 Camille Chartrand-Pleau1
Camille Chartrand-Pleau1 Emily Gallipeau-Burns1
Emily Gallipeau-Burns1 Ananya Jain2
Ananya Jain2 Victor Boddy2Patrick Fakhouri2Sarah Rehmani2Teagan Thomas1Michelle Drapeau1
Victor Boddy2Patrick Fakhouri2Sarah Rehmani2Teagan Thomas1Michelle Drapeau1 Jordan T. VanderBurgt3Arnaud Boudigou2
Jordan T. VanderBurgt3Arnaud Boudigou2 Adam M. Damry2
Adam M. Damry2 Allyson M. MacLean1*
Allyson M. MacLean1*- 1Department of Biology, University of Ottawa, Ottawa, ON, Canada
- 2Department of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, ON, Canada
- 3London Research and Development Centre, Agriculture and Agri-Food Canada, London, ON, Canada
The ability to express heterologous proteins in specific plant tissues and subcellular compartments is critical for advancing plant-based biotechnologies and biopharming research. Here, we present the “pTARGEX” series, a versatile toolkit designed to enable targeted protein expression within the leaves and roots of plants. These vectors incorporate subcellular targeting sequences to direct proteins of interest to the apoplast (pTARGEX-APO), cytoplasm (pTARGEX-CYT), chloroplast (pTARGEX-CHL), endoplasmic reticulum (pTARGEX-ER), or vacuole (pTARGEX-VAC). Streamlining workflow, the pTARGEX vectors encode an mCherry dropout cassette with a SapI restriction enzyme site to enable red/white screening in Escherichia coli, and an eGFP reporter gene that is constitutively expressed in planta to serve as an inbuilt marker for monitoring of successful transformation. This innovative vector series offers users an accessible, versatile platform for plant-based heterologous protein production, facilitating research and applications in systems biology, synthetic biology, and plant biotechnology.
Introduction
Plant molecular farming refers to the use of plants as a means of producing high-value biomolecules such as vaccines, therapeutic proteins, antibodies, and industrial enzymes. This approach has yielded several notable successes, including Medicago’s virus-like particle (VLP) vaccines against seasonal influenza and COVID-19, Kentucky BioProcessing’s ZMapp treatment for Ebola, and Protalix Biotherapeutics’ FDA-approved enzyme therapy “Elelyso” for Gaucher’s disease (Hager et al., 2022; Mor, 2015; Mulangu et al., 2019; Ward et al., 2020). Ongoing research continues to expand the utility of plant-based expression systems for the production of monoclonal antibodies, hormones, diagnostic reagents, and other peptide-based therapeutics (Eidenberger et al., 2023).
A variety of plant-based expression platforms have been developed, including in vitro cultures such as hairy roots and suspension cells, as well as whole-plant systems that rely on stable or transient gene expression. Among these, Nicotiana benthamiana has emerged as the leading host system for transient expression due to its amenability to Agrobacterium tumefaciens-mediated transformation and its capacity for rapid, high-yield protein production within days (Bally et al., 2018; Lomonossoff and D’Aoust, 2016). While stable transformation offers long-term expression and scalability, transient expression is often preferred for early-stage optimization and time-sensitive applications (Bharathi et al., 2024).
A central advantage of plant systems over microbial platforms is their ability to perform complex post-translational modifications, most notably glycosylation, which may be critical to the structure and function of many therapeutic proteins (Dammen-Brower et al., 2022). Advances in glycoengineering now allow plants to mimic mammalian glycosylation patterns, further enhancing their suitability for producing human-compatible biologics (Göritzer et al., 2022; Hanania et al., 2017; Herman et al., 2021; Jansing et al., 2019; Kogelmann et al., 2024). In addition, plants offer notable advantages in terms of biosafety and production cost. As hosts, they are inherently free of human pathogens and can be cultivated in relatively low-cost environments, such as greenhouses or growth chambers, thereby eliminating the need for sterile, high-tech fermentation infrastructure and costly growth media.
One key factor influencing the yield and functionality of heterologous proteins produced in plants is their subcellular localization, and the different environments presented by different localizations can have a profound effect on protein accumulation, stability, folding, and biological activity (Benchabane et al., 2008; Liu and Timko, 2022; Margolin et al., 2020). The cytoplasm, as the default compartment for proteins lacking targeting signals, is generally considered suboptimal for the production of complex eukaryotic proteins, as it lacks post-translational modification machinery such as glycosyltransferases and often only yields low levels of recombinant protein accumulation. Instead, targeting recombinant proteins to the endoplasmic reticulum (ER) or apoplast via the secretory pathway offers several advantages. Proteins retained in the ER may benefit from N-linked glycosylation, often accumulate at high levels (Sainsbury and Lomonossoff, 2008), and are protected from cytosolic proteases (Schouten et al., 1996; Spiegel et al., 1999). Secretion to the apoplast enables additional O-linked glycosylation and can simplify downstream protein recovery; however, it may also expose the protein to increased extracellular degradation, particularly by defense-associated proteases (Hehle et al., 2011). Chloroplast targeting has been associated with high accumulation for some proteins (Yanez et al., 2017), although outcomes are highly protein-dependent. Chloroplasts also lack glycosylation machinery, which may be preferable for the expression of prokaryotic proteins (Kaldis et al., 2023). Vacuolar targeting may confer stability due to the compartment’s storage function (Marin Viegas et al., 2017), but the acidic pH and active proteases can compromise sensitive proteins. Thus, determining the optimal compartment for the expression of any given protein is highly empirical and must account for both the biochemical properties of the protein and its intended downstream application. Despite widespread recognition of the importance of subcellular targeting in determining protein expression and accumulation levels, tools that enable rapid, parallel testing of localization strategies remain limited. To address these gaps, we developed the pTARGEX vector series (Targeted Application of Recombinant Gene EXpression), a modular, open-source toolkit that enables efficient screening of protein expression and accumulation levels across multiple subcellular compartments in plants.
The pTARGEX vectors are designed to streamline early-stage optimization of plant-based protein production. The toolkit supports Type IIS restriction enzyme-based (Golden Gate) cloning, red/white screening in Escherichia coli via an mCherry dropout cassette, and visual confirmation of successful plant transformation using a constitutively expressed eGFP reporter gene. The system includes the NSs viral RNA silencing suppressor to maximize transient expression and enables parallel assessment of protein accumulation in five subcellular locations—cytoplasm, ER, apoplast, chloroplast, and vacuole—in both N. benthamiana leaves and Medicago truncatula roots. To demonstrate the utility of the pTARGEX toolkit, we expressed the SARS-CoV-2 receptor-binding domain (RBD)—a clinically relevant model protein (Yang et al., 2020) — in N. benthamiana, and the fluorescent reporter mScarlet (Bindels et al., 2016) in both N. benthamiana and M. truncatula. Our results validated the fidelity of the subcellular localization tags and revealed substantial compartment-specific differences in RBD accumulation, highlighting the empirical nature of subcellular targeting and the trade-offs between protein yield and quality. Notably, the pTARGEX toolkit was developed through an undergraduate-led initiative and is designed to be modular, user-friendly, and fully open-source. By enabling rapid and parallel testing of subcellular localization strategies, pTARGEX serves as both a practical research platform and a valuable educational resource for training in plant synthetic biology and biopharming.
Materials and methods
Gibson assembly
The pTARGEX vector library was constructed using four-fragment Gibson Assembly (NEBuilder HiFi DNA Assembly Master Mix, NEB, Ipswich, MA, United States). Individual fragments were PCR-amplified using Q5 High-Fidelity 2X Master Mix (NEB) from either synthetic gBlocks (IDT, Leuven, Belgium) or the pHREAC plasmid (Peyret et al., 2019), incorporating subcellular targeting sequences previously developed for the pCLGG series (Kaldis et al., 2023; VanderBurgt et al., 2023). Primer sequences are provided in Supplementary Table S1. All primers were obtained from Integrated DNA Technologies (IDT).
To eliminate residual plasmid template, PCR products derived from pHREAC were treated with 1 µL (20 U) of DpnI (NEB) directly in the reaction and incubated at 37 °C for 30 min. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Venlo, the Netherlands) when a single, specific band was observed. If multiple bands were present, the target band was excised from an agarose gel and purified using the QIAquick Gel Extraction Kit. Gibson Assembly reactions were prepared according to the manufacturer’s instructions and incubated at 50 °C for 2 h. The assembled products were transformed into chemically competent E. coli DH5α cells, and transformants were selected on LB agar plates supplemented with 50 mg/L kanamycin (Fisher Bioreagents) and 0.5 mM IPTG (Omega BioTek). Colonies expressing LacP-mCherry appeared pink and were selected for overnight growth in LB-kanamycin broth. In some cases, visible pigmentation appeared only after an additional 24 h at 4 °C. Plasmid DNA was isolated using the 5-min miniprep kit (FroggaBio, Concord, ON, Canada) and whole plasmid sequencing was performed by Plasmidsaurus (Eugene, OR, United States).
Golden gate assembly
Both pTARGEX-mScarlet and pTARGEX-RBD libraries were constructed using Golden Gate Assembly with the five pTARGEX vector backbones. This technique enabled seamless excision of the LacP-mCherry cassette and replacement with either the mScarlet or SARS-CoV-2 RBD gene, using the Type IIS restriction enzyme SapI (NEB). Gene inserts were PCR-amplified using Q5 High-Fidelity DNA Polymerase (NEB) from the plasmids pCLGG-VAC-mScarlet and pHREAC-RBD-MAL (Demone et al., 2022). Primers were synthesized by Integrated DNA Technologies (IDT) and included SapI recognition sites (GCTCTTC) at both ends of the amplicons, and sequences encoding a hexa-His tag (5′ CATCACCACCATCATCAC 3′) at the N-terminus of mScarlet and a Thrombin (5′ CTTGTTCCAAGGGGATCA 3′) and octo-His tag (5′ CATCACCACCATCATCATCATCAT 3′) at the C-terminus of RBD. Following PCR, templates were digested with DpnI (NEB) to remove methylated parental plasmid DNA, and products were visualized by agarose gel electrophoresis and purified using a Qiagen PCR purification kit.
Two Golden Gate strategies were tested: (1) a one-pot digestion-ligation reaction and (2) a two-step approach with separated digestion and ligation. Constructs generated via method (1) included CYT-mScarlet, VAC-mScarlet, ER-mScarlet, and ER-RBD, while method (2) was used for CHL-mScarlet, APO-mScarlet, VAC-RBD, APO-RBD, CHL-RBD, and CYT-RBD. For the one-pot assembly, 15 units of SapI and 500 units of T4 DNA ligase (NEB) were combined with a 5:1 molar ratio of insert to backbone DNA (based on 200 ng of plasmid) in a 20 µL reaction containing equal volumes of 10× T4 DNA Ligase Buffer and 10× rCutSmart Buffer (NEB), and topped up with nuclease-free H2O. The reaction was incubated at 25 °C for 2 h, followed by enzyme deactivation at 65 °C for 20 min. A 5 µL aliquot of the reaction was used to transform chemically competent E. coli DH5α cells, which were plated on LB agar supplemented with 50 mg/L kanamycin and 0.5 mM IPTG.
