 Yiyi Hu
Yiyi Hu Zhenghong Sui*
Zhenghong Sui* Wei ZhouJinguo WangMinjie JiangHaihong ChenJingyu ZhangWei ZhangXiaoqing FengYuehua LeiBaoheng XiaoPeng Guo
Wei ZhouJinguo WangMinjie JiangHaihong ChenJingyu ZhangWei ZhangXiaoqing FengYuehua LeiBaoheng XiaoPeng Guo- Key Laboratory of Marine Genetics and Breeding (Ocean Univeristy of China), Ministry of Education, Qingdao, China
Gracilariopsis lemaneiformis is an important commercial macroalga. Whole-genome resequencing was conducted to identify single nucleotide polymorphisms (SNPs) in parental gametophytes and 60 F1 gametophytes of Gp. lemaneiformis in this study, and 9,989 SNPs located in nonrepetitive sequences were obtained. Among these SNPs, 92.02% of loci were identified as having a heterozygous genotype in at least one gametophyte, and 48.07% of loci had identical heterozygous genotypes in the 62 gametophytes. For each gametophyte, the proportions of homozygous and heterozygous loci ranged between 13.74 and 21.61% (mean of 17.04%) and between 66.36 and 83.59% (mean of 77.12%), respectively. The remainder were missing loci, representing an average 5.84%. Sources of heterozygous SNPs were free of exogenous DNA contamination, cross contamination among individuals, plastid and mitochondrial sequences, chimeras of different thallus parts or different cells, and repetitive sequences. Genotypes of heterozygous SNPs were verified by Sanger sequencing of PCR products and monoclones. Duplications of chromosomal rearrangements in the genome of Gp. lemaneiformis might explain the presence of heterozygous SNPs in haploid gametophytes.
Introduction
Gracilariopsis lemaneiformis (Bory de Saint-Vincent) E. Y. Dawson, Acleto & Foldvik is a commercially important macroalga that is mainly utilized for agar extraction and abalone aquaculture (Santelices and Doty, 1989; Mchugh, 1991). The high content of good quality agar produced by Gp. lemaneiformis represents 53% of the annual agar output worldwide (Mchugh, 1991). Gp. lemaneiformis is rich in glutamic acid and taurine, which can enhance the flavor of abalone and promote its feed intake (Chopin et al., 2001; Fei, 2004; Neori et al., 2004; Lu et al., 2011). In addition, Gp. lemaneiformis can effectively reduce the contents of N and P in the seawater and plays a role in bioremediation (Fei, 2004).
Gp. lemaneiformis is an ideal material for genetic research. Its whole life history can be completed in a laboratory culture (Plastino and de Oliveira, 1988). Mature tetrasporophytes (2n) produce tetraspores (n) through meiosis, and then tetraspores are released and develop into male and female gametophytes (n) (Engel et al., 2010). After fertilization, the zygotes develop into carposporophytes (2n), which release carpospores (2n) through mitosis (Guzmán-Urióstegui et al., 2002). Subsequently, carpospores develop into tetrasporophytes (Yamamoto and Sasaki, 1988). The reproductive modes of Gp. lemaneiformis include both sexual and asexual reproduction. In recent years, genetic studies on Gp. lemaneiformis have focused on genetic diversity analyses (Wang et al., 2007; Pang et al., 2010; Ding et al., 2012; Hu et al., 2018), functional gene studies (Liu et al., 2016; Liu et al., 2018; Hu et al., 2019), mutation research (Zhang and van der Meer, 2011; Fu et al., 2014), mitochondrial genome (Zhang et al., 2012), chloroplast genome (Du et al., 2016) and genomic studies (Zhou et al., 2013; Sun et al., 2018)
Currently, Gp. Lemaneiformis is broadly cultivated in Guangdong, Fujian, Zhejiang, Shandong, and Liaoning Provinces, representing the second most cultivated seaweed in China. Screening for new strains and developing large-scale industrialized cultivation methods are of great significance for the use of this species. Molecular marker-assisted breeding technology can be used for mapping important quantitative trait loci (QTL), allowing the selection of a trait by analyzing molecular markers that are closely linked to the target trait’s loci. It is an effective way to accelerate the breeding process (Ribaut and Hoisington, 1998). Genetic linkage maps play a fundamental role in QTL localization by providing selectable markers for traits selection (Li et al., 2015; Lu et al., 2018; Raman et al., 2018). Due to their relative abundance, high polymorphism rate, stable inheritance, and wide genomic distributions, single nucleotide polymorphism (SNP) markers have been widely applied in molecular marker assisted breeding (Berger et al., 2001; Wicks et al., 2001; Stickney et al., 2002; Chen et al., 2008). The development of SNP markers for breeding have never been conducted in Gp. lemaneiformis. With the advent of next-generation sequencing which provide rapid and low-cost methods for genotyping large populations, SNP markers can be obtained using reduced-representation sequencing (Baird et al., 2008; Kerstens et al., 2009; Wang et al., 2012; Poland et al., 2012; Fu et al., 2016) and whole-genome resequencing (Xia et al., 2009; Huang et al., 2009; Lam et al., 2010; Cao et al., 2011).
Because of the industrial importance of the species and the usefulness of SNP markers in species breeding, SNPs were exploited in the parental gametophytes and 60 F1 gametophytes of Gp. lemaneiformis in this study. If the F1 tetrasporophyte (2n) is heterozygous at one SNP locus, F1 gametophytes (n) should be homozygous and the segregation ratios of the two genotypes should be 1:1 at this locus. However, we identified abundant heterozygous SNP loci in gametophytes, and we intend to investigate where the heterozygous SNP loci originated. This study promotes investigating the genomic characteristics of Gp. lemaneiformis, which will be of great importance in guiding studies on the genomic evolution of marine algae.
Materials and Methods
Construction of Mapping Population
The life history of Gp. lemaneiformis includes a gametophyte phase, which is equal to the haploid population, and it can be used for genetic linkage mapping. In this study, the female gametophyte ♀6 and male gametophyte ♂9 were produced from Cultivar Lulong No. 1, which is tetrasporophyte strain. F1 tetrasporophytes were produced from a cross between ♀6 and ♂9. Then, mature F1 tetrasporophytes gave rise to tetraspores, which subsequently developed into F1 gametophytes. Whole-genome resequencing was carried out for the two parental gametophytes and 60 F1 gametophytes.
