 Shoichi Sakaguchi
Shoichi Sakaguchi Takashi Nakano1
Takashi Nakano1 So Nakagawa
So Nakagawa- 1Department of Microbiology and Infection Control, Faculty of Medicine, Osaka Medical and Pharmaceutical University, Osaka, Japan
- 2Department of Molecular Life Science, Tokai University School of Medicine, Isehara, Japan
- 3Division of Interdisciplinary Merging of Health Research, Micro/Nano Technology Center, Tokai University, Hiratsuka, Japan
- 4Division of Genome Sciences, Institute of Medical Sciences, Tokai University, Isehara, Japan
RNA-dependent RNA polymerase (RdRp) is a marker gene for RNA viruses; thus, it is widely used to identify RNA viruses from metatranscriptome data. However, because of the high diversity of RdRp domains, it remains difficult to identify RNA viruses using RdRp sequences. To overcome this problem, we created a NeoRdRp database containing 1,182 hidden Markov model (HMM) profiles utilizing 12,502 RdRp domain sequences. Since the development of this database, more RNA viruses have been discovered, mainly through metatranscriptome sequencing analyses. To identify RNA viruses comprehensively and specifically, we updated the NeoRdRp by incorporating recently reported RNA viruses. To this end, 557,197 RdRp-containing sequences were used as seed RdRp datasets. These sequences were processed through deduplication, clustering, alignment, and splitting, thereby generating 19,394 HMM profiles. We validated the updated NeoRdRp database, using the UniProtKB dataset and found that the recall and specificity rates were improved to 99.4% and 81.6%, from 97.2% and 76.8% in the previous version, respectively. Comparisons of eight different RdRp search tools showed that NeoRdRp2 exhibited balanced RdRp and nonspecific detection power. Expansion of the annotated RdRp datasets is expected to further accelerate the discovery of novel RNA viruses from various transcriptome datasets. The HMM profiles of NeoRdRp2 and their annotations are available at https://github.com/shoichisakaguchi/NeoRdRp.
1 Introduction
RNA viruses play pivotal roles in various environments and influence several ecosystems and organisms including humans, animals, and plants (1). Studies on RNA virus detection using metatranscriptome data have emerged as powerful tools for obtaining insights into both known and novel RNA viruses in samples (2). RNA-dependent RNA polymerase (RdRp) is a universal gene found in almost all RNA viruses except retroviruses and deltaviruses. RdRp sequences are commonly used to search for various RNA viruses from metatranscriptome data; however, because of the great diversity of RdRps, finding various RNA viruses based on homology-based searches using RdRp sequences is known to be difficult (2). To overcome this problem, we developed a bioinformatics pipeline to generate hidden Markov model (HMM) profiles from 12,502 RdRp domain sequences. We shared 1,182 of these RdRp HMM profiles as NeoRdRp 1.1, along with the associated RdRp domain sequences and RdRp domain sequences, and the bioinformatics pipeline at NeoRdRp (3): https://github.com/shoichisakaguchi/NeoRdRp. NeoRdRp was used in various RNA virus identification studies (4–6).
Around the time we reported on the NeoRdRp database, large-scale metatranscriptome studies identified a large number of undetected RNA viruses (7–10). Each study reported various novel RNA viruses containing diverse RdRps. Thus, by incorporating those RdRp sequences into the NeoRdRp database, further undetermined RNA viruses could be searched. Various bioinformatic tools targeting RdRp for RNA virus identification have been developed, including LucaProt (11), Palmscan (12), RdRpBin (13), RdRp-scan (14), Serratus Lite (8), and ViralRdRp_pHMMs (15). Additionally, RVDB-prot (16) and VirSorter2 (17) have been used to identify the RNA viruses. However, the performances of these programs for RNA virus detection have not yet been compared.
This study aimed to update the NeoRdRp by including the RdRp sequences of recently reported RNA viruses to enhance the ability of the model to identify RNA viruses from metatranscriptome data. To this end, we modified our bioinformatics pipeline and processed and annotated 557,197 amino acid sequences containing RdRp domains. Consequently, 19,394 HMM profiles of the RdRp domains were generated and named as NeoRdRp2. We evaluated the RNA detection performance of NeoRdRp2, a previous version of NeoRdRp (version 1.1), and other RNA virome bioinformatics tools. All datasets and annotations for NeoRdRp2 are available at https://github.com/shoichisakaguchi/NeoRdRp.
2 Materials and methods
2.1 Datasets
The following four RdRp sequence datasets were used: Wolf2018 (1), Zayed2022 (10), Edgar2022 (8), and Neri2022 (9). Wolf2018 consists of 4,620 RdRp sequences extracted from RNA viruses and unclassified viral sequences registered in GenBank as of April 2017 using PSI-BLAST (18) and iterative clustering and alignment. Zayed2022 is an RdRp sequence dataset from marine RNA metatranscriptomic data, consisting of 209,588 RdRp sequences, 6,238 of which are near-full-length RdRps. From Edgar2022, we employed two datasets: the rdrp1 dataset consisting of 14,680 annotated and non-annotated RdRp sequences from GenBank, and the Serratus dataset consisting of 250,799 recently detected RdRps stored in the Serratus database (https://serratus.io/). Neri2022 contains 77,510 RdRp sequences obtained from metagenome datasets registered in IMG/M [https://img.jgi.doe.gov/, (9)]. Additionally, we conducted an HMM search against the NCBI RNA Virus database and obtained 7,896 RdRp domains. A total of 565,093 amino acid sequences containing the RdRp domain were used in this study.