The second method separated the digestion and ligation steps, allowing for purification of the digested DNA prior to ligation. Digestion reactions (20 µL total volume) were prepared using 2 µL of 10× rCutSmart Buffer (NEB), 10 units of SapI (NEB), and a 5:1 molar ratio of insert to plasmid DNA (based on 200 ng of plasmid DNA). Reactions were incubated at 37 °C for 2 h, followed by heat inactivation at 65 °C for 20 min. DNA was precipitated by adding 1/10 volume of 3 M sodium acetate (pH 5.2) and 2 volumes of 99% ethanol, and stored overnight at −20 °C. Precipitated DNA was pelleted by centrifugation at 16,000 × g for 30 min, washed with chilled 70% ethanol, and air-dried. The pellet was resuspended in 8 µL of nuclease-free H2O. Ligation reactions were initiated by adding 1 µL of 10× T4 DNA ligase buffer and 400 units of T4 DNA ligase (NEB), followed by incubation at room temperature for 3 h. Subsequently, 2.5 µL of the ligation reaction was used to transform chemically competent E. coli DH5α cells by heat shock. Transformants were selected on LB agar plates containing 50 mg/L kanamycin and 0.5 mM IPTG.
After overnight incubation at 37 °C followed by an additional 24 h at 4 °C, bacterial colonies exhibiting either red or white phenotypes—conferred by the LacP-mCherry dropout cassette within the pTARGEX backbones—were readily distinguishable. White colonies, which presumably lacked the mCherry cassette due to successful insertion of the gene of interest, were selected for further screening. Colony PCR was performed, and amplicons were analyzed via agarose gel electrophoresis. The appearance of a DNA fragment at the expected size indicated proper assembly. Candidate colonies were subsequently grown overnight in LB broth containing kanamycin, and plasmid DNA was isolated using the Geneaid miniprep kit. To confirm excision of the SapI sites and insertion of the correct fragment, purified plasmids were subjected to diagnostic digestion with SapI (NEB) prior to whole plasmid sequencing (Plasmidsaurus, Eugene, OR, United States).
Agroinfiltration of N. benthamiana
Wild-type N. benthamiana plants were cultivated in a controlled greenhouse environment equipped with 400-W high-pressure sodium (HPS) lamps, operating on a 20 h light/4 h dark photoperiod at a constant temperature of 25 °C. Plants were grown for 5 weeks prior to infiltration. Fertilization was carried out weekly using a water-soluble fertilizer (Miracle-Gro® 24-8-16) at a concentration of 1.1 g/L. Seeds were germinated on a rolling basis, and seedlings were transplanted into individual pots at 2 weeks of age to ensure a continuous supply of plants at the appropriate developmental stage for experimental use.
Each of the five subcellular localization constructs for the genes of interest (SARS-CoV-2 RBD and mScarlet), along with corresponding empty vector controls, were introduced into A. tumefaciens strain AGL1 via electroporation. AGL1 carries the helper plasmid pTiBo542, which is stably maintained in the strain without the need for antibiotic selection. Following recovery, transformants were plated on LB agar supplemented with kanamycin (50 μg/mL) and rifampicin (5 μg/mL) and incubated overnight. Single colonies were used to inoculate LB broth (supplemented with 50 μg/mL kanamycin), and cultures were grown overnight with shaking at 28 °C. Cultures were harvested by centrifugation at 2,500 × g for 5 min, and the cell pellets were resuspended in infiltration buffer [10 mM MES, 10 mM MgCl2, pH 5.6] to an optical density at 600 nm (OD600) between 0.1 and 0.75, depending on experimental requirements. Acetosyringone (3′,5′-dimethoxy-4′-hydroxyacetophenone) was added to each suspension at a final dilution 1:2,000 to enhance virulence gene expression, and the mixtures were incubated at room temperature for 1 h prior to infiltration, although we note that this step is not essential for this strain. Agroinfiltration was performed on healthy, fully expanded young leaves of N. benthamiana using a 1 mL needleless syringe to apply gentle pressure to the abaxial (lower) leaf surface. Infiltrated leaves were marked for identification and returned to the greenhouse for incubation under standard growth conditions until sample collection.
Transformation of M. truncatula roots via A. rhizogenes
The five subcellular localization vectors for mScarlet expression were transformed into A. rhizogenes strain ARqua1 via electroporation. Following recovery, transformants were plated on LB agar supplemented with kanamycin (50 μg/mL) and incubated at 28 °C for 2–3 days. Single colonies were then resuspended in 50 μL of sterile H2O and spread onto solid TY medium supplemented with kanamycin (50 μg/mL). These plates were incubated at 28 °C for two additional days to establish a bacterial lawn.
M. truncatula seeds were surface scarified by immersion in concentrated sulfuric acid for 9 min, then thoroughly rinsed with sterile water. Seeds were subsequently sterilized in a 10% bleach, 0.1% Tween-20 solution for 9 min, followed by repeated sterile water washes. The sterilized seeds were wrapped in aluminum foil and shaken gently on an orbital shaker for 3 h at room temperature to imbibe water. Seeds were then placed in fresh sterile water and continued to imbibe overnight at 4 °C. The following day, approximately 30 sterilized seeds were spaced 0.5–1 cm apart on an inverted Pyrex dish wrapped in foil and incubated overnight at 28 °C for germination. Germinated seedlings with radicles approximately 0.5–1.5 cm in length were selected for transformation. Seed coats were gently removed, and seedlings were diagonally cut to remove the radicle tip. The cut surface of each seedling was then dipped into the A. rhizogenes lawn on TY media. Inoculated seedlings were transferred to slanted square plates containing Fåhraeus medium, with cut tips placed into pre-cut wells to ensure root contact with growth media. Plates were partially sealed with Parafilm that was perforated to allow gas exchange, and incubated under low light conditions at 18 °C for up to 5 days. Plates were then transferred to an environmental growth chamber and maintained for an additional 3 weeks under a photoperiod of 16 h light (25 °C), 8 h dark (22 °C) with a light intensity of 45 µM/m2s to allow for root development and transgene expression.
Protein extraction and quantification
Leaf tissue was harvested and immediately flash-frozen in liquid nitrogen. The frozen tissue was then mechanically ground into a fine powder using small pestles. The resulting powder was resuspended (1 wt/2 vol) in 1X phosphate-buffered saline (PBS) [137 mM NaCl; 2.7 mM KCl; 10 mM Na2HPO4; 1.8 mM KH2PO4] and thoroughly homogenized. The crude lysate was isolated from cellular and leaf debris by centrifugation at 15,000 x g for 10 min at 4 °C. The supernatant, containing the protein extract, was carefully collected for further analysis. Total protein concentration was determined using a Bradford assay with bovine serum albumin (BSA) standards. Protein samples were then denatured by boiling in Laemmli buffer for 10 min, after which they were used for SDS-PAGE and subsequent Western blotting.
Western blotting
Protein samples were separated by SDS-PAGE on a 12% polyacrylamide gel and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% skim milk in tris-buffered saline (TBS) [20 mM Tris; 150 mM NaCl] for 30 min at room temperature. To minimize non-specific antibody binding, 0.1% Tween-20 was added to the blocking solution. Primary anti-His (C-term) HRP-conjugated antibodies (Thermo Fisher Scientific, Waltham, MA, United States) were diluted 1:2000 in blocking buffer and incubated with the membrane overnight at 4 °C. After washing with TBS, protein bands were visualized using chemiluminescence, and images were captured using the Chemidoc Imaging System (Bio-Rad Laboratories Inc., Hercules, CA, United States).
To ensure equal protein loading, a separate gel was stained with Coomassie Brilliant Blue after electrophoresis, followed by destaining in MilliQ H2O until clear protein bands were visible. The stained gel was imaged using the Chemidoc Imaging System (Bio-Rad Laboratories Inc.).
Confocal microscopy
Three to 4 days following agroinfiltration, infiltrated areas of N. benthamiana leaves were cut into approximately 1 cm squares and placed on slides with the abaxial side up. A drop of water was added, and a cover slip (0.17 µm) was placed on top. With respect to M. truncatula, root segments showing fluorescence due to mScarlet (as visualized under a ZEISS Axio Zoom.V16 dissecting microscope) were excised from growth media and cut into 5-7 mm pieces. Root sections were then cut longitudinally using a double-edged razor blade and placed on a glass slide. A drop of water was added, followed by the placement of a cover slip (0.17 µm).
Leaf and root sections were observed and fluorescence was imaged using a Nikon A1RsiMP laser scanning confocal microscope with a 10x dry objective lens (Plan APO λ 0.45). eGFP (488 nm) and mScarlet (561 nm) were excited using an optically pumped semiconductor laser, and emitted fluorescence was captured from 500 to 550 nm for eGFP and 570–620 nm for mScarlet. Brightfield images were captured simultaneously with fluorescence using a Nikon A1 Plus camera. Maximum intensity projections were generated from z-stacks using the Galvano scanner. Images were processed using the NIS-Elements confocal package and FIJI (Schindelin et al., 2012).
Imaging of whole leaves and roots using a dissecting microscope
Three days following agroinfiltration with AGL1 carrying either pTARGEX-CYT or mScarlet-pTARGEX-CYT, N. benthamiana leaves were excised at the base of the leaf and placed directly onto the stage of ZEISS Axio Zoom V16 dissecting microscope, adaxial side up (abaxial side up, which is standard protocol yielded comparable levels of fluorescence). Fluorescence was observed and imaged using fluorescent LED lamp under “GFP” (488 nm) and “RFP” (561 nm) settings with Plan-NEOFLUAR Z 1.0x/0.25 FWD objective. “GFP” and “RFP” images were captured simultaneously with Axiocam 506. Non transformed leaves were collected and imaged in parallel. Transformed root segments expressing mScarlet encoded by pTARGEX-CYT were imaged approximately 3 weeks following transformation of A17 with A. rhizogenes by gently removing the seedlings from F plates with forceps and placing directly onto the Axio Zoom V16 microscope. Fluorescence was observed and imaged as described for N. benthamiana leaves, with the addition of images capture under “Brightfield” settings. In this instance, A17 roots transformed with pGWB405 were included alongside pTARGEX-CYT-mScarlet as a control to assess background levels of auto-fluorescence.
