 Tatiana Shaurova†
Tatiana Shaurova† David W. Goodrich
David W. Goodrich Pamela A. Hershberger
Pamela A. Hershberger- Department of Pharmacology and Therapeutics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, United States
Somatic alterations in the epidermal growth factor receptor gene (EGFR) result in aberrant activation of kinase signaling and occur in ∼15% of non-small cell lung cancers (NSCLC). Patients diagnosed with EGFR-mutant NSCLC have good initial clinical response to EGFR tyrosine kinase inhibitors (EGFR TKIs), yet tumor recurrence is common and quick to develop. Mechanisms of acquired resistance to EGFR TKIs have been studied extensively over the past decade. Great progress has been made in understanding two major routes of therapeutic failure: additional genomic alterations in the EGFR gene and activation of alternative kinase signaling (so-called “bypass activation”). Several pharmacological agents aimed at overcoming these modes of EGFR TKI resistance are FDA-approved or under clinical development. Phenotypic transformation, a less common and less well understood mechanism of EGFR TKI resistance is yet to be addressed in the clinic. In the context of acquired EGFR TKI resistance, phenotypic transformation encompasses epithelial to mesenchymal transition (EMT), transformation of adenocarcinoma of the lung (LUAD) to squamous cell carcinoma (SCC) or small cell lung cancer (SCLC). SCLC transformation, or neuroendocrine differentiation, has been linked to inactivation of TP53 and RB1 signaling. However, the exact mechanism that permits lineage switching needs further investigation. Recent reports indicate that LUAD and SCLC have a common cell of origin, and that trans-differentiation occurs under the right conditions. Options for therapeutic targeting of EGFR-mutant SCLC are limited currently to conventional genotoxic chemotherapy. Similarly, the basis of EMT-associated resistance is not clear. EMT is a complex process that can be characterized by a spectrum of intermediate states with diverse expression of epithelial and mesenchymal factors. In the context of acquired resistance to EGFR TKIs, EMT frequently co-occurs with bypass activation, making it challenging to determine the exact contribution of EMT to therapeutic failure. Reversibility of EMT-associated resistance points toward its epigenetic origin, with additional adjustments, such as genetic alterations and bypass activation, occurring later during disease progression. This review will discuss the mechanistic basis for EGFR TKI resistance linked to phenotypic transformation, as well as challenges and opportunities in addressing this type of targeted therapy resistance in EGFR-mutant NSCLC.
Introduction
Lung Cancer
Every year lung cancer is estimated to claim more lives than colorectal, breast, and prostate cancers combined, making it the deadliest cancer worldwide (Didkowska et al., 2016). Selection of treatment strategies for lung cancer patients depends on the stage, histopathologic and molecular subtype of the tumor. Patients with early stage disease are likely to be treated with surgical resection with or without adjuvant chemo- and radiation therapies (Latimer and Mott, 2015). Over 50% of patients, however, present with advanced disease, when surgical intervention is no longer an option. For these patients, accurate classification of the tumor is particularly important for selection of appropriate therapy.
Lung cancer is a heterogeneous group of malignancies, encompassing two main histologic subtypes, non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). SCLC is linked to heavy tobacco smoke exposure and comprises ∼20% of all lung cancer cases (Huang et al., 2015; Travis et al., 2015). Since these tumors express neuroendocrine markers, such as synaptophysin and chromogranin, they are thought to arise from neuroendocrine cells within the lung epithelium (Park et al., 2011; Osmani et al., 2018). At the genomic level, ∼10% of all SCLCs are mutant in PTEN, and ∼90% are mutant in both RB1 and TP53 tumor suppressor genes (Yokomizo et al., 1998; Beasley et al., 2003; Meuwissen et al., 2003; Sutherland et al., 2011). SCLC usually presents at late advanced stages and is seldom surgically resectable. Immune-based therapies in combination with systemic chemotherapy plays a critical role in management of advanced stage SCLC.
The majority of all lung cancers (80–85%) are NSCLC, of which adenocarcinoma of the lung (LUAD) is the most prevalent subtype (Latimer and Mott, 2015). Compared to SCLC, LUAD is less tightly associated with smoking (Couraud et al., 2012). Genetic alterations frequently seen in LUAD include mutations in the TP53, KRAS, and EGFR genes, as well as ALK-NPM fusion. LUAD lesions typically develop in the periphery of the lung and are usually diagnosed at a late advanced stage due to delayed occurrence of symptoms (Latimer and Mott, 2015). Before the era of precision medicine, treatment selection for advanced NSCLC was limited to systemic chemo-and radiation therapies. Recent development of immune checkpoint inhibitors and discoveries of targetable molecular drivers in NSCLC revolutionized the clinical care of these patients. Arguably, the most dramatic improvement in clinical outcomes are seen in patients whose tumors harbor one of several activating mutations in the epidermal growth factor receptor (EGFR) gene and are treated with small molecule EGFR tyrosine kinase inhibitors (EGFR TKIs) (Rosell et al., 2012; Sequist et al., 2013; Douillard et al., 2014; Mok et al., 2017; Soria et al., 2018). Immune-based therapies are approved for use in patients with no actionable molecular tumor alterations (Figure 1) but are not indicated for front-line use in EGFR-mutant NSCLC patients that are the subject of this review (Lee C. K. et al., 2017).
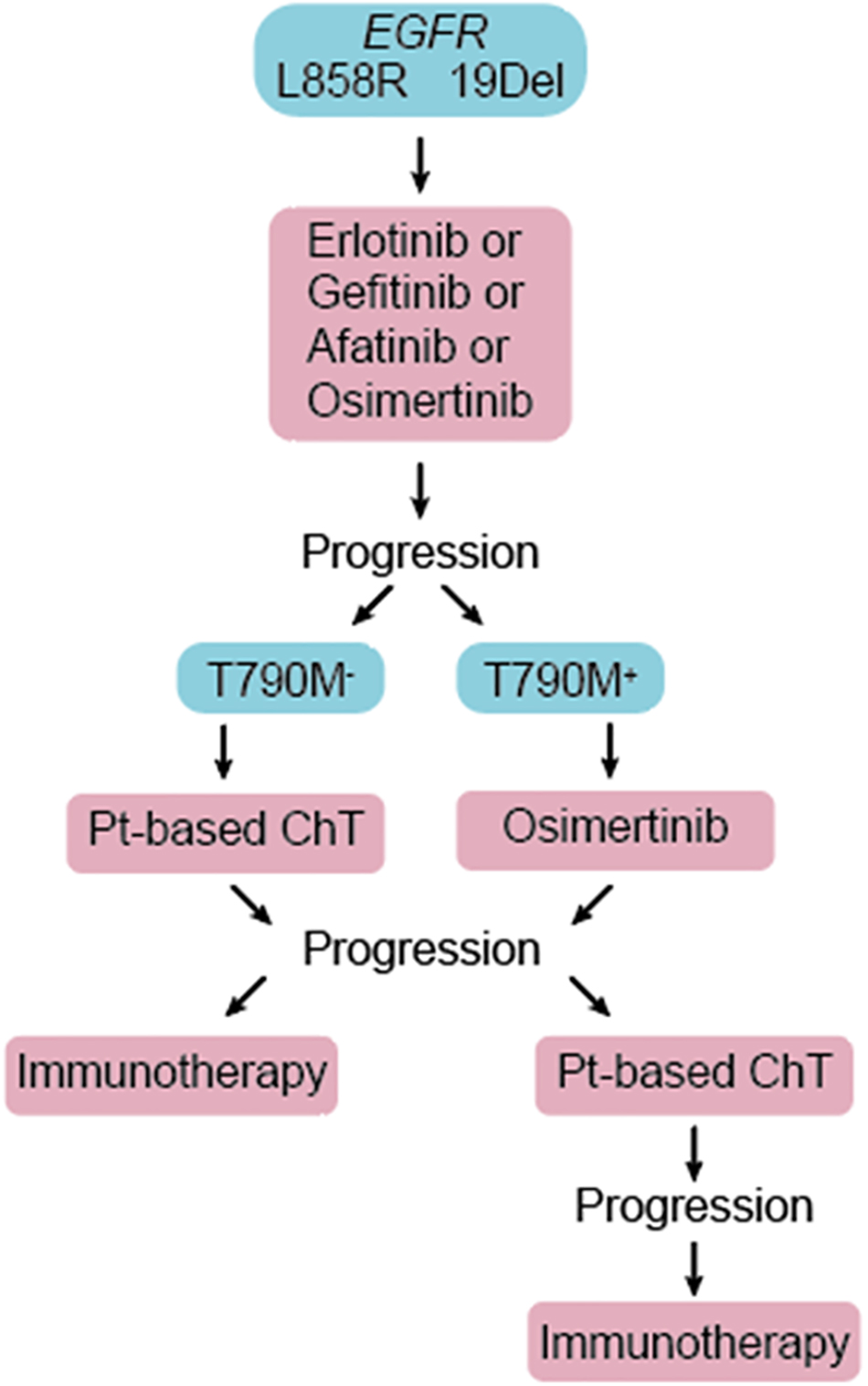
Figure 1. Patients diagnosed with advanced EGFR-mutant NSCLC are treated with one of the approved EGFR TKIs in the first line. Upon disease progression, repeat molecular testing is recommended. Patients not previously treated with osimertinib and whose tumor acquired second site T790M mutation in the EGFR gene (T790M+) are treated with 3rd generation EGFR TKI, osimertinib. Platinum-based chemotherapy (Pt-based ChT) and immunotherapies are reserved for T790M– patients, and those who progress on osimertinib.
EGFR as a Therapeutic Target in Lung Cancer
EGFR is a plasma membrane receptor tyrosine kinase that, upon ligand activation, auto phosphorylates and functions as a rapid activator of downstream signaling cascades (Ushiro and Cohen, 1980). The key targets of EGFR include mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K/AKT), and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways (Bjorge et al., 1990; Zhong et al., 1994; Franke et al., 1995). The abovementioned transduction networks link EGFR tyrosine kinase activity to increased proliferation, motility, pro-survival, and anti-apoptotic cellular responses and highlight the oncogenic potential of abnormal EGFR activation.
The initial interest in exploiting EGFR as a principal therapeutic target was prompted by the results of molecular profiling studies that determined EGFR overexpression or amplification in 5–11% of LUAD (Cancer Genome Atlas Research Network, 2014; Gupta et al., 2009). First generation small molecule tyrosine kinase inhibitors, erlotinib (Tarceva) and gefitinib (Iressa), were developed and demonstrated significant survival improvement in advanced NSCLC patients who failed chemotherapy (Shepherd et al., 2005). Although early clinical trials demonstrated objective response rates to first generation EGFR TKIs below 10%, some patients experienced dramatic and durable response to these agents, pointing toward the presence of a unique prognostic feature in tumors that were exquisitely sensitive to EGFR inhibition (Fukuoka et al., 2003; Shepherd et al., 2005; Thatcher et al., 2005).
