 Savithri Purayannur1
Savithri Purayannur1 Liliana M. Cano1,2*
Liliana M. Cano1,2* Megan J. Bowman3,4
Megan J. Bowman3,4 Kevin L. Childs3
Kevin L. Childs3 David H. Gent5
David H. Gent5 Lina M. Quesada-Ocampo1*
Lina M. Quesada-Ocampo1*- 1Department of Entomology and Plant Pathology, North Carolina State University, Raleigh, NC, United States
- 2Indian River Research and Education Center, Department of Plant Pathology, Institute of Food and Agricultural Sciences, University of Florida, Fort Pierce, FL, United States
- 3Department of Plant Biology, Michigan State University, East Lansing, MI, United States
- 4Ball Horticultural Company, West Chicago, IL, United States
- 5United States Department of Agriculture-Agricultural Research Service, Forage Seed and Cereal Research Unit, Oregon State University, Corvallis, OR, United States
Pseudoperonospora humuli is an obligate biotrophic oomycete that causes downy mildew (DM), one of the most destructive diseases of cultivated hop that can lead to 100% crop loss in susceptible cultivars. We used the published genome of P. humuli to predict the secretome and effectorome and analyze the transcriptome variation among diverse isolates and during infection of hop leaves. Mining the predicted coding genes of the sequenced isolate OR502AA of P. humuli revealed a secretome of 1,250 genes. We identified 296 RXLR and RXLR-like effector-encoding genes in the secretome. Among the predicted RXLRs, there were several WY-motif-containing effectors that lacked canonical RXLR domains. Transcriptome analysis of sporangia from 12 different isolates collected from various hop cultivars revealed 754 secreted proteins and 201 RXLR effectors that showed transcript evidence across all isolates with reads per kilobase million (RPKM) values > 0. RNA-seq analysis of OR502AA-infected hop leaf samples at different time points after infection revealed highly expressed effectors that may play a relevant role in pathogenicity. Quantitative RT-PCR analysis confirmed the differential expression of selected effectors. We identified a set of P. humuli core effectors that showed transcript evidence in all tested isolates and elevated expression during infection. These effectors are ideal candidates for functional analysis and effector-assisted breeding to develop DM resistant hop cultivars.
Introduction
Downy mildew (DM) pathogens are a group of obligate biotrophic oomycetes that belong to the Peronosporales lineage oomycetes and have caused epidemics in many agriculturally important plants including grapes (Gessler et al., 2011), spinach (Correll et al., 2011), cucumber (Holmes et al., 2015), and lettuce (Parra et al., 2016), to name a few. Despite their economic importance, DM pathogens have been relatively under-examined because they cannot be cultured on artificial media and are difficult to transform. Most DM research has been conducted on the Arabidopsis DM pathogen Hyaloperonospora arabidopsidis due to the wealth of genetic information available on the model plant, Arabidopsis (Kamoun, 2006). Advancements in genome sequencing have facilitated development of genomic resources for several economically important DM pathogens in the past decade. These include Pseudoperonospora cubensis (cucurbit DM) (Savory et al., 2012a, b), Peronospora tabacina (tobacco DM) (Derevnina et al., 2015), Plasmopara halstedii (sunflower DM) (Sharma et al., 2015), Plasmopara viticola (grape DM) (Dussert et al., 2016, 2019), Bremia lactucae (lettuce DM) (Fletcher et al., 2019), and Pseudoperonospora humuli (hop DM) (Rahman et al., 2019).
During infection on host plants, oomycetes secrete molecules called effectors that alter host cell machinery to allow colonization (Bozkurt et al., 2012). In resistant hosts, effectors can be directly or indirectly recognized by R proteins, in which case an effector acts as an avirulence factor that activates effector-triggered immunity (ETI) in host plants (Jones and Dangl, 2006). This avirulence function of effectors can be used in plant improvement programs to search for genotypes that contain R genes against core in planta expressed pathogen effectors (Jones et al., 2014; Vleeshouwers and Oliver, 2014).
Based on host cell localization, oomycete effectors are classified into apoplastic and cytoplasmic effectors, those localizing to the plant apoplast and those translocated into the host cell, respectively (Schornack et al., 2009). Apoplastic effectors are enzymes and enzyme inhibitors that bind to host-secreted defense molecules (Sperschneider et al., 2017). Oomycete cytoplasmic effectors are modular proteins that are distinguished by conserved domains. The most well-studied class of cytoplasmic effectors are the RXLRs, which possess an Arg-X-Leu-Arg (RXLR) amino acid motif positioned within 40 amino acids downstream of the secretion signal followed by a Glu-Glu-Arg (EER) motif (Bozkurt et al., 2012). The RXLR motif bears a striking similarity to the Plasmodium falciparum PEXEL proteins that contain a characteristic RXLXE/D/Q motif (Hiller et al., 2012) and the Toxoplasma gondii TEXEL motif (RRLXX) (Coffey et al., 2015). RXLR effector proteins are enriched in the Peronosporales clade, which contains Phytophthora and the DM pathogens (Thines and Kamoun, 2010; Thines, 2014). Several evolutionary scenarios such as gene duplication, recombination events and point mutation scenarios that can promote diversification of RXLR effectors within Peronosporales have been described (Jiang et al., 2008; Goss et al., 2013). A level of degeneracy can occur in the RXLR motif as evidenced by the RVRN motif in H. arabidopsidis (Bailey et al., 2011), QXLRs in P. cubensis (Tian et al., 2011), GKLR in B. lactucae (Stassen et al., 2013), and RXLKs in P. halstedii (Sharma et al., 2015).
The function of the RXLR motif is highly debated (Ellis and Dodds, 2011). This motif is thought to enable host cell targeting in a mechanism yet to be described. The role of the RXLR motif in host cell translocation was proposed in the Phytophthora infestans effector Avr3a (Whisson et al., 2007) whereas in the Phytophthora sojae effector AVR1b, the RXLR motif was shown to mediate binding to phosphatidylinositol phosphates for host cell uptake (Kale et al., 2010). However, recent research suggests that the RXLR motif is cleaved before secretion in Avr3a and is involved in secretion from the pathogen rather than translocation into the host cell (Wawra et al., 2017).
To add another level of complexity to the RXLR effector domain features, the C-termini of some RXLRs have combinations of the hydrophobic amino acids W, Y, and L forming a WY domain (Jiang et al., 2008; Haas et al., 2009). This domain forms a conserved structural feature and may also be present in tandem repeats joined by linker sequences (Boutemy et al., 2011; Chou et al., 2011). The WY domain is thought to mediate the “effector” function of RXLRs and suppress host immune signaling by various mechanisms, like suppression of cell death in the P. sojae effector Avr1b (Dou et al., 2008), interaction with the E3 ligase CMPG1 in the P. infestans effectors Avr3a (Bos et al., 2010), and interaction with MAPKKK in the P. infestans PexRD2 (King et al., 2014). Mutations in the WY domain abolish the interaction of the Avr3a (Bos et al., 2010) and PexRD2 (King et al., 2014) effectors with their targets. A recent report shows the role of the WY domain in effector dimerization and subsequent virulence activity (Guo et al., 2019).
Recently, genome and effectorome analyses of DM pathogens revealed the presence of WY domain-containing effectors that lacked a canonical RXLR motif in P. tabacina (Derevnina et al., 2015), P. halstedii (Sharma et al., 2015; Pecrix et al., 2019), P. viticola (Combier et al., 2019; Dussert et al., 2019), and B. lactucae (Fletcher et al., 2019). Similar effectors also are present in some Phytophthora species (Wood et al., 2019). Intriguingly, recent reports suggest that DM effectors lacking canonical RXLR motif but possessing EER and WY motifs (henceforth referred to as WY-EERs herein) display virulence and avirulence activities during host interaction (Combier et al., 2019; Wood et al., 2019).
