. CDK kinases play a crucial role in the regulation of the cell cycle, and this observation aligns with previous studies showing that a cell cycle arrest at G2 occurs, particularly in tubular epithelial cells, during the initial stages of kidney damage. At the same time, this is not the case anymore at a later stage (Koyano et al., 2019; Lovisa et al., 2015). This cell cycle arrest is dependent on the CDK inhibitor p21, whose presence appears protective, as p21-deficient mice exacerbate the progression of renal fibrosis (Koyano et al., 2019), demonstrating its importance in disease development. Several studies evaluate the effects of CDK inhibitors on CKD development (Pellarin et al., 2025). For example, the blockage of CDK4/6 in mice has been shown to improve renal function and reduce tubular injury and fibrosis (Osaki et al., 2022). Paradoxically, selective deletion of cyclin D1, which complexes with CDK4/6, was shown to increase tubular injury (Osaki et al., 2022), suggesting that potential therapeutic strategies should be rather specific and prioritize the inhibition of CDK4/6. Overall, CDKs play a key role in the development of CKD; however, based on our results, supported by literature, their role is very stage-dependent.
Overall, this study identified key and novel kinases that are regulated during CKD development in hyperlipidemic mice. While validating several key kinases, the deployed unbiased kinomics approach also identified several novel kinases that so far have not yet been described to play a role in CKD development. Therefore, these results are a valuable source for future studies to investigate further whether these novel identified kinases also play a causal role in CKD development.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Germany, approval number 81-02.04.2019.A364. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AB-M: Conceptualization, Data curation, Formal Analysis, Writing – original draft. SM: Formal Analysis, Methodology, Visualization, Writing – review and editing. MC-B: Data curation, Formal Analysis, Methodology, Visualization, Writing – review and editing. EV: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Corona Foundation (S199/10084/2021), and by the Deutsche Forschungsgemeinschaft (DFG) (SFB TRR219 – Project-ID 322900939; subproject M07) to E.P.C.v.d.V. Open access funding provided by the Open Access Publishing Fund of RWTH Aachen University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2025.1684982/full#supplementary-material
References
Bao Q., Wang A., Hong W., Wang Y., Li B., He L., et al. (2024). The c-Abl-RACK1-FAK signaling axis promotes renal fibrosis in mice through regulating fibroblast-myofibroblast transition. Cell Commun. Signal 22 (1), 247. doi:10.1186/s12964-024-01603-z
Blighe K., Rana S., Lewis M. (2025). EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. R. package version 1.26.0.
Borie D. C., Si M. S., Morris R. E., Reitz B. A., Changelian P. S. (2003). JAK3 inhibition as a new concept for immune suppression. Curr. Opin. Investig. Drugs 4 (11), 1297–1303.
Chalmers S. A., Glynn E., Garcia S. J., Panzenbeck M., Pelletier J., Dimock J., et al. (2018). BTK inhibition ameliorates kidney disease in spontaneous lupus nephritis. Clin. Immunol. 197, 205–218. doi:10.1016/j.clim.2018.10.008
Collaboration GBDCKD (2020). Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet 395 (10225), 709–733. doi:10.1016/S0140-6736(20)30045-3
Foreman K. J., Marquez N., Dolgert A., Fukutaki K., Fullman N., McGaughey M., et al. (2018). Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 392 (10159), 2052–2090. doi:10.1016/S0140-6736(18)31694-5
Francis A., Harhay M. N., Ong A. C. M., Tummalapalli S. L., Ortiz A., Fogo A. B., et al. (2024). Chronic kidney disease and the global public health agenda: an international consensus. Nat. Rev. Nephrol. 20 (7), 473–485. doi:10.1038/s41581-024-00820-6
Fraser S. D., Roderick P. J., May C. R., McIntyre N., McIntyre C., Fluck R. J., et al. (2015). The burden of comorbidity in people with chronic kidney disease stage 3: a cohort study. BMC Nephrol. 16, 193. doi:10.1186/s12882-015-0189-z