The subsequent discovery of somatic alterations in the tyrosine kinase domain of the EGFR gene provided an explanation as to why only a fraction of lung cancer patients had excellent response to EGFR TKIs (Shigematsu and Gazdar, 2006; Suzuki et al., 2005). The majority of tyrosine kinase domain mutations occur as deletions in exon 19 (Del19, ∼44%) and point mutations in exon 21 (L858R, ∼41%) (Shi et al., 2014; Kobayashi et al., 2015; Shigematsu et al., 2005). Mutant EGFR undergoes ligand-independent autophosphorylation to constitutively activate downstream oncogenic signaling (Okabe et al., 2007). These activating mutations confer reduced receptor affinity for ATP and allow EGFR TKIs to preferentially occupy the ATP-binding site of the receptor (Carey et al., 2006; Gazdar, 2009). Lower ATP binding affinity for the mutant receptor is the key to success of EGFR TKIs in the clinic, as it permits competitive EGFR inhibitors to achieve efficacious concentrations in vivo with a favorable toxicity profile (Shepherd et al., 2005). Not surprisingly, Del19- or L858R-positive NSCLC tumors are intrinsically sensitive to early generation EGFR targeting therapies. Today, several EGFR TKIs have gained an approval as standard of care in patients with advanced EGFR-mutant NSCLC. Hence, defining EGFR mutation status is a critical step in lung cancer diagnosis. Targetable EGFR mutations occur in ∼15% of NSCLC patients in the US and Europe, and ∼30% of NSCLC patients of Asian descent (Zhang et al., 2016).
Acquired Resistance to EGFR TKIs
As EGFR-mutant tumors are “addicted” to oncogenic EGFR signaling (Politi et al., 2006), it is not surprising that persistent blockade of EGFR forces tumors to adapt and escape the anti-proliferative effects of EGFR TKIs. Therefore, the dramatic initial response to these agents is inevitably followed by emergence of treatment resistance. Generally speaking, mechanisms of acquired resistance to EGFR TKIs can be placed into one of three categories: EGFR-dependent resistance (50–60%), activation of bypass signaling (20–30%), and histologic or phenotypic transformation (5–10%) (Sequist et al., 2011; Yu et al., 2013).
EGFR-dependent resistance is marked by appearance of additional mutations in the EGFR gene. For instance, the T790M, or “gatekeeper” mutation, increases receptor binding affinity for ATP, resulting in loss of response to early generation TKIs (Yun et al., 2008). In a series of clinical trials, third generation EGFR TKI osimertinib demonstrated a favorable toxicity profile and 70% objective response rate in EGFR-mutation positive NSCLC patients whose disease progressed on early generation EGFR TKIs (Goss et al., 2016; Mok et al., 2017). Moreover, osimertinib was proven to be superior to first generation EGFR TKIs in the first line treatment setting (Soria et al., 2018). As with early generation EGFR targeting agents, acquired resistance to osimertinib is commonplace and can be attributed to third site mutations in EGFR (such as C797S), bypass activation, or phenotypic transformation (Kim et al., 2015; Thress et al., 2015; Ham et al., 2016; Ortiz-Cuaran et al., 2016; Li et al., 2017b; Oztan et al., 2017; Ricordel et al., 2017; Yang et al., 2018).
Activation of alternative tyrosine kinases (AXL, IGFR, FGFR, FAK, etc.) leads to loss of dependency on EGFR signaling and development of resistance to EGFR blockade (Guix et al., 2008; Kim et al., 2012; Zhang et al., 2012; Cortot et al., 2013; Ichihara et al., 2017; Lee A. F. et al., 2017). Many of these kinases are known to activate MAPK, AKT, or JAK/STAT networks, ultimately replacing, or bypassing, the requirement for pro-tumorigenic EGFR signaling. Ongoing clinical trials are investigating new targeted agents that inhibit these alternative kinases for their ability to overcome resistance to EGFR TKIs as mono-treatment modalities or in combination with approved EGFR therapeutics (ClinicalTrials.gov identifiers NCT02424617, NCT03450330, NCT03255083, NCT03123484). Phenotypic transformation can occur alongside or independently of bypass-linked resistance to EGFR TKIs. In the setting of acquired EGFR TKI resistance, phenotypic transformation encompasses EMT, SCLC, and transformation to squamous cell carcinoma (AST). In the remaining sections of this review, we will discuss in detail these less well understood mechanisms of EGFR TKI failure.
Emt as a Mechanism of Resistance to Egfr Tkis
EMT is characterized by loss of intercellular adhesions and polarity, reorganization of cytoskeleton, and acquisition of invasive phenotype. At the molecular level, EMT is coordinated by increased expression of pro-mesenchymal transcription factors including Snail (SNAIL), Twist (TWIST1), and Zeb (ZEB1/2) families, and loss of a number of epithelial markers, including E-cadherin (CDH1), epithelial cell adhesion molecule (EPCAM), and claudin 4 (CLDN4) (De Craene and Berx, 2013). Loss of epithelial integrity facilitates tumor invasion of adjacent tissues, metastatic dissemination, and contributes to chemoresistance.
The Role of Epithelial Phenotype in Intrinsic Sensitivity to EGFR TKIs
As previously discussed, early clinical trials of first generation EGFR TKIs were conducted in molecularly non-selected cohorts of patients with advanced NSCLC (Fukuoka et al., 2003; Shepherd et al., 2005; Thatcher et al., 2005). Dramatic tumor response to EGFR blockade was observed only in a small proportion of patients. These findings prompted a considerable research effort to determine prognostic tumor biomarkers of intrinsic sensitivity and resistance to EGFR targeted therapeutics. Several groups reported that an EMT gene expression signature is predictive of reduced EGFR TKI efficacy in vitro and in vivo. Using panels of predominantly EGFR-wild type NSCLC cell lines, several EGFR TKI response signatures were derived (Thomson et al., 2005; Yauch et al., 2005; Witta et al., 2006; Byers et al., 2013). Common to all proposed signatures, sensitivity to first generation EGFR TKIs was linked to high expression of a well-established epithelial marker CDH1, while resistance was associated with high expression of pro-mesenchymal markers vimentin (VIM) and ZEB1. The ability of these signatures to predict clinical response to EGFR TKIs was tested in cohorts that included both EGFR-mutant and EGFR-wild type NSCLC patients (Yauch et al., 2005; Kim et al., 2011). Although EGFR TKIs were more efficacious in patients whose tumors were classified as epithelial, it is important to note that CDH1 expression since has been reported to be positively correlated with EGFR mutation status (Zhang et al., 2006; Zhao et al., 2015). Therefore, the apparent association between tumor epithelial phenotype and sensitivity to EGFR TKIs might have been driven by the presence of activating EGFR mutations in tumors with more epithelial phenotype.
The importance of EMT in predicting initial response to EGFR targeted therapies in the setting of EGFR-mutant NSCLC is still unclear. Several reports indicate a trend toward better objective response rate in patients treated with first generation EGFR TKIs whose tumors express high levels of E-cadherin and can be classified as epithelial, but the results do not reach statistical significance (Chen et al., 2013; Ren et al., 2014).
The Role of EMT in Acquired Resistance to EGFR TKIs
EMT Co-occurring With Bypass Kinase Activation
EMT is reported in a number of pre-clinical studies that investigate acquired resistance to EGFR TKIs. However, it is difficult to determine whether EMT is the sole driver of resistance in every case, since it frequently occurs alongside bypass activation. The most common indicator of bypass-associated EGFR TKI resistance is loss of TKI-induced phosphorylation blockade at the main downstream targets of EGFR, such as MAPK and AKT, in the absence of additional mutations in the EGFR gene. MAPK and AKT are critically located at the interface of multiple oncogenic signaling cascades reported to be activated as a part of EGFR TKI resistance. Therefore, alternative kinase activation is likely to involve one or both of these proteins. Recently, Src/FAK emerged as a critical node in the EGFR bypass network.
Src/FAK are non-receptor tyrosine kinases that can be activated by EGFR and multiple other plasma membrane RTKs, including AXL, IGF1R, and FGFR (Sandilands et al., 2007; Sulzmaier et al., 2014; Cox et al., 2015; Marlowe et al., 2016). The Src/FAK network converges on AKT and MAPK to sustain oncogenic signaling and induce EMT. Many of the RTK that signal through Src/FAK have been implicated in EMT-associated resistance to EGFR targeting agents (Tang et al., 2016; Ichihara et al., 2017; Nukaga et al., 2017; Weng et al., 2018).
Activation of AXL tyrosine kinase was one of the first identified bypass-associated mechanisms of resistance to EGFR TKIs (Zhang et al., 2012; Okimoto and Bivona, 2015). AXL is a member of TAM family of receptors that is capable of activating AKT and MAPK signaling cascades. Hence, in the setting of persistent blockade of the EGFR pathway, AXL may play a critical role in cell survival, providing an alternative axis of pro-tumorigenic signal propagation. Moreover, AXL is known to govern cytoskeletal rearrangement and invasive capacity in lung cancer (Shieh et al., 2005). Early reports indicated that in murine xenograft- and cell line-based models of acquired resistance to first generation EGFR TKIs, AXL was among the most upregulated genes compared to control, erlotinib-naïve specimens (Zhang et al., 2012). Strikingly, resistance was also accompanied by a dramatic upregulation of VIM and near complete loss of CDH1 expression. Inhibition of AXL kinase activity restored both CDH1 expression and sensitivity to EGFR TKIs. Overexpression of AXL is also linked to EMT-associated resistance to 3rd generation EGFR TKI, osimertinib (McGowan et al., 2017). Therefore, AXL remains a promising next line therapeutic target for EMT-associated resistance, with several pharmacological inhibitors in early clinical development (ClinicalTrials.gov identifiers NCT03255083, NCT02424617, NCT02729298).
Activation of Insulin-like growth factor 1 receptor (IGF1R) as an alternative to EGFR signaling can also occur as a part of EMT-linked resistance. Similar to EGFR, IGF1R signal transduction primarily involves MAPK and AKT pathways and may lead to mesenchymal transdifferentiation (Vazquez-Martin et al., 2013). Elevated levels of IGF1R have been documented in pre-clinical models of acquired resistance to EGFR TKIs, where its expression inversely correlates with epithelial phenotype (Vazquez-Martin et al., 2013; Zhou et al., 2015; Hussmann et al., 2017; Li et al., 2017a). Intriguingly, tumor IGF1R expression is associated with lymphatic invasion and predicts shorter progression-free survival in EGFR-mutant but not EGFR–wild type NSCLC, suggesting a unique interplay between EGFR and IGF1R pathways in this type of malignancy (Park et al., 2015). The above observations point toward therapeutic relevance of IGF1R. However, the results of early clinical trials of anti–IGF-1R monoclonal antibodies R1507 or figitumumab in combination with erlotinib demonstrated unfavorable toxicity profiles and lack of improved response (Ramalingam et al., 2011; Belani et al., 2012). It is important to note that the abovementioned clinical trials were conducted in molecularly non-selected cohorts of lung cancer patients. Therefore, it is possible that IGF1R remains an important target in EGFR-mutant NSCLC.
To compensate for consistent EGFR blockade, some tumor cells activate fibroblast growth factor family of receptors (FGFR). FGFR activation promotes mesenchymal transdifferentiation and reduces sensitivity to EGFR TKIs. Indeed, it appears that in the context of acquired resistance to EGFR inhibitors, FGFR activation is almost inevitably accompanied by gain of mesenchymal features (by phenotypic transformation toward more mesenchymal phenotype) (Jakobsen et al., 2017). To address causative relationship between FGFR1 activation and onset of EMT, either FGFR1 or ZEB1 were overexpressed in EGFR-mutant, treatment-naïve cells (Vad-Nielsen et al., 2019). Cells expressing high levels of ZEB1 underwent EMT and upregulated FGFR1 expression. Surprisingly, no induction of EMT was noted in cells overexpressing FGFR1. These observations suggest that the FGFR1-associated kinase switch occurs as a consequence of EMT and not the other way around. Nevertheless, targeted inhibition of FGFR may be a viable strategy to overcome resistance to EGFR TKIs linked to EMT (Raoof et al., 2019).