Whole-Genome Resequencing Protocol
Genomic DNA was extracted using a Plant Genomic DNA Kit (Tiangen Biotech, Beijing, China) following the manufacturer’s instructions, and at least 1 μg qualified genomic DNA was used for constructing a whole-genome resequencing library. Genomic DNA was sheared randomly into fragments, and 300–500-bp fragments were purified using gel electrophoresis. The sequencing library was prepared using a TruSeq™ DNA Sample Prep Kit and a TruSeq PE Cluster Kit. The prepared library was sequenced with paired end reads (2 × 150 bp) on an Illumina HiSeq 4000 sequencing platform. The whole-genome resequencing data of 62 gametophytes have been deposited in Short Read Archive (SRA) database with Project number PRJNA574029, under the accession number SRP223151.
Raw sequence reads were processed using custom PERL scripts to separate them into individual fastq format files based on the barcode sequence of each sample. Contamination reads, like those containing adaptors or primers, were identified using SeqPrep (https://github.com/jstjohn/SeqPrep) with the following parameters: “'-q 20 -L 75 -B AGATCGGAAGAGCGTCGTGT -A AGATCGGAAGAGCACACGTC.” Sickle (https://github.com/najoshi/sickle) was applied to perform read data trimming with default parameters to produce clean data (high quality data) in this study. The high quality sequencing reads were aligned to the reference genome sequence using BWA (http://bio-bwa.sourceforge.net/) software in “bwa aln” mode. The genome survey sequencing of Gp. lemaneiformis had been conducted previously (Zhou et al., 2013), and the assembled scaffolds were used as reference genome sequences in this study. After removing PCR-duplication reads using SAMtools (http://samtools.sourceforge.net/) software, the sequencing depth and coverage were calculated based on alignments performed by custom PERL scripts.
SNP Calling
The valid BAM file was used to detect SNPs using the GATK “UnifiedGenotyper” function (http://www.broadinstitute.org/gatk/). Then, variant call format (VCF) files were generated by quality filtering (VariantFiltration with parameters: QD < 2.0 || FS > 60.0 || MQ <40.0 || SOR > 10.0). Furthermore, VCF files were filtered using VCFtools (version 0.1.11; parameters: –minQ 20 –minDP 4).
Verification of Heterozygous SNP Loci
The SNP loci that had identical heterozygous genotypes in the two parents, were selected to determine whether they belonged to exogenous DNA contamination. Sequences of 400 bp in which a heterozygous SNP locus was located were obtained from reference genome sequences, and served as target sequences. BLASTN in NCBI with default parameters was used for sequence annotation. The SNP loci that had a genotype that was homozygous (or missing) in one parent but heterozygous in the other, were suspected to result from cytoplasmic inheritance. Sequences of 400 bp containing the target SNP were aligned to the complete plastid and mitochondrial sequences.
SNP primers were designed using Primer 6.0 and synthesized by Sangon Biotech (Shanghai, China). The reaction solution for PCR amplification contained 20 ng genomic DNA, 1× PCR buffer, 1.5 mm Mg2+, 0.2 mm dNTPs, 0.5 μm of each primer, 0.5 U Taq DNA polymerase (Fermentas, Burlington, Canada) and autoclaved distilled water to a total volume of 20 μl. The PCR program performed was as follows: initial denaturation at 94°C for 5 min; 35 cycles of denaturation for 1 min at 94°C, annealing for 1 min at 60°C and extension for 1 min at 72°C; a final extension step for 5 min at 72°C. Sanger sequencing of PCR products and monoclones were completed by TsingKe Biological Technology (Qingdao, China)
Chimera Testing
To determine whether heterozygous SNPs resulted from chimerism, a single gametophyte of Gp. lemaneiformis was cut from the base to the top, obtaining 22 1 cm segments. The segments included nine representative parts: base, middle part, and tip of primary/secondary branch; junctions of primary and secondary branches; junctions of secondary and tertiary branches; tertiary branches. The genomic DNA of every segment was extracted and amplified using primers for SNP locus scaffold1837_730, scaffold7339_502 and scaffold7339_533, and then, the PCR products were analyzed by Sanger sequencing.
SNP Detection in Single Cells
A gametophyte thallus was used to produce a cell suspension by grinding in liquid nitrogen and filtering. Then, single cells were isolated in a glass capillary tube under a microscope. Genomic DNA of each cell was extracted and amplified using a Single Cell Whole Genome Amplification Kit (Yikon Genomics, Suzhou, China) following the manufacturer’s instructions. Then, heterozygous SNP loci were identified using PCR amplification and Sanger sequencing.
Results
Development of SNPs Using Whole-Genome Resequencing Technology
The statistical data of the whole-genome resequencing are listed in Table S1. The total data for the female gametophyte parent, male gametophyte parent and the mean of the F1 gametophytes were 1.21, 1.36 and 1.62 Gb, respectively. The GC content range per individual was between 45.03 and 52.53%, with a mean of 48.92%, and all three values were in the normal range. The mean Q20 value was greater than 95%, and the mean Q30 value was greater than 91%, revealing the high quality of sequencing. The depth of the female, male gametophyte parents, and the mean of the F1 gametophytes, were 12×, 14× and 16×, respectively. Each gametophyte was sequenced to an average 80.15% coverage of the genome. The base distribution and base quality distribution of whole-genome resequencing data were shown in Figures S1 and S2.
The high quality sequencing reads of each individual were aligned to the reference genome sequences, and the mapping results are displayed in Table S2. The mapping rate of the female parent was 84.35% and that of the male parent was 49.15%. The percentage of mapped reads among the 60 F1 gametophytes was between 30.20 and 83.75%, with an average of 57.33%. The differences in the mapping rates among individuals might be caused by exogenous DNA contamination or unprovided reference genome sequences of Gp. lemaneiformis.
As a result, the miss rate of SNP genotypes was less than 15% in 62 gametophytes, and 46,499 SNPs were identified, representing 1.83% homozygous loci (no homozygous genotypes occurred in the 62 gametophytes) and 98.17% heterozygous loci (heterozygous genotypes occurred in at least one individual). Further statistical analyses revealed that 22,640 SNP loci had identical heterozygous genotypes in the 62 gametophytes, accounting for 48.69% of the total SNPs.
Discarding SNPs Located in Repetitive Sequences
The annotation of repetitive sequences had been conducted previously (Zhou et al., 2013). SNP loci that were located in repetitive sequences were discarded, and 9,989 SNP markers remained. In these SNPs, the number of loci that were identified as having a heterozygous genotype in at least one individual accounted for 92.02%, and 48.07% loci had identical heterozygous genotypes in the 62 gametophytes. In addition, the number of homozygous SNPs with different genotypes in the two parents was only 25, and these loci did not have 1:1 segregation ratios in the 60 F1 gametophytes. Genotyping results of representative SNP loci in two parents and partial gametophytes are displayed in Table 1. Complete genotype information for the 9,989 SNPs is shown in Appendix S1.