The UniProtKB database, known for its curated protein sequences and high reliability, was used to evaluate RdRp searches (19). This database contains 565,254 protein sequences and was downloaded on October 11, 2021, from https://www.uniprot.org/uniprotkb. The sequences in this dataset were obtained from multiple organisms and included 836 RdRps (3). In addition to the UniProtKB database, we utilized a metatranscriptomic assembly dataset obtained using a fragmented and primer-ligated double-stranded RNA (dsRNA) sequencing (FLDS) method, named “FLDS-data” (20). This dataset was derived from marine samples and comprised 228 RdRp domains and 20 capsids in 1,143 assembled transcripts. This dataset was used to assess the efficacy of various tools for accurately detecting RdRp sequences, inadvertently identifying capsid proteins, and probing potentially unidentified RdRps. This dataset contains appropriately annotated data and is a suitable evaluation platform.
2.2 Construction of RdRp HMM profiles
The bioinformatics programs for HMM construction are summarized in Table 1, and the procedure is illustrated in Figure 1. To construct the RdRp HMM profile, amino acid sequences of the five RdRp datasets were merged and clustered using CD-HIT with a 99% threshold and word size of 5 to exclude sequences that exist redundantly within the datasets. This process was also aimed at removing sequences in which part of one sequence was contained entirely within a longer sequence. After clustering, a representative sequence from each cluster was retained as a deduplicated RdRp dataset. Based on the deduplicated RdRp dataset, HMM profiles of the RdRp domains were constructed as shown in Figure 1, which was modified from the procedure used in our previous study (3). The deduplicated RdRp seed datasets were clustered using CD-HIT; parameters for CD-HIT were tested with 40% to 60% similarity (Supplementary Table 1). Clusters containing more than three sequences were aligned using MAFFT L-INS-i. Following the alignment, we employed our script “cutgap.py” (available at https://github.com/shoichisakaguchi/NeoRdRp/blob/main/script/archive/v2.0/cutgap.py) to delineate and split the sequences based on the gappy regions. Specifically, the script scrutinized each aligned sequence column and identified regions where the gap occurrence rate was greater than 25%, which was decided by testing three different gap occurrence rates: 15%, 25%, and 35% (Supplementary Table 2). Consecutive regions with more than 25% gaps spanning eight or more alignment positions, which were decided by comparing six different spanning lengths (2, 4, 6, 8, 10, and 12), were identified as potential split points (Supplementary Table 1). After identifying these gappy regions, the sequences were split and saved in distinct FASTA files, with file names reflecting gap thresholds and split positions. Finally, HMM profiles were created for each conserved domain using the HMMER hmmbulid program. The constructed HMM profiles were designated NeoRdRp2.
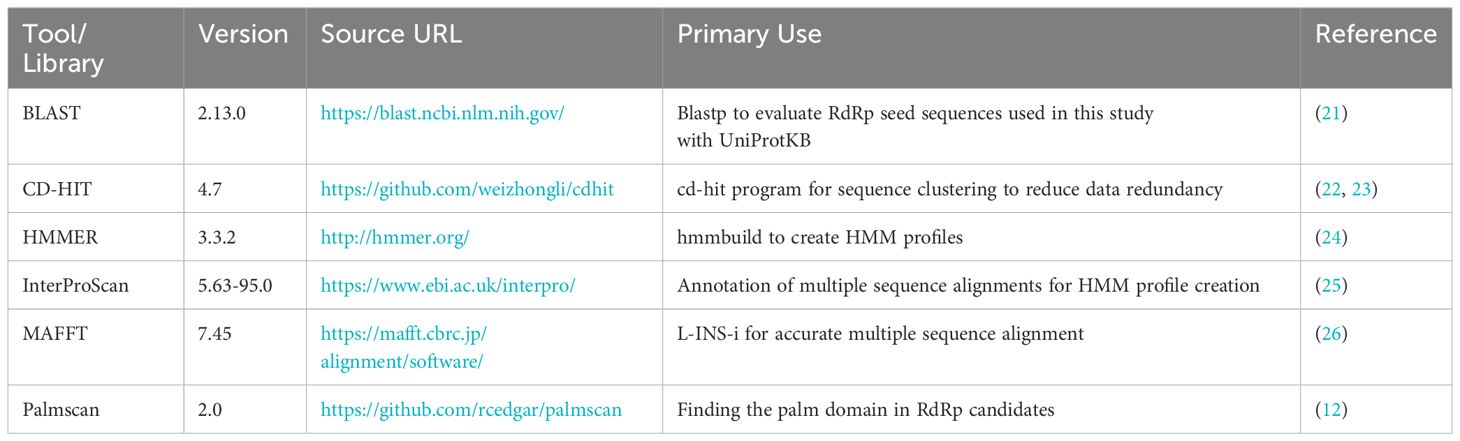
Table 1 Bioinformatics program for constructing HMM.