EMT in the Absence of Bypass Activation
EMT as the sole driver of acquired resistance to EGFR TKIs was initially identified in a series of clinical case studies (Uramoto et al., 2010; Chung et al., 2011; Sequist et al., 2011; Uramoto et al., 2011). The first report of a trend toward EMT in EGFR-mutant NSCLC with acquired resistance to EGFR TKI gefitinib came in 2010, when biospecimens from 9 cases of EGFR-mutant lung adenocarcinoma pre- and post-acquisition of resistance to gefitinib were analyzed for expression of epithelial (E-cadherin, γ-catenin) and mesenchymal (vimentin, fibronectin) markers (Uramoto et al., 2010). Although statistical significance was not achieved, possibly due to small sample size, the report indicated that loss of E-cadherin and gain of mesenchymal markers may be associated not only with intrinsic, but also with acquired resistance to EGFR targeting agents. Results from a larger study suggested that EMT may be a relatively rare mechanism of EGFR TKI failure, affecting ∼5% of patients (Sequist et al., 2011). EMT occurred in the absence of any other mechanisms of resistance known at the time, and therefore was determined to be the basis for therapeutic failure. It is important to note that early studies investigated the most common mechanisms of resistance known at the time, mainly involving additional genomic alterations in the EGFR gene and several other known oncogenic drivers, such as amplification of MET and PIK3CA mutations. Therefore, activation of an alternative tyrosine kinase might have been easily missed.
Nevertheless, involvement of EMT in acquired resistance to EGFR TKIs is supported by a number of studies conducted in pre-clinical models of EGFR-mutant NSCLC. Many of them, as discussed earlier, suggested that transition toward mesenchymal phenotype co-occurs with a kinase switch. However, in some cases, bypass activation was not identified, suggesting that EMT itself might be responsible for the development of therapy-refractory disease (Lee A. F. et al., 2017; Song et al., 2018; Yochum et al., 2019).
The mechanistic basis for the role of E-cadherin in the efficacy to EGFR TKIs may be placed in its ability to activate cell-intrinsic apoptosis. Cell intrinsic apoptosis is initiated by binding of tumor necrosis factor family ligands, such as Apo2L/TRAIL to death receptors, TRAILR1/2. This binding event ultimately leads to assembly of the death-inducing signaling complex and caspase-8 activation (Wilson et al., 2009). E-cadherin has been shown to physically interact with ligated TRAIL1/2, couple them to the actin cytoskeleton, and promote activation of cellular caspases. Intriguingly, EGFR-mutant cells with acquired EMT-associated resistance to erlotinib develop resistance to Apo2L (Lu et al., 2014).
Although a link between intrinsic sensitivity to EGFR TKIs and tumor E-cadherin expression has been proposed, the direct role of E-cadherin loss in acquisition of resistance to this class of drugs is not universally agreed upon. A dramatic reduction in E-cadherin levels is a common feature of EMT-associated EGFR TKI resistance. However, shRNA –dependent depletion of CDH1 in EGFR-mutant therapy-naïve NSCLC cells does not necessarily lead to loss of EGFR TKI efficacy (Lee A. F. et al., 2017). It is possible that, in addition to low expression of epithelial molecules, upregulation of mesenchymal markers is necessary to drive EGFR TKI resistance.
Ectopic expression of the Snail family of transcription factors represses CDH1 expression to induce EMT and resistance to EGFR TKIs in EGFR-mutant NSCLC (Lee A. F. et al., 2017). Additionally, expression of pro-mesenchymal factor TWIST in EGFR-mutant NSCLC has been linked to induction of EMT and acquired resistance to EGFR TKIs. Some studies suggest that TWIST can inhibit transcriptional activation of pro-apoptotic protein BIM either by directly binding to its promoter, or by inducing ZEB1, which then acts as a repressor of BIM transcription (Song et al., 2018; Yochum et al., 2019). Therefore, BH3 mimetics, a class of drugs that target BCL family of proteins to promote apoptosis, may be beneficial for treating EMT-associated EGFR TKI resistance. Navitoclax, a novel BH3 mimetic compound, is currently under early phase clinical investigation in EGFR-mutation positive NSCLC patients who relapsed on previous EGFR TKI therapy (CilicalTrials.gov identifier NCT02520778).
Activation of Notch-1 has also been linked to EMT-associated EGFR TKI resistance. In cell-based and murine models, acquired resistance to gefitinib was accompanied not only by increased expression of mesenchymal markers and diminished E-cadherin, but also upregulation of Notch-1 (Xie et al., 2012, 2013). Notch-1 silencing resulted in E-cadherin re-expression and partial restoration of sensitivity to gefitinib. Pharmacological inhibition of Notch-1 signaling pathway is currently in early clinical development (NCT01158404, NCT01653470).
The Use of Epigenetic Agents in the Context of Acquired Resistance to EGFR TKIs
Development of drug “tolerance,” a reversible state of reduced sensitivity to treatment associated with mesenchymal phenotype, precedes the onset of more permanent resistance (Sharma et al., 2010). Drug tolerant persisters (DTPs) harbor an altered chromatin state with high expression levels of KDM5A, a H3K4 histone demethylase, and other chromatin-modifying enzymes. Therefore, the use of epigenetic agents may be beneficial to combat phenotypic plasticity as part of acquired drug resistance. Indeed, several histone deacetylate inhibitors (HDACi), including entinostat and trichostatin A, have been shown to restore expression of CDH1 and promote sensitivity to EGFR TKIs in pre-clinical models of EGFR-mutant NSCLC (Sharma et al., 2010; Suda et al., 2011; Weng et al., 2018).
Combination of HDACi vorinostat and erlotinib was investigated in a phase I/II clinical trial, where it demonstrated a favorable toxicity profile but failed to provide survival benefit in EGFR-mutant NSCLC patients with prior progression on erlotinib (Reguart et al., 2014). However, out of 19 patients included in the phase II part of the study, 7 were positive for the T790M mutation in the EGFR gene and therefore were unlikely to derive any benefit from erlotinib-based combination treatment. It is worth noting, that a set of patients achieved long-term stable disease on the vorinostat/erlotinib regimen. No additional molecular profiling was performed to pinpoint the features associated with susceptible tumors. In a larger phase II trial conducted in molecularly non-selected advanced stage NSCLC, combination of erlotinib and entinostat produced a significant survival benefit only in patients with high tumor E-cadherin expression (Witta et al., 2012). Further investigation of epigenetic modifiers in combination with contemporary 3rd generation EGFR TKI osimertinib in EGFR-mutant NSCLC are needed to determine the utility of epigenetic drugs in the context of acquired resistance to EGFR targeted therapeutics that is associated with phenotypic transformation.
Challenges in Addressing EMT-Associated Resistance to EGFR TKIs
EMT emerges with acquired resistance to EGFR TKIs. However, the direct cause and effect relationship between EMT and EGFR TKI resistance appears to be context dependent. The majority of pre-clinical reports that link loss of sensitivity to anti-EGFR drugs to mesenchymal phenotype demonstrate the co-occurrence of a kinase switch. Hence, it is a challenging to determine whether EMT is truly a driver of therapeutic failure or simply a passenger to some other primary mechanism of resistance (i.e., bypass activation). The notion that epithelial phenotype directly correlates with the efficacy of EGFR targeted therapies was put forward when early clinical trials performed in non-selected NSCLC patients identified a link between tumor E-cadherin levels and response to first generation EGFR TKI erlotinib (Richardson et al., 2012). The research was focused on identifying EMT signatures that could predict sensitivity to early generation TKIs in patients with undefined EGFR mutation status. As a result, epithelial phenotype in general, and E-cadherin expression specifically, were determined to be important contributors to intrinsic sensitivity to EGFR TKIs. However, current knowledge dictates that activating mutations within tyrosine kinase domain of the EGFR predict clinical relevance of TKIs, and their use remains limited to the EGFR-mutant subset of NSCLC. Moreover, EGFR signaling appears to be critical for epithelial lineage differentiation and it is now clear that EGFR-mutant tumors are more likely to express high levels of E-cadherin than tumors with wild type receptor (Miettinen et al., 1995). Therefore, in the context of molecularly non-selected patients and cell lines, the sensitivity to EGFR TKIs may be driven not by the epithelial phenotype per se, but rather by the positive EGFR mutation status that is linked to such phenotype. Consequently, it remains unclear whether the EMT signatures derived from cell panels that included both EGFR-wild type and -mutant cell lines and validated in predominantly EGFR-wild type clinical cohorts are optimal for the study of EMT as a basis for acquired resistance to EGFR TKIs in EGFR-mutant NSCLC.
EMT is characterized as a complex multistep process that involves molecular changes that are often context dependent. Therefore, a pleotropic agent that is capable of initiating a broad-spectrum epithelial differentiation program could be of exceptional therapeutic value. We previously reported that 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], an active metabolite of vitamin D, is uniquely suitable for applications in EGFR-mutant NSCLC (Upadhyay et al., 2013). Vitamin D receptor (VDR), a critical player in vitamin D signaling axis, is highly expressed in EGFR-mutant lung cancer cells, making them intrinsically sensitive to 1,25(OH)2D3. Moreover, 1,25(OH)2D3 restores expression of E-cadherin in models of EMT-associated resistance to EGFR TKIs, suggesting its capacity to promote sensitivity to this class of therapeutics (Liu et al., 2018). Studies to elucidate clinical relevance of 1,25(OH)2D3-based combination therapies in the context of acquired resistance to EGFR TKIs are currently under way in our laboratory.
To effectively address EMT-associated resistance, its onset must be reliably detected in clinical samples. Traditional or liquid biopsy is commonly performed in EGFR-mutation positive NSCLC patients who failed first line EGFR TKI therapy. Generally, contemporary tests allow for detection of additional mutations in the EGFR and genomic alterations in a limited number of resistance-associated genes, such as amplification of MET. However, EMT as a potential mechanism of resistance is not routinely investigated. Lack of a clinically validated panel of biomarkers that identify the transition toward mesenchymal phenotype in tumor biopsies and in circulating tumor cells, is a hurdle yet to be overcome.
The frequency of EMT as a part of acquired resistance to EGFR TKIs may be greatly underestimated not only in the clinic, but also at the laboratory bench. In many cases, primary investigation into the mechanisms underlying therapeutic failure is aimed at determining acquisition of genetic alterations in the EGFR gene and activation of alternative tyrosine kinases. Often, phenotypic transformation as a factor contributing to loss of sensitivity is considered only in the absence of the abovementioned mechanisms. However, it is becoming abundantly clear that EMT may occur concomitantly with activation of bypass signaling. Therefore, whether EMT is a driver of the resistance or a passenger to the primary mechanism, i.e., bypass activation, may be difficult to establish. It is possible that the phenotypic state of the tumor cells dictates dependency on a particular set of growth factors. Thus, while epithelial cells depend on EGFR signaling, cells that undergo EMT may depend instead upon RTKs such as AXL, IGF1R, or FGFR, while EGFR signaling becomes less relevant. In this context, the abovementioned RTKs not only allow the cells to retain mesenchymal properties but also serve as markers of the adoption of an alternative phenotypic state. For instance, similar to EGFR TKI-resistant tumor cells, normal lung fibroblasts express high levels of FGFR and PDGFR, indicating the critical importance of these growth factor signals in survival of mesenchymal cells (Green et al., 2016; Danopoulos et al., 2019). Therefore, in contrast to the conventional paradigm that separates EMT-associated resistance to EGFR TKIs from bypass-dependent disease, these phenomena can be united as two sides of a phenotypic switch driven by chronic EGFR inhibition (Figure 2). The implication of such a model is that EMT-associated resistance may be targeted either by using epigenetic drugs [or 1,25(OH)2D3] to maintain/restore epithelial phenotype and EGFR TKI sensitivity or inhibiting the alternative receptor dependencies of mesenchymal variants.