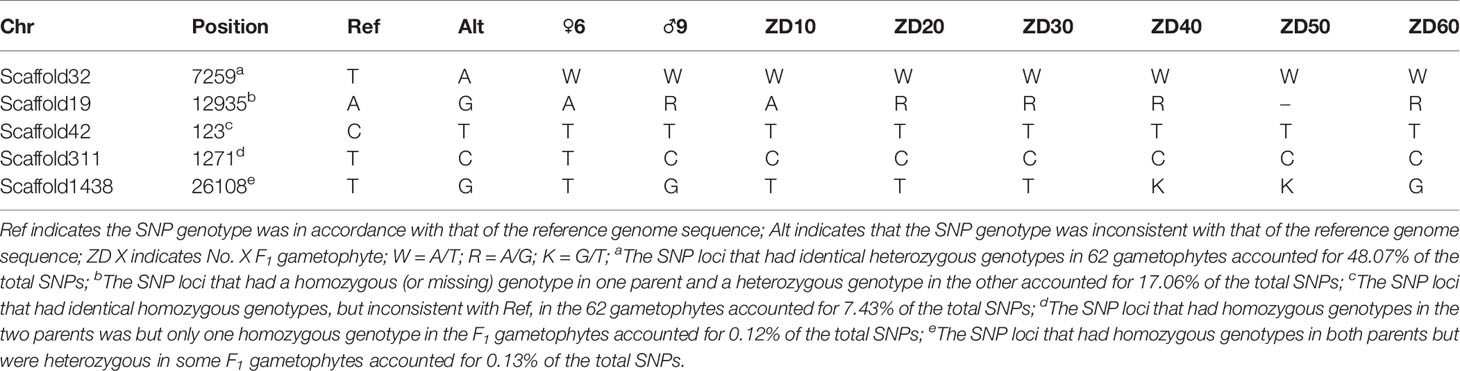
Table 1 Genotyping results of representative SNP loci in two parents and partial F1 gametophytes using whole-genome resequencing technology.
For each gametophyte, the proportions of homozygous and heterozygous loci ranged between 13.74 and 21.61% (mean of 17.04%) and between 66.36 and 83.59% (mean of 77.12%), respectively. The remainder were missing loci, representing an average 5.84% (Figure 1). An overwhelming number of heterozygous loci existed in all the gametophytes.
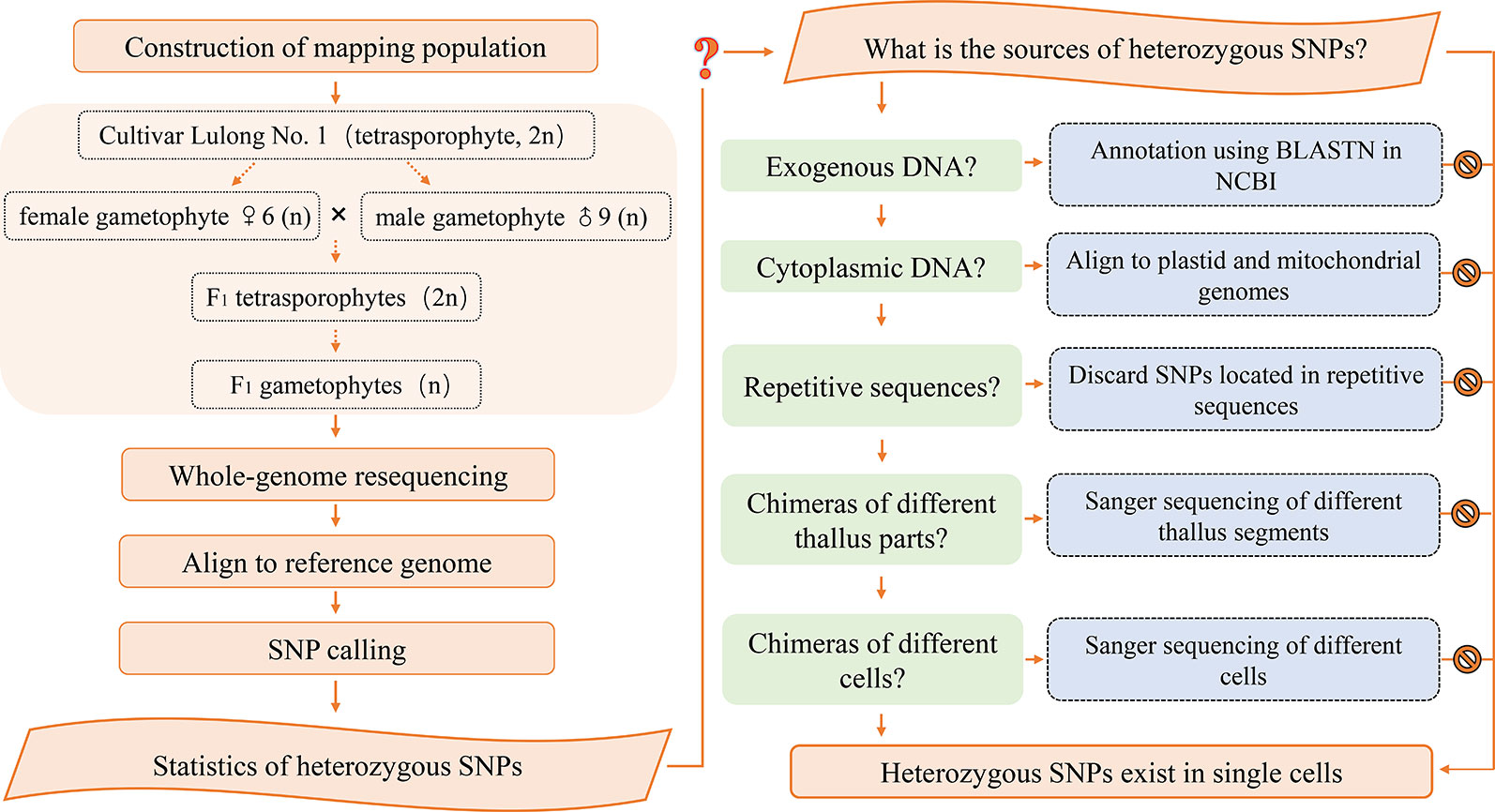
Figure 1 Flowchart of the research methodology.
Verification of Heterozygous SNPs
The unmapped reads obtained from whole-genome resequencing were removed to filter out exogenous DNA contamination. However, because the reference genome was the result of a preliminary genome survey assembly, it was possible that exogenous DNA existed in the assembled reference genome. The GC content and average sequencing depth of the genomic data of Gp. lemaneiformis had been reported previously (Zhou et al., 2013) and are shown in Figure S3. The density points were concentrated in the 40–60% range, revealing no exogenous DNA contamination. For further verification, 6,755 SNPs that had identical heterozygous genotypes in the two parental gametophytes were annotated using BLASTN in NCBI. As shown in Table 2, 6,687 sequences were not annotated to any species, representing a proportion of 98.99%. In total, 68 sequences were mapped to similar sequences in the NT database, with matched species from four classes, bacteria, fungi, red alga, and animal, accounting for only 1%. The annotation results reflected the unlikelihood that the heterozygous SNPs were derived from exogenous DNA.