It has been suggested that RXLRs from haustoria-forming Peronosporaceae species may be an adaptation to facilitate biotrophy because their expression is induced during pre-infection and biotrophic phases of infection (Whisson et al., 2007; Thines, 2014; Fawke et al., 2015). Therefore, follow up studies that can facilitate the identification of in planta expressed effector genes during interaction with the host are essential. To date, there are only two reported studies of gene expression profiling during pathogen-host interactions of haustoria-forming Peronosporaceae species, Pseudoperonospora cubensis infecting cucumber (Savory et al., 2012a) and H. arabidopsidis infecting Arabidopsis (Asai et al., 2014).
Pseudoperonospora humuli is an oomycete pathogen that causes DM on hop (Humulus lupulus). Symptoms including arrest in shoot development, abortion of developing cones, and defoliation, result in reduced yield, decrease in bittering acids, and even plant death in some cultivars (Ojiambo et al., 2015; Purayannur et al., 2020). DM is perhaps the most important and destructive disease that threatens cone yield and quality in certain production regions (Gent et al., 2009). Management of hop DM is mainly achieved through cultural practices and regular application of fungicides (Gent et al., 2010, 2012). Nonetheless, the pathogen can develop resistance to fungicides and cultivars with resistance to P. humuli are rare (Gent et al., 2008, 2020; Gent and Ocamb, 2009). Host resistance is the ideal control method for plant diseases, however, breeding in perennial crops such as hop is a long-term effort (Natsume et al., 2014) and a complicated one because of the narrow genetic base of resistant germplasm. Identification and utilization of effectors to identify germplasm containing R genes can accelerate breeding for resistance to DM (Vleeshouwers and Oliver, 2014). Recently, the genome sequence of P. humuli was published thus expanding the resources in this pathogen (Rahman et al., 2019). In this study we describe the effector repertoire of P. humuli with emphasis on the RXLR family. The presence of effector transcripts across different isolates and the expression of core effectors during infection are also presented.
Materials and Methods
Pathogen and Plant Material
Pseudoperonospora humuli isolate OR502AA, originally isolated from hop in Oregon, was used in this study. For RNA-Seq experiments, the susceptible hop cultivar, Pacific Gem was used. Hop leaves were placed inside square petri dishes (245 mm × 245 mm × 26 mm, Corning, Cat No. 06-443-22) and over wet sterile paper towels. Leaves were drop inoculated using 10 μl of a zoospores solution and incubated at 25°C with a 12 h light/dark cycle in a precision plant growth chamber (Thermo Fisher Scientific, Cat No. PR505755L). Twenty leaf disks were collected with a sterile core borer size 4 (Humboldt Mfg., Co., H9664, Cat No. S50166D) at 2, 3, and 4 days post inoculation (DPI). Leaf samples were ground in liquid nitrogen with a mortar and pestle to a fine powder and stored at −80°C for later RNA extraction.
RNA Extraction, Library Preparation, and Sequencing of Pseudoperonospora humuli
RNA was extracted from a fresh pellet of collected sporangia and from frozen fine leaf powder samples using the Qiagen RNeasy Plant Mini Kit (Qiagen, Cat No. 74904) and submitted to The Genomic Sciences Laboratory at North Carolina State University for library preparation and sequencing in an Illumina NextSeq 500 platform (Illumina, Inc). cDNA libraries of 350 bp insert size were prepared and sequenced as described in Withers et al. (2016). All data generated in this study is available at the Sequence Read Archive (SRA) database under accession PRJNA354153: SRX2363032 (2 DPI), SRX2363027 (3 DPI), SRX2363031 (4 DPI). The predicted coding genes from P. cubensis isolate MSU1 (study number SRP011018) reported by Savory et al. (2012b) were used for comparative sequence analysis with P. humuli OR502AA. The nuclear genome assembly and annotation of P. humuli OR502AA (Rahman et al., 2019) can be found under GenBank accession PRJNA354153, and figshare https://figshare.com/s/5cfeda89bd3d29f3d259 and https://figshare.com/s/35951fc4569554efdc34, respectively.
Phylogenetic Analysis of Nuclear Genes
A phylogenetic analysis was conducted with a concatenated set of 362 Core Eukaryotic Genes (CEGs) obtained by CEGMA version 2.5 (Parra et al., 2007) following methods by Sharma et al. (2015) with modifications. The 362 single-copy CEG genes from P. humuli OR502AA were used to extract homologs shared among 11 oomycete species The oomycete species included: P. cubensis MSU1 (Savory et al., 2012b), P. tabacina 968-J2 (Derevnina et al., 2015), P. halstedii BLA4 (Sharma et al., 2015), H. arabidopsidis EMOY2 (Baxter et al., 2010), P. infestans T30-4 (Haas et al., 2009), Phytophthora ramorum PR102a (Tyler et al., 2006), P. sojae P6497 (Tyler et al., 2006), Pythium ultimum BR144 (Levesque et al., 2010), Albugo laibachii NC14 (Kemen et al., 2011), and Saprolegnia parasitica CBS223.65 (Jiang et al., 2013). All genomes, except for P. tabacina, are available at the Fungi and Oomycete Genomics Resources Database1 and Ensembl2. The P. tabacina genome data was obtained by courtesy of R. Michelmore. Multiple sequence alignments were performed using MUSCLE version 3.8.31 (Edgar, 2004) and subsequently concatenated using custom scripts. Maximum likelihood phylogenetic analysis was done with RAxML version 8.2.4 (Stamatakis et al., 2005), with 1000 bootstrap replicates.
Identification of the Secretome
The 18,656 predicted proteins of P. humuli from published genome assembly by Rahman et al., 2019 were evaluated for the presence of signal peptides using SignalP version 2.0 (Nielsen et al., 1997) and for the absence of transmembrane domains with TMHMM version 2.0 (Krogh et al., 2001). Secreted proteins with a SignalP HMM score >0.9, NN cleavage site within 10 and 40 amino acids (aa), and no transmembrane domains found at >40 amino acids away from the starting amino acid methionine, were selected. The gene calls originally inferred from genome assembly for P. cubensis MSU1 reported by Savory et al. (2012b) were revised in this study using a BLASTN search against the NCBI nucleotide database with a cutoff sequence coverage of 90% and a percentage of identity of 90% to remove contaminating sequences. The remaining filtered gene calls of P. cubensis MSU1 were then analyzed for potential secreted proteins as described above for P. humuli. Apoplastic effectors were predicted using ApoplastP (Sperschneider et al., 2017). CAZymes were predicted using the dbCAN2 metaserver (Zhang et al., 2018). Effectors were functionally annotated using Blast2GO and BlastP searches against NCBI nr databases using an e-value of 1e-0.5.
RXLR Effector Annotation
The presence of RXLR motifs on secreted proteins was determined with custom scripts as described by Win et al. (2007) with some modifications. RXLR effectors were annotated as such when: the RXLR motif was present within 25 and 110 amino acids, the RXLR position was higher than the NN cleavage site, and the signal peptide length was 10–40 amino acids. We also used the WY domain hidden markov model (HMM) model described by Boutemy et al. (2011) to predict proteins carrying this motif downstream of the RXLR, RXLR-like motif, EER, and EER-like motifs. Proteins were permuted using the script published by Ai et al. (2020) and each script was run separately on the permuted proteins and the total number of motifs predicted was averaged. Proteins were clustered using OrthoFinder version 2.3.3 (Emms and Kelly, 2019). Sub-cellular localization of RXLRs was predicted using DeepLoc version 1.0 (Almagro Armenteros et al., 2017).