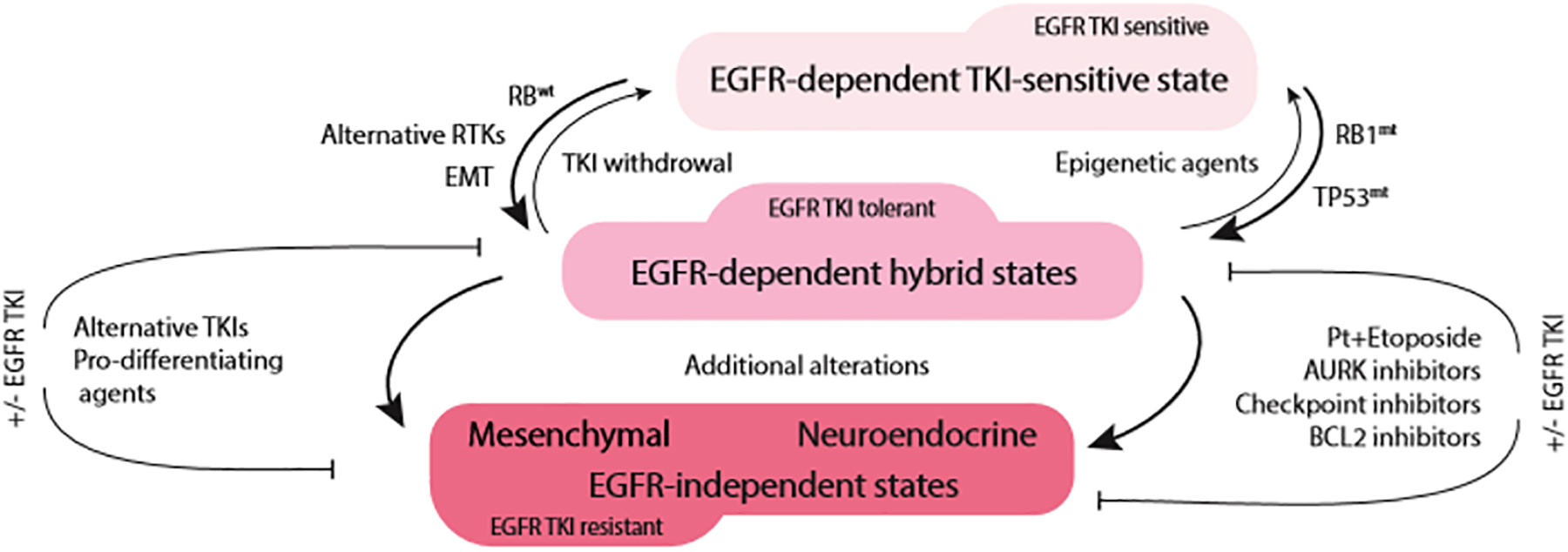
Figure 2. Acute EGFR TKI exposure drives phenotypic plasticity and TKI tolerance in EGFR-mutant NSCLC. The route to therapeutic resistance appears to be dependent on RB1 status. In RB1-wild type tumors (RB1wt), transition toward mesenchymal phenotype with activation of alternative tyrosine kinases (RTKs) is likely to occur (left side of the diagram). On the other hand, RB1-mutant/TP53-mutant (RB1mt, TP53mt) tumors are likely to undergo neuroendocrine transformation (right side of the diagram). This hybrid, EGFR TKI-tolerant state appears to be still dependent on EGFR signaling and may revert to EGFR TKI-sensitive phenotype upon EGFR TKI withdrawal or epigenetic therapies. With prolonged treatment, due to additional genomic alterations, tumors lose their dependence on EGFR signaling and lock in the terminal state of EGFR TKI resistance.
Phenotypic Transformation to Sclc as a Mechanism of Resistance to Egfr Tkis
SCLC Is Diagnosed in a Subset of EGFR-Mutant Patients at the Time of EGFR TKI Failure
The transformation of EGFR-mutant LUAD to SCLC (referred to as SCLC transformation or lineage transformation) accounts for up to 14% of acquired resistance to EGFR TKIs (Sequist et al., 2011). Pre-clinical models that faithfully recapitulate SCLC transformation have yet to be developed. Thus, our understanding of this important mechanism of resistance to date has come exclusively from the analysis of clinical specimens. In an early case report, a 45-year-old female patient who never smoked was diagnosed with LUAD and treated with erlotinib (Zakowski et al., 2006). She responded well for 18 months, but then her disease progressed. A biopsy performed at the time of relapse showed evidence of SCLC. SCLC was identified by morphology and confirmed with immunohistochemical staining for synaptophysin. Typically, SCLC cells are positive for neuroendocrine markers including synaptophysin, chromogranin, or CD56. Since this initial observation, numerous additional cases have been reported in which SCLC is absent in initial diagnostic samples of EGFR-mutant LUAD but detected at the time of EGFR TKI failure (Oser et al., 2015). Therapy associated SCLCs have reduced expression of EGFR protein compared to corresponding baseline tumors (Niederst et al., 2015), which could partially explain why they fail to respond to EGFR TKIs (Zakowski et al., 2006; Popat et al., 2013). Treatment of therapy associated SCLC follows classical SCLC guidelines, typically a combination of platinum and etoposide (Ham et al., 2016).
EGFR TKI Resistant SCLC Arises From the Original EGFR-Mutant LUAD
In theory, SCLC detected at the time of EGFR TKI failure could arise de novo or via transformation of the original EGFR-mutant LUAD. Two complementary lines of evidence support the latter mechanism. First, SCLC detected at relapse harbors the same EGFR mutations as the original LUAD, indicating a shared clonal origin (Niederst et al., 2015; Lee J. K. et al., 2017). Second, EGFR activating mutations are rarely detected in de novo SCLC (frequency ranges from 0 to 4% in different case series). Interestingly, SCLC is also observed at the time of relapse in a subset of patients diagnosed with ALK-positive LUAD who receive targeted therapy with ALK TKIs (Levacq et al., 2016). However, SCLC is not observed in patients with EGFR wild-type LUAD that develop resistance to chemotherapy (Sequist et al., 2011). Cumulatively, these observations suggest that SCLC transformation is not a generic mechanism of therapeutic resistance. Rather, tumor transformation occurs in response to targeted inhibition of oncogenic drivers. As improved molecularly targeted therapies are integrated into clinical practice, the frequency of SCLC transformation is predicted to rise. This highlights the need to understand the molecular mediators of treatment associated SCLC transformation so that appropriate interventions can be developed.
Molecular Predictors of SCLC Transformation
To identify candidate mediators of SCLC transformation, Niederst et al. used genomic sequencing to compare the molecular profiles of EGFR TKI resistant-NSCLC and EGFR TKI resistant-SCLC samples that were obtained from a single patient who was initially diagnosed with EGFR-mutant LUAD (Niederst et al., 2015). The SCLC transformed samples contained the original EGFR mutation plus additional mutations in TP53 and PIK3CA. SCLC transformed tumors also uniquely showed evidence of RB1 loss. Subsequent immunohistochemistry assays performed on 10 additional cases of transformed SCLC demonstrated that loss of RB1 protein (pRb) is ubiquitous. Notably, TP53 and RB1 mutation are also ubiquitous in classical SCLC. Thus, transformed SCLC recapitulates the molecular features of classical SCLC. In contrast, pRb was retained in 10/11 EGFR-mutant LUADs that developed EGFR TKI resistance without SCLC transformation. These data implicate RB1 loss as a driver of SCLC transformation.
A causal role for RB1 is further supported by the observation that patients who present with EGFR/TP53/RB1 triple mutant lung cancers (approximately 5% of all EGFR-mutant cases) are at a sixfold higher risk for SCLC transformation compared to patients with EGFR-mutant lung cancer who do not harbor concurrent alterations in TP53 and RB1 (Offin et al., 2019). However, it is important to acknowledge that not all EGFR/TP53/RB1 triple mutant lung cancers transform as disease progresses. We can infer from these data that TP53/RB1 mutation may be necessary, but is not sufficient, for SCLC transformation. Consistent with this idea, RB1 knockdown in EGFR-mutant LUAD cell lines is also not sufficient for transdifferentiation into SCLC (Niederst et al., 2015).
An existing mouse model of classical SCLC provides some insight into other factors that may be required. In this model, TP53 and RB1 were inactivated in pulmonary epithelial cells (Meuwissen et al., 2003). Aggressive lung tumors developed. Cell lines developed from resultant tumors displayed mixed (neuroendocrine and non-neuroendocrine) phenotypes. Molecular characterization of these cell lines showed that neuroendocrine differentiation was observed only in cells that also harbor amplification of LMYC and increased expression of Ascl-1 (Calbo et al., 2005). The requirement for multiple genetic hits to generate SCLC/neuroendocrine tumors may explain, at least in part, why pre-clinical models of EGFR-mutant transformed SCLC are still lacking.
Role of the RB1 Encoded Protein (pRb) in SCLC Transformation
How might RB1 loss facilitate SCLC transformation? RB1 was the first tumor suppressor gene to be discovered, molecularly isolated in 1986 as the gene responsible for both hereditary and sporadic forms of the childhood cancer retinoblastoma. The canonical function of the RB1 encoded protein (pRb) is to enforce cell cycle arrest in response to anti-proliferative signals. pRb mediates cell cycle control through its physical interaction with E2F transcription factors, recruiting chromatin modifying factors to repress transcription of E2F target genes. pRb activity is negatively regulated by cyclin dependent kinase (CDK) phosphorylation which inhibits pRb/E2F interaction leading to de-repression of a highly conserved gene expression program encompassing factors critical for cell cycle progression. It has become apparent in recent years that pRb has functions that extend beyond cell cycle control (Knudsen et al., 2019). Pertinent to the topic of SCLC transformation is the discovery that pRb suppresses the expression of pluripotency gene networks. RB1 deletion in mouse and human fibroblasts increases the efficiency of induced pluripotent stem cell reprogramming (Kareta et al., 2015). It appears to involve de-repression of pluripotency regulatory factors like SOX2 and OCT4. We reasoned that RB1 loss in the context of cancer would have similar effects. Indeed, we found that RB1 deletion in mouse and human experimental models of prostate cancer also facilitated lineage plasticity with transformation of prostate adenocarcinoma to neuroendocrine (small cell) variants. This neuroendocrine transformation is accompanied by acquired resistance to androgen receptor targeted therapy. Lineage transformation and therapeutic resistance in our prostate cancer models were dependent on increased expression of stem cell reprogramming factors like SOX2 and EZH2. In an analogous manner, RB1 loss may mediate upregulation of epigenetic and stem cell reprogramming factors in EGFR-mutant LUAD, facilitating SCLC transformation and acquired EGFR TKI resistance. Because reprogramming is reversible (at least until the phenotypic switch becomes genetically fixed), an opportunity may exist to combat SCLC transformation by therapeutic intervention with epigenetic modulating therapy.