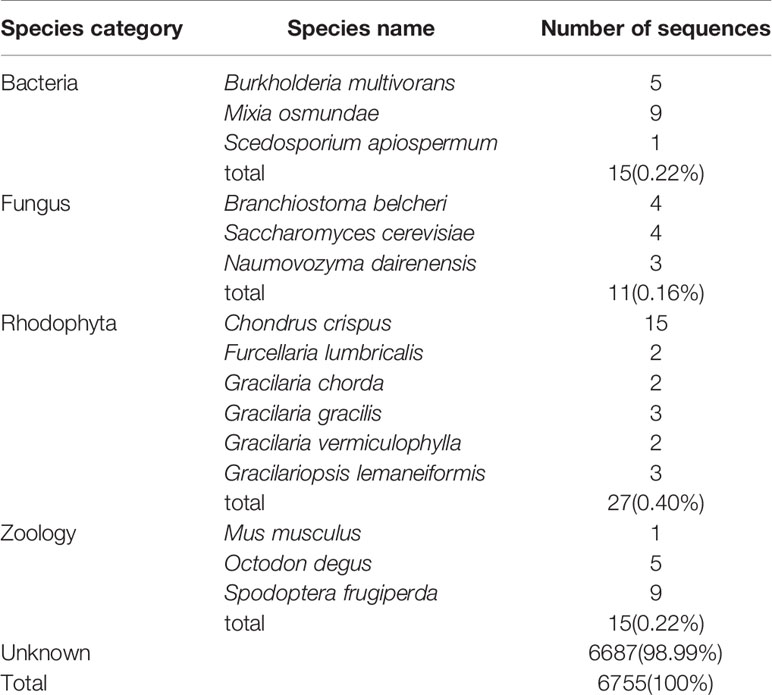
Table 2 Annotation results of the sequences containing heterozygous SNP loci in Gracilariopsis lemaneiformis.
In this study, each gametophyte was cultivated in a separate bottle and the genomic DNA was extracted independently, which should eliminate the possibility of cross contamination. Moreover, a high proportion of heterozygous sites existed in each individual, as shown in Figure 2, which further indicated that the heterozygous SNPs did not result from cross contamination among certain individuals.
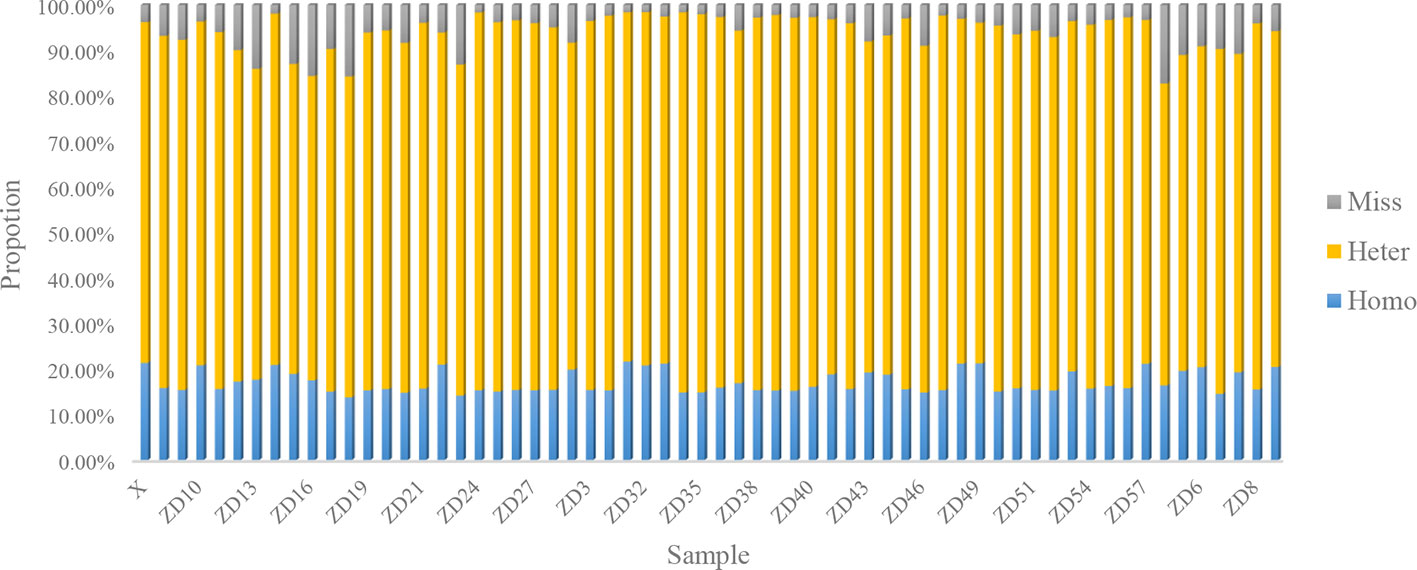
Figure 2 Statistics of SNP type in parents and 60 gametophytes.
In total, 1,704 SNP loci that had homozygous (or missing) genotypes in one parent, but heterozygous genotypes in the other parent, were aligned to the complete plastid and mitochondrial sequences of Gp. lemaneiformis. However, none of the sequences containing SNPs were mapped to plastid or mitochondrial sequences, excluding the possibility that sequence contamination occurred from plastids or mitochondria.
Information regarding primers that were used to amplify SNP loci are listed in Table 3. Sanger sequencing results for three SNP loci in one F1 gametophyte are displayed in Figure 3A. The genotyping results of the SNP locus scaffold1837_730 was A/G, and the Sanger sequencing of PCR products showed two peaks in this site, with peak A being higher than peak G. In addition, Sanger sequencing results of SNP loci scaffold7339_502 and scaffold7339_533 in this gametophyte also showed two peaks of SNPs that were consistent with the genotyping results obtained from whole-genome resequencing. To demonstrate the existence of the lower peak, monoclonal sequencing of the locus scaffold1837_730 was carried out in this gametophyte. In total, 54 monoclones were selected for sequencing. Of these, 53 monoclones had the single peak A and one monoclone had the single peak G, which verified the accuracy of the genotyping results.