RNA-Seq Analysis
To predict transcript evidence in different isolates of P. humuli, Illumina RNA-Seq read data obtained from sporangia for OR502AA and 11 other isolates used by Rahman et al. (2019) were aligned to the coding genes of P. humuli OR502AA using TopHat2 version 2.0.9 (Trapnell et al., 2009) with 200 bp as the insertion length parameter. Alignments in SAM format were obtained from TopHat2 for gene expression analysis. Absolute read counts were calculated for each gene by using the htseq-count script part of the HTSeq python module (Anders et al., 2014). The reads per kilobase million (RPKM) values were then estimated according to a published method (Mortazavi et al., 2008). To analyze expression data of P. humuli isolate OR502AA infecting hop leaf samples at 2, 3, and 4 DPI, SAM alignments from TopHat2 and RPKM values from HTSeq were obtained as described above. To estimate the expression in planta, RPKM absolute values were transformed into log2 fold values by dividing the RPKM data from infected leaf tissue by the RPKM values from sporangia of OR502AA (Wagner et al., 2012). The log2 values were used to generate heatmaps using the R package pheatmap. Upset plots were generated using the R package UpsetR (Conway et al., 2017).
Quantitative RT-PCR
Quantitative RT-PCR was performed using gene-specific oligonucleotides. Tissue was collected and RNA was extracted as described in Sections “Pathogen and Plant Material” and “RNA Extraction, Library Preparation, and Sequencing of Pseudoperonospora humuli.” PCR was performed in a CFX96 TouchTM Real-time system (Bio-Rad Laboratories, Inc.) using a PowerUpTM SYBRTM Green Master Mix (Applied Biosystems). Gene-specific primers (Supplementary Table S1) were generated using Primer3 (Untergasser et al., 2012). P. humuli actin (Phum_OR502AA_v1_g_08820), Glycerol-3-phosphate dehydrogenase (Phum_OR502AA_v1_g_18083), and ubiquitin-conjugating enzyme E2 (Phum_OR502AA_v1_g_00443) were used as reference genes and the values were averaged for calculation. ΔΔCt values were calculated for each time-point using the expression in sporangia as a control by the method published by Livak and Schmittgen (2001) with the slight modification of the initial ΔCt being calculated as ΔCT = CT(reference gene)-CT(target gene) in order to obtain positive values. RT-qPCR was performed three times with independent biological replicates. P-values were calculated by a paired t-test using the GraphPad Prism 8.00 software for Mac.
Results
Phylogenetic Relationship of Pseudoperonospora humuli With Other Oomycetes
The genome of P. humuli showed a 97.8% complete and 98.8% partial completeness based on the coverage of eukaryotic genes (Rahman et al., 2019) described in the CEGMA pipeline (Parra et al., 2007). We generated a nuclear phylogenetic tree of P. humuli with the 10 other oomycete species listed in Section “Phylogenetic Analysis of Nuclear Genes” using 362 of the core eukaryotic housekeeping genes described by Parra et al. (2007), P. humuli and its sister species P. cubensis were clustered in a group with maximum bootstrap support confirming their close relationship (Figure 1). Other DM pathogens such as P. tabacina, and the Arabidopsis DM H. arabidopsidis formed sister groups and separated themselves from the sunflower DM pathogen P. halstedii and Phytophthora species.
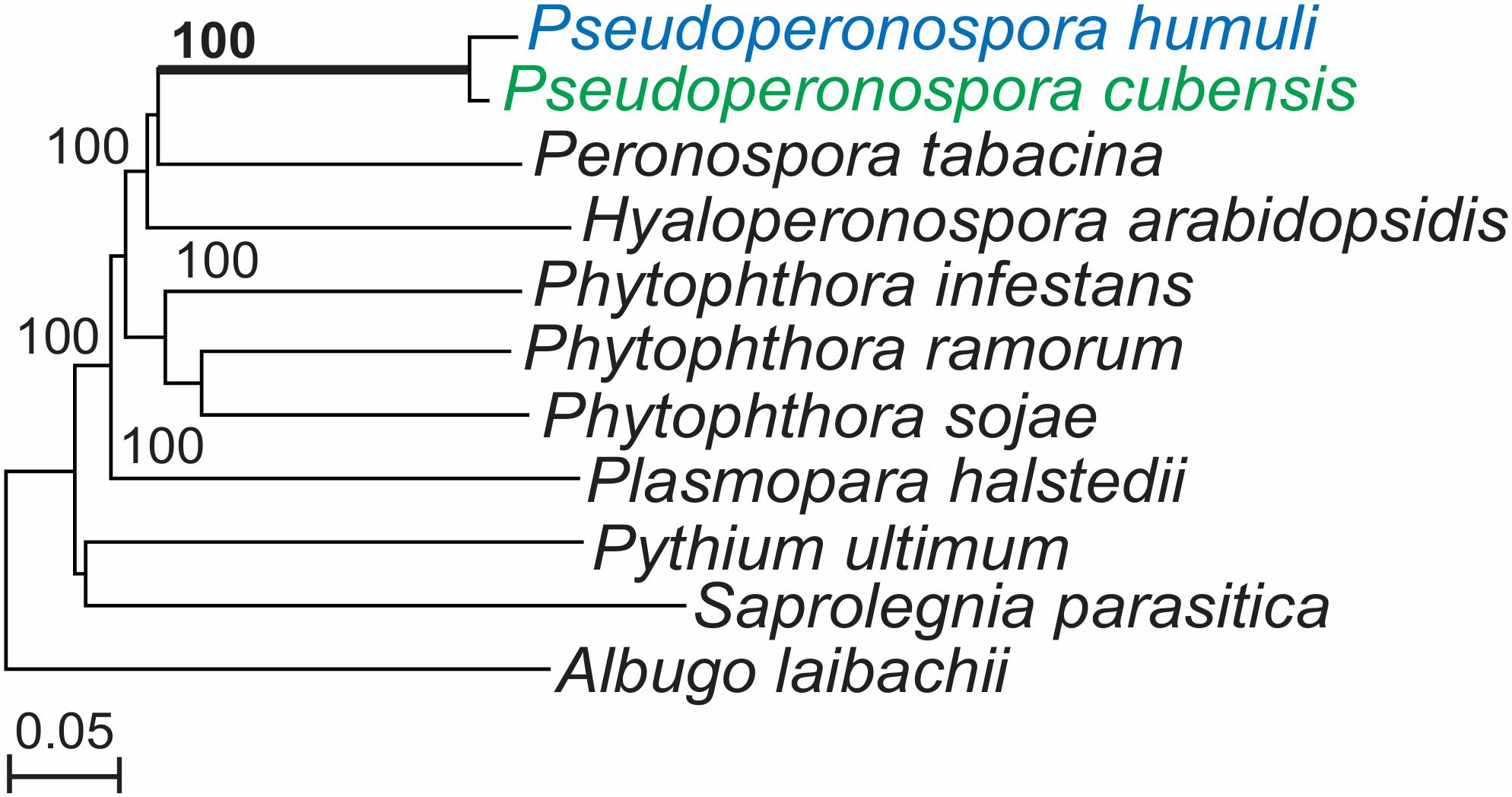
Figure 1. Pseudoperonospora humuli and Pseudoperonospora cubensis phylogenetic clade generated from conserved nuclear genes. Phylogenetic tree constructed with 362 nuclear core eukaryotic genes (CEGs) sharing homology among 11 oomycetes. CEG genes were aligned with Muscle (Edgar, 2004) and the maximum likelihood tree was generated with RAxML program (Stamatakis et al., 2005), with 1,000 bootstrap replicates.