Imaging of whole leaves using a Chemidoc
Three days following agroinfiltration with AGL1 carrying pTARGEX-CYT or pTARGEX-CYT-mScarlet, N. benthamiana leaves were excised at the base of the leaf and placed adaxial side upwards directly onto a Blue Sample Tray in a BioRad Chemidoc MP Imaging System, with images captured under the “ethidium bromide” gel default settings with auto optimal exposure.
Results
Design and assembly of a modular plant expression toolkit: the pTARGEX series
As part of an undergraduate research initiative conducted through participation in the international Genetically Engineered Machine (iGEM) competition, our team aimed to develop a suite of plant expression vectors capable of supporting high-level heterologous protein expression with targeted subcellular localization in planta. We constructed five binary plasmids, collectively named the pTARGEX series, with each vector specifically engineered to direct recombinant protein accumulation to one of five subcellular compartments: the cytoplasm, endoplasmic reticulum, vacuole, apoplast, or chloroplast. Cytoplasmic accumulation was achieved using a construct lacking any targeting sequences (pTARGEX-CYT), thereby allowing recombinant proteins to remain in the cytosol following translation. For endoplasmic reticular localization (pTARGEX-ER), we incorporated an N-terminal signal peptide derived from the Nicotiana tabacum pathogenesis-related protein 1b (PR1b) and a canonical C-terminal ER-retrieval motif (KDEL) (Denecke et al., 1991). To target proteins to the vacuole (pTARGEX-VAC), we employed the same PR1b signal peptide to direct entry into the secretory pathway, in combination with a C-terminal propeptide (CTPP) derived from N. tabacum chitinase A, which serves as a vacuolar sorting signal (Neuhaus et al., 1991). For apoplastic secretion (pTARGEX-APO), constructs included the PR1b signal peptide without additional retention or sorting motifs, permitting default secretion to the extracellular space (Lund and Dunsmuir, 1992). Chloroplast targeting (pTARGEX-CHL) was accomplished through the inclusion of an N-terminal transit peptide derived from the small subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase (RbcS) from N. tabacum (Kuntz et al., 1986), enabling post-translational import into chloroplasts via the TOC/TIC translocon system (Bédard and Jarvis, 2005). Each of these localization tags has a well-established precedent for directing heterologous recombinant proteins to the corresponding organelles (Carlsson et al., 2020; Chin-Fatt et al., 2021; Conley et al., 2009; Harding and Sainsbury, 2025; Kaldis et al., 2023; Liénard et al., 2007; Ma et al., 2019; Menassa et al., 2004; Pereira et al., 2014).
We based our new vector series upon a backbone that was derived from the pHREAC vector (Peyret et al., 2019), using a Gibson Assembly approach to integrate subcellular targeting sequences into each vector (Figure 1A). To enable red/white visual screening of recombinant clones, we also incorporated an expression cassette in which the mCherry reporter gene is placed under the control of the lac promoter-operator system and flanked by recognition sites for the Type IIS restriction enzyme SapI. This configuration allows directional cloning and seamless replacement of the mCherry coding sequence with a target gene of interest, resulting in loss of red colony colouration and fluorescence (Figure 1B) when successfully assembled plasmids are transformed into Escherichia coli DH5α and plated on IPTG-containing media.
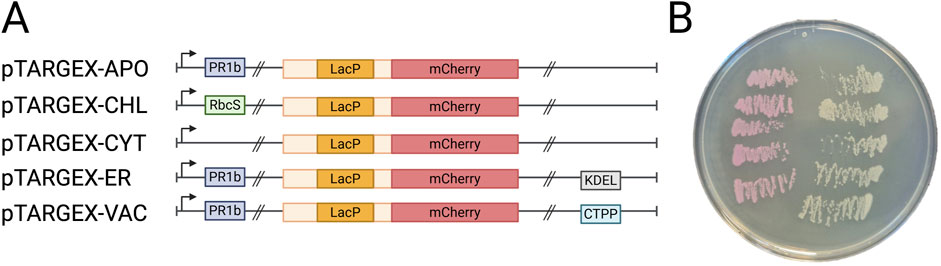
Figure 1. Schematic overview of the pTARGEX vector series. (A) Diagram of the pTARGEX targeting and cloning cassette, illustrating localization sequences for directing genes of interest to specific subcellular compartments and a LacP-mCherry cassette enabling Red/White screening of recombinant plasmids. Up-right arrow, CaMV 35S promoter. PR1b, Tobacco (Nicotiana tabacum) PR1b signal peptide. RbcS, Tobacco (N. tabacum) transit peptide derived from the small subunit of RuBisCO. KDEL, C-terminal endoplasmic reticular retrieval motif. CTPP, C-terminal ProPeptide vacuolar sorting signal of Tobacco (N. tabacum) chitinase A. Double slashes indicate locations of SapI restriction sites that flank the LacP-mCherry dropout cassette. Light yellow boxes indicate sequences flanking LacP promoter. Images not drawn to scale. (B) E. coli DH5α colonies on a Petri dish following transformation with pTARGEX vectors. Pink colonies (left) were transformed with the relevant pTARGEX empty vector plasmid, while white colonies (right) carry recombinant plasmids derived from each pTARGEX vector containing a cloned gene of interest.
Although N. benthamiana leaves are most frequently used for heterologous protein expression, plant roots also represent a valuable alternative expression platform (Drake et al., 2009; Lonoce et al., 2016; Madeira et al., 2016a; Madeira et al., 2016b). Accordingly, we aimed to design an expression vector optimized for both stable transformation of root systems and transient expression in N. benthamiana leaves. The nptII selectable marker gene present in the pHREAC vector is useful for generating stable transgenic plant lines, but it does not confer any advantage during transient agroinfiltration of leaves. Because we wished to explore the utility of an antibiotic-free method for identifying transformed roots, we replaced nptII with a gene encoding enhanced green fluorescent protein (eGFP), expressed under the same regulatory elements as the original nptII gene. This design was conceived to enable visual selection of transformed root systems via fluorescence, similar to the RedRoot system which utilizes DsRed to identify root tissues containing integrated transgenes (Ivanov and Harrison, 2014). Additionally, we hypothesized that eGFP expression, assessed via fluorescence under UV or filtered light sources or through immunoblot analysis, could serve as an inbuilt marker to confirm successful transient transformation in N. benthamiana leaves, independently of the recombinant protein of interest.
Validation of subcellular localization of target proteins in planta
Each vector in our expression toolbox was designed to direct the accumulation of a recombinant protein to one of five specific subcellular compartments: the cytoplasm, endoplasmic reticulum, vacuole, apoplast, and chloroplast/plastid (Figure 1A). To evaluate our red/white screening system and verify the functionality of these targeting sequences in planta, the mScarlet gene was cloned into each expression vector using SapI restriction sites, placing mScarlet under control of the plant-specific CaMV 35S promoter. Putative transformants that contained a cloned gene were selected based on loss of red pigmentation on IPTG-containing media, due to loss of the LacP-mCherry cassette that confers red colouration in E. coli. Colony PCR performed on white colonies confirmed the presence of the gene of interest, indicating that the loss of colouration correlated to successfully cloned plasmids (Figure 1B). For all vectors, an additional 24-h incubation at 4 °C markedly improved the distinction between red and white colonies, particularly enhancing colour development in smaller colonies.
To assess localization profiles, sequence-verified constructs were transformed into A. tumefaciens strain AGL1, which was subsequently used to transiently transform N. benthamiana leaves via agroinfection. Leaf tissues were collected 3–4 days post-infiltration and examined by confocal scanning laser microscopy (Figure 2). Excluding the chloroplast-targeted construct, fluorescence patterns observed in N. benthamiana were consistent with the expected subcellular localization profiles, confirming the functionality of the targeting sequences in this transient expression system. To more closely assess whether the pTARGEX-APO vector effectively directs protein accumulation to the apoplast, we treated infiltrated tissues with a hypertonic solution (0.8 M mannitol) for 30–60 min prior to microscopic visualization, to induce plasmolysis. Following such treatment, mScarlet fluorescence was consistently observed in the extracellular spaces that formed between the retracted plasma membrane and the cell wall (Figure 2, pTARGEX-APO), consistent with apoplastic localization. Expression of the chloroplast-targeted mScarlet resulted in a mixed localization pattern, with fluorescence detected in chloroplasts (Figure 2, pTARGEX-CHL) and also within the nucleus and cytosol (as indicated in ‘Image Annotation’ panel).
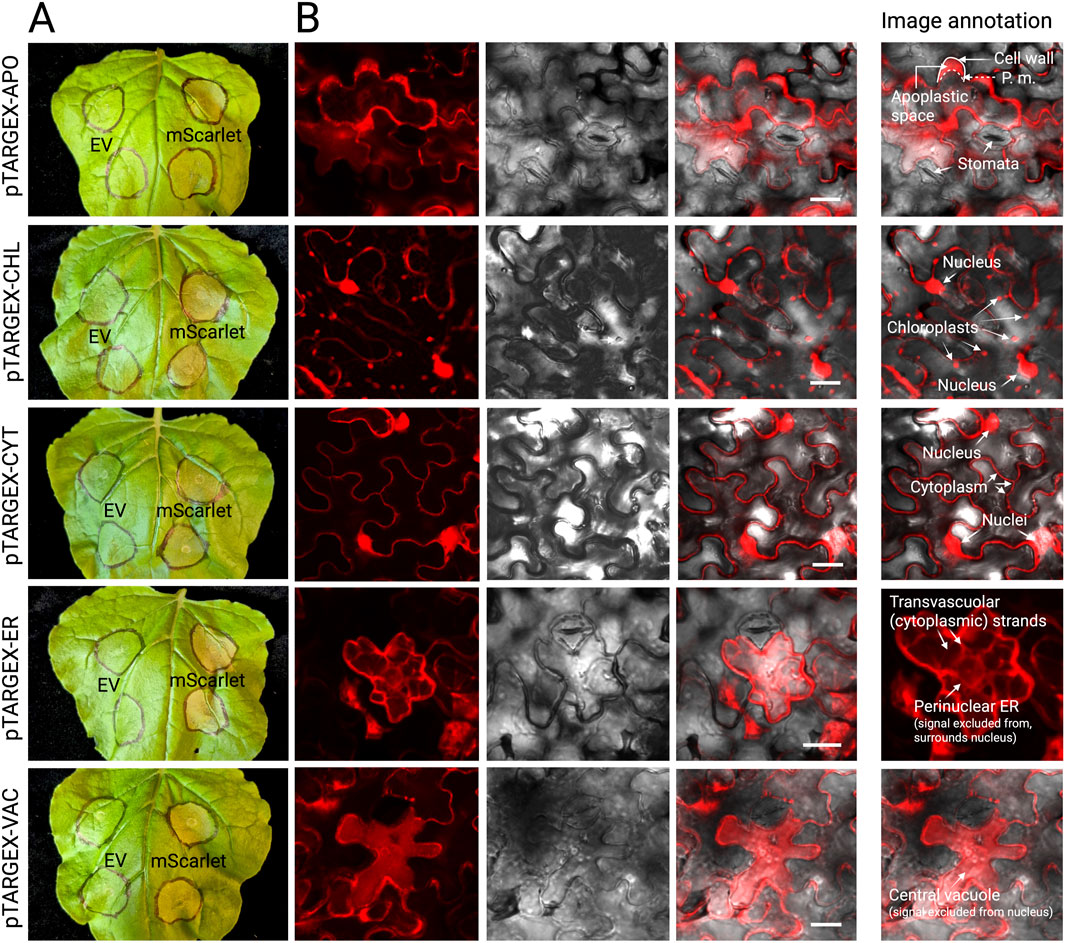
Figure 2. Expression and subcellular targeting of mScarlet via pTARGEX expression constructs in N. benthamiana. (A) N. benthamiana leaves imaged 4 days post-agroinfiltration with either the relevant pTARGEX empty vector control (EV) or the corresponding constructs encoding the fluorescent protein mScarlet. Infiltration zones are circled in black marker. (B) Confocal laser scanning microscopy images of infiltrated N. benthamiana tissues showing subcellular localization profiles of mScarlet for each pTARGEX vector. To aid interpretation, an annotated version of the image has been included in panel (B). Scale bars: 20 µm.