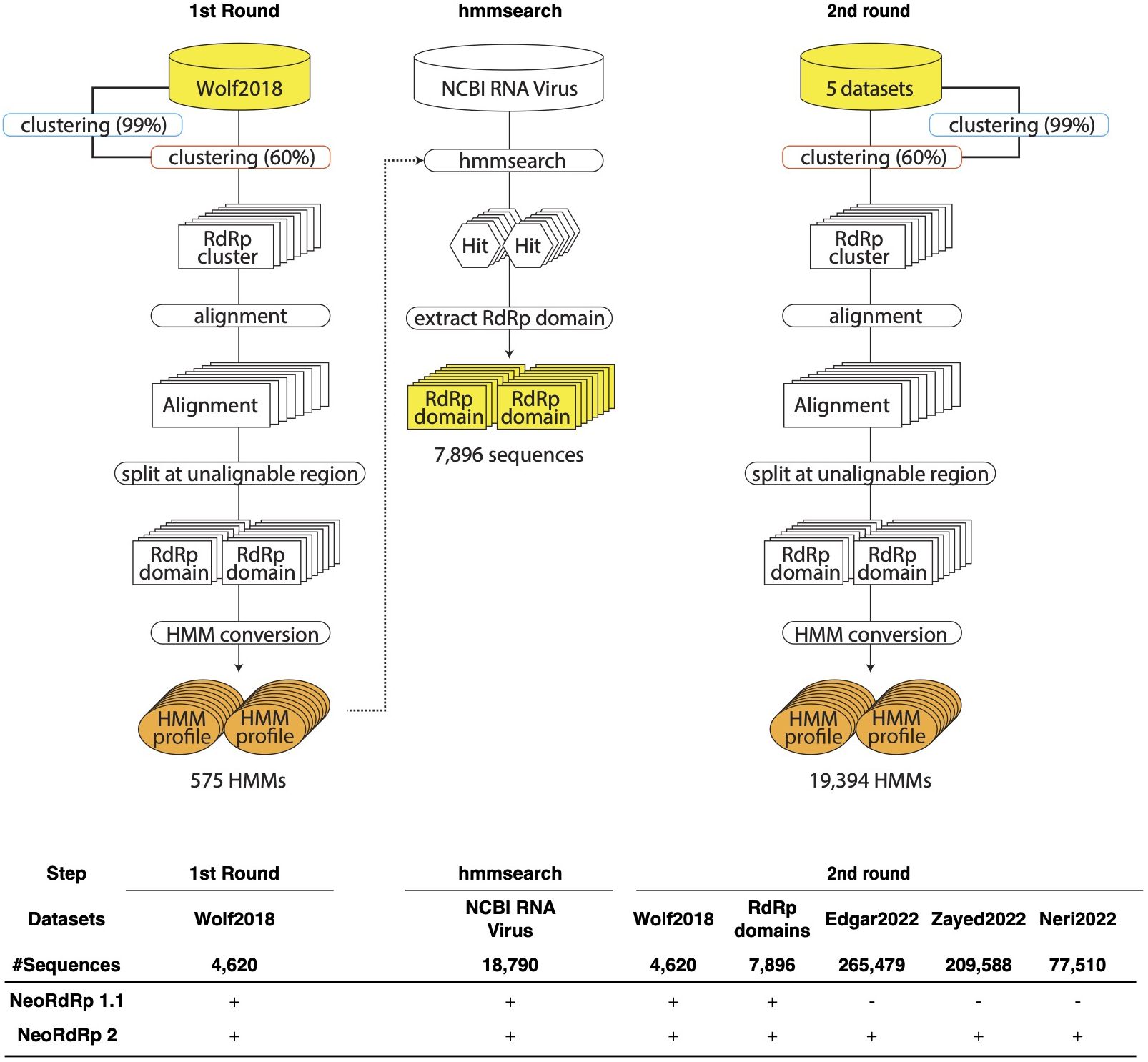
Figure 1 Schematic illustration of the pipelines for the construction of RNA-dependent RNA polymerase (RdRp) hidden Markov model (HMM) profiles. In the first round, Wolf2018 was inputted as the seed RdRp into this pipeline. The resulting orange HMM profiles were used to hmmsearch for NCBI RNA Virus dataset. In the second round, Wolf2018, the RdRp domains obtained by hmmsearch, and the recently acquired RdRp domain data were merged with the initial RdRp seed datasets and re-input into the pipeline. The resulting HMM profiles were consolidated into a single HMM profile database, referred to as NeoRdRp2. In the bottom, input datasets used for NeoRdRp 1.1 and 2 were shown for each step.
2.3 Evaluation of RdRp HMM profiles and RdRp detection tools
To demonstrate the advancements in the RdRp detection capabilities of NeoRdRp2 over 1.1, searches against the UniProtKB database using HMM and BLASTP were conducted, with a threshold E-value of ≤1E-10 and default parameters, respectively. Subsequently, to benchmark our HMM profiles for identifying RdRp sequences, the following HMM profiles were employed: RVDB-prot (version 26.0, https://rvdb-prot.pasteur.fr/) (16), RdRp-scan (version 0.90, https://github.com/JustineCharon/RdRp-scan) (14), and ViralRdRp_pHMMs (version 1.0.1, https://github.com/ingridole/ViralRdRp_pHMMs) (15). An HMMER hmmsearch with an e-value of 1E-10 (i.e., -E 1E-10) was applied for each search. In addition, the RdRp detection powers of LucaProt, RdRpBin, Serratus (Serratus Lite), Palmscan, and VirSorter2 were examined (Table 2). Notably, RVDB-prot and VirSorter2 contain not only RdRps but also other various viral proteins.