Pseudoperonospora humuli Secretome and Apoplastic Effectors
For in silico prediction of the P. humuli secretome, we used the 18,656 predicted proteins published by Rahman et al. (2019). SignalP v2 was used for the prediction of proteins containing signal peptides since it was noted to be more sensitive for oomycete effectors in comparison to newer versions (Sperschneider et al., 2015b). Using the presence of signal peptide and the absence of a transmembrane domain as criteria, 1,250 secreted proteins were predicted in P. humuli (Supplementary Table S2). Since apoplastic effectors do not have distinguishing motifs, the machine learning tool ApoplastP (Sperschneider et al., 2017) was used to predict apoplastic effector candidates in the P. humuli secretome. Among the predicted secreted proteins, 321 proteins were predicted as apoplastic proteins (Supplementary Table S3).
The predicted apoplastome was classified into known classes of effectors (Figure 2A). CAZymes formed the largest class of apoplastic effectors in P. humuli (Figure 2A and Supplementary Table S3). Of the five known classes of CAZymes: carbohydrate esterases (CE), glycoside hydrolases (GH), glycosyltransferases (GT), polysaccharide lyases (PL), and auxiliary activity (AA) (Levasseur et al., 2013), P. humuli had only CEs, GHs, and AAs (Supplementary Table S3). The P. humuli apoplastic effector suite also consisted of 32 enzyme inhibitors of which five were protease inhibitors (Figure 2A). All the identified protease inhibitors in P. humuli belonged to the Kazal-like serine protease inhibitor family. Four of the predicted protease inhibitors in P. humuli had a single Kazal-like domain (Supplementary Figures S1A,B). The remaining protease inhibitor had five Kazal-like domains (Supplementary Figure S1B). Glucanase inhibitors formed the other abundant class of enzyme inhibitors of which there were 27 proteins in P. humuli. The P. humuli glucanase inhibitors had degenerate amino acids at the catalytic triad positions H57, D102, and S195 (Supplementary Table S3).
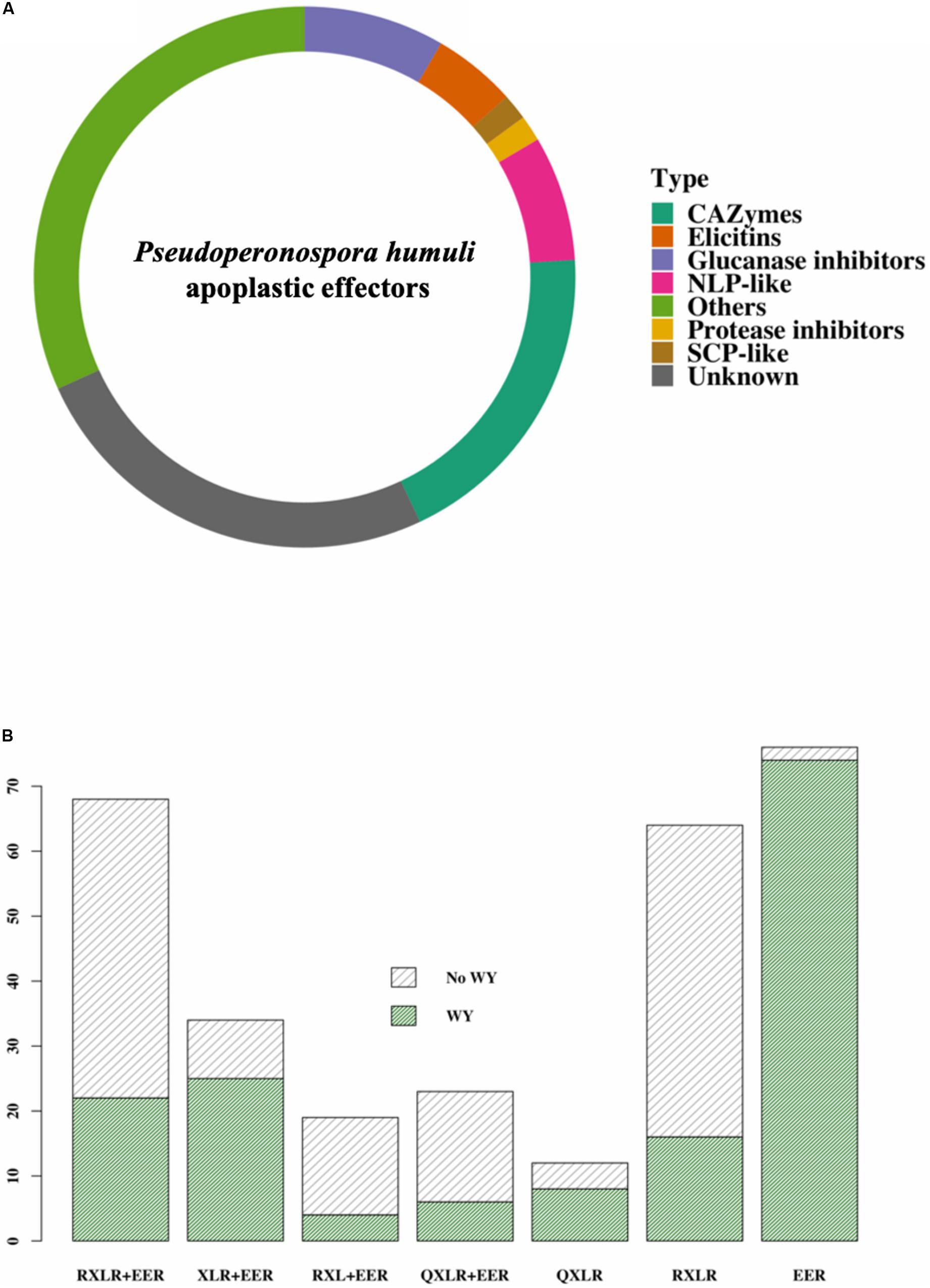
Figure 2. Effectors in Pseudoperonospora humuli. (A) Classes of apoplastic effectors. Effectors were identified from the secretome by ApoplastP (Sperschneider et al., 2017) and annotated using Blast2Go and BlastP searches. Carbohydrate active enzymes (CAZymes) were identified using the dbCAN2 metaserver (Zhang et al., 2018). [SCP, sperm coat protein; NLPs, necrosis and ethylene-inducing peptide 1 (Nep1)-like proteins]. (B) Classes of cytoplasmic effectors. RXLRs were identified using the method described by Win et al. (2007) and WY motifs were predicted by a hidden markov model (HMM) search in HMMER 3.1 (hmmer.org) using a published HMM model (Boutemy et al., 2011).
The P. humuli apoplastic effector suite had 24 necrosis and ethylene-inducing peptide 1 (Nep1)-like proteins (NLPs). However, the NLPs in P. humuli had highly degenerate heptapeptide “GHRHDWE” motifs (Supplementary Figure S2). Other predicted P. humuli apoplastic effector classes were elicitin proteins, sperm coat protein (SCP)-like extracellular proteins, and a myriad of proteins of unknown function (Figure 2A and Supplementary Table S3).
Since P. cubensis and P. humuli are sister species, the secretome of P. cubensis was also analyzed. The published proteome of P. cubensis (Savory et al., 2012b) was revised using a BLASTN search to remove genes of contaminating microorganisms. Bacterial genes with similarity to common prokaryotes found in the phyllosphere such as Xanthomonadales, Burkholderiales, and Pseudomonadales were found in the P. cubensis secretome and were removed. Other eukaryotic metazoan contaminants (Laurence et al., 2014) were also removed. Overall, we removed 5,849 gene contaminants out of 23,522 (Supplementary Table S4) coding genes predicted for P. cubensis, leaving the final P. cubensis protein count as 17,673. Of these, 941 genes were predicted to be secreted proteins using the same pipeline that was employed for P. humuli secretome prediction (Supplementary Table S5). ApoplastP predicted 216 proteins as putative apoplastic effectors (Supplementary Table S6). The numbers of the apoplastic effector classes were reduced in P. cubensis as compared to P. humuli (Table 1). Cluster analysis of apoplastic effectors of the two species showed that there were 122 clusters common to P. humuli and P. cubensis. P. humuli had more singletons than P. cubensis (Supplementary Figure S3A).