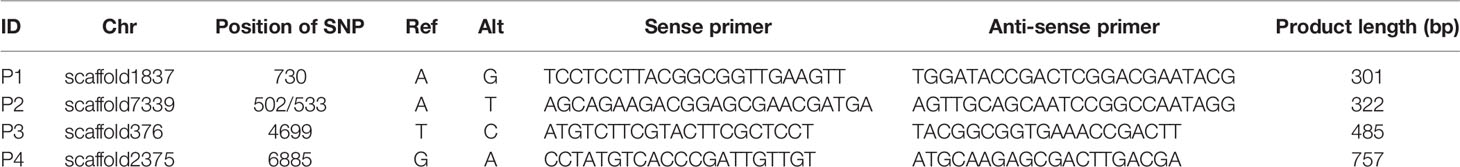
Table 3 PCR primers used for Sanger sequencing.
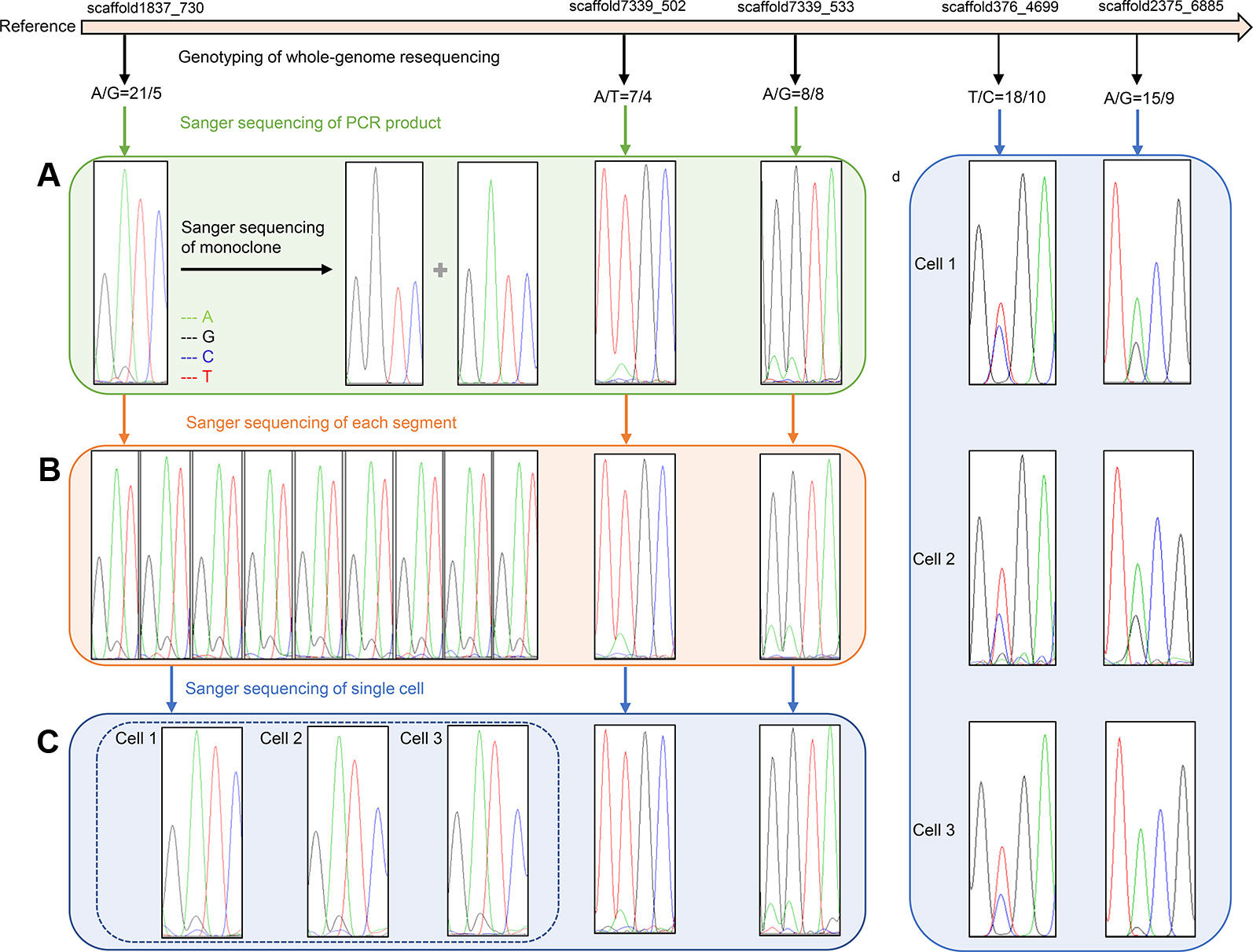
Figure 3 Verification results of heterozygous SNPs (A) Sanger sequencing results of three SNP loci (scaffold1837_730, scaffold7339_502 and scaffold7339_533) in one F1 gametophyte. Two peaks of each SNP cite are consistent with the genotyping results of whole-genome resequencing. Monoclonal sequencing results of the locus scaffold1837_730 show both single peak A and single peak G, which completely verified the accuracy of the genotyping results A/G. (B) Sanger sequencing results of three SNP loci in different segments of one F1 gametophyte. The Sanger sequencing results of nine representative parts at locus scaffold1837_730 are completely coincident, showing two peak A and G, and are consistent with genotyping results A/G. (C) Sanger sequencing results of three SNP loci in three cells of one F1 gametophytes. The Sanger sequencing results of three cells at locus scaffold1837_730 are completely coincident, showing two peaks A and G, and are consistent with the genotyping results A/G. (D) Sanger sequencing results of three cells at two SNP sites (scaffold376_4699 and scaffold2375_6885) located in gene regions. Two Sanger sequencing peaks of each SNP site located in gene region still existed, which are consistent with the genotyping results.
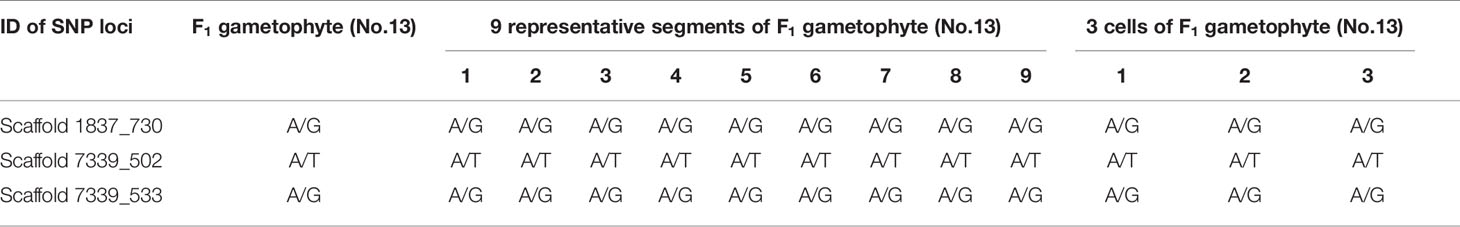
Table 4 Genotyping results of 3 SNP loci in F1 gametophytes (No.13) using whole-genome resequencing technology and Sanger sequencing results in different segments and single cells.