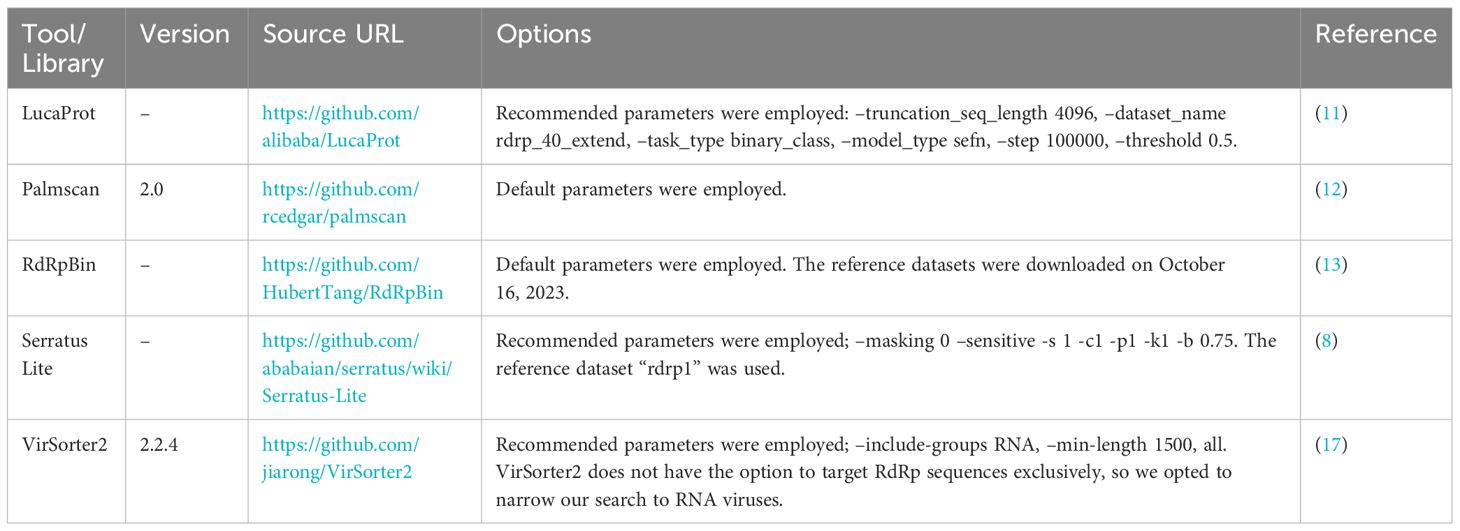
Table 2 Bioinformatic database program used for RdRp detection.
2.4 Re-annotation of seed RdRp datasets
We conducted a comprehensive reannotation of the RdRp seed datasets used for NeoRdRp2 to enable NeoRdRp users to evaluate their search results. Specifically, we used InterProScan, a tool that aggregates data from various databases to provide detailed insights into protein domains and functional annotations. The datasets were subjected to searches against the following databases: Conserved Domain Database (CDD) for identifying conserved domains; Coils for predicting coiled-coil regions; FunFam for classifying proteins into functional families; Gene3D for categorizing proteins based on their 3D structures; Pfam for protein family classification; ProSiteProfiles for domain and key site identification; SUPERFAMILY for superfamily-based classification; MobiDBLite for predicting protein mobility regions; PRINTS for protein fingerprint classification; PANTHER for protein family and subfamily classification; SMART for domain identification and annotation; ProSitePatterns for identifying protein sequence patterns and motifs; NCBIfam, PIRSF, Hamap, and AntiFam for further classification and annotation based on protein families, phylogeny, and false predictions filtering. RdRp-scan was conducted with default parameters for sequence similarity searches to identify the characteristics and origin of the RNA viruses. Additionally, we conducted Palmscan and BLASTp searches with an e-value of ≤1E-10 and default parameters using each RdRp seed dataset sequence as a query against the UniProtKB database.
3 Results
3.1 RdRp HMM profiles
For the construction of our RdRp-HMM (Figure 1), the first stringent clustering, indicated by the blue curved rectangle in Figure 1, yielded a refined dataset comprising 328,977 unique sequences from the original pool of 565,093 sequences. Secondly, a relatively loose clustering approach (orange curved rectangle in Figure 1) grouped the unique sequences into 68,118 clusters, revealing a diverse spectrum of RdRp sequences. The large number of RdRp clusters indicates the extensive diversity of the RNA virus families. Multiple sequence alignments were performed for each cluster containing more than three sequences, using MAFFT. Subsequently, consecutive regions with more than 25% gaps spanning eight or more alignment positions were identified and removed, and the sequence was split at these regions. Finally, 17,452 clusters were obtained and an RdRp HMM profile was generated for each cluster using the HMMER suite hmmbulid program. This set of 19,394 HMM profiles was named NeoRdRp2.
3.2 Evaluation of NeoRdRp2 using UniProtKB
To evaluate the performance of NeoRdRp2, we used the same database, program, and parameters as previously used to assess NeoRdRp 1.1 (3). To this end, the UniProtKB database with hmmsearch and BLASTp with an e-value of 1E-10 was analyzed using NeoRdRp 1.1 and 2 datasets. The results, including the number of amino acid sequences and HMM profiles, are summarized in Table 3. NeoRdRp2 detected 831 out of 836 RdRps in the UniProtKB database, improving the recall rate (99.4% from 97.2%) compared to that of NeoRdRp 1.1. NeoRdRp2 detected 12 genera, which NeoRdRp 1.1 could not detect, and failed to detect three genera, which 1.1 detected (Supplementary Table 3). As a result of the BLASTp search using the NeoRdRp2 seed sequences, the recall and precision rates were 99.9% and 11.5%, respectively. Three genera, Livivirus, Emvecovirus, and Orthopenumovirus, were not detected by hmmsearch using NeoRdRp2 but were detected by BLASTp. Of the 564,418 non-RdRp sequences, 188 were incorrectly identified as RdRp-containing sequences. The precision rate of NeoRdRp2 was 81.6%, which was also improved compared to that of NeoRdRp 1.1 (76.8%). These results indicate that increasing the number of RdRp sequences improves the accuracy of the RdRp search.
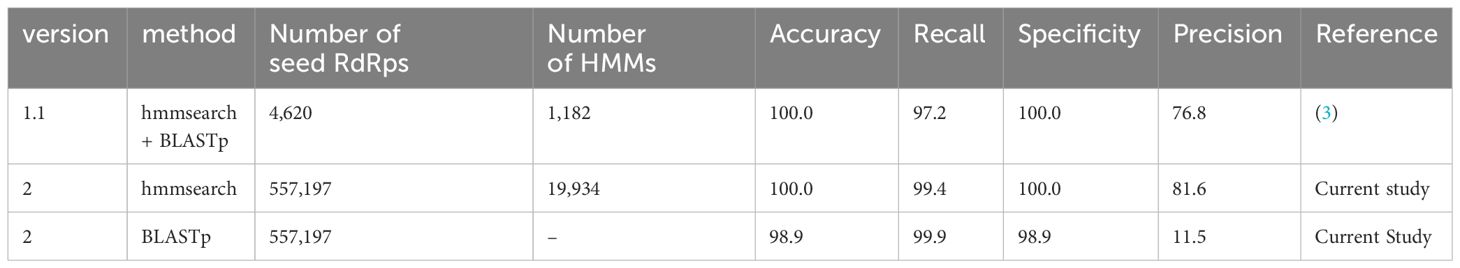
Table 3 Statistics of NeoRdRp detection power estimated using UniProtKB.
