 Lei Xu
Lei Xu Guangmin Liang1*
Guangmin Liang1*- 1School of Electronic and Communication Engineering, Shenzhen Polytechnic, Shenzhen, China
- 2Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen, China
- 3Department of Obstetrics and Gynecology, Chung Shan Medical University Hospital, Taichung, Taiwan
- 4School of Medical Informatics, Chung Shan Medical University, Taichung, Taiwan
- 5IT Office, Chung Shan Medical University Hospital, Taichung, Taiwan
In this paper, a computational method based on machine learning technique for identifying Alzheimer's disease genes is proposed. Compared with most existing machine learning based methods, existing methods predict Alzheimer's disease genes by using structural magnetic resonance imaging (MRI) technique. Most methods have attained acceptable results, but the cost is expensive and time consuming. Thus, we proposed a computational method for identifying Alzheimer disease genes by use of the sequence information of proteins, and classify the feature vectors by random forest. In the proposed method, the gene protein information is extracted by adaptive k-skip-n-gram features. The proposed method can attain the accuracy to 85.5% on the selected UniProt dataset, which has been demonstrated by the experimental results.
Introduction
Alzheimer's disease (AD) is a common cause of dementia, and it can lead a degeneration of brain. The research shows that more than 35 million people have been affected by Alzheimer's disease all over the world. It is predicted that there will be over 70 million people diagnosed by Alzheimer's disease in 2030, and the number will be increased by 50% in 2050 (Brookmeyer et al., 2007).
Until now, there is no treatment for AD. As the status becoming worse, it will destroy the ability of speak and think. At last, AD will lead to die. So, it is meaningful to predict AD at an early stage. Machine learning methods have been extensively used in multiple fields of bioinformatics (Zeng et al., 2014; Wang et al., 2016; Liu Y. et al., 2017; Zhang et al., 2017a; Cheng et al., 2018; Fu et al., 2018; Liu et al., 2018; Peng et al., 2018a; Song et al., 2018), such as anticancer peptides prediction (Xu et al., 2018b), identification of antioxidant proteins (Xu et al., 2018a), disease gene identification (Jiang et al., 2017; Liu G. et al., 2017; Peng et al., 2017; Zeng et al., 2017a; Zhang et al., 2017b; Cheng et al., 2018,a,b; Liu et al., 2018a,b; Zhu et al., 2018), microRNA classification (Wei et al., 2014; Chen et al., 2016; Zeng et al., 2018; Zhang et al., 2018), protein remote homology detection (Liu et al., 2014b; Liu and Li, 2018), drug-induced hepatotoxicity prediction (Li et al., 2018; Su et al., 2018), DNA binding protein identification (Zhang and Liu, 2017; Liu, 2018), protein interaction identification (Guo et al., 2011, 2012, 2014; Ding et al., 2016, 2017a,b; Peng et al., 2018b) and so on (Li et al., 2016, 2017; Zou et al., 2016; Zeng et al., 2017a,b; Hu et al., 2018; Xue et al., 2018; Zhang and Liu, 2018; Zhang et al., 2018). In this paper, machine learning method is used to identify AD.
Because the structural features of brain is related to AD, the structural brain information is described by structural magnetic resonance imaging (MRI) data. Most existing works use machine learning methods, such as ensemble classifier, deep learning method to classify AD and non-AD samples. However, most existing works are limited by the expensive cost on money and time. For the purpose of identifying AD efficiently and effectively, a method called k-skip-n-gramRF, which is based on gene coding information of proteins, is proposed to recognize AD samples. In this paper, adaptive k-skip-2-gram is used to extract the information from the protein sequences, and then the samples are classified by random forest (RF) classifier. Consequently the classification accuracy can attain the accuracy to 85.5% using the select data set from Uniprot database. In our proposed method, adaptive k-skip-n-gram describes the correlation information of both adjacent and non-adjacent residues based on traditional n-gram model (Wei et al., 2017a). The idea of our proposed method is shown in Figure 1. As Figure 1 shown, the protein peptides are extracted by k-skip-n-gram method. Each sequence is transferred into a vector. The training vectors are used to train the parameters of random forest. The performance of methods is evaluated by testing vectors. The testing vectors are labeled by trained random forest classifier.