To test the same set of constructs in a root-based system, the mScarlet-encoding vectors were next individually expressed in genetically transformed M. truncatula roots, following transformation with A. rhizogenes strain ARqua1. Confocal laser scanning microscopy of these root tissues revealed discrete subcellular fluorescence patterns consistent with the expected localization signals encoded by each construct (Figure 3). Notably, expression of mScarlet encoded by pTARGEX-CHL in M. truncatula roots resulted in exclusive plastidial localization, characterized by the presence of rounded, lens-shaped organelles consistent with plastid morphology as previously described (Ivanov and Harrison, 2014). This localization pattern contrasts with that observed in N. benthamiana leaves, where the same construct exhibited mScarlet fluorescence in chloroplasts as well as in the cytoplasm and nucleus. Collectively, these results (Figures 2, 3, with additional images offered in Supplementary Figure S1) demonstrate that the pTARGEX vectors are effective in both shoot and root transformation systems, supporting transgene expression and accurate subcellular targeting across diverse plant tissues.
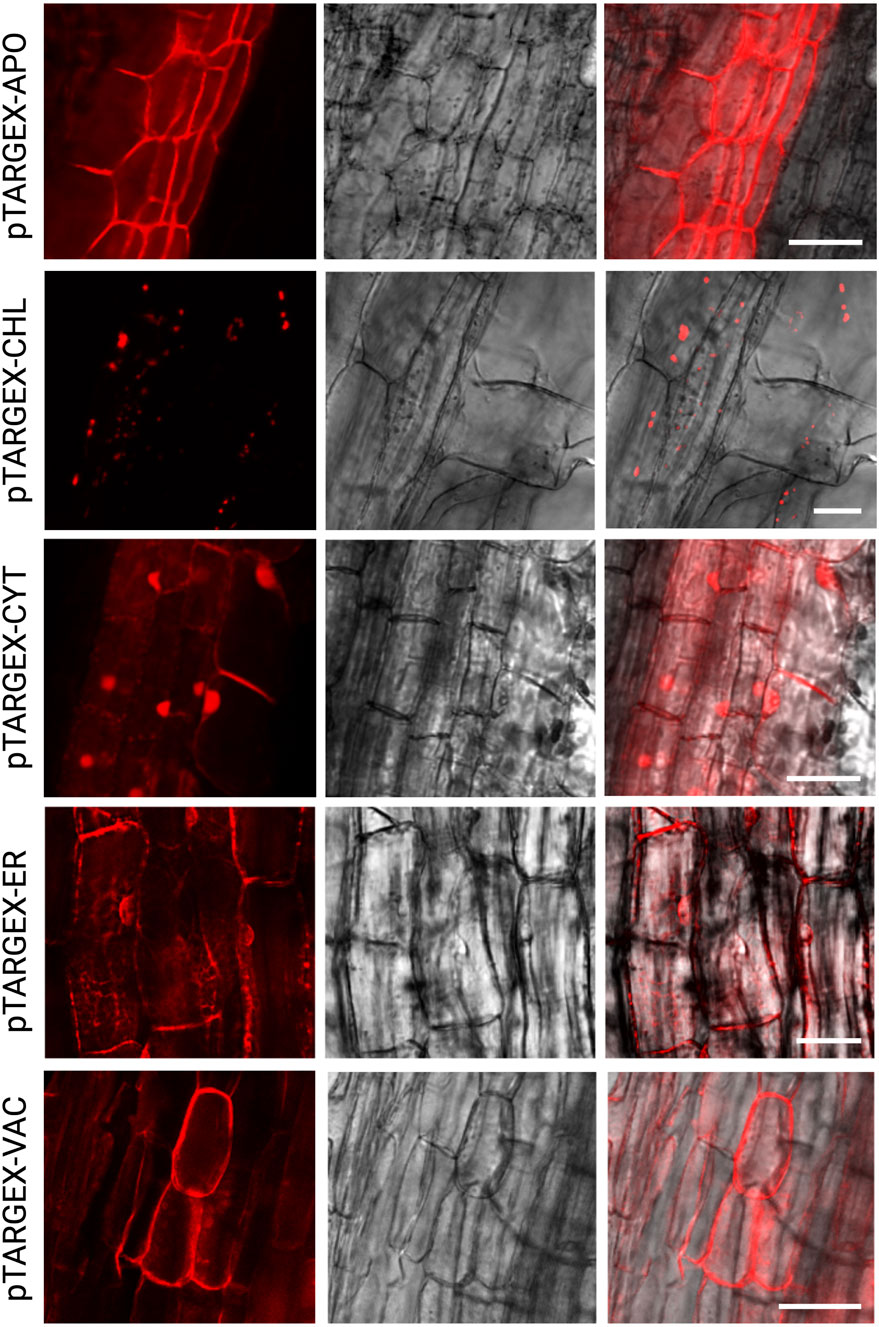
Figure 3. Subcellular targeting of mScarlet in M. truncatula root cells via pTARGEX expression constructs. Confocal laser scanning microscopy images of M. truncatula A17 root cells transformed with pTARGEX vectors expressing mScarlet, highlighting subcellular localization patterns. Scale bars: 50 µm.
Assessment of recombinant protein accumulation in N. benthamiana using pTARGEX vectors
To further assess the ability of the pTARGEX vector series to drive heterologous protein expression and accumulation of recombinant proteins in planta, we focused on transient expression in N. benthamiana, the gold standard in biopharming. Transiently expressed constructs encoding either mScarlet (Bindels et al., 2016) or the receptor-binding domain (RBD) of the SARS-CoV-2 Spike protein (Shang et al., 2020) were introduced into N. benthamiana via agroinfiltration using A. tumefaciens strain AGL1. Leaf tissue was harvested three- or 4-days post-infiltration and total protein extracts were analyzed via SDS-PAGE and immunoblotting. Infiltration zones expressing mScarlet constructs (mS) exhibited readily observable pink colouration compared to empty vector (EV) controls (Figure 4A), indicating strong expression of the fluorescent protein. This pigmentation extended to cell-free crude protein extracts, which were similarly tinted pink (Figure 4B), further reflecting high levels of mScarlet accumulation in planta. Immunoblot analysis using an anti-His antibody revealed that mScarlet was consistently expressed across all five subcellular targeting contexts specified by the pTARGEX series, with relatively uniform band intensities observed for each construct (Figure 4C), although we noted the presence of a higher mass band in the pTARGEX-CHL-mScarlet sample of approximately 35 kDa. In contrast, Western blot detection of RBD expression demonstrated strong and reproducible differences in protein accumulation across subcellular compartments (Figure 4D). Highest accumulation was observed when RBD was targeted to the cytoplasm (pTARGEX-CYT), followed by ER-retained expression (pTARGEX-ER). Substantially lower levels were detected in constructs targeting the apoplast, vacuole, and chloroplast. Overall, these findings suggest that the pTARGEX vectors support robust expression and detection of recombinant proteins across multiple subcellular locations.
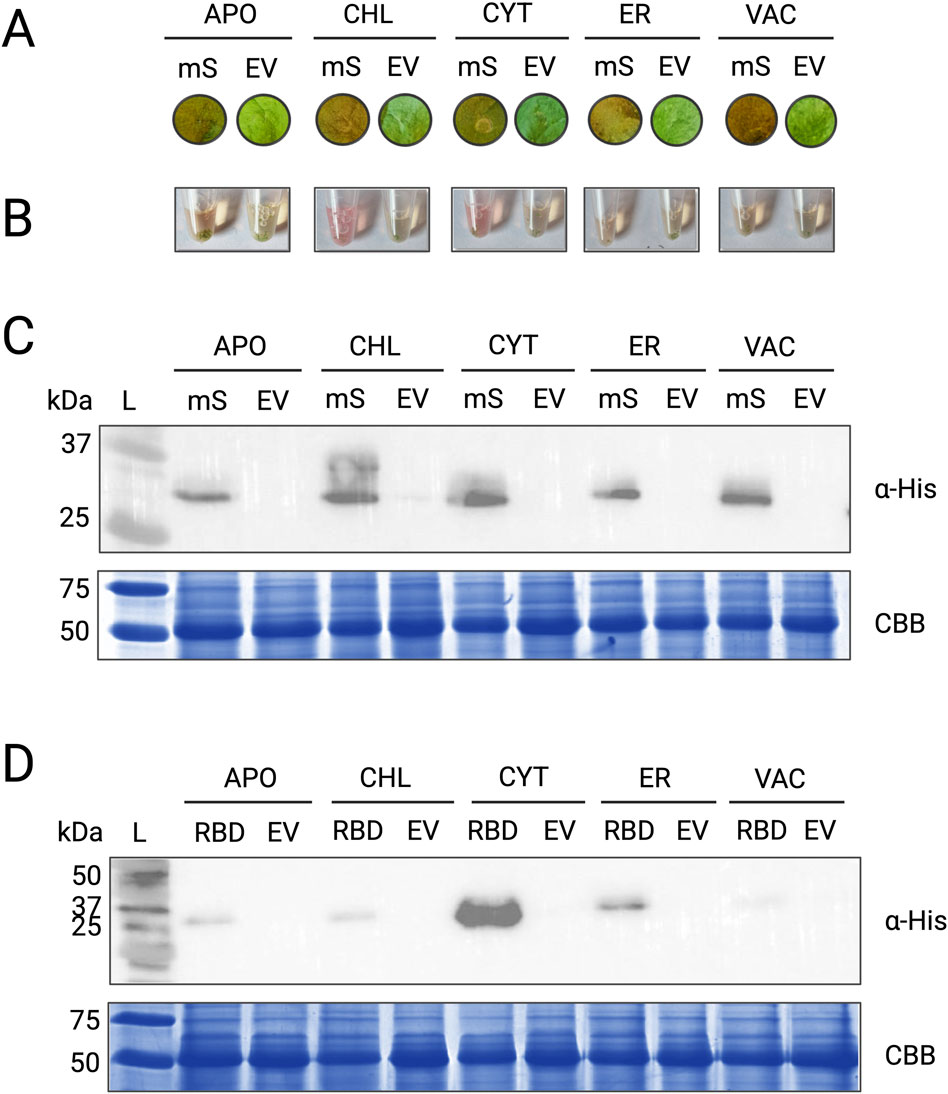
Figure 4. Expression of heterologous proteins mScarlet and RBD in N. benthamiana. (A) Infiltration spots on N. benthamiana leaves showing visible colouration in tissues infiltrated with pTARGEX vectors carrying the mScarlet gene (mS) compared to empty vector controls (EV). (B) Crude protein lysates extracted from infiltrated leaf tissues. Samples expressing mScarlet (mS) exhibit a distinct pink colouration compared to the EV controls. (C) Western blot analysis using anti-His antibody (α-His) to detect mScarlet protein in tissues infiltrated with pTARGEX-mScarlet constructs. (D) Western blot analysis of N. benthamiana tissues infiltrated with pTARGEX-RBD constructs encoding the SARS-CoV-2 receptor-binding domain (RBD) gene. All protein extracts were obtained 3 or 4 days post-agroinfiltration. mScarlet constructs were expressed in three independent experiments, and RBD constructs were expressed in two independent experiments. The images shown in the final figures are representative of these results. Protein loading was assessed by SDS-PAGE (15%) followed by Coomassie Brilliant Blue (CBB) staining.