Potential Treatment Strategies for Those at Risk or Diagnosed With EGFR-TKI Therapy Associated SCLC
Use of Bcl-2 Inhibitors
Currently, TP53 and RB1 status does not inform clinical decision making for patients diagnosed with advanced EGFR-mutant NSCLC. Alteration of RB1 and/or TP53 is likely to increase the likelihood that EGFR-mutant adenocarcinoma will relapse as an SCLC variant. These variants have distinct therapeutic sensitivities and are generally lethal because they rapidly develop resistance (Offin et al., 2019). Thus, routine determination of RB1/TP53 mutation status merits consideration, especially if therapeutic intervention can prevent or delay SCLC transformation. Once such at-risk patients are identified, several therapeutic options might be considered. In patients who present with triple mutant cancers, time to treatment failure could be extended by combining EGFR TKIs upfront with conventional SCLC therapy (platinum/etoposide). As suggested by Offin and colleagues, this combination is expected to be effective because EGFR TKIs will eradicate the bulk of EGFR-mutant LUAD cells. Platinum/etoposide chemotherapy will inhibit EGFR/TP53/RB1 triple mutant subclones that escape EGFR TKI treatment. Clinical trials have been initiated to test this approach (NCT03567642). It is worth noting here that if platinum/etoposide is not given upfront but instead is used after SCLC transformation, a response rate of 54% is obtained (Marcoux et al., 2019). However, response is not durable (progression free survival under 4 months) (Marcoux et al., 2019). In this situation, Bcl-2 family inhibitors might be used to block progression. ABT-263, an orally bioavailable Bcl-2 family protein inhibitor, is effective in SCLC models (Shoemaker et al., 2008). EGFR-mutant, transformed SCLC cells are more sensitive to ABT-263 than EGFR-TKI-resistant NSCLC cell lines (Niederst et al., 2015). However, clinical trials on transformed SCLC patients will be needed to test its safety and efficacy.
Targeting Unique Cell Cycle Vulnerabilities Created by RB1 Loss
Another potential therapeutic option to counter the emergence of transformed SCLC is to target cell cycle vulnerabilities that are created upon RB1 loss. In triple negative breast cancers (lack estrogen receptor, progesterone receptor, and Her2), RB1 loss places stress on the DNA replication machinery, which results in sensitivity to CHK1 and PLK1 inhibitors that target DNA damage checkpoints (Witkiewicz et al., 2018). It is possible that RB1 loss in EGFR mutant lung cancer cells will yield a similar, synthetically lethal interaction with these inhibitors.
Recent studies implicate Aurora kinases (AURKA or AURKB) that regulate mitotic spindle assembly and chromosome segregation as alternative therapeutic targets. Independent CRISPR/Cas9 (Oser et al., 2019) and drug screening assays (Gong et al., 2019) using Rb1-deficient classical SCLC cell lines simultaneously identified a unique dependence upon AURK for survival. In these studies, AURK inhibitors potently suppressed the growth of Rb1-negative SCLC cells but had no effect on Rb1-positive lung cancer cells (including the EGFR mutant cell lines H1975 and PC9). One might conclude based on these results that single agent AURK inhibitors will be useful in targeting the subpopulation of EGFR mutant cells that lack Rb1 and are prone to SCLC transformation. However, the utility of AURKA inhibitors in EGFR mutant lung cancer exceeds this specific molecular context. This is because in Rb1-proficient models (like PC9 and H1975), third generation EGFR TKIs induce AURKA activation (Shah et al., 2019). Upon induction, AURKA blocks EGFR TKI-induced apoptosis and promotes drug resistance. In this scenario, AURKA inhibitors can be used in combination with EGFR TKIs to counter the emergence of drug resistance. AURK inhibitors thus hold considerable promise for prolonging the efficacy of third generation EGFR TKIs via their ability to target both Rb1-deficient clones that are prone to SCLC transformation and bulk Rb1-proficient cells that upregulate AURKA during EGFR TKI treatment. A Phase 1/1b clinical trial of AURKA inhibitor Alisertib with osimertinib in metastatic EGFR-mutant lung cancer is currently recruiting participants (NCT04085315).
Use of Epigenetic Therapies
A third approach to block SCLC transformation involves the application of epigenetic therapy. As discussed above, RB1 loss promotes lineage transformation in prostate cancer via the upregulation of epigenetic and reprogramming factors including EZH2 (Ku et al., 2017). Pharmacologic inhibitors of EZH2 are currently being evaluated as cancer therapies, often in combination with other therapies (NCT03460977, NCT03525795, NCT04104776). We have determined that EZH2 inhibitors can suppress prostate cancer lineage plasticity, neuroendocrine transformation, and resistance to androgen receptor targeted therapies in pre-clinical mouse and human experimental models (Ku et al., 2017). We anticipate analogous results may be obtained in EGFR-mutant lung cancers that lose RB1 since these variants are likely to up regulate EZH2 (Coe et al., 2013; Ishak et al., 2016). Supporting this idea, classical SCLCs have been found to overexpress EZH2 protein (Byers et al., 2012). Although it is conceivable that EZH2 inhibitors may reverse lineage transformation, based on pre-clinical experimental models, in the clinic it may be most beneficial to use them upfront in combination with EGFR-TKIs to prevent SCLC transformation, particularly in EGFR/TP53/RB1 triple mutant lung cancers. The identity of other epigenetic factors that become dysregulated in EGFR/TP53/RB1 mutant lung cancers and their effects on mutant EGFR lung cancer lineage plasticity and therapeutic resistance is an area of active research that may yield additional therapeutic targets.
Targeting New RTK Dependencies in SCLC Variants
Analogous to mesenchymal variants of EGFR mutant lung cancer, transformed SCLC variants may possess novel RTK dependencies that can be therapeutically targeted. Matsumura and colleagues found that c-Kit was the only RTK that is significantly expressed in high-grade neuroendocrine carcinomas of the lung, including both large cell neuroendocrine carcinoma (LCNEC) and SCLCs, but not in NSCLC (Matsumura et al., 2015). IGF1R and KDR also showed higher expression scores in SCLC and LCNEC than in LUAD. FGFR1 amplification is also reported in SCLC and potentially may be used as a therapeutic target (Peifer et al., 2012). Another large group of tyrosine kinase receptors is vascular endothelial growth factor receptor (VEGFR) family. Many neuroendocrine tumors are hyper-vascular and histologically show a rich, well-developed vascular network. VEGF-A has been demonstrated to play a critical role in tumorigenesis and neuroendocrine tumor progression. Sunitinib can irreversibly inhibit VEGFR family and is effective in several solid tumors and pancreatic neuroendocrine tumors (Capozzi et al., 2016). It is possible that during the process of acquired EGFR TKI resistance, RB1 loss could facilitate a shift in the signaling pathways activated, like the FGFR1 pathway, reducing dependency on EGFR. This shift in signaling pathway activation may, in turn, contribute to SCLC transformation. Many knowledge gaps will need to be filled to identify and rationally implement such approaches, but this is made difficult by the dearth of experimental models mimicking treatment associated SCLC transformation.
Phenotypic Transformation to Scc as a Mechanism of Resistance to Egfr Tkis
Approximately one percent of EGFR-mutant lung cancer patients develop resistance to EGFR targeting therapeutics that is accompanied by a phenotypic switch from ADC to SCC (Clery et al., 2017; Park et al., 2019). Although SCC is a distinct histological subtype of NSCLC, lung tumors, including those positive for activating mutations in the EGFR gene with mixed adenosquamous histology, are known to occur (Ricciuti et al., 2018). Hence, some tumors may possess high potential for AST when certain genomic alterations are acquired. ADC to SCC lineage transformation remains the least common in the setting of acquired resistance to EGFR TKIs, and the exact molecular traits involved in the process are not well-characterized. Available evidence point toward critical involvement of the PI3K/AKT/mTOR signaling axis, where mTOR inhibitors may have an important clinical utility (Park et al., 2019).
AST is also observed in KRAS-mutant lung cancers upon loss of the LKB1 tumor suppressor (Han et al., 2014). Genetically engineered mouse models have revealed the mechanistic underpinnings of AST in this molecular context (Chen et al., 2019). Loss of LKB1 promotes lineage plasticity via a multi-step process that involves remodeling of the extracellular matrix, inactivation of YAP1 and consequent de-repression of the squamous lineage promoting factor, DNp63 (Gao et al., 2014). LKB1 loss also promotes metabolic reprogramming that leads to an increase in the generation of reactive oxygen species (ROS). Agents that either increase or decrease oxidative stress in the KRAS mutant/LKB1- model modify AST and thus link redox imbalance to plasticity (Li et al., 2015).
LKB1 loss is rarely detected in EGFR-mutant NSCLC (Koivunen et al., 2008; Gao et al., 2010), but metabolic reprogramming and redox imbalance nonetheless occur and contribute to EGFR TKI resistance (Wang et al., 2019). Chronic, sub lethal EGFR TKI exposure results in increased expression of branched-chain amino acid aminotransferase (BCAT1) via an epigenetically controlled mechanism. When expressed, BCAT1 attenuates ROS production that is required for EGFR TKI-induced cytotoxicity. In metabolically reprogrammed cells, phenethyl isothiocyanate can be used to stimulate ROS production and restore EGFR TKI efficacy. Although the relationship between BCAT1-induced metabolic reprogramming and AST remains to be established, these contemporary studies support the idea that dysregulated metabolic programs may be targeted in EGFR-mutant cells to overcome EGFR TKI resistance.
Conclusion and Remaining Challenges
Advanced EGFR-mutant NSCLC can be effectively treated using contemporary EGFR TKIs. However, disease progression is inevitable due to acquired drug resistance. Drug resistance develops by several mechanisms, the least well understood of which is lineage plasticity, that results in both mesenchymal, SCLC/neuroendocrine, and squamous transformation. Routine evaluation of EMT/AST signatures and TP53/RB1 mutation status in patients newly diagnosed with EGFR-mutant LUAD could be implemented to facilitate the early identification of those at risk for phenotypic transformation and aid in treatment planning.
In this review, we present several options to counter drug resistance that results from phenotypic transformation (Table 1). Several questions remain regarding the best therapeutic approach to use. Existing data support an underlying epigenetic basis for lineage plasticity and EMT, SCLC transformation, or AST. This affords an opportunity to intervene with epigenetic therapies. However, several issues will need to be addressed to optimally implement these approaches. First, a number of epigenetic alterations likely co-occur in cells that undergo EMT or SCLC transformation. Indeed, KDM5A, HDAC, and SWI/SNF complexes are all dysregulated in models of EMT-associated EGFR TKI resistance that are available in our labs (unpublished data). It is as yet unclear which epigenetic therapy will prove most efficacious in this context, or whether different individuals will require different interventions. Second, the epigenetic changes that dispose toward SCLC transformation and EMT are rarely studied together and likely to be distinct. Given that EGFR-mutant lung tumors are heterogeneous, an individual EGFR-mutant lung cancer patient in theory may have subclones that are disposed to EMT and others that are disposed to SCLC. It will therefore be important to understand how a particular epigenetic therapy dynamically influences both processes. Third, the best schedule for administering epigenetic therapy needs to be determined. Epigenetic therapy may be applied early to block lineage plasticity and prevent the emergence of variant phenotypes. If the transformed phenotype has not been fixed, epigenetic therapies may also be used after phenotypic variants appear to restore EGFR dependency and EGFR TKI sensitivity. The benefit of using epigenetic therapy versus taking the alternative approach of targeting the new dependencies of transformed variants (such as FGFR for mesenchymal variants or AURKA for Rb1-deficient, transformed SCLC) also needs to be clarified. Ultimately, factors such as tumor heterogeneity, tolerability of combination regimens, patient comorbidities, and accessibility of therapy may all play a role in dictating the right treatment for each patient.

Table 1. Targets to control phenotypic plasticity in EGFR-mutant NSCLC.
Author Contributions
TS, LZ, DG, and PH jointly conceived of content. TS and LZ drafted the review, which was edited by DG and PH.