3.3 Comparison of RdRp identification
We next evaluated NeoRdRp2, using the FLDS-data containing 228 RdRps, 20 capsids, and 895 unannotated amino acid sequences (20), which were used in our previous analysis (3). For comparison, we used other RdRp and RNA virus detection tools, including RVDB-prot, RdRp-scan, ViralRdRp_pHMMs, LucaProt, Serratus, Palmscan, RdRpBin, and VirSorter2. See Materials and Methods for further details. Using these HMM models and RdRp and RNA virus detection programs, the total number of identified RdRp sequences, false positives in capsid protein identification, and detection of unannotated sequences were assessed (Table 4). Among the 228 RdRp-annotated FLDS sequences, NeoRdRp2 showed the highest count of RdRp detection with 216 sequences, closely followed by LucaProt with 212 sequences. RdRp-scan and RVDB-prot also exhibited substantial RdRp detection, with 201 and 199 sequences, respectively (Figure 2A). On the lower end, VirSorter2 identified 67 RdRp sequences, indicating relatively low sensitivity for RdRp detection. Notably, there were cases in which other tools detected RdRp, whereas NeoRdRp2 did not (Supplementary Table 4). In the HMM searches, there were ten sequences that NeoRdRp2 could not detect, but RdRp-scan and/or RVDB-prot detected two sequences. One RdRp sequence was only detected by non-HMM-based searches for Serratus and RdRpBin. Interestingly, the ML-based RdRpBin alone detected eight RdRps. The taxonomy of the RNA viruses that could not be detected by NeoRdRp2 was as follows: three of 24 Narnaviridae, one of 22 Partitiviridae, and one of 3 Endornaviridae.

Table 4 RdRp search against FLDS data.
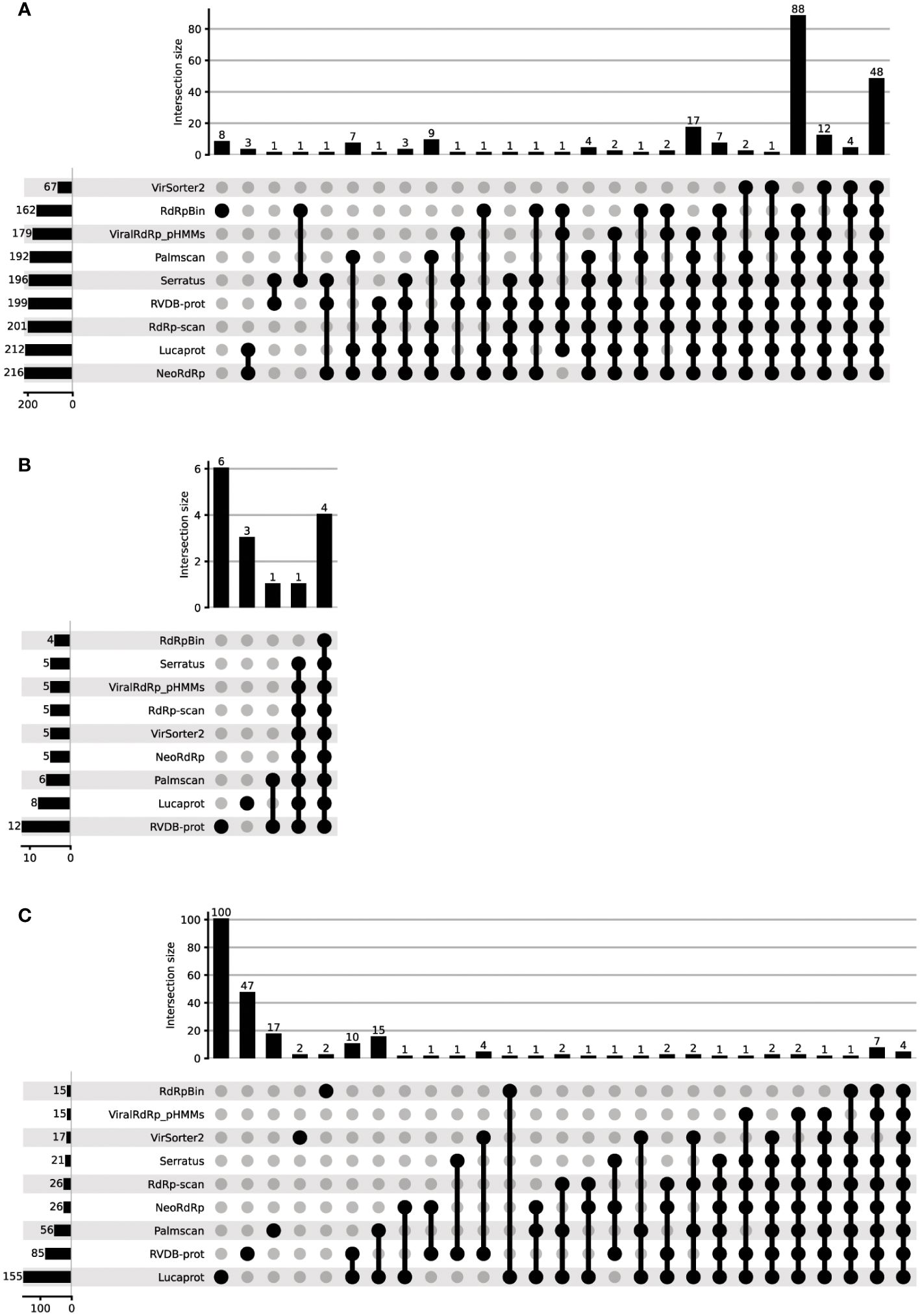
Figure 2 The performance of various RdRp detection tools in detecting the unannotated FLDS data. The performance of various RdRp detection tools was evaluated using FLDS data containing 228 RdRps, 20 capsids, and 895 unannotated amino acid sequences. (A) RdRp, (B) capsid, and (C) non-RdRp detection. The detection ability of each tool was plotted to evaluate its ability to identify RdRp sequences.