Integration of eGFP for fluorescence-based tracking of transformed root systems
To enhance our ability to selectively propagate transformed root cultures without the use of antibiotic selection, we integrated an eGFP fluorescent protein gene into the pTARGEX vector series under the control of the nopaline synthase (NOS) promoter (Peyret et al., 2019) and terminator, replacing the nptII kanamycin resistance gene at that position. The eGFP gene did not encode any localization tags, thus the fluorescent protein is expected to accumulate in the cytosol. To evaluate the functionality of this fluorescent tracking feature, we first performed confocal microscopy on N. benthamiana leaves 3–4 days following agroinfiltration with each of the pTARGEX series vectors (Figure 5A) and M. truncatula roots (Figure 5B). In N. benthamiana leaves, fluorescence was readily observed under a confocal microscope, confirming successful expression of eGFP under the control of the NOS transcriptional elements. In contrast, fluorescence in M. truncatula roots transformed with pTARGEX-CYT was noticeably less intense, suggesting that eGFP expression and/or protein accumulation may be less robust in these tissues. Western blotting with anti-GFP antibody confirmed consistent and uniform accumulation of eGFP in N. benthamiana leaves when expressed from each of the five pTARGEX vectors (Figure 5C) corroborating observations made under the confocal microscope.
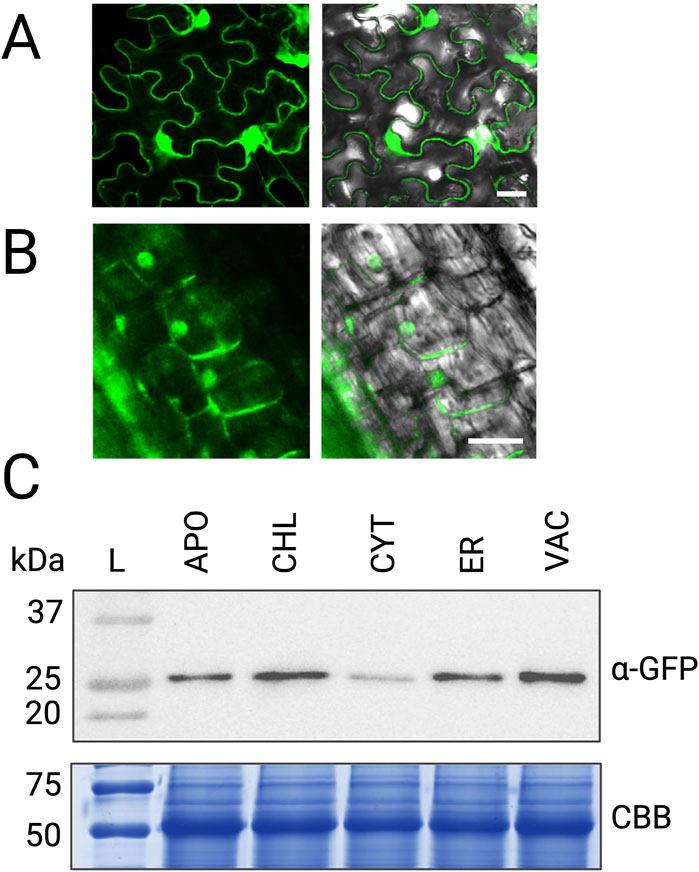
Figure 5. eGFP expression driven by pTARGEX vectors in plant tissues. (A) Confocal laser scanning microscopy of N. benthamiana leaves infiltrated with A. tumefaciens strain AGL1 harboring pTARGEX-CYT, showing eGFP fluorescence with nuclear and cytoplasmic localization. (B) Confocal microscopy of M. truncatula roots transformed with pTARGEX-CYT, also displaying nuclear and cytoplasmic distribution of eGFP. (C) Western blot analysis of protein extracts from N. benthamiana leaves collected 4 days post-agroinfiltration with the indicated pTARGEX constructs. Blots were probed with anti-GFP antibody (α-GFP). Protein loading was assessed by SDS-PAGE (15%) followed by Coomassie Brilliant Blue (CBB) staining. Scale bars: 20 µm (A), 25 µm (B).
Because our goal was to incorporate eGFP into the backbone of our expression vectors to permit facile screening of successful transformation using minimally specialized equipment, we also evaluated N. benthamiana leaves for eGFP-based fluorescence under UV illumination and low-magnification dissection microscopy (Figure 6). Following transient expression with the pTARGEX-CYT construct, low-level eGFP fluorescence was detectable when whole, excised N. benthamiana leaves were imaged using a Chemidoc transilluminator system (Figure 6A). In contrast, the same tissues did not exhibit readily discernible fluorescence when examined under a fluorescence-equipped dissecting microscope using a GFP filter set (Figure 6B), although moderate autofluorescence was observed in agroinfiltrated regions when viewed under the ‘RFP’ filter. In comparison, expression of mScarlet from the pTARGEX-CYT construct in N. benthamiana resulted in markedly higher fluorescence levels compared to the empty vector, which were readily detectable under both UV illumination and fluorescence stereomicroscopy. Unfortunately, while we could easily identify transformed root tissues that expressed red fluorescence from mScarlet-tagged constructs expressed in M. truncatula, confirming successful integration of the T-DNA, we were unable to use eGFP fluorescence to identify transformed root sections under the dissecting microscope (Figure 6C).
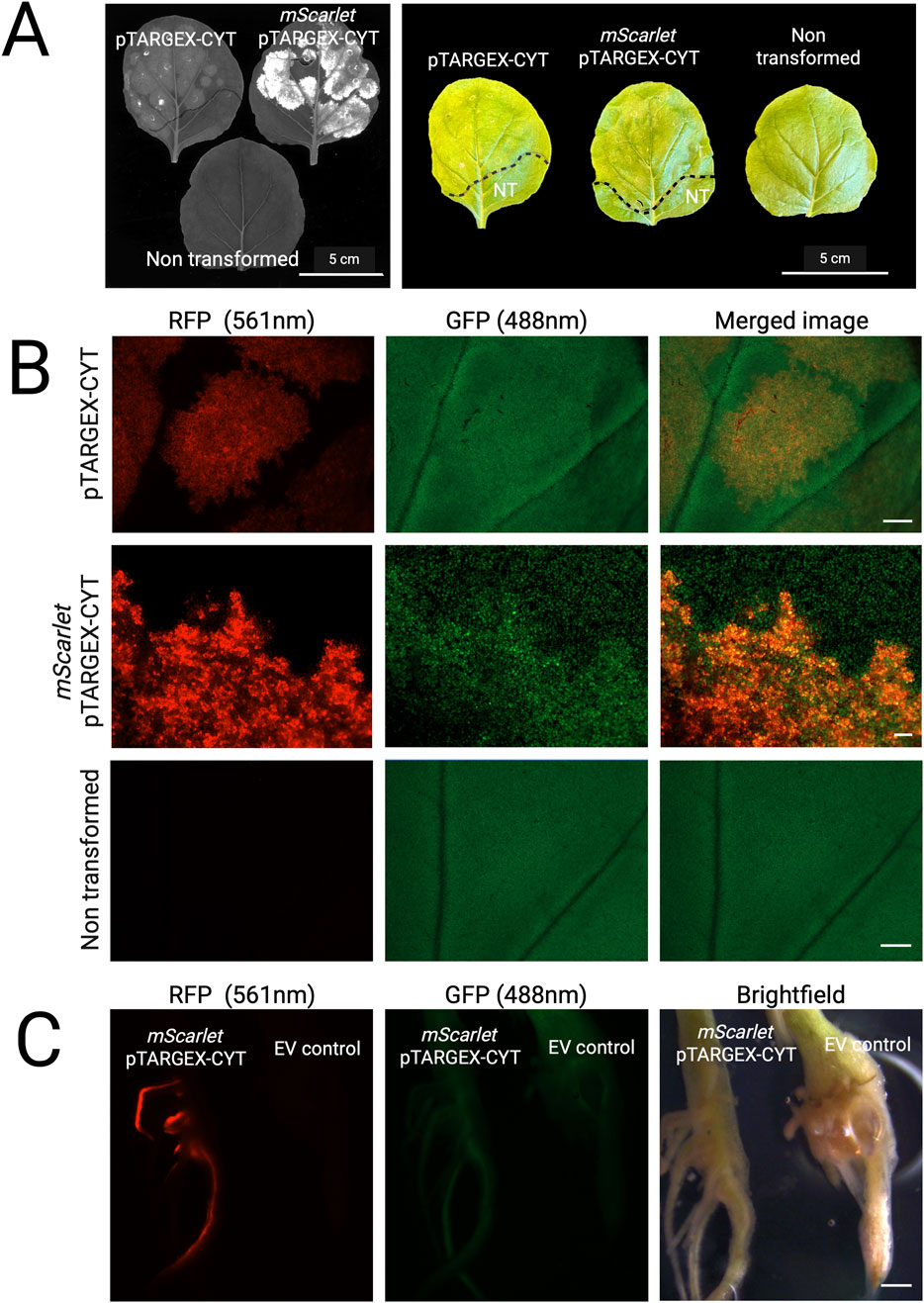
Figure 6. Visualization of pTARGEX-based fluorescence in whole tissues. (A) Left: Image of excised N. benthamiana leaves acquired under UV illumination using a Chemidoc Imaging System. Right: Corresponding photographs of the same leaves captured using an iPhone. Images were taken 3 days post-agroinfiltration with pTARGEX-CYT constructs, as labelled. A leaf from a non-transformed plant was included as a negative control. (B) Images of leaf tissues examined under a dissecting microscope using RFP or GFP filters. Images were obtained from leaves imaged in panel (A). Scale bars: 2,000 µm (pTARGEX-CYT and Non transformed panels); 200 µm (pTARGEX-mScarlet panel) (C) Images of transformed M. truncatula root systems approximately 3 weeks post transformation. EV control refers to root systems transformed with pGWB405. Scale bar: 2,000 µm.