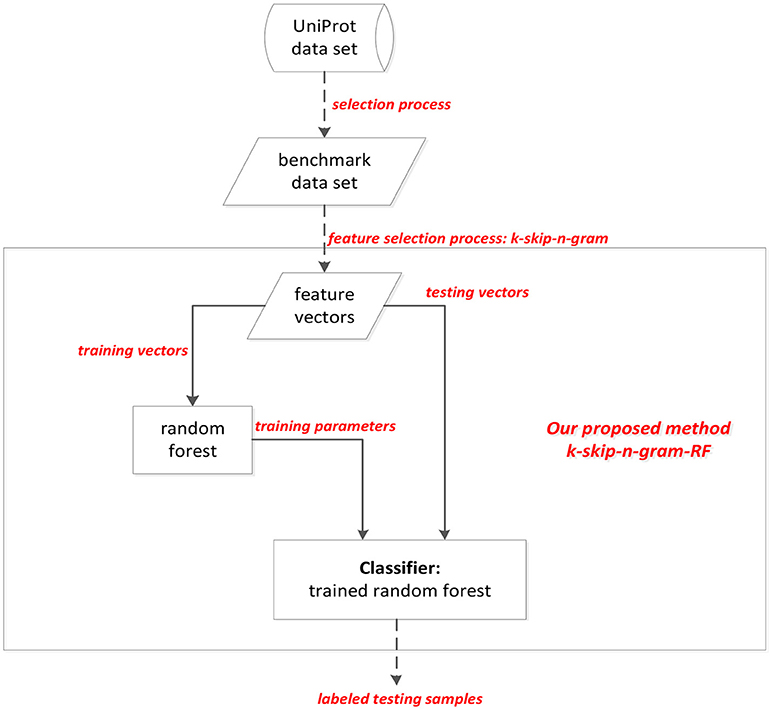
Figure 1. The flow chart of proposed method.
In the proposed method, adaptive n-gram-k-skip model is used to represent the gene coding information by a 400-dimensional vector. Then an ensemble classifier named random forest (RF) is used to classify the samples. In the experiments, the accuracy of the proposed classifier is 85.5%, which is competitive to existing works with low cost. In other words, the experimental results demonstrated that the proposed methods can be utilized to identify AD samples. The contributions of our work include:
• A computational model for predicting Alzheimer's disease is proposed in the paper. The experimental results demonstrate that the classification accuracy of the prediction model is 85.5%, which is competitive to some existing works with low cost and fast speed.
• Different from previous work using MRI data, the gene coding information of proteins is considered to identify Alzheimer's disease protein. Each protein sequence is represented by a 400-dimensional vector, which the information of distance is considered.
• In our work, random forest is used to classify the AD protein peptides and non-AD protein peptides. Random forest is an ensemble classification method based on bagging, which is used to predict AD peptides in the work.
The rest of the paper is organized as follows. Section Materials and Methods introduces the dataset and the proposed method (k-skip-n-gram-RF) for identifying AD peptides. The results of AD prediction are described in Section Results and Discussions. The conclusion is made in Section Conclusions.
Materials and Methods
Benchmark Dataset
The used data is selected from the UniProt database. The data set S is composed of positive samples S+ and negative samples S−. The positive sample set is represented by Alzheimeir's disease (AD) samples, and the negative sample set is represented by non-AD samples.
Positive Data Set
The positive data set contains AD samples. The samples are built by the sequences which are labeled by “Alzheimer's disease.” As a result, 310 AD samples are selected from the UniProt database. To avoid the overestimation of the performance, the sequences with more than 60% similarity are removed. Thus, there are 279 positive samples left.
Negative Data Set
The data labeled with “non Alzheimer's disease” are chosen, then there are 312 non-AD samples. The proteins which are confirmed as non Alzheimer's disease are also selected in the negative data set. After CD-HIT program (Fu et al., 2012), 1,743 negative samples are left in the benchmark data set for experiments.
In the experiments, the benchmark data set is divided into training data set and testing data set. The training data are used for train the classifier, and the testing data are used for the performance evaluation.
Random Forest
Random forest (Ho, 1995) is an ensemble classifier by combining decision trees together. Due to its effectiveness, random forest has been widely used in many bioinformatics problems (Deng and Chen, 2015; Liu, 2018). The key idea will be introduced briefly here.