Funding
This study was supported by grants CA207757 and CA234162 to DG, and CA249411 and American Lung Cancer Association Discovery Award to PH. Additionally, the authorship team was supported by funding from the Herd of Hope Campaign of the Roswell Park Alliance Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all of the donors.
Abbreviations
ALK, anaplastic lymphoma kinase; ALK-NPM, fusion of ALK with nucleophosmin; AST, adenosquamous transformation; AURK, Aurora kinase; AXL, Axl receptor tyrosine kinase; BCAT1, branched-chain amino acid aminotransferase 1; CDH1, gene that encodes E-cadherin; CHK1, checkpoint kinase 1; EGFR, epidermal growth factor receptor; EMT, epithelial mesenchymal transition; FAK, focal adhesion kinase; FGFR, fibroblast growth factor receptor; HDACi, histone deacetylase inhibitor; IGFR, insulin growth factor receptor; JAK, Janus kinase; LCNEC, large cell neuroendocrine carcinoma; LKB1, serine/threonine kinase 11 or liver kinase B1; LUAD, adenocarcinoma of the lung; MAPK, mitogen-activated protein kinase; MET, MET proto-oncogene; NSCLC, non-small cell lung cancer; PI3K, phosphatidylinositol 3-kinase; PIK3CA, Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha; PLK1, polo like kinase 1; PTEN, phosphatase and tensin homolog; ROS, reactive oxygen species; Rb1, tumor suppressor Retinoblastoma 1; SCC, squamous cell carcinoma; SCLC, small cell lung cancer; STAT, signal transducer and activator of transcription; TP53, tumor protein p53; TKI, tyrosine kinase inhibitor; VEGFR, vascular endothelial growth factor receptor; 1,25(OH)2D3, 1,25-dihydroxyvitamin D3.
References
Beasley, M. B., Lantuejoul, S., Abbondanzo, S., Chu, W. S., Hasleton, P. S., Travis, W. D., et al. (2003). The P16/cyclin D1/Rb pathway in neuroendocrine tumors of the lung. Hum. Pathol. 34, 136–142. doi: 10.1053/hupa.2003.8
Belani, C. P., Goss, G., and Blumenschein, G. Jr. (2012). Recent clinical developments and rationale for combining targeted agents in non-small cell lung cancer (NSCLC). Cancer Treat. Rev. 38, 173–184. doi: 10.1016/j.ctrv.2011.05.009
Bjorge, J. D., Chan, T. O., Antczak, M., Kung, H. J., and Fujita, D. J. (1990). Activated type I phosphatidylinositol kinase is associated with the epidermal growth factor (EGF) receptor following EGF stimulation. Proc. Natl. Acad. Sci. U.S.A. 87, 3816–3820. doi: 10.1073/pnas.87.10.3816
Byers, L. A., Diao, L., Wang, J., Saintigny, P., Girard, L., Peyton, M., et al. (2013). An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 19, 279–290. doi: 10.1158/1078-0432.ccr-12-1558
Byers, L. A., Wang, J., Nilsson, M. B., Fujimoto, J., Saintigny, P., Yordy, J., et al. (2012). Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2, 798–811. doi: 10.1158/2159-8290.cd-12-0112
Calbo, J., Meuwissen, R., van Montfort, E., van Tellingen, O., and Berns, A. (2005). Genotype-phenotype relationships in a mouse model for human small-cell lung cancer. Cold Spring Harb. Symp. Quant. Biol. 70, 225–232. doi: 10.1101/sqb.2005.70.026
Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. doi: 10.1038/nature13385
Capozzi, M., Von, A. C., Divitiis, C., Ottaiano, A., Tatangelo, F., Romano, G. M., et al. (2016). Antiangiogenic therapy in pancreatic neuroendocrine tumors. Anticancer Res. 36, 5025–5030. doi: 10.21873/anticanres.11071
Carey, K. D., Garton, A. J., Romero, M. S., Kahler, J., Thomson, S., Ross, S., et al. (2006). Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 66, 8163–8171. doi: 10.1158/0008-5472.can-06-0453
Chen, B., Xiao, F., Li, B., Xie, B., Zhou, J., Zheng, J., et al. (2013). The role of epithelial-mesenchymal transition and IGF-1R expression in prediction of gefitinib activity as the second-line treatment for advanced nonsmall-cell lung cancer. Cancer Invest. 31, 454–460. doi: 10.3109/07357907.2013.820315
Chen, Y., Tang, W. Y., Tong, X., and Ji, H. (2019). Pathological transition as the arising mechanism for drug resistance in lung cancer. Cancer Commun. 39:53. doi: 10.1186/s40880-019-0402-8
Chung, J. H., Rho, J. K., Xu, X., Lee, J. S., Yoon, H. I., Lee, C. T., et al. (2011). Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 73, 176–182. doi: 10.1016/j.lungcan.2010.11.011
Clery, E., Pisapia, P., Feliciano, S., Vigliar, E., Marano, A., De Luca, C., et al. (2017). There is still a role for cytology in the ‘liquid biopsy’ era. A lesson from a TKI-treated patient showing adenocarcinoma to squamous cell carcinoma transition during disease progression. J. Clin. Pathol. 70, 798–802. doi: 10.1136/jclinpath-2017-204370
Coe, B. P., Thu, K. L., Aviel-Ronen, S., Vucic, E. A., Gazdar, A. F., Lam, S., et al. (2013). Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS One 8:e71670. doi: 10.1371/journal.pone.0071670
Cortot, A. B., Repellin, C. E., Shimamura, T., Capelletti, M., Zejnullahu, K., Ercan, D., et al. (2013). Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 73, 834–843. doi: 10.1158/0008-5472.can-12-2066
Couraud, S., Zalcman, G., Milleron, B., Morin, F., and Souquet, P. J. (2012). Lung cancer in never smokers–a review. Eur. J. Cancer 48, 1299–1311.
Cox, O. T., O’Shea, S., Tresse, E., Bustamante-Garrido, M., Kiran-Deevi, R., and O’Connor, R. (2015). IGF-1 receptor and adhesion signaling: an important axis in determining cancer cell phenotype and therapy resistance. Front. Endocrinol. 6:106. doi: 10.3389/fendo.2015.00106
Danopoulos, S., Shiosaki, J., and Al Alam, D. (2019). FGF signaling in lung development and disease: human versus mouse. Front. Genet. 10:170. doi: 10.3389/fgene.2019.00170
De Craene, B., and Berx, G. (2013). Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 13, 97–110. doi: 10.1038/nrc3447
Didkowska, J., Wojciechowska, U., Manczuk, M., and Lobaszewski, J. (2016). Lung cancer epidemiology: contemporary and future challenges worldwide. Ann. Transl. Med. 4:150. doi: 10.21037/atm.2016.03.11
Douillard, J. Y., Ostoros, G., Cobo, M., Ciuleanu, T., McCormack, R., Webster, A., et al. (2014). First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br. J. Cancer 110, 55–62. doi: 10.1038/bjc.2013.721
Franke, T. F., Yang, S. I., Chan, T. O., Datta, K., Kazlauskas, A., Morrison, D. K., et al. (1995). The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81, 727–736. doi: 10.1016/0092-8674(95)90534-0
Fukuoka, M., Yano, S., Giaccone, G., Tamura, T., Nakagawa, K., Douillard, J. Y., et al. (2003). Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J. Clin. Oncol. 21, 2237–2246. doi: 10.1200/jco.2003.10.038
Gao, B., Sun, Y., Zhang, J., Ren, Y., Fang, R., Han, X., et al. (2010). Spectrum of LKB1, EGFR, and KRAS mutations in chinese lung adenocarcinomas. J. Thorac. Oncol. 5, 1130–1135. doi: 10.1097/jto.0b013e3181e05016
Gao, Y., Zhang, W., Han, X., Li, F., Wang, X., Wang, R., et al. (2014). YAP inhibits squamous transdifferentiation of Lkb1-deficient lung adenocarcinoma through ZEB2-dependent DNp63 repression. Nat. Commun. 5:4629.
Gazdar, A. F. (2009). Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 28(Suppl. 1), S24–S31.
Gong, X., Du, J., Parsons, S. H., Merzoug, F. F., Webster, Y., Iversen, P. W., et al. (2019). Aurora a kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 9, 248–263.
Goss, G., Tsai, C. M., Shepherd, F. A., Bazhenova, L., Lee, J. S., Chang, G. C., et al. (2016). Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 17, 1643–1652. doi: 10.1016/s1470-2045(16)30508-3
Green, J., Endale, M., Auer, H., and Perl, A. K. (2016). Diversity of interstitial lung fibroblasts is regulated by platelet-derived growth factor receptor alpha kinase activity. Am. J. Respir. Cell Mol. Biol. 54, 532–545. doi: 10.1165/rcmb.2015-0095oc
Guix, M., Faber, A. C., Wang, S. E., Olivares, M. G., Song, Y., Qu, S., et al. (2008). Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Invest. 118, 2609–2619.
Gupta, R., Dastane, A. M., Forozan, F., Riley-Portuguez, A., Chung, F., Lopategui, J., et al. (2009). Evaluation of EGFR abnormalities in patients with pulmonary adenocarcinoma: the need to test neoplasms with more than one method. Mod. Pathol. 22, 128–133. doi: 10.1038/modpathol.2008.182
Ham, J. S., Kim, S., Kim, H. K., Byeon, S., Sun, J. M., Lee, S. H., et al. (2016). Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J. Thorac. Oncol. 11, e1–e4. doi: 10.1016/j.jtho.2015.09.013
Han, X., Li, F., Fang, Z., Gao, Y., Li, F., Fang, R., et al. (2014). Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat. Commun. 5:3261.
Huang, R., Wei, Y., Hung, R. J., Liu, G., Su, L., Zhang, R., et al. (2015). Associated links among smoking, chronic obstructive pulmonary disease, and small cell lung cancer: a pooled analysis in the international lung cancer consortium. EBioMedicine 2, 1677–1685.
Hussmann, D., Madsen, A. T., Jakobsen, K. R., Luo, Y., Sorensen, B. S., and Nielsen, A. L. (2017). IGF1R depletion facilitates MET-amplification as mechanism of acquired resistance to erlotinib in HCC827 NSCLC cells. Oncotarget 8, 33300–33315.
Ichihara, E., Westover, D., Meador, C. B., Yan, Y., Bauer, J. A., Lu, P., et al. (2017). SFK/FAK signaling attenuates osimertinib efficacy in both drug-sensitive and drug-resistant models of egfr-mutant lung cancer. Cancer Res. 77, 2990–3000. doi: 10.1158/0008-5472.can-16-2300
Ishak, C. A., Marshall, A. E., Passos, D. T., White, C. R., Kim, S. J., Cecchini, M. J., et al. (2016). An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell. 64, 1074–1087. doi: 10.1016/j.molcel.2016.10.021
Jakobsen, K. R., Demuth, C., Madsen, A. T., Hussmann, D., Vad-Nielsen, J., Nielsen, A. L., et al. (2017). MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells. Oncogenesis 6:e307. doi: 10.1038/oncsis.2017.17
Kareta, M. S., Gorges, L. L., Hafeez, S., Benayoun, B. A., Marro, S., Zmoos, A. F., et al. (2015). Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16, 39–50. doi: 10.1016/j.stem.2014.10.019
Kim, E. S., Herbst, R. S., Wistuba, I. I., Lee, J. J., Blumenschein, G. R. Jr., Tsao, A., et al. (2011). The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 1, 44–53.