Discussion
In this study, we present the design, construction, and functional validation of the pTARGEX series, a modular toolkit of plant expression vectors developed as part of an undergraduate research initiative through the 2024 iGEM competition. Our goal was to provide a versatile and accessible platform to support high-level heterologous protein expression in plants, with the added functionality of targeting recombinant proteins to distinct subcellular compartments. The resulting plasmids (pTARGEX-CYT, -ER, -VAC, -APO, and -CHL) enable protein accumulation in the cytoplasm, ER, vacuole, apoplast, or chloroplast, respectively, expanding the utility of plant-based expression systems in both research and applied contexts. Importantly, the pTARGEX vector series is designed to facilitate modular, directional, and seamless cloning of a gene of interest into all five constructs using a unified primer set. This streamlined strategy allows the potential to rapidly screen the accumulation and yield of a given protein targeted to multiple subcellular compartments using a standardized cloning workflow. Collectively, the pTARGEX system provides a versatile and efficient platform for optimizing the expression of recombinant proteins in plants.
Subcellular targeting and protein accumulation
Validation of the pTARGEX constructs using the fluorescent protein mScarlet revealed successful and specific localization to all five target compartments in both N. benthamiana leaves and M. truncatula roots (Figures 2, 3; Supplementary Figure S1), discussed here in more detail. mScarlet is a synthetically-derived monomeric red fluorescent protein with a pKa of 5.3, indicating that it should be tolerant of moderately acidic environments such as the apoplast and central vacuole (Bindels et al., 2016). Moreover, mScarlet has been shown to outperform other fluorescent proteins in terms of signal-to-noise ratio and overall effectiveness during transient expression in N. benthamiana (Khosla et al., 2020). These considerations guided our selection of this fluorescent protein for our proof-of-concept. As the specific tags incorporated in our vectors have been extensively characterized previously (Carlsson et al., 2020; Chin-Fatt et al., 2021; Conley et al., 2009; Harding and Sainsbury, 2025; Kaldis et al., 2023; Liénard et al., 2007; Ma et al., 2019; Menassa et al., 2004; Pereira et al., 2014), co-localization with established markers was not performed in this study.
In plants, the apoplast is an extracellular compartment that includes the cell walls, and spacing between the plant cell walls and plasma membranes of individual cells. mScarlet targeted to the apoplastic space via expression from pTARGEX-APO yielded signal outlining the cell borders, and was entirely excluded from cell nuclei, and is consistent with a pattern to be expected of an apoplastic protein (Trusova et al., 2019). To more precisely evaluate subcellular localization, mannitol-induced plasmolysis was performed on epidermal and mesophyll cells, a treatment that promotes the detachment of the plasma membrane from the cell wall and thereby enlarges the apoplastic space (Ma et al., 2012). In plasmolysed cells, fluorescence was clearly observed within the expanded regions between the plasma membrane and cell wall, consistent with apoplastic localization of the fluorescent signal. This approach was not applied to M. truncatula roots, as it has not, to the best of our knowledge, been previously implemented in this system, and we anticipate its effectiveness to be limited by challenges associated with achieving effective infiltration of mannitol into root tissues.
When expressed from the pTARGEX-CYT construct, mScarlet exhibits a characteristic nuclear and cytoplasmic distribution, with strong fluorescence observed within the nuclei as well as along the cell periphery. In epidermal and mesophyll cells of N. benthamiana leaves, and root cortical cells of M truncatula, the presence of a central vacuole displaces the cytoplasm of these cells into a thin peripheral layer that is pressed up against the plasma membrane. As a result, cytosolic fluorescent proteins typically produce a signal that outlines the cell shape in confocal images (Figures 2, 3). Owing to their small size and ability to pass through nuclear pores, fluorescent proteins such as mScarlet also accumulate in the nucleoplasm when expressed without a specific localization signal in these same cells, giving rise to a typical nuclear/cytoplasmic profile, as observed when mScarlet is expressed via pTARGEX-CYT in N. benthamiana and M. truncatula. When retained in the ER, fluorescent proteins give rise to signal patterns that reflect the intricate web-like network of ER tubules and cisternae distributed within the cytoplasm, with perinuclear ER forming a halo that encompasses (but does not penetrate) the nucleus, as may be observed in Figures 2, 3 (pTARGEX-ER). Vacuole-targeted proteins (as exemplified by mScarlet in pTARGEX-VAC) accumulate within the vacuolar lumen, resulting in a relatively uniform signal that is dispersed across a large proportion of the cell volume, and an absence of fluorescence in the nucleus or peripheral cytoplasm.
However, a variation from the expected localization pattern was observed for the chloroplast-targeted construct (pTARGEX-CHL) when expressed in N. benthamiana, where fluorescence was unexpectedly detected in both the cytoplasm and nucleus in addition to chloroplasts (Figure 2; Supplementary Figure S1). We interpret this mixed localization profile as a potential consequence of very high levels of transient expression overwhelming the chloroplast import machinery, which relies on energy-dependent transport through the TOC/TIC translocon complex (Bédard and Jarvis, 2005). In contrast, the same construct yielded exclusive plastid localization in M. truncatula roots (Figure 3), where overall expression levels were likely lower, allowing the import machinery to function properly.
A recent study investigated the efficiency of various chloroplast-targeting peptides (cTPs) in directing cTP-tagged GFP to chloroplasts when expressed in tobacco (N. tabacum) leaves (Thagun et al., 2024). The study reported several cTPs to be more effective than the comparator AtRbcS cTP (a homologue of the tobacco cTP in pTARGEX-CHL) in targeting GFP specifically to chloroplasts. However, none of the 88 cTPs that were examined yielded nuclear accumulation of the protein. This finding, in combination with the M. truncatula root data, supports a hypothesis that the strong nuclear and cytoplasmic fluorescence observed in our experiment may be due to an overload of the chloroplast-targeting machinery, rather than a failure of the pTARGEX-CHL design. Further supporting this hypothesis, we observed a distinct ∼35 kDa band unique to the pTARGEX-CHL sample expressed in N. benthamiana (Figure 4C), consistent with the predicted size of mScarlet retaining an uncleaved chloroplast transit peptide. This suggests that the recombinant protein may be only partially translocated into chloroplasts, resulting in the accumulation of uncleaved precursor in the cytosol. Moreover, it is well known that overloading translocon machinery in bacteria can similarly lead to the cytoplasmic accumulation of heterologously expressed proteins tagged for localized expression (Schlegel et al., 2013). These findings highlight the importance of regulating protein expression levels when precise chloroplast targeting is important and indicate that the fidelity of localization may vary with the host system and the strength of the promoter.
Expression and accumulation of mScarlet and RBD in N. benthamiana
While mScarlet exhibited relatively uniform accumulation across all five compartments when expressed in N. benthamiana leaves, accumulation of the RBD of SARS-CoV-2 Spike protein varied significantly. Expression was highest when RBD was expressed in the cytoplasm, followed by protein localized to the ER, while accumulation in the vacuole, apoplast, and chloroplast was markedly reduced in comparison (Figure 4). As the extraction buffer lacked detergent, protease inhibitors, and chelators, it is important to acknowledge that these Western blots serve as proof-of-function rather than allowing quantitative comparisons between compartments. Quantification would require compartment-specific isolation or a general extraction buffer capable of fully releasing all compartments. Although we did not perform quantitative analyses here, future users of these vectors may wish to apply ratiometric quantification—for example, using a co-expressed control protein such as the GFP encoded within the backbone of the pTARGEX vectors—to enable accurate comparisons of expression levels.
As a general rule, accumulation of recombinant proteins to high levels in specific subcellular compartments can be highly variable and must be empirically determined for each protein of interest. This variability is influenced by the intrinsic biochemical properties of the expressed protein, including its structural stability, folding dynamics, and susceptibility to proteolytic degradation in different cellular environments. In our study, the SARS-CoV-2 RBD accumulated to the highest levels when retained in the cytoplasm. Although cytosolic expression of recombinant proteins is often associated with suboptimal expression levels, exceptions to this trend have been noted (e.g., Pêra et al., 2015). However, despite this enhanced accumulation, the cytosol may not be the most suitable compartment for in planta production of RBD, depending upon downstream application. Proteins expressed in the cytoplasm will not undergo glycosylation, an important post-translational modification that contributes to proper folding, structural stability, and high-affinity binding of the RBD to the ACE2 receptor (Bouwman et al., 2021; Huang et al., 2021; Ives et al., 2024; Shin et al., 2021). As glycosylation occurs within the ER and Golgi apparatus, effective glycosylation requires trafficking of the recombinant protein through the secretory pathway. This can be achieved in plants by targeting the protein to the apoplast or by retaining it within the endoplasmic reticulum, should N-linked glycosylation be sufficient to support proper function. These considerations underscore the importance of evaluating both yield and functional quality when optimizing subcellular targeting strategies for heterologous protein production in plants. Nevertheless, the flexibility of the pTARGEX vectors offers researchers the ability to empirically determine the most suitable compartment for each target protein.