Among the 20 capsid FLDS sequences, a few possible misidentifications were found using all the tools (Table 4; Figure 2B). RVDB-prot had the highest number of capsid identifications (12 sequences), followed by LucaProt with eight sequences. Note that RVDB-prot contains the HMM profiles of all RNA viral proteins, not just RdRp (16). In contrast, the lowest number (4) was found using RdRpBin, followed by 5 with NeoRdRp2, VirSorter2, RdRp-scan, ViralRdRp_pHMMs, and Serratus Lite.
The detection of unannotated sequences varied significantly among the tools (Table 4). LucaProt identified a notably large number of unannotated sequences (155). The fourth largest number was 26, for both NeoRdRp2 and RdRp-scan. We compared the overlapping numbers of the detected sequences using different tools (Figure 2C). Multiple tools detected 62 sequences: four sequences were detected by all tools used in this study, whereas 168 sequences were detected using only one tool. The machine-learning-based searches Lucaprot and RdRpBin uniquely detected 100 and 2 unannotated datasets, respectively. In contrast, NeoRdRp2 and RdRp-scan did not detect any RdRp sequences that were annotated only by these tools, although they did not entirely overlap (i.e., 22 of 26 overlapped). To provide a fair comparison, it is important to note that both RVDB-prot and VirSorter2 include HMM profiles for various viral proteins, in addition to RdRp. Consequently, the detection numbers reported in these databases may reflect a broader range of viral protein identifications.
3.4 Re-annotation of seed RdRp datasets
NeoRdRp is aimed to provide a comprehensive dataset of RdRp sequences. To this end, we collected as many RdRp sequences as possible. We cannot exclude the possibility that some RdRp sequences used in NeoRdRp may not function as RdRp. Therefore, we evaluated the 557,197 RdRp seed sequences used in this study using InterProScan, Palmscan, and BLASTP searches against UniProtKB and hmmsearch with RdRp-scan, as summarized in the Supplementary Data.
However, even if a sequence was not determined to be RdRp by the programs, this may simply be due to the low sequence similarity; we did not remove such unmatched sequences in this study. Users can check the degree of certainty of a given HMM profile based on the annotation results. InterProScan analysis facilitated the identification of a wide range of conserved domains, motifs, and protein families, thereby enriching our understanding of the functional landscape of RdRp. Palmscan and RdRp-scan annotated 453,579 and 246,017 sequences in the seed RdRp datasets, respectively. Although there were viruses that NeoRdRp2 failed to detect, the seed RdRp datasets contained at least 717 Partitiviruses, 122 Narnaviruses, 101 Endornaviruses, 9 Morbilliviruses, 89 Leviviruses, and 29 Orthopneumoviruses. While Embecovirus was not found in the seed RdRp datasets, 61 beta Coronaviruses were included, and Embecoviruses were their subgenera.
4 Discussion
A comparison using the UniProtKB dataset showed improved performance of NeoRdRp2 from version 1.1; introducing expanded seed RdRp datasets enhanced the detection of non-mammalian virus sequences missed in version 1.1. This improvement stems from the enriched metatranscriptome data of environmental origin in the dataset. In contrast, 188 non-RdRp sequences in UniProtKB were detected by hmmsearch using NeoRdRp2, which appears to be a cautionary point for NeoRdRp that is not limited to the core motifs of RdRp. However, because the NeoRdRp HMM profiles were generated based on conserved regions (see Materials and Methods), non-RdRp domains could rarely be merged in the HMM profiles. Indeed, NeoRdRp detected a relatively small number of possible false-positive RdRp hits in the capsid or unannotated sequences compared with other estimation programs (Figure 2; Table 4). As we previously reported, a BLASTp search of each RdRp amino acid sequence stored in the NeoRdRp 1.1 dataset could further identify RdRp candidates. However, this method can also detect many false positives (3). Similar trends were observed in the NeoRdRp2 dataset because the amino acid sequences used for NeoRdRp2 also contained non-RdRp regions. NeoRdRp2 showed high performance using hmmsearch alone compared to its version 1.1, as well as other RNA virus detention tools, which promise a highly specific RdRp search. Additionally, the possibility that these 188 non-RdRp sequences contained non-annotated RdRps should be considered. Despite employing the curated and reliable UniProtKB dataset, which we consider a robust benchmark dataset, the possibility of misannotations cannot be ruled out. Therefore, a solid benchmark dataset that is well annotated and comprehensive is required for the variation in detection capabilities across software versions and tools. The methodology for accurate annotation of RdRp and the need for consensus benchmark datasets were discussed at the RdRp summit in 2023 and summarized in a consensus statement (27). Establishing such a benchmark dataset is crucial for evaluating and improving RdRp detection tools, including future NeoRdRp iterations.