Kim, S. M., Kwon, O. J., Hong, Y. K., Kim, J. H., Solca, F., Ha, S. J., et al. (2012). Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther. 11, 2254–2264. doi: 10.1158/1535-7163.mct-12-0311
Kim, T. M., Song, A., Kim, D. W., Kim, S., Ahn, Y. O., Keam, B., et al. (2015). Mechanisms of acquired resistance to AZD9291: a mutation-selective, irreversible EGFR inhibitor. J. Thorac. Oncol. 10, 1736–1744.
Knudsen, E. S., Pruitt, S. C., Hershberger, P. A., Witkiewicz, A. K., and Goodrich, D. W. (2019). Cell cycle and beyond: exploiting new RB1 controlled mechanisms for cancer therapy. Trends Cancer 5, 308–324. doi: 10.1016/j.trecan.2019.03.005
Kobayashi, Y., Togashi, Y., Yatabe, Y., Mizuuchi, H., Jangchul, P., Kondo, C., et al. (2015). EGFR Exon 18 mutations in lung cancer: molecular predictors of augmented sensitivity to afatinib or neratinib as compared with first- or third-generation TKIs. Clin. Cancer Res. 21, 5305–5313. doi: 10.1158/1078-0432.ccr-15-1046
Koivunen, J. P., Kim, J., Lee, J., Rogers, A. M., Park, J. O., Zhao, X., et al. (2008). Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br. J. Cancer 99, 245–252. doi: 10.1038/sj.bjc.6604469
Ku, S. Y., Rosario, S., Wang, Y., Mu, P., Seshadri, M., Goodrich, Z. W., et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. doi: 10.1126/science.aah4199
Latimer, K. M., and Mott, T. F. (2015). Lung cancer: diagnosis, treatment principles, and screening. Am. Fam. Phys. 91, 250–256.
Lee, A. F., Chen, M. C., Chen, C. J., Yang, C. J., Huang, M. S., and Liu, Y. P. (2017). Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS One 12:e0180383. doi: 10.1371/journal.pone.0180383
Lee, C. K., Man, J., Lord, S., Links, M., Gebski, V., Mok, T., et al. (2017). Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J. Thorac. Oncol. 12, 403–407. doi: 10.1016/j.jtho.2016.10.007
Lee, J. K., Lee, J., Kim, S., Kim, S., Youk, J., Park, S., et al. (2017). Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J. Clin. Oncol. 35, 3065–3074. doi: 10.1200/jco.2016.71.9096
Levacq, D., D’Haene, N., de Wind, R., Remmelink, M., and Berghmans, T. (2016). Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: a new mechanism of resistance to ALK inhibitors. Lung Cancer 102, 38–41. doi: 10.1016/j.lungcan.2016.10.012
Li, F., Han, X., Li, F., Wang, R., Wang, H., Gao, Y., et al. (2015). LKB1 inactivation elicits a redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer Cell 27, 698–711. doi: 10.1016/j.ccell.2015.04.001
Li, L., Gu, X., Yue, J., Zhao, Q., Lv, D., Chen, H., et al. (2017a). Acquisition of EGFR TKI resistance and EMT phenotype is linked with activation of IGF1R/NF-kappaB pathway in EGFR-mutant NSCLC. Oncotarget 8, 92240–92253.
Li, L., Wang, H., Li, C., Wang, Z., Zhang, P., and Yan, X. (2017b). Transformation to small-cell carcinoma as an acquired resistance mechanism to AZD9291: a case report. Oncotarget 8, 18609–18614.
Liu, C., Shaurova, T., Shoemaker, S., Petkovich, M., Hershberger, P. A., and Wu, Y. (2018). Tumor-targeted nanoparticles deliver a vitamin D-based drug payload for the treatment of EGFR tyrosine kinase inhibitor-resistant lung cancer. Mol. Pharm. 15, 3216–3226. doi: 10.1021/acs.molpharmaceut.8b00307
Lu, M., Marsters, S., Ye, X., Luis, E., Gonzalez, L., and Ashkenazi, A. (2014). E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol. Cell 54, 987–998. doi: 10.1016/j.molcel.2014.04.029
Marcoux, N., Gettinger, S. N., O’Kane, G., Arbour, K. C., Neal, J. W., Husain, H., et al. (2019). EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J. Clin. Oncol. 37, 278–285. doi: 10.1200/jco.18.01585
Marlowe, T. A., Lenzo, F. L., Figel, S. A., Grapes, A. T., and Cance, W. G. (2016). Oncogenic receptor tyrosine kinases directly phosphorylate focal adhesion kinase (FAK) as a resistance mechanism to FAK-kinase inhibitors. Mol. Cancer Ther. 15, 3028–3039. doi: 10.1158/1535-7163.mct-16-0366
Matsumura, Y., Umemura, S., Ishii, G., Tsuta, K., Matsumoto, S., Aokage, K., et al. (2015). Expression profiling of receptor tyrosine kinases in high-grade neuroendocrine carcinoma of the lung: a comparative analysis with adenocarcinoma and squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 141, 2159–2170. doi: 10.1007/s00432-015-1989-z
McGowan, M., Kleinberg, L., Halvorsen, A. R., Helland, A., and Brustugun, O. T. (2017). NSCLC depend upon YAP expression and nuclear localization after acquiring resistance to EGFR inhibitors. Genes Cancer 8, 497–504.
Meuwissen, R., Linn, S. C., Linnoila, R. I., Zevenhoven, J., Mooi, W. J., and Berns, A. (2003). Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189. doi: 10.1016/s1535-6108(03)00220-4
Miettinen, P. J., Berger, J. E., Meneses, J., Phung, Y., Pedersen, R. A., Werb, Z., et al. (1995). Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376, 337–341. doi: 10.1038/376337a0
Mok, T. S., Wu, Y. L., Ahn, M. J., Garassino, M. C., Kim, H. R., Ramalingam, S. S., et al. (2017). Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 376, 629–640.
Niederst, M. J., Sequist, L. V., Poirier, J. T., Mermel, C. H., Lockerman, E. L., Garcia, A. R., et al. (2015). RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 6:6377.
Nukaga, S., Yasuda, H., Tsuchihara, K., Hamamoto, J., Masuzawa, K., Kawada, I., et al. (2017). Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 77, 2078–2089. doi: 10.1158/0008-5472.can-16-2359
Offin, M., Chan, J. M., Tenet, M., Rizvi, H. A., Shen, R., Riely, G. J., et al. (2019). Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J. Thorac. Oncol. 14, 1784–1793. doi: 10.1016/j.jtho.2019.06.002
Okabe, T., Okamoto, I., Tamura, K., Terashima, M., Yoshida, T., Satoh, T., et al. (2007). Differential constitutive activation of the epidermal growth factor receptor in non-small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res. 67, 2046–2053. doi: 10.1158/0008-5472.can-06-3339
Okimoto, R. A., and Bivona, T. G. (2015). AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer 6, 27–34.
Ortiz-Cuaran, S., Scheffler, M., Plenker, D., Dahmen, L., Scheel, A. H., Fernandez-Cuesta, L., et al. (2016). Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin. Cancer Res. 22, 4837–4847.
Oser, M. G., Fonseca, R., Chakraborty, A. A., Brough, R., Spektor, A., Jennings, R. B., et al. (2019). Cells lacking the RB1 tumor suppressor gene are hyperdependent on aurora B kinase for survival. Cancer Discov. 9, 230–247. doi: 10.1158/2159-8290.cd-18-0389
Oser, M. G., Niederst, M. J., Sequist, L. V., and Engelman, J. A. (2015). Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 16, e165–e172. doi: 10.1016/s1470-2045(14)71180-5
Osmani, L., Askin, F., Gabrielson, E., and Li, Q. K. (2018). Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): moving from targeted therapy to immunotherapy. Semin. Cancer Biol. 52(Pt 1), 103–109. doi: 10.1016/j.semcancer.2017.11.019
Oztan, A., Fischer, S., Schrock, A. B., Erlich, R. L., Lovly, C. M., Stephens, P. J., et al. (2017). Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer 111, 84–87. doi: 10.1016/j.lungcan.2017.07.002
Park, E., Park, S. Y., Kim, H., Sun, P. L., Jin, Y., Cho, S. K., et al. (2015). Membranous insulin-like growth factor-1 receptor (IGF1R) expression is predictive of poor prognosis in patients with epidermal growth factor receptor (EGFR)-mutant lung adenocarcinoma. J. Pathol. Transl. Med. 49, 382–388. doi: 10.4132/jptm.2015.07.10
Park, K. S., Liang, M. C., Raiser, D. M., Zamponi, R., Roach, R. R., Curtis, S. J., et al. (2011). Characterization of the cell of origin for small cell lung cancer. Cell Cycle 10, 2806–2815.
Park, S., Shim, J. H., Lee, B., Cho, I., Park, W. Y., Kim, Y., et al. (2019). Paired genomic analysis of squamous cell carcinoma transformed from EGFR-mutated lung adenocarcinoma. Lung Cancer 134, 7–15. doi: 10.1016/j.lungcan.2019.05.024
Peifer, M., Fernandez-Cuesta, L., Sos, M. L., George, J., Seidel, D., Kasper, L. H., et al. (2012). Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 44, 1104–1110.
Politi, K., Zakowski, M. F., Fan, P. D., Schonfeld, E. A., Pao, W., and Varmus, H. E. (2006). Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 20, 1496–1510. doi: 10.1101/gad.1417406
Popat, S., Wotherspoon, A., Nutting, C. M., Gonzalez, D., Nicholson, A. G., and O’Brien, M. (2013). Transformation to “high grade” neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer 80, 1–4. doi: 10.1016/j.lungcan.2012.12.019
Ramalingam, S. S., Spigel, D. R., Chen, D., Steins, M. B., Engelman, J. A., Schneider, C. P., et al. (2011). Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 29, 4574–4580. doi: 10.1200/jco.2011.36.6799
Raoof, S., Mulford, I. J., Frisco-Cabanos, H., Nangia, V., Timonina, D., Labrot, E., et al. (2019). Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 38, 6399–6413.
Reguart, N., Rosell, R., Cardenal, F., Cardona, A. F., Isla, D., Palmero, R., et al. (2014). Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer 84, 161–167. doi: 10.1016/j.lungcan.2014.02.011
Ren, S., Su, C., Wang, Z., Li, J., Fan, L., Li, B., et al. (2014). Epithelial phenotype as a predictive marker for response to EGFR-TKIs in non-small cell lung cancer patients with wild-type EGFR. Int. J. Cancer 135, 2962–2971. doi: 10.1002/ijc.28925
Ricciuti, B., Metro, G., Brambilla, M., Ludovini, V., Baglivo, S., Siggillino, A., et al. (2018). Acquired resistance to afatinib due to T790M-positive squamous progression in EGFR-mutant adenosquamous lung carcinoma. J. Thorac. Oncol. 13, e9–e12. doi: 10.1016/j.jtho.2017.08.025
Richardson, F., Young, G. D., Sennello, R., Wolf, J., Argast, G. M., Mercado, P., et al. (2012). The evaluation of E-Cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small cell lung cancer treated with erlotinib as second- or third-line therapy. Anticancer Res. 32, 537–552.
Ricordel, C., Llamas-Gutierrez, F., Chiforeanu, D., Lena, H., and Corre, R. (2017). Large cell neuroendocrine lung carcinoma transformation as an acquired resistance mechanism to osimertinib. J. Thorac. Oncol. 12, e184–e186. doi: 10.1016/j.jtho.2017.07.019
Rosell, R., Carcereny, E., Gervais, R., Vergnenegre, A., Massuti, B., Felip, E., et al. (2012). Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 13, 239–246.