The key element in random forest is decision tree. The decision trees are built based on bagging. Bagging is a sampling method. The used samples will be put back into the data set for reusing. In other words, a sample may be used more than one time for building data set. For example, there is a data set with n samples. If m decision trees are needed, m data sets will be built by bagging for training. Each node on the decision tree is represented by a feature used for classification (Quinlan, 1986). The features used on different levels of the tree are selected in sequence by the entropy value. Entropy is considered as information gain, and the entropy is calculated as Equation (1). The information gain is calculated as Equation (2)
The attribute with the maximum entropy gain is selected. Random forest is built based on the decision tree, so the features with larger information gains will be selected first in the training process. Because random forest is an ensemble classifier, the decision is made by voting process shown as Figure 2. As Figure 2 shown, the sample will be assigned to the class with the maximum votes. In our problem, the decision trees are trained by protein sequences, and the input of Figure 2 is protein peptide.
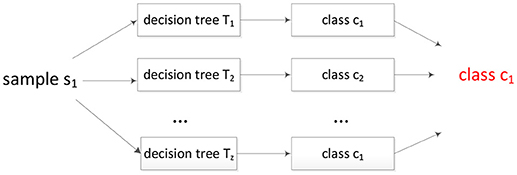
Figure 2. The voting process of random forest.
Sequence Representation
The sequence information of each protein peptides is encoded into a 400-dimensional feature vector by adaptive k-skip-2-gram model (Wei et al., 2017a). The k-skip-2-gram method is proposed based on k-skip-n-gram. The key idea of k-skip-n-gram is the distance information integrated into traditional n-gram model (Liu et al., 2014a). However, the maximum value of k is the length of the shortest amino acid, which is small for long peptides. Thus, for the purpose of mining more relation between peptides, adaptive k-skip-n-gram method is proposed in and used in our method.
There is a peptide sequence S, denoted by R1R2 … Rn, where n is the length of the sequence. In n-gram model, the occurrence frequencies of any n consecutive amino acids are measured. The amino acid set is denoted as L, where Li is the ith element in L. The n-gram features can be calculated as follows:
Where N(Ts) is denoted as the number of all items in the set Ts, and Ts represents the number of segments with n consecutive amino acids in the peptide S. Each position has 20 possible amino acids, so there are 20n dimensions with the length of n peptides in n-gram model. It is obvious that the features are sparse. Thus, n-gram-k-skip is proposed to overcome the sparse problem of n-gram model. The distance information is considered in k-skip-n-gram model.
In k-skip-n-gram model, distance between residues Ri and Rj is calculated as Equation (3). For example, if there is i = 2 and j = 3, the distance between A2 and A3 is 0.The distance between A4 and A2 is 1, because A2 and A4 is separated by A3.
In the k-skip-n-gram model, the sequence information of n residues within distance k is calculated, which means that the only residues within distance k are considered. The calculation of k-skip-n-gram is shown as Equation (4). In Equation (4), N(TSkipG) is denoted as all the elements in TSkipG. The calculation of TSkipG is shown as Equation (5). In Equation (5), Skip(DT = z) = {AiAi+z+1…Ai+z+n−1|1 ≤ z ≤ L−1, 1 ≤ z ≤ k}. When n equals 1, the model is reduced to n-gram model. For the purpose of avoiding overfitting problem, n is constrained less than 3. Thus, only the case of n equals to 2 is analyzed. The model is considered as k-skip-2-gram. The elements of k-skip-2-gram include R1R2,R2R3,…,Rn−1Rn,R1R3,… Rn−2Rn,… R1Rn, which all of them are two amino acids pair within distance k. The number of the 2-item combination is 400. Thus, the number of features extracted by k-skip-2-gram is 400. The 20n dimensional vector is reduced to a 400 dimensional vector.
The amino acids distance within k is calculated. K is the minimum sequence length of the peptides. The length of some sequences is sometimes short. If k is small, the features will be limited in local information. In adaptive k-skip-n-gram, k is the length of each sequence. When the information of varying distances of sequences is described, adaptive k-skip-n-gram is more flexible than k-skip-n-gram.
Performance Evaluation
In the literature of bioinformatics, accuracy(Acc), specificity(Sp), sensitivity(Sn) are frequently used for evaluating the performance of classification methods (Chou, 2001a,b). The performance of the method is measured by the above metrics. Specificity is used to measure the rate of retrieved true positive samples of the real positive samples, which is represented by Equation (6). Sensitivity is the metric for measuring the rate of real non-AD samples identified as non-AD samples of real non-AD samples, which is calculated by Equation (7). Accuracy is the rate that the samples are classified into the correct class, shown as Equation (8).