As shown in Figure 2; Table 4, the number of detected RdRp sequences varied significantly depending on the program used. NeoRdRp2 showed the highest detection in the search of FLDS data using multiple HMMs for RNA virus detection. This highlights the benefits of developing a comprehensive HMM profile to ensure broader sequence detection. The present results, which show that adding newly found RdRp sequences can increase detection and maintain accuracy, demonstrate the advantages of RNA Virome studies using HMM. In addition, the detection of many RdRps by LucaProt and eight RdRps by RdRpBin alone indicates that machine-learning-based RdRp searches can also detect comprehensive and low-similarity RdRp sequences. However, the possibility that a large fraction of false positives was included cannot be ruled out, and extra care must be exercised when using machine-learning-based methods.
In recent years, the field of viral discovery and the molecular analysis of novel viruses has experienced significant advancements, leading to substantial enrichment of the viral sequence database and enhanced annotation accuracy (28). Therefore, tools that use newer databases can potentially identify undiscovered viral sequences in published data. In this regard, while the FLDS data that we used for the evaluation of RNA virus detection tools is well-annotated metatranscriptome data from marine samples (29), there might be potential RdRp sequences. Originally, annotations for this dataset were performed using BLAST, along with manual curation, leaving several sequences without comprehensive annotations. Our findings indicate that with the advent of improved bioinformatics tools, there is a significant opportunity to identify RdRp sequences in previously unannotated sequences. Although this study did not examine the unannotated FLDS data employed in the benchmark dataset, several unannotated sequences were detected using multiple programs that can be undiscovered RdRp sequences (Supplementary Table 5), and this should be assessed in future research. This highlights the need for a standardized approach to annotation in virology and the importance of revisiting existing datasets using contemporary methodologies to discover potential novel RNA viruses (27).
The core domains A, B, and C are crucial motifs in RdRp proteins and act as the principal catalytic sites (30). As a popular approach, RdRp search tools have focused on these domains to generate HMMs that encapsulate all core domains to ensure the detection of complete RdRp sequences (8, 14, 15). In contrast, our approach broadens the search scope to include conserved regions outside the core domains identified through the alignment of input sequences. While innovative, this strategy introduces the potential risk of inadvertent splitting of the core domain during HMM creation. Acknowledging this, we performed a comprehensive evaluation using the UniProtKB dataset to refine our threshold parameters, aiming to effectively balance sensitivity and specificity. However, our method does not explicitly prevent the division of the core domains. Instead, it leverages the presence of less conserved, yet important, regions, as demonstrated in our previous work and confirmed by the performance of NeoRdRp2 in this study. These regions, although not always encapsulating complete ABC domains, contributed to enhancing the accuracy of the tool. This approach may lead to the inclusion of partial RdRp sequences, potentially increasing the risk of false positives. However, even if the domains are split into different profiles, each may be hit as an RdRp domain in a given input sequence; they may also be properly identified. Indeed, based on our benchmarks, the risk of NeoRdRp2 remained within acceptable limits. Future updates will focus on refining our validation steps and incorporating new seed RdRp datasets to improve the specificity and reduce the likelihood of false positives.
In conclusion, NeoRdRp2, which contains 19,394 HMM profiles of the RdRp domain, improves the detection of RdRp in various RNA viruses. Compared with other RNA search tools, NeoRdRp2 exhibited a good balance between sensitivity and specificity of the RdRp domains. NeoRdRp2 achieved more precise RdRp identification in various RNA virome studies.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: Edgar2022 (rdrp1): https://github.com/ababaian/serratus/wiki/Serratus-Lite Edgar 2022 (serratus): https://github.com/rcedgar/palmdb/tree/main Zayed2022: https://datacommons.cyverse.org/browse/iplant/home/shared/iVirus/ZayedWainainaDominguez-Huerta_RNAevolution_Dec2021 Neri2022: https://zenodo.org/records/6553771 Wolf2018: https://doi.org/10.1128/mbio.02329-18. The NeoRdRp2 is available on https://github.com/shoichisakaguchi/NeoRdRp.
Author contributions
SS: Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. TN: Supervision, Writing – original draft, Writing – review & editing. SN: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a KAKENHI Grant-in-Aid for Scientific Research (C) 20K06775 (to SS and SN), Early-Career Scientists 22K14999 (to SS), Scientific Research on Innovative Areas 16H06429 (to SN), 16K21723 (to SN), 19H04843 (to SN), AMED under grant number JP21wm0325004 (to SN), and 2024 Core research fund of the Institute of Medical Sciences, Tokai University (to SN).