Gao C., He J., Wang Y., Shao G., Lin S., Liu J., et al. (2025). The role of FAK in renal collecting duct in the progression from acute kidney injury to chronic kidney disease. Transl. Res. 282, 41–55. doi:10.1016/j.trsl.2025.06.002
Jankowski J., Floege J., Fliser D., Bohm M., Marx N. (2021). Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation 143 (11), 1157–1172. doi:10.1161/CIRCULATIONAHA.120.050686
Jha V., Al-Ghamdi S. M. G., Li G., Wu M. S., Stafylas P., Retat L., et al. (2023). Global economic burden associated with chronic kidney disease: a pragmatic review of medical costs for the inside CKD research programme. Adv. Ther. 40 (10), 4405–4420. doi:10.1007/s12325-023-02608-9
Junho C. V. C., Caio-Silva W., Trentin-Sonoda M., Carneiro-Ramos M. S. (2020). An overview of the role of Calcium/calmodulin-dependent protein kinase in cardiorenal syndrome. Front. Physiol. 11, 735. doi:10.3389/fphys.2020.00735
Kidney Disease: Improving Global Outcomes CKDWG (2024). KDIGO 2024 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 105 (4S), S117–S314. doi:10.1016/j.kint.2023.10.018
Kim I. Y., Song S. H., Seong E. Y., Lee D. W., Bae S. S., Lee S. B. (2023). Akt1 is involved in renal fibrosis and tubular apoptosis in a murine model of acute kidney injury-to-chronic kidney disease transition. Exp. Cell Res. 424 (2), 113509. doi:10.1016/j.yexcr.2023.113509
Koyano T., Namba M., Kobayashi T., Nakakuni K., Nakano D., Fukushima M., et al. (2019). The p21 dependent G2 arrest of the cell cycle in epithelial tubular cells links to the early stage of renal fibrosis. Sci. Rep. 9 (1), 12059. doi:10.1038/s41598-019-48557-8
Kroller S., Wissuwa B., Dennhardt S., Krieg N., Thiemermann C., Daniel C., et al. (2023). Bruton's tyrosine kinase inhibition attenuates disease progression by reducing renal immune cell invasion in mice with hemolytic-uremic syndrome. Front. Immunol. 14, 1105181. doi:10.3389/fimmu.2023.1105181
Kumar U., Wettersten N., Garimella P. S. (2019). Cardiorenal syndrome: pathophysiology. Cardiol. Clin. 37 (3), 251–265. doi:10.1016/j.ccl.2019.04.001
Lan A., Du J. (2015). Potential role of akt signaling in chronic kidney disease. Nephrol. Dial. Transpl. 30 (3), 385–394. doi:10.1093/ndt/gfu196
Levin A., Tonelli M., Bonventre J., Coresh J., Donner J. A., Fogo A. B., et al. (2017). Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 390 (10105), 1888–1917. doi:10.1016/S0140-6736(17)30788-2
Lovisa S., LeBleu V. S., Tampe B., Sugimoto H., Vadnagara K., Carstens J. L., et al. (2015). Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 21 (9), 998–1009. doi:10.1038/nm.3902
Lv J., Li Y., Shi S., Liu S., Xu X., Wu H., et al. (2023). Frontier and hotspot evolution in cardiorenal syndrome: a bibliometric analysis from 2003 to 2022. Curr. Probl. Cardiol. 48 (8), 101238. doi:10.1016/j.cpcardiol.2022.101238
Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002). The protein kinase complement of the human genome. Science. 298 (5600), 1912–1934. doi:10.1126/science.1075762
Marx-Schutt K., Cherney D. Z. I., Jankowski J., Matsushita K., Nardone M., Marx N. (2025). Cardiovascular disease in chronic kidney disease. Eur. Heart J. 46 (23), 2148–2160. doi:10.1093/eurheartj/ehaf167
Noels H., van der Vorst E. P. C., Rubin S., Emmett A., Marx N., Tomaszewski M., et al. (2025). Renal-cardiac crosstalk in the pathogenesis and progression of heart failure. Circ. Res. 136 (11), 1306–1334. doi:10.1161/CIRCRESAHA.124.325488
Osaki Y., Manolopoulou M., Ivanova A. V., Vartanian N., Mignemi M. P., Kern J., et al. (2022). Blocking cell cycle progression through CDK4/6 protects against chronic kidney disease. JCI Insight 7 (12), e158754. doi:10.1172/jci.insight.158754
Pellarin I., Dall'Acqua A., Favero A., Segatto I., Rossi V., Crestan N., et al. (2025). Cyclin-dependent protein kinases and cell cycle regulation in biology and disease. Signal Transduct. Target Ther. 10 (1), 11. doi:10.1038/s41392-024-02080-z
Qi X. M., Chen G. (2023). p38γ MAPK inflammatory and metabolic signaling in physiology and disease. Cells 12 (13), 1674. doi:10.3390/cells12131674