Where P+ is the number of AD samples, and P− is the number of non-AD samples. TP is denoted as the number of AD samples recognized as AD samples. TN is represented by the number of non-AD samples labeled by non-AD samples by the classifier.
Results and Discussions
In the experiments, the data set is divided into training set and testing set. The training set is used for learning parameters, and the testing set is for performance evaluation. The performance of our proposed method is reported in Section The Performance of Proposed Method. The performance of our method compared with other feature selection methods is described in Section The Comparison of Performance Evaluation on Feature Extraction Methods. We also compared random forest with other classifiers, and the performance evaluation comparison is shown in Section The Comparison of Performance Evaluation on Other Classifiers.
The Performance of Proposed Method
The experimental results of our proposed method are reported in Table 1. Table 1 shows that the accuracy of our proposed method is 0.855, which means that the proposed method can classified the 85.5% samples correctly in the benchmark data set. Sp(specificity) describes the performance for identifying AD samples. 85.5% AD samples of all the positive samples in the data set will be recognized in the experiment. Moreover, 85.5% non-AD samples of the negative samples can be classified correctly by the proposed method. The experimental results demonstrated that the method is practical.

Table 1. The performance evaluation of k-skip-n-gram-RF.
The Comparison of Performance Evaluation on Feature Extraction Methods
For the purpose of showing the effective performance of our proposed method, the feature select method of our proposed method is compared with information theory. Information theory is a feature selection method representing.
The comparison results are shown in Table 2. The metrics of accuracy, sp and sn of k-skip-n-gram method performs are better than that of information theory. The accuracy of the proposed method (k-skip-n-gram-RF) is better than that of information theory based random forest. The accuracy of information theory method is 0.715, while the accuracy of k-skip-n-gram is 0.855. For the problem of Alzheimer's disease protein prediction, k-skip-n-gram performs better than information theory when random forest is used.
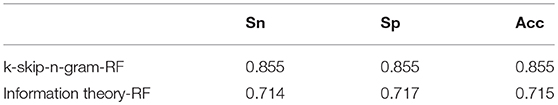
Table 2. Comparison of our features with other methods on Sn.
The Comparison of Performance Evaluation on Other Classifiers
To demonstrate the performance of our classifier, the classification methods are compared with other classification method, such as naive bayes (Peter Norvig, 1995), LibD3C (Lin et al., 2014), Adaboost (Rojas, 2009), and bagging. The mentioned methods are shown as followings.
Naive bayes is probability method. The sample is labeled by the class with the maximum probability.
LibD3C is an ensemble based method. k-means is integrated into the method for classifier selection.
Adaboost and bagging are ensemble classification algorithms. The difference between then is the building strategy of sample set. Bagging reuses the samples during classification. In Adaboost, the samples classified to the wrong class, the weight will be increased. The samples which are classified into the correct class, the weight will be decreased.
The comparison of our proposed method with other classifiers on Sn, Sp and accuracy is shown in Figure 3. Random forest performs better than other classifiers on Sn, Sp and Acc. Random forest is an ensemble classifier with competitive performance. The accuracy of naïve bayes and is 0.801 and 0.812. The accuracy of bagging and LibD3C is 0.83 and 0.837. When the features of proteins are extracted by k-skip-n-gram method, the accuracy of random forest is 0.855, which is better than that of other methods. As the results shown, LibD3C preforms better than naïve bayes, Adaboost and bagging.
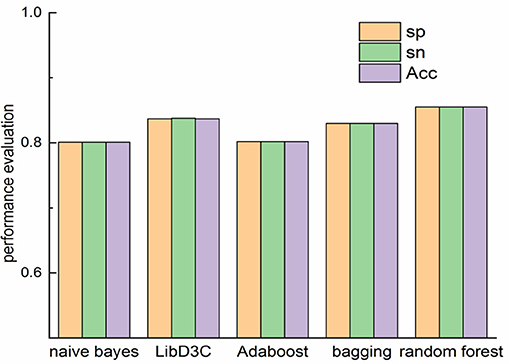
Figure 3. Comparison of performance evaluation on different classifiers.
The feature selection method and classifiers are compared. The k-skip-n-gram method can represent more accurately than information theory. Random forest performs better than other classifiers. Thus, the experimental results demonstrate that our proposed method can provide competitive results.