Sandilands, E., Akbarzadeh, S., Vecchione, A., McEwan, D. G., Frame, M. C., and Heath, J. K. (2007). Src kinase modulates the activation, transport and signalling dynamics of fibroblast growth factor receptors. EMBO Rep. 8, 1162–1169. doi: 10.1038/sj.embor.7401097
Sequist, L. V., Waltman, B. A., Dias-Santagata, D., Digumarthy, S., Turke, A. B., Fidias, P., et al. (2011). Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3:75ra26. doi: 10.1126/scitranslmed.3002003
Sequist, L. V., Yang, J. C., Yamamoto, N., O’Byrne, K., Hirsh, V., Mok, T., et al. (2013). Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 31, 3327–3334. doi: 10.1200/jco.2012.44.2806
Shah, K. N., Bhatt, R., Rotow, J., Rohrberg, J., Olivas, V., Wang, V. E., et al. (2019). Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 25, 111–118.
Sharma, S. V., Lee, D. Y., Li, B., Quinlan, M. P., Takahashi, F., Maheswaran, S., et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80.
Shepherd, F. A., Rodrigues Pereira, J., Ciuleanu, T., Tan, E. H., Hirsh, V., Thongprasert, S., et al. (2005). Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 353, 123–132.
Shi, Y., Au, J. S., Thongprasert, S., Srinivasan, S., Tsai, C. M., Khoa, M. T., et al. (2014). A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J. Thorac. Oncol. 9, 154–162. doi: 10.1097/jto.0000000000000033
Shieh, Y. S., Lai, C. Y., Kao, Y. R., Shiah, S. G., Chu, Y. W., Lee, H. S., et al. (2005). Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 7, 1058–1064. doi: 10.1593/neo.05640
Shigematsu, H., and Gazdar, A. F. (2006). Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 118, 257–262. doi: 10.1002/ijc.21496
Shigematsu, H., Takahashi, T., Nomura, M., Majmudar, K., Suzuki, M., Lee, H., et al. (2005). Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 65, 1642–1646.
Shoemaker, A. R., Mitten, M. J., Adickes, J., Ackler, S., Refici, M., Ferguson, D., et al. (2008). Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin. Cancer Res. 14, 3268–3277. doi: 10.1158/1078-0432.ccr-07-4622
Song, K. A., Niederst, M. J., Lochmann, T. L., Hata, A. N., Kitai, H., Ham, J., et al. (2018). Epithelial-to-mesenchymal transition antagonizes response to targeted therapies in lung cancer by suppressing BIM. Clin Cancer Res. 24, 197–208. doi: 10.1158/1078-0432.ccr-17-1577
Soria, J. C., Ohe, Y., Vansteenkiste, J., Reungwetwattana, T., Chewaskulyong, B., Lee, K. H., et al. (2018). Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125.
Suda, K., Tomizawa, K., Fujii, M., Murakami, H., Osada, H., Maehara, Y., et al. (2011). Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. 6, 1152–1161. doi: 10.1097/jto.0b013e318216ee52
Sulzmaier, F. J., Jean, C., and Schlaepfer, D. D. (2014). FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 14, 598–610. doi: 10.1038/nrc3792
Sutherland, K. D., Proost, N., Brouns, I., Adriaensen, D., Song, J. Y., and Berns, A. (2011). Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764. doi: 10.1016/j.ccr.2011.04.019
Suzuki, M., Shigematsu, H., Hiroshima, K., Iizasa, T., Nakatani, Y., Minna, J. D., et al. (2005). Epidermal growth factor receptor expression status in lung cancer correlates with its mutation. Hum. Pathol. 36, 1127–1134. doi: 10.1016/j.humpath.2005.08.007
Tang, Z. H., Jiang, X. M., Guo, X., Fong, C. M., Chen, X., and Lu, J. J. (2016). Characterization of osimertinib (AZD9291)-resistant non-small cell lung cancer NCI-H1975/OSIR cell line. Oncotarget 7, 81598–81610.
Thatcher, N., Chang, A., Parikh, P., Rodrigues Pereira, J., Ciuleanu, T., von Pawel, J., et al. (2005). Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa survival evaluation in lung cancer). Lancet 366, 1527–1537. doi: 10.1016/s0140-6736(05)67625-8
Thomson, S., Buck, E., Petti, F., Griffin, G., Brown, E., Ramnarine, N., et al. (2005). Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 65, 9455–9462. doi: 10.1158/0008-5472.can-05-1058
Thress, K. S., Paweletz, C. P., Felip, E., Cho, B. C., Stetson, D., Dougherty, B., et al. (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562. doi: 10.1038/nm.3854
Travis, W. D., Brambilla, E., Nicholson, A. G., Yatabe, Y., Austin, J. H. M., Beasley, M. B., et al. (2015). The 2015 world health organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260.
Upadhyay, S. K., Verone, A., Shoemaker, S., Qin, M., Liu, S., Campbell, M., et al. (2013). 1,25-Dihydroxyvitamin D3 (1,25(OH)2D3) signaling capacity and the epithelial-mesenchymal transition in non-small cell lung cancer (NSCLC): implications for use of 1,25(OH)2D3 in NSCLC treatment. Cancers 5, 1504–1521. doi: 10.3390/cancers5041504
Uramoto, H., Iwata, T., Onitsuka, T., Shimokawa, H., Hanagiri, T., and Oyama, T. (2010). Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 30, 2513–2517.
Uramoto, H., Shimokawa, H., Hanagiri, T., Kuwano, M., and Ono, M. (2011). Expression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinoma. Lung Cancer 73, 361–365. doi: 10.1016/j.lungcan.2011.01.008
Ushiro, H., and Cohen, S. (1980). Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J. Biol. Chem. 255, 8363–8365.
Vad-Nielsen, J., Gammelgaard, K. R., Daugaard, T. F., and Nielsen, A. L. (2019). Cause-and-Effect relationship between FGFR1 expression and epithelial-mesenchymal transition in EGFR-mutated non-small cell lung cancer cells. Lung Cancer 132, 132–140. doi: 10.1016/j.lungcan.2019.04.023
Vazquez-Martin, A., Cufi, S., Oliveras-Ferraros, C., Torres-Garcia, V. Z., Corominas-Faja, B., Cuyas, E., et al. (2013). IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci Rep. 3:2560.
Wang, Y., Zhang, J., Ren, S., Sun, D., Huang, H. Y., Wang, H., et al. (2019). Branched-chain amino acid metabolic reprogramming orchestrates drug resistance to EGFR tyrosine kinase inhibitors. Cell Rep. 28, 512.e6–525.e6.
Weng, C. H., Chen, L. Y., Lin, Y. C., Shih, J. Y., Lin, Y. C., Tseng, R. Y., et al. (2018). Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 38, 455–468. doi: 10.1038/s41388-018-0454-2
Wilson, N. S., Dixit, V., and Ashkenazi, A. (2009). Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat. Immunol. 10, 348–355. doi: 10.1038/ni.1714
Witkiewicz, A. K., Chung, S., Brough, R., Vail, P., Franco, J., Lord, C. J., et al. (2018). Targeting the vulnerability of RB tumor suppressor loss in triple-negative breast cancer. Cell Rep. 22, 1185–1199. doi: 10.1016/j.celrep.2018.01.022
Witta, S. E., Gemmill, R. M., Hirsch, F. R., Coldren, C. D., Hedman, K., Ravdel, L., et al. (2006). Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 66, 944–950. doi: 10.1158/0008-5472.can-05-1988
Witta, S. E., Jotte, R. M., Konduri, K., Neubauer, M. A., Spira, A. I., Ruxer, R. L., et al. (2012). Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 30, 2248–2255. doi: 10.1200/jco.2011.38.9411
Xie, M., He, C. S., Wei, S. H., and Zhang, L. (2013). Notch-1 contributes to epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance in non-small cell lung cancer in vitro and in vivo. Eur. J. Cancer 49, 3559–3572. doi: 10.1016/j.ejca.2013.07.007
Xie, M., Zhang, L., He, C. S., Xu, F., Liu, J. L., Hu, Z. H., et al. (2012). Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell Biochem. 113, 1501–1513.
Yang, Z., Yang, N., Ou, Q., Xiang, Y., Jiang, T., Wu, X., et al. (2018). Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin. Cancer Res. 24, 3097–3107. doi: 10.1158/1078-0432.ccr-17-2310
Yauch, R. L., Januario, T., Eberhard, D. A., Cavet, G., Zhu, W., Fu, L., et al. (2005). Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 11(24 Pt 1), 8686–8698. doi: 10.1158/1078-0432.ccr-05-1492
Yochum, Z. A., Cades, J., Wang, H., Chatterjee, S., Simons, B. W., O’Brien, J. P., et al. (2019). Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 38, 656–670. doi: 10.1038/s41388-018-0482-y
Yokomizo, A., Tindall, D. J., Drabkin, H., Gemmill, R., Franklin, W., Yang, P., et al. (1998). PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene 17, 475–479. doi: 10.1038/sj.onc.1201956
Yu, H. A., Arcila, M. E., Rekhtman, N., Sima, C. S., Zakowski, M. F., Pao, W., et al. (2013). Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19, 2240–2247. doi: 10.1158/1078-0432.ccr-12-2246
Yun, C. H., Mengwasser, K. E., Toms, A. V., Woo, M. S., Greulich, H., Wong, K. K., et al. (2008). The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U.S.A. 105, 2070–2075. doi: 10.1073/pnas.0709662105
Zakowski, M. F., Ladanyi, M., and Kris, M. G. (2006). Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome G. EGFR mutations in small-cell lung cancers in patients who have never smoked. N. Engl. J. Med. 355, 213–215. doi: 10.1056/nejmc053610
Zhang, W., Stabile, L. P., Keohavong, P., Romkes, M., Grandis, J. R., Traynor, A. M., et al. (2006). Mutation and polymorphism in the EGFR-TK domain associated with lung cancer. J. Thorac. Oncol. 1, 635–647. doi: 10.1097/01243894-200609000-00007
Zhang, Y. L., Yuan, J. Q., Wang, K. F., Fu, X. H., Han, X. R., Threapleton, D., et al. (2016). The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget 7, 78985–78993.
Zhang, Z., Lee, J. C., Lin, L., Olivas, V., Au, V., LaFramboise, T., et al. (2012). Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 44, 852–860.
Zhao, C., Li, X., Su, C., Li, J., Cheng, N., Ren, S., et al. (2015). High expression of E-cadherin in pleural effusion cells predicts better prognosis in lung adenocarcinoma patients. Int. J. Clin. Exp. Pathol. 8, 3104–3109.
Zhong, Z., Wen, Z., and Darnell, J. E. Jr. (1994). Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98. doi: 10.1126/science.8140422
Keywords: EGFR mutant lung cancer, EGFR tyrosine kinase inhibitors, acquired resistance, lineage plasticity, epithelial-mesenchymal transition, neuroendocrine transformation, Rb1
Citation: Shaurova T, Zhang L, Goodrich DW and Hershberger PA (2020) Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front. Genet. 11:281. doi: 10.3389/fgene.2020.00281
Received: 28 October 2019; Accepted: 09 March 2020;
Published: 27 March 2020.
Edited by:
Shaochun Bai, GeneDx, United StatesReviewed by:
Rodrigo Fernandez-Valdivia, Wayne State University, United StatesMargarida Bernardo, Wayne State University, United States
Copyright © 2020 Shaurova, Zhang, Goodrich and Hershberger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela A. Hershberger, cGFtZWxhLmhlcnNoYmVyZ2VyQHJvc3dlbGxwYXJrLm9yZw==
†These authors have contributed equally to this work