Excluding the Possibility of Chimerism
All 22 segments that were cut from one gametophyte were amplified using primers for SNP locus scaffold1837_730. The Sanger sequencing results of each segment were completely coincident, showing two peaks, A and G, and even the height differences between peaks A and G were identical (Figure 3B).
Additionally, SNP loci scaffold7339_502 and scaffold7339_533 were detected in each part of the gametophyte. For each SNP locus, the Sanger sequencing results for each part were completely coincident and were consistent with the genotyping results derived from whole-genome resequencing, as shown in Figure 3B and Table 4.
Detection of Heterozygous SNPs in a Single Cell
Single cells were isolated and selected using glass capillary tube (Figure S4). Sanger sequencing results for three SNP loci (scaffold1837_730s, scaffold7339_502 and scaffold7339_533) in three cells from one F1 gametophyte are displayed in Figure 3C. The results were consistent with the genotyping results, as shown in Table 4. Thus, heterozygous SNP sites existed in single cells.
To eliminate background interference, high fidelity DNA polymerase was used to conduct the PCR amplification of three cells. Two SNP loci, scaffold376_4699 and scaffold2375_6885, which were located in genic regions, were further used to test single cells. Sanger sequencing results revealed that the two peaks of each SNP locus located in the genomic region still existed, which was consistent with the genotyping results of the whole-genome resequencing (Figure 3D). Thus, the SNP genotypes were shown to be heterozygous in individual cells.
Inference That Heterozygous SNPs Exist in Haploid Gametophytes of Gp. lemaneiformis
Based on our results, exogenous DNA contamination, cross contamination among individuals, plastid and mitochondrial sequences, chimeras of different thallus parts, or different cells, and repetitive sequences were excluded as being the source of heterozygous SNPs. However, the reference sequence we used to identify SNPs in this study, was a preliminary assembly version, with a scaffold N50 of 20 kb and a scaffold number of 125,685. If a chromosomal rearrangement event, for instance, a duplication and deletion, occurred in the genome of Gp. lemaneiformis, the length of a scaffold might not cover the large area of duplication. Consequently, the unique scaffold may actually exist in multiple copies in the genome. This could explain why heterozygous SNPs existed in haploid gametophyte genomes.
Discussion
In this study, whole-genome resequencing was conducted to develop SNP markers, which have been widely applied in many plants. Whole-genome resequencing of two parental lines Brassica oleracea L. C1184 and C1234 was performed to develop genome-wide SNP markers (Lee et al., 2015), and the sequencing data represented approximately 18-fold genome coverage for both parental lines. After reference-based genome-guided mapping, 82.1 and 77.6% of reads from C1184 and C1234, respectively, were successfully aligned to the reference genome. As a result, a total of 674,521 SNPs was detected between C1184 and C1234. In Capsicum baccatum L. (Lee et al., 2016), the parental genomes were sequenced using the whole-genome resequencing method. After trimming the raw data, the parental sequences corresponded to approximately 3.1 and 9.6 times, respectively, the size of the reference genome. Then, 48.5% of the trimmed reads were successfully mapped to the reference sequences. Finally, 97,085 SNPs were selected as homozygous SNPs between the parents. Whole-genome resequencing was performed on 96 F6 recombinant inbred lines (RILs) of a cross between safflower Carthamus tinctorius L. and its wild progenitor C. palaestinus Eig (Bowers et al., 2016). Each RIL was sequenced to an average 2.26 × coverage of the estimated genome, while the parents were sequenced to a greater depth. Sequence reads from the RILs were mapped to the reference genome, and 2,008,196 SNPs were identified.
In our result, the number of SNPs (46,499) identified by whole-genome resequencing in Gp. lemaneiformis was smaller than in other plants, which might result from the small genome and population sizes. However, as C-value paradox indicates that there is no absolute correlation between the size of a genome and evolutionary status of an organism. Although the genome size of Gp. lemaneiformis is only 97.02 Mb (Zhou et al., 2013), its complexity level may be high.
The reference genome sequences selected in this study were from the first genome version, which consisted of 125,685 scaffolds with an N50 length of 20 kb (Zhou et al., 2013) and not from the second version, which consisted of 13,825 scaffolds with an N50 length of 30.59 kb (Sun et al., 2018). The main reason for the selection was that first genome sequencing was a wild female gametophyte, as our resequencing gametophyte, while in the second version, the material was the 981 strain, which is a diploid tetrasporophyte.
SNP markers were first developed in F1 gametophytes of Gp. lemaneiformis using whole-genome resequencing technology, laying a foundation for construction of genetic linkage map and localization of quantitative trait locus. We further identified 2,386 SNPs which were located in 818 genes. For example, SNPs (scaffold2671_ 31163,31176,31189,31266) were located in the gene region of LEM_GLEAN_10003235 whose function was annotated as carotenoid isomerase, which is an important enzyme in synthesis pathway of carotenoids. Moreover, LEM_GLEAN_0001204, the gene encoding phosphoglucose isomerase in metabolic pathways of agar, contains one SNP locus (scaffold1981_ 16188). These SNPs could be potentially used as genetic markers for the selection and provided the theoretical basis of molecular marker assisted breeding
An overwhelming number of heterozygous SNP loci were verified to exist in the 62 gametophytes, which might result from chromosomal duplication events in the genome of Gp. lemaneiformis. There are several kinds of chromosomal rearrangement, including the duplication or deletion of a chromosomal segment, the inversion of a segment and the translocation of segments between non-homologous chromosomes (Rieseberg 2001). Each chromosomal rearrangement event can be caused by the breakage of DNA double-helices at two different locations, followed by rejoining or by crossing-over between repetitive DNA (Griffiths et al., 1999). Chromosomal rearrangements in the genome can produce duplicated genes, which may differ in DNA sequence, gene structure and even function. One genetic effect of duplication is that the gene expression may be enhanced as the gene copies increase in number.
A chromosome level of assembly is required to verify the source of the heterozygous SNPs. This study promoted the assembly of a Gp. lemaneiformis genomic map of using single molecule real-time sequencing (SMRT), or high-throughput chromosome conformation capture. SMRT sequencing can produce considerably longer and highly accurate reads from single unamplified molecules. The average read length of PacBio RS II platform is about 15 kb. The long SMRT sequencing reads will span complex repeats and missing bases, which will substantially improve genome complete assembly. A complete genome has the potential to facilitate genome-assisted breeding and will be useful to investigate genomic patterns in the evolution of marine algae.
Data Availability Statement
The datasets generated for this study can be found in the Short Read Archive (SRA) database with Project number PRJNA574029, under the accession number SRP223151 (SRR10177194 – SRR10177255).