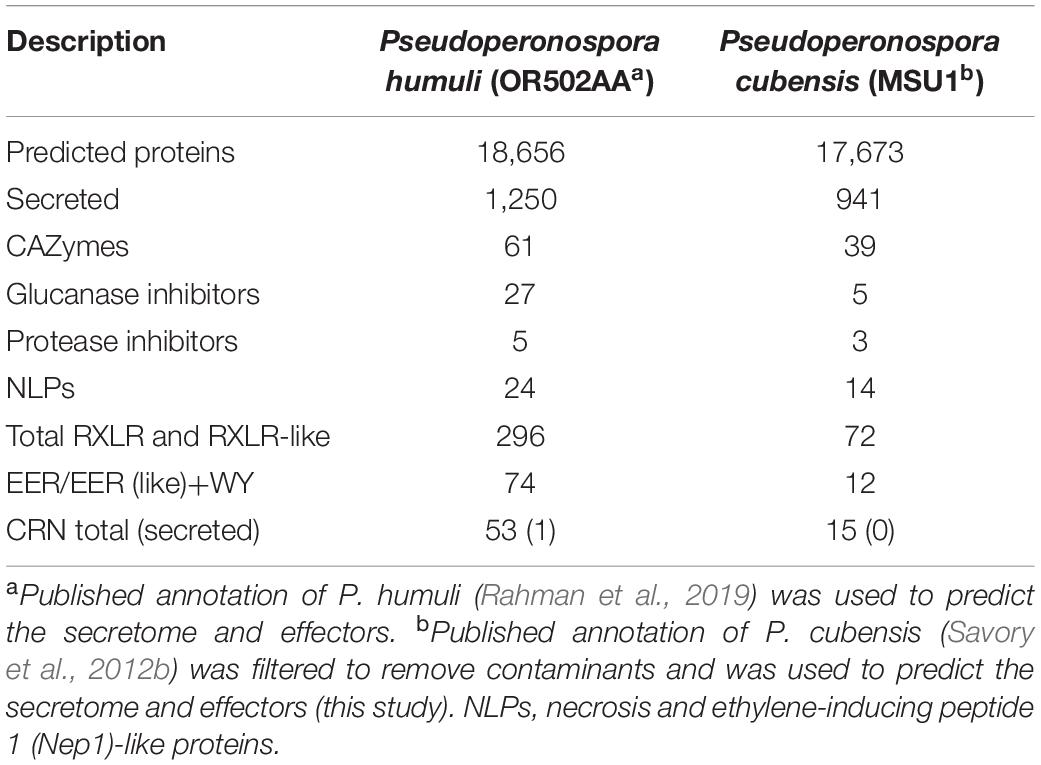
Table 1. Comparison of secretome and effector families in Pseudoperonospora humuli and Pseudoperonospora cubensis.
Cytoplasmic Effectors in Pseudoperonospora humuli
Rahman et al. (2019) predicted a total of 189 putative RXLR-like effector candidates and 49 CRinkling and Necrosis (CRN)-like candidates (with or without signal peptides) from P. humuli. We re-examined the genome of P. humuli using a comprehensive in silico string search and similarity analysis to expand the P. humuli cytoplasmic effector repertoire. Our analysis revealed a total of 296 RXLR-EER and RXLR-EER-like cytoplasmic effector candidates in the P. humuli secretome, including those proteins that contained non-canonical RXLR and/or EER motifs (Figure 2B and Supplementary Table S7). Among the predicted RXLR-like effector candidates, a large number of these effectors contained RXLR and EER motifs (Figure 2B). Several of these effectors contained non-canonical EER motifs (Supplementary Table S7). There were 35 QXLR motif-containing effectors in P. humuli including some that contained only a QXLR motif and lacked an EER (Figure 2B). Degeneracy in the RXLR motif was observed in some effectors, with changes in the first and the second arginine residues. Proline (P) and lysine (K) were common substitutes for R in the first position while glutamine (G) and K were common in the second position (Supplementary Table S7).
We used the previously published HMM model (Boutemy et al., 2011) to search for WY domains in the identified RXLRs in P. humuli. 154 out of the identified 296 RXLRs had one or more WY domains. An interesting observation was the high number of WY-EERs in P. humuli (Figure 2B). Of the 154 WY domain-containing effectors, 74 were WY-EERs (Figure 2B and Supplementary Table S7). It has been proposed previously (Wood et al., 2019) that an HMM search for WY domains in the whole secretome is an important step in oomycete effector prediction to identify additional effectors that are not revealed in RXLR-EER string searches. However, an HMM search of the P. humuli secretome failed to reveal additional WY effector candidates.
The sub-cellular localization of the 296 RXLR candidates was predicted using Deeploc v1 (Almagro Armenteros et al., 2017). There were 140 proteins that were predicted to be localized to the cytoplasm and 98 that were predicted to localize to the nucleus. There were a few proteins that had predicted localization to mitochondria, plastid, and peroxisome (Supplementary Table S8). In contrast to P. humuli, the secretome of the sister species P. cubensis had only 72 RXLR and RXLR-like proteins (Supplementary Table S9). There was a considerable reduction in number of RXLR effectors in P. cubensis as compared to P. humuli (Table 1). Gene orthology analysis of identified effector candidates in P. humuli and P. cubensis revealed 20 clusters that were common to both species (Supplementary Figure S3B).
A permutation test was performed to estimate the false discovery rate (FDR) of scripts used for analysis. The first 150 residues of the P. humuli and P. cubensis secretomes were permuted 150 times and the average number of motifs predicted was calculated. The FDRs for N-terminal single-motif scripts were higher than those for double-motif scripts as expected. FDRs were higher in P. cubensis than P. humuli with the scripts for QXLR motifs showing a higher FDR (Supplementary Table S10).
To identify members of the CRN (CRinkling and Necrosis) family of effectors in P. humuli, an HMM search was performed using a previously published HMM model (Haas et al., 2009). The P. humuli secretome had a single candidate that possessed an LFLAK-like (LYLARK) and an HVLVXXP-like motif. On the other hand, an HMM search on the 941 P. cubensis secreted proteins produced no hits. Since the low number of CRN effectors was surprising, an HMM search was performed on all 18,656 predicted P. humuli proteins, which resulted in 62 hits of which nine had no LFLAK or HVLVXXP motifs. Among the remaining 53 CRNs, 20 had both LFLAK-like and HVLVXXP-like motifs (Table 1 and Supplementary Table S11). Apart from these, there were 24 proteins with only an LFLAK-like domain and nine with only an HVLVXXP-like motif. However, only one of them had a SignalP HMM probability >0.9. P. cubensis, on the other hand had only 15 CRNs with an LFLAK-like and/or or an HVLVXXP-like motif but none had a predicted signal peptide (Table 1 and Supplementary Table S12).
Sporangial Expression of Effectors in Different Pseudoperonospora humuli Isolates
Core effectors that are expressed across different isolates may be good candidates for effector-assisted breeding. For this reason, we looked for RXLRs that have conserved transcript evidence in different P. humuli isolates. RNA-Seq performed on sporangia of the P. humuli isolate OR502AA and eight others used by Withers et al. (2016) were used to study the presence of transcripts of the predicted effector-encoding genes. In addition, we also used the three isolates NY507570BC, NC18668CAS, and NC18668GAL used by Rahman et al. (2019). Of the total 18,656 predicted genes in P. humuli, we found transcript evidence with an RPKM value > 0 for 10,536 genes in all the sequenced isolates (Supplementary Figure S4A and Supplementary Table S13). Of the 1,250 predicted secreted proteins, 754 showed transcript evidence (Supplementary Figure S4B), out of which, 171 were apoplastic effectors (Supplementary Figure S5). Two hundred one out of the predicted 296 RXLR genes exhibited transcript evidence in all the tested isolates (Figure 3). The expression level of the 201 common effectors was analyzed across the isolates (Supplementary Figure S6). Drastic differences in sporangial expression were not observed across the isolates tested. We investigated differences in transcript presence/absence based on the geographical location from which sporangia were collected. There were 85 genes that did not show transcript evidence in the two isolates collected from North Carolina (Supplementary Figure S4A). Out of these, two were genes coding for secreted apoplastic effectors (Supplementary Figures S4A, S5). Apart from this, we did not observe any notable similarities or differences in isolates collected from different cultivars or geographical locations.