Utility of eGFP for antibiotic-free selection of transformed root cultures
To facilitate non-antibiotic-based selection of transformed plant tissues, we replaced the nptII selection marker in the pHREAC backbone with an eGFP gene, driven by the same NOS promoter and terminator previously used to express nptII. Although the NOS promoter is considered weaker than commonly used viral promoters such as CaMV 35S, this characteristic was initially considered as an advantage in our system. A weaker promoter limits the metabolic burden on transformed cells that might otherwise compromise expression of the recombinant protein of interest. In this context, eGFP expression driven by the NOS promoter was meant to serve as a non-intrusive visual marker of transformation success, without significantly competing for the cellular resources required by the primary expression cassette. Unfortunately, eGFP accumulation in M. truncatula roots transformed with pTARGEX vectors does not appear to be sufficient to permit facile identification of transformed root segments in intact, living roots when examined under a dissection microscope. Nevertheless, the eGFP module offers potential as an inbuilt marker to assess transient transformation in N. benthamiana leaves, enabling researchers to quickly determine whether agroinfiltration has been successful—an especially valuable feature for proteins of interest that yield poor or undetectable expression levels. In such cases, visible eGFP fluorescence could help distinguish between expression failures caused by inherent challenges with the recombinant protein, and issues related to infiltration efficiency or susceptibility of plants to agroinfection. The presence of eGFP fluorescence can be confirmed by confocal microscopy, immunoblotting, or simple UV illumination, allowing for rapid, non-destructive screening of living plant tissues. As such, the pTARGEX vectors offer an internal control that can enhance troubleshooting, streamline workflow, and support reproducibility across experiments.
Concluding remarks
A key strength of this work lies in its origin: the pTARGEX vectors were designed, built, and tested by a team of undergraduate students participating in the iGEM competition, under the mentorship of faculty members and senior graduate students. This initiative not only yielded a useful molecular toolkit but also demonstrated the feasibility of engaging students in high-impact synthetic biology projects. The pTARGEX series is intended to be user-friendly and adaptable, making it well-suited for further optimization and use in both academic and teaching laboratories. To ensure broad accessibility and facilitate further research, these vectors are available to the scientific community through deposition in the Addgene plasmid bank. Future directions might potentially include the integration of inducible expression systems and the systematic testing of different promoters to further optimize the eGFP reporter system. Importantly, the open-source nature of iGEM projects position the pTARGEX vectors as a sharable and modifiable platform for the broader scientific community.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
CC-P: Investigation, Writing – original draft, Visualization, Conceptualization, Validation, Writing – review and editing. EG-B: Visualization, Validation, Writing – review and editing, Investigation. AJ: Writing – review and editing, Writing – original draft, Investigation. VB: Writing – original draft, Resources, Funding acquisition, Writing – review and editing. PF: Writing – review and editing, Investigation. SR: Investigation, Writing – review and editing. TT: Writing – review and editing. MD: Investigation, Writing – review and editing. JV: Writing – review and editing, Resources. AB: Writing – review and editing, Supervision. AD: Funding acquisition, Resources, Supervision, Conceptualization, Writing – review and editing. AM: Visualization, Resources, Validation, Conceptualization, Project administration, Writing – review and editing, Writing – original draft, Funding acquisition, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was financially supported by grants from the Faculty of Science–Science Students Association Joint Grant Fund at the University of Ottawa, the Faculty of Engineering Endowment fund, and through the University of Ottawa Students’ Union. Additionally, we thank the Office of the Dean of Science, the Vice Dean of Strategic Partnerships and Innovation, the Chairs of the Departments of Chemistry and Biology, and supporting faculty members for their assistance in securing funding for the project through the NSERC Undergraduate Student Research Awards program and a full teaching assistant contract.
Acknowledgments
We gratefully acknowledge the bioGARAGE team for generously hosting us and providing access to their research space, and we particularly thank François-Xavier Campbell-Valois for his valuable assistance. We also acknowledge François Carrier from the Office of International Research and Experiential Learning for facilitating additional allocations for NSERC USRA applications. Finally, we thank Andrew Ochalski, Manager of the Research Imaging Facility, and Chloë van Oostende-Triplet, Core Manager and Senior Microscopy Specialist at the University of Ottawa, for their assistance in obtaining early preliminary imaging data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. ChatGPT was used to rephrase select sentences to suggest options for improving grammar and readability, only.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsybi.2025.1619871/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Additional confocal images showing subcellular localization profiles of mScarlet expressed via pTARGEX constructs in N. benthamiana. Supplementary confocal laser scanning microscopy images corresponding to Figure 2, depicting mScarlet localization patterns for each pTARGEX vector in infiltrated N. benthamiana tissues. Scale bars: 20 µm.
References
Bally, J., Jung, H., Mortimer, C., Naim, F., Philips, J. G., Hellens, R., et al. (2018). The rise and rise of nicotiana benthamiana: a plant for all reasons. Annu. Rev. Phytopathol. 56, 405–426. doi:10.1146/annurev-phyto-080417-050141
Bédard, J., and Jarvis, P. (2005). Recognition and envelope translocation of chloroplast preproteins. J. Exp. Bot. 56, 2287–2320. doi:10.1093/jxb/eri243
Benchabane, M., Goulet, C., Rivard, D., Faye, L., Gomord, V., and Michaud, D. (2008). Preventing unintended proteolysis in plant protein biofactories. Plant Biotechnol. J. 6, 633–648. doi:10.1111/j.1467-7652.2008.00344.x
Bharathi, J. K., Suresh, P., Prakash, M. A. S., and Muneer, S. (2024). Exploring recent progress of molecular farming for therapeutic and recombinant molecules in plant systems. Heliyon 10, e37634. doi:10.1016/j.heliyon.2024.e37634
Bindels, D. S., Haarbosch, L., Van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., et al. (2016). MScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56. doi:10.1038/nmeth.4074
Bouwman, K. M., Tomris, I., Turner, H. L., van der Woude, R., Shamorkina, T. M., Bosman, G. P., et al. (2021). Multimerization- and glycosylation-dependent receptor binding of SARS-CoV-2 spike proteins. PLoS Pathog. 17, e1009282. doi:10.1371/JOURNAL.PPAT.1009282
Carlsson, M. L. R., Kristiansson, A., Bergwik, J., Kanagarajan, S., Bülow, L., Åkerström, B., et al. (2020). Expression, purification and initial characterization of functional α1-Microglobulin (A1M) in nicotiana benthamiana. Front. Plant Sci. 11, 593773. doi:10.3389/fpls.2020.593773
Chin-Fatt, A., Saberianfar, R., and Menassa, R. (2021). A rationally designed bovine IgA Fc scaffold enhances in planta accumulation of a VHH-Fc fusion without compromising binding to enterohemorrhagic E. coli. Front. Plant Sci. 12, 651262. doi:10.3389/fpls.2021.651262
Conley, A. J., Mohib, K., Jevnikar, A. M., and Brandle, J. E. (2009). Plant recombinant erythropoietin attenuates inflammatory kidney cell injury. Plant Biotechnol. J. 7, 183–199. doi:10.1111/j.1467-7652.2008.00389.x
Dammen-Brower, K., Epler, P., Zhu, S., Bernstein, Z. J., Stabach, P. R., Braddock, D. T., et al. (2022). Strategies for glycoengineering therapeutic proteins. Front. Chem. 10, 863118. doi:10.3389/fchem.2022.863118
Demone, J., Maltseva, M., Nourimand, M., Nasr-Sharif, M., Galipeau, Y., Alarcon, E. I., et al. (2022). Scalable agroinfiltration-based production of SARS-CoV-2 antigens for use in diagnostic assays and subunit vaccines, PLOS ONE, 17 (12), e0277668. doi:10.1371/journal.pone.0277668
Denecke, J., Goldman, M. H. S., Demolder, J., Seurinck, J., and Botterman, J. (1991). The tobacco luminal binding protein is encoded by a multigene family. Plant Cell 3, 1025–1035. doi:10.1105/tpc.3.9.1025
Drake, P. M. W., Barbi, T., Sexton, A., McGowan, E., Stadlmann, J., Navarre, C., et al. (2009). Development of rhizosecretion as a production system for recombinant proteins from hydroponic cultivated tobacco. FASEB J. 23, 3581–3589. doi:10.1096/fj.09-131771
Eidenberger, L., Kogelmann, B., and Steinkellner, H. (2023). Plant-based biopharmaceutical engineering. Nat. Rev. Bioeng. 1, 426–439. doi:10.1038/s44222-023-00044-6
Göritzer, K., Grandits, M., Grünwald-Gruber, C., Figl, R., Mercx, S., Navarre, C., et al. (2022). Engineering the N-glycosylation pathway of Nicotiana tabacum for molecular pharming using CRISPR/Cas9. Front. Plant Sci. 13, 1003065. doi:10.3389/fpls.2022.1003065
Hager, K. J., Pérez Marc, G., Gobeil, P., Diaz, R. S., Heizer, G., Llapur, C., et al. (2022). Efficacy and safety of a recombinant plant-based adjuvanted Covid-19 vaccine. N. Engl. J. Med. 386, 2084–2096. doi:10.1056/nejmoa2201300
Hanania, U., Ariel, T., Tekoah, Y., Fux, L., Sheva, M., Gubbay, Y., et al. (2017). Establishment of a tobacco BY2 cell line devoid of plant-specific xylose and fucose as a platform for the production of biotherapeutic proteins. Plant Biotechnol. J. 15, 1120–1129. doi:10.1111/pbi.12702
Harding, M. D., and Sainsbury, F. (2025). Protein engineering strategies to optimise recombinant product synthesis and accumulation in Nicotiana benthamiana. Plant Biotechnol. J. doi:10.1111/pbi.70232
Hehle, V. K., Paul, M. J., Drake, P. M., Ma, J. K. C., and van Dolleweerd, C. J. (2011). Antibody degradation in tobacco plants: a predominantly apoplastic process. BMC Biotechnol. 11, 128. doi:10.1186/1472-6750-11-128