Conclusions
In this paper, we study the problem of Alzheimer's disease prediction by using gene coding information. MRI data are usually used to identifying Alzheimer's disease, but the data are complex and with expensive cost. Different from previous work, the information is extracted from the protein peptides by k-skip-n-gram model for Alzheimer's disease prediction. Finally, random forest is used to classify Alzheimer's disease (AD) samples and non-AD samples. The accuracy of our proposed method is 85.5%, which means that it is meaningful for Alzheimer's disease detection with low cost. In the literature, most work have provided web server for protein classification, and we will develop our web server for Alzheimer's disease proteins classification. Moreover, to further improve the prediction performance, there are still many aspects can be explored. For example, other effective machine learning algorithms, such as ensemble learning algorithms and deep learning algorithms, have recently showed they can achieve better performance than traditional algorithms (Mrozek et al., 2009a; Wei et al., 2017b, 2018a; Wang et al., 2018). On the other hand, feature representation learning has demonstrated that it can exploit more informative features and improve the performance in multiple bioinformatics problems (Mrozek et al., 2009b; Momot et al., 2010; Liu et al., 2015; Wei et al., 2018b, 2019; Tang et al., 2019).
Author Contributions
LX initially drafted the manuscript and did most of the codes work and the experiments. CL and G-DC collected the features, analyzed the experiments and revised the paper. GL and C-CC revised to draft the manuscript. All authors designed the work, read and approved the final manuscript and are agree to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the natural science foundation of Guangdong province (grant no. 2018A0303130084), the Science and Technology Innovation Commission of Shenzhen (grant no. JCYJ20170818100431895, JCYJ20160523113602609), the Grant of Shenzhen Polytechnic (grant no. 601822K19011), and National Nature Science Foundation of China (grant nos. 61575128, 61771331).
References
Brookmeyer, R., Johnson, E., Ziegler-Graham, K., and Arrighi, H. M. (2007). O1-02-01: Forecasting the global prevalence and burden of Alzheimer's disease. Alzheimers Dement. 3:S168. doi: 10.1016/j.jalz.2007.04.381
Chen, J., Wang, X., and Liu, B. (2016). iMiRNA-SSF: improving the identification of microRNA precursors by combining negative sets with different distributions. Sci. Rep. 6:19062. doi: 10.1038/srep19062
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018a). DincRNA: a comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics. 34, 1953–1956. doi: 10.1093/bioinformatics/bty002
Cheng, L., Jiang, Y., Ju, H., Sun, J., Peng, J., Zhou, M., et al. (2018b). InfAcrOnt: calculating cross-ontology term similarities using information flow by a random walk. BMC Genomics 19(Suppl, 1):919. doi: 10.1186/s12864-017-4338-6
Cheng, L., Wang, P., Tian, R., Wang, S., Guo, Q., Luo, M., et al. (2018). LncRNA2Target v2.0: a comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 47, D140–D144. doi: 10.1093/nar/gky1051
Chou, K.C. (2001a). Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 44, 246–255. doi: 10.1002/prot.1035
Chou, K. C. (2001b). Using subsite coupling to predict signal peptides. Protein Eng. 14, 75–79. doi: 10.1093/protein/14.2.75
Deng, L., and Chen, Z. (2015). An integrated framework for functional annotation of protein structural domains. IEEE/ACM Trans. Comput. Biol. Bioinform. 12, 902–913. doi: 10.1109/TCBB.2015.2389213
Ding, Y., Tang, J., and Guo, F. (2016). Identification of protein-protein interactions via a novel matrix-based sequence representation model with amino acid contact information. Int. J. Mol. Sci. 17:1623. doi: 10.3390/ijms17101623
Ding, Y., Tang, J., and Guo, F. (2017a). Identification of drug-target interactions via multiple information integration. Inf. Sci. 418, 546–560. doi: 10.1016/j.ins.2017.08.045
Ding, Y., Tang, J., and Guo, F. (2017b). Identification of protein-ligand binding sites by sequence information and ensemble classifier. J. Chem. Inf. Model. 57, 3149–3161. doi: 10.1021/acs.jcim.7b00307
Fu, J., Tang, J., Wang, Y., Cui, X., Yang, Q., Hong, J., et al. (2018). Discovery of the consistently well-performed analysis chain for SWATH-MS based pharmacoproteomic quantification. Front. Pharmacol. 9:681. doi: 10.3389/fphar.2018.00681
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Guo, F., Li, S. C., Du, P., and Wang, L. (2014). Probabilistic models for capturing more physicochemical properties on protein-protein interface. J. Chem. Inf. Model. 54, 1798–1809. doi: 10.1021/ci5002372
Guo, F., Li, S. C., and Wang, L. (2011). Protein-protein binding sites prediction by 3D structural similarities. J. Chem. Inf. Model. 51, 3287–3294. doi: 10.1021/ci200206n
Guo, F., Li, S. C., Wang, L., and Zhu, D. (2012). Protein-protein binding site identification by enumerating the configurations. BMC Bioinformatics 13:158. doi: 10.1186/1471-2105-13-158
Ho, T. K. (1995). “Random Decision Forests,” in International Conference on Document Analysis and Recognition (Montreal, QC).