Author Contributions
ZS and YH contributed conception and design of the study. YH, WZho and JW constructed the mapping population. YH performed the experiments. YH and ZS analyzed the data. MJ, HC, JZ, WZha, XF, YL, BX and PG contributed reagents/materials/analysis tools. YH wrote the manuscript and ZS made revision. All authors read and approved the submitted version.
Funding
This work was supported by the China Agriculture Research System (CARS-50), the National Natural Science Foundation of China (No. 31372529) and the Fundamental Research Funds for the Central Universities (No. 201762016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01256/full#supplementary-material
References
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS One 3, e3376. doi: 10.1371/journal.pone.0003376
Berger, J., Suzuki, T., Senti, K., Stubbs, J., Schaffner, G., Dickson, B. J. (2001). Genetic mapping with SNP markers in Drosophila. Nat. Genet. 29, 475–481. doi: 10.1038/ng773
Bowers, J. E., Pearl, S. A., Burke, J. M. (2016). Genetic mapping of millions of SNPs in safflower (Carthamus tinctorius L.) via whole genome resequencing. G3: Genes Genom. Genet. 6, 2203–2211. doi: 10.1534/g3.115.026690
Cao, J., Schneeberger, K., Ossowski, S., Gunther, T., Bender, S., Fitz, J., et al. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963. doi: 10.1038/ng.911
Chen, D., Ahlford, A., Schnorrer, F., Kalchhauser, I., Fellner, M., Viragh, E., et al. (2008). High-resolution, high-throughput SNP mapping in Drosophila melanogaster. Nat. Methods 5, 323–329. doi: 10.1038/nmeth.1191
Chopin, T., Buschmann, A. H., Halling, C., Troell, M., Kautsky, N., Neori, A., et al. (2001). Integrating seaweeds into marine aquaculture systems: a key toward sustainability. J. Phycol. 37, 975–986. doi: 10.1046/j.1529-8817.2001.01137.x
Ding, H. Y., Sui, Z. H., Zhong, J., Zhou, W., Wang, Z. X. (2012). Analysis and comparison on genetic diversity of haploid and diploid Gracilaria lemaneiformis populations from different places of Qingdao by AFLP. Period Ocean Univ. China 42, 99–105. doi: 10.1007/s11783-011-0280-z
Du, Q. W., Bi, G. Q., Mao, Y. X., Sui, Z. H. (2016). The complete chloroplast genome of Gracilariopsis lemaneiformis (Rhodophyta) gives new insight into the evolution of family Gracilariaceae. J. Phycol. 52, 441–450. doi: 10.1111/jpy.12406
Engel, C., Aberg, P., Gaggiotti, O. E., Destombe, C., Valero, M. (2010). Population dynamics and stage structure in a haploid-diploid red seaweed, Gracilaria gracilis. J. Ecol. 89, 436–450. doi: 10.1046/j.1365-2745.2001.00567.x
Fei, X. G. (2004). Solving the coastal eutrophication problem by large scale seaweed cultivation. Hydrobiologia 512, 145–151. doi: 10.1023/b:hydr.0000020320.68331.ce
Fu, F., Sui, Z. H., Zhou, W., Wang, J. G., Chang, L. P., Ci, S. F. (2014). UV-irradiation mutation of tetraspores of Gracilariopsis lemaneiformis and screening of thermotolerant strains. J. Appl. Phycol. 26, 647–656. doi: 10.1007/s10811-013-0087-3
Fu, B., Liu, H., Yu, X., Tong, J. (2016). A high-density genetic map and growth related QTL mapping in bighead carp (Hypophthalmichthys nobilis). Sci. Rep. 6, 28679. doi: 10.1038/srep28679
Griffiths, A. J. F., Gelbart, W. M., Miller, J. H., Lewontin., R. C. (1999). Modern Genetic Analysis (New York: W. H. Freeman), 319 pp.
Guzmán-Urióstegui, A., García-Jiménez, P., Marián, F., Robledo, D., Robaina, R. (2002). Polyamines influence maturation in reproductive structures of Gracilaria cornea (Gracilariales, Rhodophyta). J. Phycol. 38, 1169–1175. doi: 10.1046/j.1529-8817.2002.01202.x
Hu, Y. Y., Sui, Z. H., Zhou, W., Wang, J. G., Chang, L. P., Guo, W. H., et al. (2018). Development of genomic simple sequence repeat markers and genetic diversity analysis of Gracilariopsis lemaneiformis (Rhodophyta). J. Appl. Phycol. 30, 707–716. doi: 10.1007/s10811-017-1237-9
Hu, Y. Y., Du, Q. W., Mi, P., Shang, E. L., Sui, Z. H. (2019). Gene cloning and expression regulation in the pathway of agar and floridean starch synthesis of Gracilariopsis lemaneiformis (Rhodophyta). J. Appl. Phycol. 31, 1889–1896. doi: 10.1007/s10811-018-1690-0
Huang, X., Feng, Q., Qian, Q., Zhao, Q., Wang, L., Wang, A., et al. (2009). High-throughput genotyping by whole-genome resequencing. Genome Res. 19, 1068–1076. doi: 10.1101/gr.089516.108
Kerstens, H. H., Crooijmans, R. P., Veenendaal, A., Dibbits, B. W., Chin-A-Woeng, T. F., Dunnen, J. T. D., et al. (2009). Large scale single nucleotide polymorphism discovery in unsequenced genomes using second generation high throughput sequencing technology: applied to turkey. BMC Genomics 10, 479. doi: 10.1186/1471-2164-10-479
Lam, H. M., Xu, X., Liu, X., Chen, W. B., Yang, G. H., Wong, F. L., et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Lee, J., Izzah, N. K., Jayakodi, M., Perumal, S., Joh, H. J., Lee, H. J., et al. (2015). Genome-wide SNP identification and QTL mapping for black rot resistance in cabbage. BMC Plant Biol. 15, 32. doi: 10.1186/s12870-015-0424-6
Lee, Y. R., Yoon, J. B., Lee, J. (2016). A SNP-based genetic linkage map of Capsicum baccatum and its comparison to the Capsicum annuum reference physical map. Mol. Breed. 36, 61. doi: 10.1007/s11032-016-0485-8
Li, C. L., Bai, G. H., Carver, B. F., Chao, S. M., Wang, Z. H. (2015). Single nucleotide polymorphism markers linked to QTL for wheat yield traits. Euphytica 206, 89–101. doi: 10.1007/s10681-015-1475-3