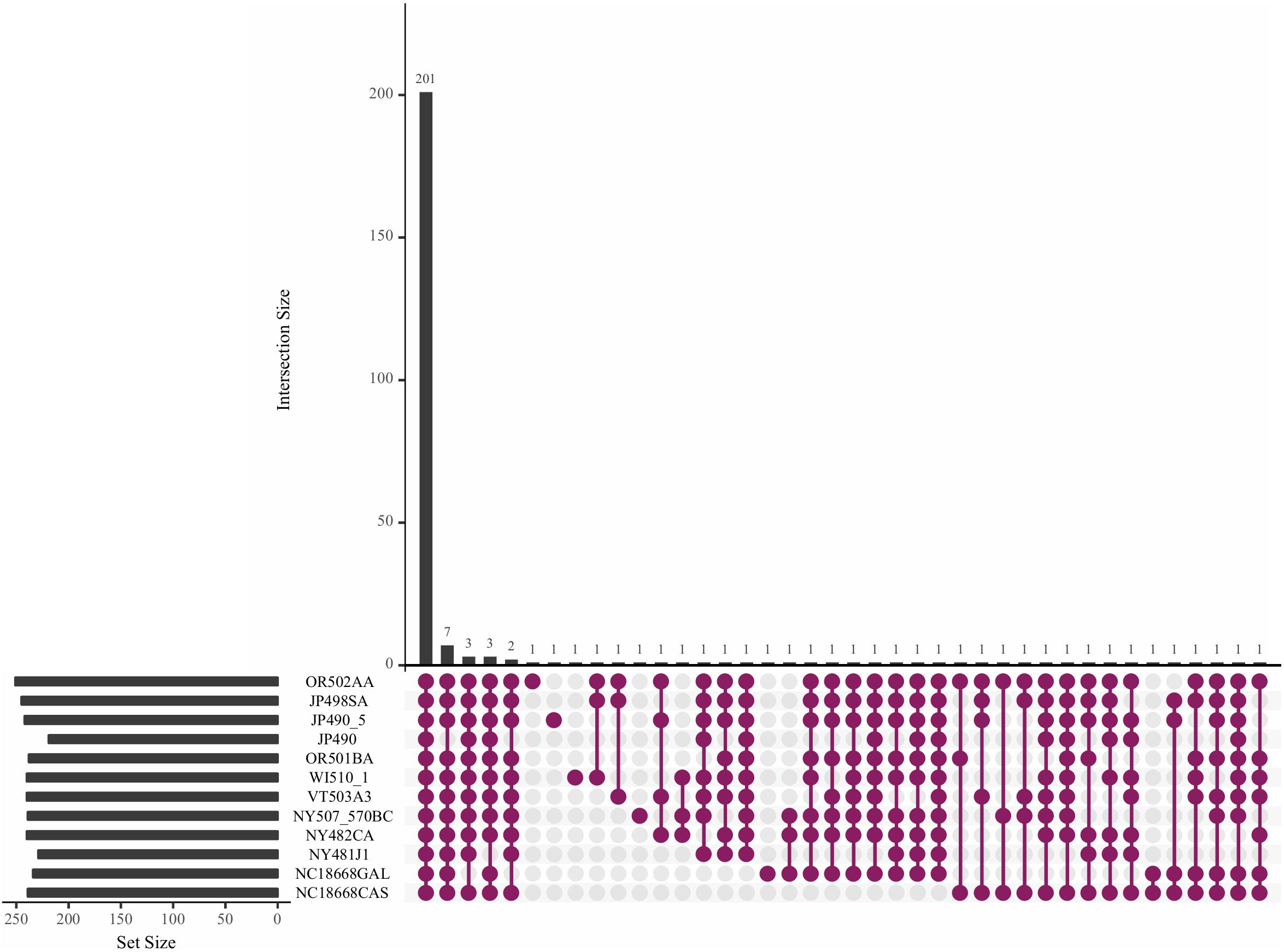
Figure 3. Upset plot showing the trend of effector transcripts in the sporangia of Pseudoperonospora humuli isolates. RNAseq reads from sequenced sporangia of different isolates were mapped to P. humuli gene calls (Rahman et al., 2019) using TopHat version 2.0.6 (Trapnell et al., 2009). Reads were counted using the HTSeq python module (Anders et al., 2014) and reads per kilobase million (RPKM) values were calculated. An RPKM > 0 was used to indicate the presence of a transcript. The plot was generated using the R package UpsetR (Conway et al., 2017).
Pseudoperonospora humuli Effectors That Are Expressed During Infection
The susceptible hop variety Pacific Gem was infected with the P. humuli isolate OR502AA and tissue was harvested at 2, 3, and 4 DPI and sequenced for expression profiling. Out of the 18,656 predicted genes, 1,381 were upregulated (log2 fold value ≥ 2) at all time-points post inoculation (Supplementary Table S14). There were 197 genes that were upregulated only at 2 DPI out of which 23 were uniquely expressed (log2 value 0 at 3 DPI and 4 DPI). Likewise, out of the 168 genes upregulated at 3 DPI, 57 were uniquely expressed and of the 190 genes upregulated at 4 DPI, 82 were uniquely expressed. Among the 321 predicted apoplastic effectors, 64 showed upregulation at all time-points (Supplementary Figure S7 and Supplementary Table S14). Glucanase inhibitors as a class were notable among the upregulated apoplastic effectors. It was also interesting to note that seven among the 11 upregulated glucanase inhibitors showed transcript presence in all the P. humuli isolates tested, suggesting the importance of this class of apoplastic effectors in infection (Supplementary Figure S7 and Supplementary Table S14).
The expression pattern of cytoplasmic effectors at different time-points was also analyzed. The single CRN gene with a predicted signal peptide (Phum_OR502AA_v1_g_03069) did not show upregulation at any time-point. Out of the 296 predicted RXLR effectors, 62 were upregulated (log2 values ≥ 2) at all time-points. It was interesting to observe that most of the expressed effectors showed consistent levels of expression across the time-points tested (Supplementary Figure S8). There were 6, 4 and 6 upregulated effectors each at 2 DPI, 3 DPI, and 4 DPI, respectively (Supplementary Figure S8 and Supplementary Table S14). The gene Phum_OR502AA_v1_g_16070 was uniquely expressed at 4 DPI (Supplementary Figure S8 and Supplementary Table S14). Since P. humuli showed an expansion of WY-motif containing effectors with EER or EER-like domains, we were specifically interested in the expression profile of that class. Of the 74 EER-like effectors with one or more WY motifs, 10 were upregulated at all time-points (Supplementary Table S14).
There were 75 core effectors in P. humuli that were upregulated during infection and were present in all the isolates tested (Supplementary Table S15). This list included both apoplastic and cytoplasmic effectors. RXLR-EERs with no WY domains and glucanase inhibitors were the largest class in this list.
The expression patterns of selected genes were validated by quantitative RT-PCR. Randomly selected RXLR genes and apoplastic effector-encoding genes were analyzed for their expression patterns. CRN genes were not selected for qPCR since the only CRN with a predicted signal peptide did not show elevated expression in RNA-seq data. All genes showed significant upregulation at all time-points with patterns comparable to those seen in the RNA-seq data (Figure 4).