Herman, X., Far, J., Courtoy, A., Bouhon, L., Quinton, L., De Pauw, E., et al. (2021). Inactivation of N-Acetylglucosaminyltransferase I and α1,3-Fucosyltransferase genes in nicotiana tabacum BY-2 cells results in glycoproteins with highly homogeneous, high-mannose N-glycans. Front. Plant Sci. 12, 634023. doi:10.3389/fpls.2021.634023
Huang, H. C., Lai, Y. J., Liao, C. C., Yang, W. F., Huang, K. B., Lee, I. J., et al. (2021). Targeting conserved N-glycosylation blocks SARS-CoV-2 variant infection in vitro. EBioMedicine 74, 103712. doi:10.1016/j.ebiom.2021.103712
Ivanov, S., and Harrison, M. J. (2014). A set of fluorescent protein-based markers expressed from constitutive and arbuscular mycorrhiza-inducible promoters to label organelles, membranes and cytoskeletal elements in Medicago truncatula. Plant J. 80, 1151–1163. doi:10.1111/tpj.12706
Ives, C. M., Nguyen, L., Fogarty, C. A., Harbison, A. M., Durocher, Y., Klassen, J., et al. (2024). Role of N343 glycosylation on the SARS-CoV-2 S RBD structure and co-receptor binding across variants of concern. Elife 13, RP95708. doi:10.7554/eLife.95708
Jansing, J., Sack, M., Augustine, S. M., Fischer, R., and Bortesi, L. (2019). CRISPR/Cas9-mediated knockout of six glycosyltransferase genes in Nicotiana benthamiana for the production of recombinant proteins lacking β-1,2-xylose and core α-1,3-fucose. Plant Biotechnol. J. 17, 350–361. doi:10.1111/pbi.12981
Kaldis, A., Uddin, M. S., Guluarte, J. O., Martin, C., Alexander, T. W., and Menassa, R. (2023). Development of a plant-based oral vaccine candidate against the bovine respiratory pathogen Mannheimia haemolytica. Front. Plant Sci. 14, 1251046. doi:10.3389/fpls.2023.1251046
Khosla, A., Rodriguez-Furlan, C., Kapoor, S., Van Norman, J. M., and Nelson, D. C. (2020). A series of dual-reporter vectors for ratiometric analysis of protein abundance in plants. Plant Direct 4, e00231. doi:10.1002/pld3.231
Kogelmann, B., Melnik, S., Bogner, M., Kallolimath, S., Stöger, E., Sun, L., et al. (2024). A genome-edited N. benthamiana line for industrial-scale production of recombinant glycoproteins with targeted N-glycosylation. Biotechnol. J. 19, e2300323. doi:10.1002/biot.202300323
Kuntz, M., Simons, A., Schell, J., and Schreier, P. H. (1986). Targeting of protein to chloroplasts in transgenic tobacco by fusion to mutated transit peptide. MGG Mol. General Genet. 205, 454–460. doi:10.1007/BF00338082
Liénard, D., Sourrouille, C., Gomord, V., and Faye, L. (2007). Pharming and transgenic plants. Biotechnol. Annu. Rev. 13, 115–147. doi:10.1016/S1387-2656(07)13006-4
Liu, H., and Timko, M. P. (2022). Improving protein quantity and quality-the next level of plant molecular farming. Int. J. Mol. Sci. 23, 1326. doi:10.3390/ijms23031326
Lomonossoff, G. P., and D’Aoust, M. A. (2016). Plant-produced biopharmaceuticals: a case of technical developments driving clinical deployment. Sci. (1979) 353, 1237–1240. doi:10.1126/science.aaf6638
Lonoce, C., Salem, R., Marusic, C., Jutras, P. V., Scaloni, A., Salzano, A. M., et al. (2016). Production of a tumour-targeting antibody with a human-compatible glycosylation profile in N. benthamiana hairy root cultures. Biotechnol. J. 11, 1209–1220. doi:10.1002/biot.201500628
Lund, P., and Dunsmuir, P. (1992). A plant signal sequence enhances the secretion of bacterial ChiA in transgenic tobacco. Plant Mol. Biol. 18, 47–53. doi:10.1007/BF00018455
Ma, L., Lukasik, E., Gawehns, F., and Takken, F. L. W. (2012). The use of agroinfiltration for transient expression of plant resistance and fungal effector proteins in nicotiana benthamiana leaves. Methods Mol. Biol. 835, 61–74. doi:10.1007/978-1-61779-501-5_4
Ma, T., Li, Z., and Wang, S. (2019). Production of bioactive recombinant reteplase by virus-based transient expression system in nicotiana benthamiana. Front. Plant Sci. 10, 1225. doi:10.3389/fpls.2019.01225
Madeira, L. M., Szeto, T. H., Henquet, M., Raven, N., Runions, J., Huddleston, J., et al. (2016a). High-yield production of a human monoclonal IgG by rhizosecretion in hydroponic tobacco cultures. Plant Biotechnol. J. 14, 615–624. doi:10.1111/pbi.12407
Madeira, L. M., Szeto, T. H., Ma, J. K. C., and Drake, P. M. W. (2016b). Rhizosecretion improves the production of Cyanovirin-N in Nicotiana tabacum through simplified downstream processing. Biotechnol. J. 11, 910–919. doi:10.1002/biot.201500371
Margolin, E. A., Strasser, R., Chapman, R., Williamson, A. L., Rybicki, E. P., and Meyers, A. E. (2020). Engineering the plant secretory pathway for the production of next-generation pharmaceuticals. Trends Biotechnol. 38, 1034–1044. doi:10.1016/j.tibtech.2020.03.004
Marin Viegas, V. S., Ocampo, C. G., and Petruccelli, S. (2017). Vacuolar deposition of recombinant proteins in plant vegetative organs as a strategy to increase yields. Bioengineered 8, 203–211. doi:10.1080/21655979.2016.1222994
Menassa, R., Zhu, H., Karatzas, C. N., Lazaris, A., Richman, A., and Brandle, J. (2004). Spider dragline silk proteins in transgenic tobacco leaves: accumulation and field production. Plant Biotechnol. J. 2, 431–438. doi:10.1111/j.1467-7652.2004.00087.x
Mor, T. S. (2015). Molecular pharming’s foot in the FDA’s door: Protalix’s trailblazing story. Biotechnol. Lett. 37, 2147–2150. doi:10.1007/s10529-015-1908-z
Mulangu, S., Dodd, L. E., Davey, R. T., Tshiani Mbaya, O., Proschan, M., Mukadi, D., et al. (2019). A randomized, controlled trial of Ebola virus disease therapeutics. N. Engl. J. Med. 381, 2293–2303. doi:10.1056/nejmoa1910993
Neuhaus, J. M., Sticher, L., Meins, F., and Boller, T. (1991). A short C-terminal sequence is necessary and sufficient for the targeting of chitinases to the plant vacuole. Proc. Natl. Acad. Sci. U. S. A. 88, 10362–10366. doi:10.1073/pnas.88.22.10362
Pêra, F. F. P. G., Mutepfa, D. L. R., Khan, A. M., Els, J. H., Mbewana, S., van Dijk, A. A. A., et al. (2015). Engineering and expression of a human rotavirus candidate vaccine in Nicotiana benthamiana. Virol. J. 12, 205. doi:10.1186/s12985-015-0436-8
Pereira, E. O., Kolotilin, I., Conley, A. J., and Menassa, R. (2014). Production and characterization of in planta transiently produced polygalacturanase from Aspergillus niger and its fusions with hydrophobin or ELP tags. BMC Biotechnol. 14, 59. doi:10.1186/1472-6750-14-59
Peyret, H., Brown, J. K. M., and Lomonossoff, G. P. (2019). Improving plant transient expression through the rational design of synthetic 5′ and 3′ untranslated regions. Plant Methods 15, 108. doi:10.1186/s13007-019-0494-9
Sainsbury, F., and Lomonossoff, G. P. (2008). Extremely high-level and rapid transient protein production in plants without the use of viral replication. Plant Physiol. 148, 1212–1218. doi:10.1104/pp.108.126284
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi:10.1038/nmeth.2019
Schlegel, S., Rujas, E., Ytterberg, A. J., Zubarev, R. A., Luirink, J., and de Gier, J. W. (2013). Optimizing heterologous protein production in the periplasm of E. coli by regulating gene expression levels. Microb. Cell Fact. 12, 24. doi:10.1186/1475-2859-12-24
Schouten, A., Rossien, J., Van Engelen, F. A., De Jong, G. A., Borst-Vrenssen, A. W., Zilverentant, J. F., et al. (1996). The C-terminal KDEL sequence increases the expression level of a single-chain antibody designed to be targeted to both the cytosol and the secretory pathway in transgenic tobacco. Plant Mol. Biol. 30, 781–793. doi:10.1007/BF00019011
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., et al. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224. doi:10.1038/s41586-020-2179-y
Shin, Y.-J., König-Beihammer, J., Vavra, U., Schwestka, J., Kienzl, N. F., Klausberger, M., et al. (2021). N-glycosylation of the SARS-CoV-2 receptor-binding domain is important for functional expression in plants. Front. Plant Sci. 12, 689104. doi:10.3389/fpls.2021.689104
Spiegel, H., Schillberg, S., Sack, M., Holzem, A., Nähring, J., Monecke, M., et al. (1999). Accumulation of antibody fusion proteins in the cytoplasm and ER of plant cells. Plant Sci. 149, 63–71. doi:10.1016/S0168-9452(99)00145-4
Thagun, C., Odahara, M., Kodama, Y., and Numata, K. (2024). Identification of a highly efficient chloroplast-targeting peptide for plastid engineering. PLoS Biol. 22, e3002785. doi:10.1371/journal.pbio.3002785
Trusova, S. V., Teplova, A. D., Golyshev, S. A., Galiullina, R. A., Morozova, E. A., Chichkova, N. V., et al. (2019). Clathrin-mediated endocytosis delivers proteolytically active phytaspases into plant cells. Front. Plant Sci. 10, 873. doi:10.3389/fpls.2019.00873
VanderBurgt, J. T., Harper, O., Garnham, C. P., Kohalmi, S. E., and Menassa, R. (2023). Plant production of a virus-like particle-based vaccine candidate against porcine reproductive and respiratory syndrome. Front. Plant Sci. 14, 1044675. doi:10.3389/fpls.2023.1044675
Ward, B. J., Makarkov, A., Séguin, A., Pillet, S., Trépanier, S., Dhaliwall, J., et al. (2020). Efficacy, immunogenicity, and safety of a plant-derived, quadrivalent, virus-like particle influenza vaccine in adults (18–64 years) and older adults (≥65 years): two multicentre, randomised phase 3 trials. Lancet 396, 1491–1503. doi:10.1016/S0140-6736(20)32014-6
Yanez, R. J. R., Lamprecht, R., Granadillo, M., Weber, B., Torrens, I., Rybicki, E. P., et al. (2017). Expression optimization of a cell membrane-penetrating human papillomavirus type 16 therapeutic vaccine candidate in Nicotiana benthamiana. PLoS One 12, e0183177. doi:10.1371/journal.pone.0183177
Keywords: biopharming, gibson assembly, golden gate, transgenic plants, molecular farming, transient expression, N. benthamiana
Citation: Chartrand-Pleau C, Gallipeau-Burns E, Jain A, Boddy V, Fakhouri P, Rehmani S, Thomas T, Drapeau M, VanderBurgt JT, Boudigou A, Damry AM and MacLean AM (2025) pTARGEX vectors: a versatile toolbox for plant-based protein expression. Front. Synth. Biol. 3:1619871. doi: 10.3389/fsybi.2025.1619871
Received: 28 April 2025; Accepted: 15 August 2025;
Published: 28 August 2025.
Edited by:
James Jordan Steel, United States Air Force Academy, United StatesReviewed by:
Simon Scofield, Cardiff University, United KingdomRyan Coates, Cardiff University, United Kingdom
Copyright © 2025 Chartrand-Pleau, Gallipeau-Burns, Jain, Boddy, Fakhouri, Rehmani, Thomas, Drapeau, VanderBurgt, Boudigou, Damry and MacLean. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Allyson M. MacLean, YW1hY2xlYTNAdW90dGF3YS5jYQ==