Rangaswami J., Bhalla V., Blair J. E. A., Chang T. I., Costa S., Lentine K. L., et al. (2019). Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American heart association. Circulation 139 (16), e840–e878. doi:10.1161/CIR.0000000000000664
Reiss A. B., Voloshyna I., De Leon J., Miyawaki N., Mattana J. (2015). Cholesterol metabolism in CKD. Am. J. Kidney Dis. 66 (6), 1071–1082. doi:10.1053/j.ajkd.2015.06.028
Seo H. Y., Jeon J. H., Jung Y. A., Jung G. S., Lee E. J., Choi Y. K., et al. (2016). Fyn deficiency attenuates renal fibrosis by inhibition of phospho-STAT3. Kidney Int. 90 (6), 1285–1297. doi:10.1016/j.kint.2016.06.038
Stevens P. E., Levin A. (2013). Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med 158(11):825–830. doi:10.7326/0003-4819-158-11-201306040-00007
Stewart S., Kalra P. A., Blakeman T., Kontopantelis E., Cranmer-Gordon H., Sinha S. (2024). Chronic kidney disease: detect, diagnose, disclose-a UK primary care perspective of barriers and enablers to effective kidney care. BMC Med. 22 (1), 331. doi:10.1186/s12916-024-03555-0
Team R. C. R. (2013). A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Ubersax J. A., Ferrell J. E. (2007). Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8 (7), 530–541. doi:10.1038/nrm2203
Vaduganathan M., Mensah G. A., Turco J. V., Fuster V., Roth G. A. (2022). The global burden of cardiovascular diseases and risk: a compass for future health. J. Am. Coll. Cardiol. 80 (25), 2361–2371. doi:10.1016/j.jacc.2022.11.005
Visconti L., Benvenga S., Lacquaniti A., Cernaro V., Bruzzese A., Conti G., et al. (2016). Lipid disorders in patients with renal failure: role in cardiovascular events and progression of chronic kidney disease. J. Clin. Transl. Endocrinol. 6, 8–14. doi:10.1016/j.jcte.2016.08.002
Wang J., Zhuang S. (2017). Src family kinases in chronic kidney disease. Am. J. Physiol. Ren. Physiol. 313 (3), F721–F728. doi:10.1152/ajprenal.00141.2017
WangH. H., Gao L., Zhao C., Fang F., Liu J., Wang Z., et al. (2024). The role of PI3K/Akt signaling pathway in chronic kidney disease. Int. Urol. Nephrol. 56 (8), 2623–2633. doi:10.1007/s11255-024-03989-8
Wang D., Li Y., Li G., Liu M., Zhou Z., Wu M., et al. (2024). Inhibition of PKC-delta retards kidney fibrosis via inhibiting cGAS-STING signaling pathway in mice. Cell Death Discov. 10 (1), 314. doi:10.1038/s41420-024-02087-z
Weber A. N. R., Bittner Z., Liu X., Dang T. M., Radsak M. P., Brunner C. (2017). Bruton's tyrosine kinase: an emerging key player in innate immunity. Front. Immunol. 8, 1454. doi:10.3389/fimmu.2017.01454
Webster A. C., Nagler E. V., Morton R. L., Masson P. (2017). Chronic kidney disease. Lancet 389 (10075), 1238–1252. doi:10.1016/S0140-6736(16)32064-5
Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., et al. (2021). clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. (Camb) 2 (3), 100141. doi:10.1016/j.xinn.2021.100141
Xu D., Xie L., Cheng C., Xue F., Sun C. (2024). Triglyceride-rich lipoproteins and cardiovascular diseases. Front. Endocrinol. (Lausanne) 15, 1409653. doi:10.3389/fendo.2024.1409653
Yan Y., Ma L., Zhou X., Ponnusamy M., Tang J., Zhuang M. A., et al. (2016). Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 89 (1), 68–81. doi:10.1038/ki.2015.293
Yuan Q., Tang B., Zhang C. (2022). Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target Ther. 7 (1), 182. doi:10.1038/s41392-022-01036-5
Keywords: chronic kidney disease, hyperlipidemia, kinases, kinomics, cardiorenal
Citation: Bonnin-Marquez A, Maas SL, Corcini-Berndt M and Van der Vorst EPC (2025) Unbiased kinome profiling identifies key and novel mediators of chronic kidney disease in hyperlipidemic mice. Front. Physiol. 16:1684982. doi: 10.3389/fphys.2025.1684982
Received: 13 August 2025; Accepted: 15 September 2025;
Published: 02 October 2025.
Edited by:
Bin Wang, Southeast University, ChinaReviewed by:
Masashi Suzuki, Tokyo Gakugei University, JapanMarta Riera, Hospital del Mar Medical Research Institute (IMIM), Spain
Copyright © 2025 Bonnin-Marquez, Maas, Corcini-Berndt and Van der Vorst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emiel P.C. Van der Vorst, ZXZhbmRlcnZvcnN0QHVrYWFjaGVuLmRl
†These authors have contributed equally to this work