Acknowledgments
We would like to thank Editage (www.editage.jp) for the English language editing. Computing resources were partially provided by the NIG supercomputer at the ROIS National Institute of Genetics, Japan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2024.1378695/full#supplementary-material
References
1. Wolf YI, Kazlauskas D, Iranzo J, Lucía-Sanz A, Kuhn JH, Krupovic M, et al. Origins and evolution of the global RNA virome. mBio. (2018) 9:e02329–18. doi: 10.1128/mBio.02329-18
2. Nakagawa S, Sakaguchi S, Ogura A, Mineta K, Endo T, Suzuki Y, et al. Current trends in RNA virus detection through metatranscriptome sequencing data. FEBS Open Bio. (2023) 13:992–1000. doi: 10.1002/2211-5463.13626
3. Sakaguchi S, Urayama S-I, Takaki Y, Hirosuna K, Wu H, Suzuki Y, et al. NeoRdRp: A comprehensive dataset for identifying RNA-dependent RNA polymerases of various RNA viruses from metatranscriptomic data. Microbes Environ. (2022) 37:ME22001. doi: 10.1264/jsme2.ME22001
4. Chiba Y, Yabuki A, Takaki Y, Nunoura T, Urayama S-I, Hagiwara D. The first identification of a narnavirus in bigyra, a marine protist. Microbes Environ. (2023) 38:ME22077. doi: 10.1264/jsme2.ME22077
5. Nweze JE, Schweichhart JS, Angel R. Viral communities in millipede guts: Insights into diversity and the potential role in modulating the microbiome. Environ Microbiol. (2024) 26:e16586. doi: 10.1111/1462-2920.16586
6. Urayama S-I, Fukudome A, Hirai M, Okumura T, Nishimura Y, Takaki Y, et al. Double-stranded RNA sequencing reveals distinct riboviruses associated with thermoacidophilic bacteria from hot springs in Japan. Nat Microbiol. (2024) 9:514–23. doi: 10.1038/s41564-023-01579-5
7. Dominguez-Huerta G, Zayed AA, Wainaina JM, Guo J, Tian F, Pratama AA, et al. Diversity and ecological footprint of Global Ocean RNA viruses. Science. (2022) 376:1202–8. doi: 10.1126/science.abn6358
8. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature. (2022) 602:142–7. doi: 10.1038/s41586-021-04332-2
9. Neri U, Wolf YI, Roux S, Camargo AP, Lee B, Kazlauskas D, et al. Expansion of the global RNA virome reveals diverse clades of bacteriophages. Cell. (2022) 185:4023–4037.e18. doi: 10.1016/j.cell.2022.08.023
10. Zayed AA, Wainaina JM, Dominguez-Huerta G, Pelletier E, Guo J, Mohssen M, et al. Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science. (2022) 376:156–62. doi: 10.1126/science.abm5847
11. Hou X, He Y, Fang P, Mei S-Q, Xu Z, Wu W-C, et al. Artificial intelligence redefines RNA virus discovery. bioRxiv. (2023). doi: 10.1101/2023.04.18.537342
12. Babaian A, Edgar R. Ribovirus classification by a polymerase barcode sequence. PeerJ. (2022) 10:e14055. doi: 10.7717/peerj.14055
13. Tang X, Shang J, Sun Y. RdRp-based sensitive taxonomic classification of RNA viruses for metagenomic data. Brief Bioinform. (2022) 23:bbac011. doi: 10.1093/bib/bbac011
14. Charon J, Buchmann JP, Sadiq S, Holmes EC. RdRp-scan: A bioinformatic resource to identify and annotate divergent RNA viruses in metagenomic sequence data. Virus Evol. (2022) 8:veac082. doi: 10.1093/ve/veac082
15. Olendraite I, Brown K, Firth AE. Identification of RNA virus-derived RdRp sequences in publicly available transcriptomic datasets. Mol Biol Evol. (2023) 40:msad060. doi: 10.1093/molbev/msad060
16. Bigot T, Temmam S, Pérot P, Eloit M. RVDB-prot, a reference viral protein database and its HMM profiles. F1000Res. (2019) 8:530. doi: 10.12688/f1000research.18776.2
17. Guo J, Bolduc B, Zayed AA, Varsani A, Dominguez-Huerta G, Delmont TO, et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome. (2021) 9:37. doi: 10.1186/s40168-020-00990-y
18. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. (1997) 25:3389–402. doi: 10.1093/nar/25.17.3389
19. UniProt Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. (2023) 51:D523–31. doi: 10.1093/nar/gkac1052
20. Urayama S-I, Takaki Y, Nishi S, Yoshida-Takashima Y, Deguchi S, Takai K, et al. Unveiling the RNA virosphere associated with marine microorganisms. Mol Ecol Resour. (2018) 18:1444–55. doi: 10.1111/1755-0998.12936
21. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinform. (2009) 10:421. doi: 10.1186/1471-2105-10-421
22. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. (2006) 22:1658–9. doi: 10.1093/bioinformatics/btl158
23. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. (2012) 28:3150–2. doi: 10.1093/bioinformatics/bts565
24. Eddy SR. Accelerated profile HMM searches. PloS Comput Biol. (2011) 7:e1002195. doi: 10.1371/journal.pcbi.1002195
25. Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar GA, et al. InterPro in 2022. Nucleic Acids Res. (2023) 51:D418–27. doi: 10.1093/nar/gkac993
26. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
27. Charon J, Olendraite I, Forgia M, Chong CL, Hillary SL, Roux S, et al. Consensus statement from the first RdRp Summit: advancing RNA virus discovery at scale across communities. Front Virol. (2024) 4:1371958. doi: 10.3389/fviro.2024.1371958
28. Brister JR, Ako-Adjei D, Bao Y, Blinkova O. NCBI viral genomes resource. Nucleic Acids Res. (2015) 43:D571–7. doi: 10.1093/nar/gku1207
29. Urayama S, Takaki Y, Nunoura T. FLDS: A comprehensive dsRNA sequencing method for intracellular RNA virus surveillance. Microbes Environ. (2016) 31:33–40. doi: 10.1264/jsme2.ME15171
Keywords: RNA-dependent RNA polymerase, RNA-directed RNA polymerase, RdRp, virome, RNA virus
Citation: Sakaguchi S, Nakano T and Nakagawa S (2024) NeoRdRp2 with improved seed data, annotations, and scoring. Front. Virol. 4:1378695. doi: 10.3389/fviro.2024.1378695
Received: 30 January 2024; Accepted: 08 April 2024;
Published: 23 April 2024.
Edited by:
Masayuki Horie, Osaka Prefecture University, JapanReviewed by:
Mai Kishimoto, Osaka Metropolitan University, JapanAnne Kupczok, Wageningen University and Research, Netherlands
Copyright © 2024 Sakaguchi, Nakano and Nakagawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shoichi Sakaguchi, c2hvaWNoaS5zYWthZ3VjaGlAb21wdS5hYy5qcA==; So Nakagawa, c29AdG9rYWkuYWMuanA=