Hu, Y., Zhao, T., Zhang, N., Zang, T., Zhang, J., and Cheng, L. (2018). Identifying diseases-related metabolites using random walk. BMC Bioinformatics 19 (Suppl. 5):116. doi: 10.1186/s12859-018-2098-1
Jiang, Q., Jin, S., Jiang, Y., Liao, M., Feng, R., Zhang, L., et al. (2017). Alzheimer's disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol. Neurobiol. 54, 594–600. doi: 10.1007/s12035-015-9670-8
Li, B., Tang, J., Yang, Q., Cui, X., Li, S., Chen, S., et al. (2016). Performance evaluation and online realization of data-driven normalization methods used in LC/MS based untargeted metabolomics analysis. Sci. Rep. 6:38881. doi: 10.1038/srep38881
Li, B., Tang, J., Yang, Q., Li, S., Cui, X., Li, Y., et al. (2017). NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 45, W162–W170. doi: 10.1093/nar/gkx449
Li, X. X., Yin, J., Tang, J., Li, Y., Yang, Q., Xiao, Z., et al. (2018). Determining the balance between drug efficacy and safety by the network and biological system profile of its therapeutic target. Front. Pharmacol. 9:1245. doi: 10.3389/fphar.2018.01245
Lin, C., Chen, W., Qiu, C., Wu, Y., Krishnan, S., and Zou, Q. (2014). LibD3C: ensemble classifiers with a clustering and dynamic selection strategy. Neurocomputing 123, 424–435. doi: 10.1016/j.neucom.2013.08.004
Liu, B. (2018). BioSeq-Analysis: a platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinform. doi: 10.1093/bib/bbx165. [Epub ahead of print].
Liu, B., Jiang, S., and Zou, Q. (2018). HITS-PR-HHblits: protein remote homology detection by combining pagerank and hyperlink-induced topic search. Brief. Bioinform. doi: 10.1093/bib/bby104. [Epub ahead of print].
Liu, B., and Li, S. (2018). ProtDet-CCH: protein remote homology detection by combining long short-term memory and ranking methods. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/TCBB.2018.2789880. [Epub ahead of print].
Liu, B., Liu, F., Wang, X., Chen, J., Fang, L., and Chou, K. C. (2015). Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 43, W65–W71. doi: 10.1093/nar/gkv458
Liu, B., Xu, J., Zou, Q., Xu, R., Wang, X., and Chen, Q. (2014a). Using distances between Top-n-gram and residue pairs for protein remote homology detection. BMC Bioinformatics 15(Suppl. 2):S3. doi: 10.1186/1471-2105-15-S2-S3
Liu, B., Zhang, D., Xu, R., Xu, J., Wang, X., Chen, Q., et al. (2014b). Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 30, 472–479. doi: 10.1093/bioinformatics/btt709
Liu, G., Jin, S., Hu, Y., and Jiang, Q. (2018a). Disease status affects the association between rs4813620 and the expression of Alzheimer's disease susceptibility gene TRIB3. Proc. Natl. Acad. Sci. U S A. 115, E10519–E10520. doi: 10.1073/pnas.1812975115
Liu, G., Xu, Y., Jiang, Y., Zhang, L., Feng, R., and Jiang, Q. (2017). PICALM rs3851179 variant confers susceptibility to Alzheimer's disease in Chinese population. Mol. Neurobiol. 54, 3131–3136. doi: 10.1007/s12035-016-9886-2
Liu, G., Zhang, Y., Wang, L., Xu, J., Chen, X., Bao, Y., et al. (2018b). Alzheimer's disease rs11767557 variant regulates EPHA1 gene expression specifically in human whole blood. J. Alzheimers. Dis. 61, 1077–1088. doi: 10.3233/JAD-170468
Liu, Y., Wang, X., and Liu, B. (2017). A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief. Bioinform. 20, 330–346. doi: 10.1093/bib/bbx126