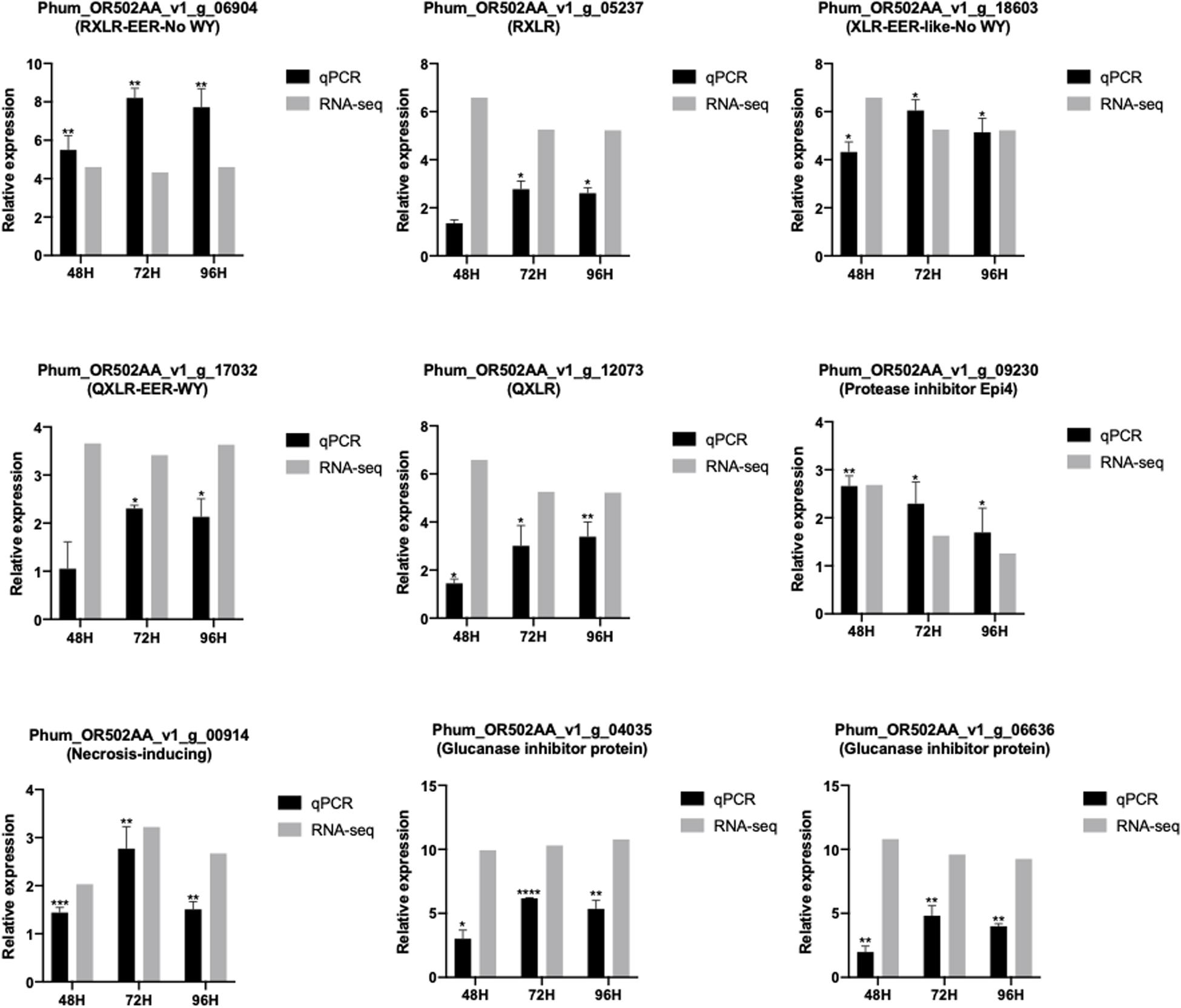
Figure 4. Comparison of RNAseq and RT-qPCR expression values of selected effector genes in different time-points of Pseudoperonospora humuli infection of hop. RNA-seq expression levels are depicted in log2 values of reads per kilobase million (RPKM) values. RT-qPCR expression levels are represented as mean ΔΔCT values from three independent biological replicates. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001, ****P-value < 0.0001. P-values were calculated using a paired t-test in the GraphPad Prism 8.00.
Discussion
Effector Content in Pseudoperonospora humuli and Pseudoperonospora cubensis
Our analysis predicted that 1,250 out of 18,656 (6.69%) proteins in P. humuli were secreted as compared to 941 out of 17,673 (5.32%) in P. cubensis. Despite similar secretome sizes between P. humuli and P. cubensis, there was an increased number of predicted effector proteins in P. humuli. The apoplastic effector content in P. humuli was much higher than in P. cubensis with a notable increase in glucanase inhibitors and CAZymes. The most astonishing increase in the number of effectors between P. humuli and P. cubensis, however, was observed in the RXLR class of effectors, especially in the number of WY-EERS. Also, despite the close phylogenetic relationship between P. humuli and P. cubensis, the number of orthologous RXLRs were few. This lack of orthology between species of the same genus is not surprising considering the high divergence in the RXLR family that has previously been reported in Phytophthora spp., with P. sojae and P. ramorum having a low number of shared orthologs (Jiang et al., 2008). Moreover, P. cubensis has most likely arisen due to a host jump from P. humuli and it has been suggested that loss of effectors required for hop colonization has possibly occurred in P. cubensis following the pathogen’s adaptation to a new host (Runge and Thines, 2012). It is also interesting to note that gene loss associated with host jumps may be due to the loss of genes that do not have targets in the new host (Sharma et al., 2014; Thines, 2019). There was a large reduction in the number of RXLRs in P. cubensis in our analysis from the earlier predicted 271 RXLRs (Savory et al., 2012a). However, differences in the pipelines used for genome annotation and effector prediction in the two taxa cannot be ruled out. Moreover, oomycete effectors are known to reside in repeat-rich regions (Raffaele and Kamoun, 2012). Due to the inherent difficulties associated with assembling repeat-rich regions with short-read sequencing technologies like Illumina, some effector genes may be missing from the prediction performed in this analysis. Nevertheless, the number of effectors predicted in P. humuli is comparable to the numbers found in other sequenced DM pathogens (Derevnina et al., 2015; Sharma et al., 2015; Dussert et al., 2019).
The variation in numbers between P. humuli and P. cubensis could also be attributed to our initial filtering for contaminants in the P. cubensis proteome. The presence of contaminants is not surprising in next generation sequencing data from biotrophic pathogens like P. cubensis and P. humuli (Laurence et al., 2014; Rahman et al., 2019). The finding of bacterial sequences in the P. cubensis proteome affirms the need for specialized laboratory protocols that account for the phyllosphere microbiome and stringent bioinformatics filtering in initial steps when dealing with the genomic data of biotrophic pathogens (Klein et al., 2019). With the range of sequencing technologies now available, resequencing the P. cubensis genome with clean material and the use of in silico methods to remove contaminants might result in more accurate RXLR predictions.
The permutation test revealed a higher FDR for proteins with positional constrained N-terminal single-motif scripts, especially for proteins with the prediction of QXLRs motifs. It is therefore important to validate the veracity of effector proteins containing degenerate RXLR motifs such as QXLR that lack a downstream EER motif with experiments such as quantitative RT-PCR before they are used for further functional studies.