Liu, Y. T., Zhang, X., Sun, H. Y., Yang, Q., Zang, X. N., Zhang, X. C., et al. (2016). Cloning and transcription analysis of six members of the calmodulin family in Gracilaria lemaneiformis under heat shock. J. Appl. Phycol. 28, 643–651. doi: 10.1007/s10811-015-0575-8
Liu, Y. T., Sun, H. Y., Ding, Y., Zang, X. N., Zhang, X. C. (2018). A novel heat shock protein from Gracilariopsis lemaneiformis: gene cloning and transcription analysis in response to heat stress. J. Appl. Phycol. 30, 3623–3631. doi: 10.1007/s10811-018-1494-2
Lu, H., Xie, H., Gong, Y., Wang, Q., Yang, Y. (2011). Secondary metabolites from the seaweed Gracilaria lemaneiformis and their allelopathic effects on Skeletonema costatum. Biochem. Syst. Ecol. 39, 397–400. doi: 10.1016/j.bse.2011.05.015
Lu, J. J., Liu, Y. Y., Xu, J., Mei, Z. W., Ziwei, M., Shi, Y. J., et al. (2018). High-density genetic map construction and stem total polysaccharide content-related qtl exploration for chinese endemic Dendrobium (Orchidaceae). Front. Plant Sci. 9, 398. doi: 10.3389/fpls.2018.00398
Mchugh, D. J. (1991). Worldwide distribution of commercial resources of seaweeds including Gelidium. Hydrobiologia 221, 19–29. doi: 10.1007/BF00028359
Neori, A., Chopin, T., Troell, M., Buschmann, A. H., Kraemer, G. P., Halling, C., et al. (2004). Integrated aquaculture: rationale, evolution and state of the art emphasizing seaweed biofiltration in modern mariculture. Aquaculture 231, 361–391. doi: 10.1016/j.aquaculture.2003.11.015
Pang, Q., Sui, Z., Kang, K. H., Kong, F., Zhang, X. (2010). Application of SSR and AFLP to the analysis of genetic diversity in Gracilariopsis lemaneiformis (Rhodophyta). J. Appl. Phycol. 22, 607–612. doi: 10.1007/s10811-009-9500-3
Plastino, E. M., de Oliveira, F. E. C. (1988). Deviations in the life-history of Gracilaria sp. (Rhodophyta, Gigartinales), from Coquimbo, Chile, under different culture conditions. Hydrobiologia 164, 67–74. doi: 10.1007/bf00014350
Poland, J., Brown, P. J., Sorrells, M. E., Jannink, J. (2012). Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PloS One 7, e32253. doi: 10.1371/journal.pone.0032253
Raman, D., George, F., Yves, D., Curtis, P., Laroche, A., Eudes, F., et al. (2018). High density single nucleotide polymorphism (SNP) mapping and quantitative trait loci (QTL) analysis in a biparental spring triticale population localized major and minor effect fusarium head blight resistance and associated traits QTL. Genes 9, 19. doi: 10.3390/genes9010019
Ribaut, J. M., Hoisington, D. A. (1998). Marker-assisted selection: new tools and strategies. Trend Plant Sci. 3, 236–239. doi: 10.1016/s1360-1385(98)01240-0
Rieseberg, L. (2001). Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358. doi: 10.1016/S0169-5347(01)02187-5
Santelices, B., Doty, M. S. (1989). A review of Gacilaria farming. Aquaculture 78, 95–133. doi: 10.1016/0044-8486(89)90026-4
Stickney, H. L., Schmutz JWoods, I. G., Holtzer, C. C., Dickson, M. C., Kelly, P. D., Myers, R. M., et al. (2002). Rapid mapping of zebrafish mutations with SNPs and oligonucleotide microarrays. Genome Res. 12, 1929–1934. doi: 10.1101/gr.777302
Sun, X., Wu, J., Wang, G., Kang, Y., Ooi, H. S., Shen, T., et al. (2018). Genomic analyses of unique carbohydrate and phytohormone metabolism in the macroalga Gracilariopsis lemaneiformis (Rhodophyta). BMC Plant Biol. 18, 94. doi: 10.1186/s12870-018-1309-2
Wang, W. J., Wang, Z. Q., Lin, X. Z., Xu, P. (2007). Characterization of Gracilaria lemaneiformis Bory (Gracilariaceae, Rhodophyta) cultivars in China using the total soluble proteins and RAPD analysis. Bot. Mar. 50, 177–184. doi: 10.1515/bot.2007.021
Wang, S., Meyer, E., Mckay, J. K., Matz, M. V. (2012). 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat. Methods 9, 808–810. doi: 10.1038/nmeth.2023
Wicks, S. R., Yeh, R. T., Gish, W. R., Waterston, R. H., Plasterk, R. H. A. (2001). Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 28, 160–164. doi: 10.1038/88878
Xia, Q., Guo, Y., Zhang, Z., Li, D., Xuan, Z., Li, Z., et al. (2009). Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 326, 433–436. doi: 10.1126/science.1176620
Yamamoto, H., Sasaki, J. (1988). Interfertility between so-called Gracilaria verrucosa (Huds.) Papenfuss and G. vermiculophylla (Ohmi) Papenfuss in Japan. Bull. Fac. Fish. Hokkaido Univ. 39, 1–3.
Zhang, X. C., van der Meer, J. P. (2011). A genetic study on Gracilaria sjoestedtii. Can. J. Bot. 66, 2022–2026. doi: 10.1139/b88-276
Zhang, L., Wang, X. M., Qian, H., Chi, S., Liu, C., Liu, T. (2012). Complete sequences of the mitochondrial DNA of the wild Gracilariopsis lemaneiformis and two mutagenic cultivated breeds (Gracilariaceae, Rhodophyta). PloS One 7, e40241. doi: 10.1371/journal.pone.0040241
Keywords: Gracilariopsis lemaneiformis, haploid gametophyte, single nucleotide polymorphism, heterozygous genotype, whole-genome resequencing
Citation: Hu Y, Sui Z, Zhou W, Wang J, Jiang M, Chen H, Zhang J, Zhang W, Feng X, Lei Y, Xiao B and Guo P (2019) Heterozygous Single Nucleotide Polymorphic Loci in Haploid Gametophytes of Gracilariopsis lemaneiformis (Rhodophyta). Front. Genet. 10:1256. doi: 10.3389/fgene.2019.01256
Received: 13 May 2019; Accepted: 14 November 2019;
Published: 06 December 2019.
Edited by:
Youri I. Pavlov, University of Nebraska Medical Center, United StatesReviewed by:
Prashanth N. Suravajhala, Birla Institute of Scientific Research, IndiaWei Xu, Texas A&M University Corpus Christi, United States
Copyright © 2019 Hu, Sui, Zhou, Wang, Jiang, Chen, Zhang, Zhang, Feng, Lei, Xiao and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenghong Sui, c3VpemhlbmdoQG91Yy5lZHUuY24=