Momot, A., Małysiak-Mrozek, B., Kozielski, S., Mrozek, D., Hera, Ł., Górczynska-Kosiorz, S., et al. (2010). “Improving performance of protein structure similarity searching by distributing computations in hierarchical multi-agent system,” in Computational Collective Intelligence. Technologies and Applications - Second International Conference, ICCCI 2010, Proceedings, Part I (Kaohsiung: Springer-Verlag). 320–329. doi: 10.1007/978-3-642-16693-8_34
Mrozek, D., Malysiak-Mrozek, B., and Kozielski, S. (2009a). “Alignment of protein structure energy patterns represented as sequences of Fuzzy Numbers,” in Fuzzy Information Processing Society, Nafips 2009 Meeting of the North American (Cincinnati, OH: IEEE). doi: 10.1109/NAFIPS.2009.5156391
Mrozek, D., Malysiak-Mrozek, B., Kozielski, S., and Swierniak, A. (2009b). “The Energy Distribution Data Bank: Collecting Energy Features of Protein Molecular Structures,” in IEEE International Conference on Bioinformatics and Bioengineering (Taichung: IEEE). doi: 10.1109/BIBE.2009.40
Peng, J., Hui, W., and Shang, X. (2018a). Measuring phenotype-phenotype similarity through the interactome. BMC Bioinform. 19:114. doi: 10.1186/s12859-018-2102-9
Peng, J., Zhang, X., Hui, W., Lu, J., Li, Q., Liu, S., et al. (2018b). Improving the measurement of semantic similarity by combining gene ontology and co-functional network: a random walk based approach. BMC Syst. Biol. 12(Suppl. 2):18. doi: 10.1186/s12918-018-0539-0
Peng, J.J., Xue, H., Shao, Y., Shang, X., Wang, Y., and Chen, J. (2017). A novel method to measure the semantic similarity of HPO terms. Int. J. Data Min. Bioinform. 17, 173–188. doi: 10.1504/IJDMB.2017.084268
Quinlan, J. R. (1986). Induction of decision trees[J]. Mach. Learn. 1, 81–106. doi: 10.1007/BF00116251
Rojas, R. (2009). AdaBoost and the Super Bowl of Classifiers - A Tutorial Introduction to Adaptive Boosting Freie Universiy. Berlin.
Song, T., Rodríguez-Patón, A., Zheng, P., and Zeng, X. (2018). Spiking neural P systems with colored spikes. IEEE Trans. Cogn. Dev. Syst. 10, 1106–1115. doi: 10.1109/TCDS.2017.2785332
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2018). Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/TCBB.2018.2858756. [Epub ahead of print].
Tang, J., Fu, J., Wang, Y., Li, B., Li, Y., Yang, Q., et al. (2019). ANPELA: analysis and performance-assessment of the label-free quantification workflow for metaproteomic studies. Brief. Bioinform. doi: 10.1093/bib/bby127. [Epub ahead of print].
Wang, H., Liu, C., and Deng, L. (2018). Enhanced prediction of hot spots at protein-protein interfaces using extreme gradient boosting. Sci. Rep. 8:14285. doi: 10.1038/s41598-018-32511-1
Wang, X., Zeng, X., Ju, Y., Jiang, Y., Zhang, Z., and Chen, W. (2016). A classification method for microarrays based on diversity. Curr. Bioinform. 11, 590–597. doi: 10.2174/1574893609666140820224436
Wei, L., Ding, Y., Su, R., Tang, J., and Zou, Q. (2018a). Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 117, 212–217. doi: 10.1016/j.jpdc.2017.08.009
Wei, L., Hu, J., Li, F., Song, J., Su, R., and Zou, Q. (2018b). Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief. Bioinform. doi: 10.1093/bib/bby107. [Epub ahead of print].
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and promising identification of human microRNAs by incorporating a high-quality negative set. IEEE/ACM Trans. Comput. Biol. Bioinform. 11, 192–201 doi: 10.1109/TCBB.2013.146
Wei, L., Su, R., Wang, B., Li, X., and Zou, Q. (2019). Integration of deep feature representations and handcrafted features to improve the prediction of N6-methyladenosine sites. Neurocomputing 324, 3–9. doi: 10.1016/j.neucom.2018.04.082
Wei, L., Tang, J., and Zou, Q. (2017a). SkipCPP-Pred: an improved and promising sequence-based predictor for predicting cell-penetrating peptides. BMC Genomics 18:1. doi: 10.1186/s12864-017-4128-1
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017b). Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Xu, L., Liang, G., Shi, S., and Liao, C. (2018a). SeqSVM: a sequence-based support vector machine method for identifying antioxidant proteins. Int. J. Mol. Sci. 19:1773. doi: 10.3390/ijms19061773
Xu, L., Liang, G., Wang, L., and Liao, C. (2018b). A novel hybrid sequence-based model for identifying anticancer peptides. Genes. 9:158. doi: 10.3390/genes9030158
Xue, W., Yang, F., Wang, P., Zheng, G., Chen, Y., Yao, X., et al. (2018). What contributes to serotonin-norepinephrine reuptake inhibitors' dual-targeting mechanism? The key role of transmembrane domain 6 in human serotonin and norepinephrine transporters revealed by molecular dynamics simulation. ACS Chem. Neurosci. 9, 1128–1140. doi: 10.1021/acschemneuro.7b00490
Zeng, X., Ding, N., Rodríguez-Patón, A., and Zou, Q. (2017a). Probability-based collaborative filtering model for predicting gene–disease associations. BMC Med. Genomics 10:76. doi: 10.1186/s12920-017-0313-y
Zeng, X., Lin, W., Guo, M., and Zou, Q. (2017b). A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 13:e1005420. doi: 10.1371/journal.pcbi.1005420
Zeng, X., Liu, L., Lü, L., and Zou, Q. (2018). Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 34, 2425–2432. doi: 10.1093/bioinformatics/bty112
Zeng, X., Pan, L, and Pérez-Jiménez, M. J (2014). Small universal simple spiking neural P systems with weights. Sci. China Inform. Sci. 57, 1–11. doi: 10.1007/s11432-013-4848-z
Zhang, J., and Liu, B. (2017). PSFM-DBT: identifying DNA-binding proteins by combing position specific frequency matrix and distance-bigram transformation. Int. J. Mol. Sci. 18:1856. doi: 10.3390/ijms18091856
Zhang, J., and Liu, B. (2018). Identification of DNA-binding proteins via a voting strategy. Curr. Proteomics. 15, 363–373. doi: 10.2174/1570164615666180718150317
Zhang, J., Zhang, Z., Chen, Z., and Deng, L. (2017b). Integrating multiple heterogeneous networks for novel LncRNA-disease association inference. IEEE/ACM Trans Comput Biol Bioinform. doi: 10.1109/TCBB.2017.2701379. [Epub ahead of print].
Zhang, X., Zou, Q., Rodruguez-Paton, A., and Zeng, X. (2018). Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 283–291. doi: 10.1109/TCBB.2017.2776280
Zhang, Z., Zhang, J., Fan, C., Tang, Y., and Deng, L. (2017a). KATZLGO: large-scale prediction of LncRNA functions by using the KATZ measure based on multiple networks. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/TCBB.2017.2704587. [Epub ahead of print].
Zhu, F., Li, X. X., Yang, S. Y., and Chen, Y. Z. (2018). Clinical success of drug targets prospectively predicted by in silico study. Trends Pharmacol. Sci. 39, 229–231. doi: 10.1016/j.tips.2017.12.002
Keywords: Alzheimer's disease, n-gram model, random forest, gene coding, sequence information
Citation: Xu L, Liang G, Liao C, Chen G-D and Chang C-C (2019) k-Skip-n-Gram-RF: A Random Forest Based Method for Alzheimer's Disease Protein Identification. Front. Genet. 10:33. doi: 10.3389/fgene.2019.00033
Received: 10 October 2018; Accepted: 17 January 2019;
Published: 12 February 2019.
Edited by:
Qinghua Jiang, Harbin Institute of Technology, ChinaReviewed by:
Leyi Wei, Tianjin University, ChinaGuiyou Liu, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, China
Copyright © 2019 Xu, Liang, Liao, Chen and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangmin Liang, Z21saWFuZ0BzenB0LmVkdS5jbg==
Chi-Chang Chang, Y2hhbmdpbnR3QGdtYWlsLmNvbQ==