Apoplastic Effectors of Pseudoperonospora humuli
In this study, we predicted the putative apoplastic effectors in the P. humuli secretome using the tool ApoplastP (Sperschneider et al., 2017). Unlike oomycete cytoplasmic effectors such as RXLRs and CRNs, apoplastic effectors do not have conserved motifs that can be used for in silico prediction. Hence, apoplastic effectors have been traditionally discovered through experimental methods like proteomics (Delaunois et al., 2014) and microscopic analysis (Doehlemann and Hemetsberger, 2013), which are time consuming. ApoplastP has limitations as an in silico prediction tool, hence, predicted apoplastic effectors need to be experimentally verified before downstream applications.
Pseudoperonospora humuli proteins predicted to be secreted to the host apoplast included known enhancers of necrotrophy and elicitors of microbe associated molecular pattern (MAMP) responses. For example, NLPs belong to a gene family that induces cell death and their role has been indicated in the switch from the biotrophic to necrotrophic phase in P. infestans (Qutob et al., 2002). The presence of NLPs in a biotrophic pathogen was surprising, however, they have been previously predicted in other DM pathogens like H. arabidopsidis (Baxter et al., 2010; Cabral et al., 2012), P. tabacina (Derevnina et al., 2015), and P. halstedii (Sharma et al., 2015). Similar to NLPs in H. arabidopsidis, the P. humuli NLPs also had highly degenerate catalytic domains suggesting that they are non-cytotoxic proteins (Cabral et al., 2012). Unlike NLPs in H. arabidopsidis, which are expressed during infection (Cabral et al., 2012), only three of the 24 P. humuli genes (Phum_OR502AA_v1_g_00914, Phum_OR502AA_v1_g_18234, and Phum_OR502AA_v1_g_19197) showed elevated expression in planta. However, variation in the time-points tested could have contributed to this discrepancy since the highest expression in H. arabidopsidis was observed during the early stages of infection (Cabral et al., 2012). Even though the importance of NLPs in DM pathogens is yet to be defined, their conservation among the different species suggests a possible role in pathogenicity. In H. arabidopsidis, NLPs also act as MAMPs and trigger immune response (Oome et al., 2014), which could be a possible reason for their predicted secretion and localization to the apoplast. Elicitins are another class of pathogen-secreted proteins that act as MAMPs (Derevnina et al., 2015; Raaymakers and den Ackerveken, 2016). Despite their negative role in pathogenicity, MAMPs like elicitins are important in disease resistance as evidenced by the successful transfer of elicitin-induced resistance against late blight to cultivated potato (Du et al., 2015). For this reason, we deemed it necessary to include proteins like NLPs and elicitins in the P. humuli effectorome, keeping with the definition of effectors as any pathogen-secreted molecules that induce or suppress plant responses (Vleeshouwers and Oliver, 2014).
CAZymes have been observed in several species of Phytophthora (Ospina-Giraldo et al., 2010; Brouwer et al., 2014) and Pythium (Zerillo et al., 2013) and their role in pathogenicity by the degradation of the cell-wall has been discussed in oomycete (Ospina-Giraldo et al., 2010; Zerillo et al., 2013) and fungal (Lyu et al., 2015) pathogens. The presence of CAZymes in the genome of pathogens alone does not implicate their role in pathogenesis since they could also be involved in the degradation and/or modification of the pathogen cell wall. However, the inclusion of CAZymes in the predicted apoplastic effector repertoire of P. humuli and the elevated expression of some members of the class indicates their possible role in pathogenesis. It is interesting to note that in previous studies CAZymes had shown elevated expression in P. cubensis during different stages of infection (Savory et al., 2012a).
The plant apoplast is a barrier that the pathogen has to overcome in order to establish infection. Host-secreted endo-β-1,4-glucanases induce the release of glucan elicitors that are recognized by host cell-surface receptors to activate immunity (Rose et al., 2002). Glucanase inhibitors, which are serine protease homologs that inhibit secretion of the plant endo-β-1,4-glucanases, are abundant in P. infestans (Damasceno et al., 2008) but have not been well-described in other sequenced DMs. However, the high number of glucanase inhibitors in P. humuli and their elevated expression during infection implicates their role in pathogenesis. Protease inhibitors were also identified in P. humuli. Three of the predicted protease inhibitors in P. humuli had a single Kazal-like domain that shared similarity to the EPI1b domain of the P. infestans EPI1 protein (Tian et al., 2004). However, none of these three proteins showed elevated expression during infection. The protease inhibitor with five Kazal-like domains, on the other hand, showed transcript evidence in all the isolates and elevated expressions during infection. The other two protease inhibitors showed elevated expression during infection and the expression pattern of Phum_OR502AA_v1_g_09230 was confirmed using quantitative RT-PCR as well.
Cytoplasmic Effectors in Pseudoperonospora humuli
The oomycete cytoplasmic RXLR and CRN effector classes are well-documented mainly due to their modular nature. CRNs are an ancient class of effectors that have been identified across phylogenetically diverse oomycete species (Schornack et al., 2010). The CRNs are a large family of effectors in P. infestans (Haas et al., 2009). In P. humuli, however, only a single CRN was identified in the secretome. A search for CRNs in the total proteome yielded more CRN candidates that contained LFLAK-like and/or HVLVXXP motifs, however these did not contain signal peptides. This trend has been observed in other DM CRNs (Derevnina et al., 2015; Sharma et al., 2015; Fletcher et al., 2018). The fewer CRN content in DM pathogens could indicate an adaptation to biotrophy since CRNs are known to induce necrosis, a trait that is not conducive for biotrophy. Intriguingly, oomycetes are known to use unconventional secretion pathways to secrete CRNs lacking a classical signal peptide (Meijer et al., 2014), suggesting that the identified CRNs in P. humuli might be secreted even though they lack a classical secretion signal. Unfortunately, there are no existing reliable algorithms for the prediction of unconventionally secreted proteins in fungi and oomycetes (Sperschneider et al., 2015a).
Effectors containing the N-terminal RXLR-EER motif are the best-understood pathogenicity factors of Phytophthora spp. and DM pathogens. P. humuli had a total of 296 RXLR and/or EER-domain containing proteins. This number includes proteins containing degenerate RXLR or EER domains. A non-canonical QXLR motif has been observed among cytoplasmic effectors in P. cubensis (Tian et al., 2011; Savory et al., 2012a) and B. lactucae (Wood et al., 2019). The high number of QXLRs in P. humuli reiterates their significance in DM pathogens. Some RXLRs are distinguished by the presence of a C-terminal α-helical fold known as the WY domain (Jiang et al., 2008; Boutemy et al., 2011). Traditionally, prediction of RXLR effectors had been based on the presence of the RXLR and the EER motif. However, recently, the presence of WY domain containing EER effectors have been identified in DM pathogens (Derevnina et al., 2015; Combier et al., 2019; Wood et al., 2019). The expansion of WY-EER effectors in P. humuli further affirms the importance of this class in DM pathogens. Intriguingly, WY-EERs form the largest class of effectors in P. humuli outnumbering even the classical RXLR-EERs. In the light of the overrepresentation of WY-EERs in P. humuli, we reiterate previous recommendations to include HMM searches for WY domains as a key criterion to identify effectors in oomycetes and especially DM pathogens (Derevnina et al., 2015; Wood et al., 2019).
The P. humuli RXLR-EER gene Phum_OR502AA_v1_g_06904 showed transcript evidence in all the 12 isolates and elevated expression in RNA-seq and RT-qPCR. A BLASTp search did not show obvious homology to any known protein except for a weak similarity (25.8 percentage identity) to a bacterial shikimate kinase. The lack of homology to known RXLRs is not surprising since RXLRs are a class of diverse proteins that show a high level of sequence divergence outside of the conserved N-terminal RXLR and EER motifs (Win et al., 2012). Despite the lack of an apparent function, the core nature of Phum_OR502AA_v1_g_06904 across the P. humuli isolates and its high expression during infection may indicate a role during the host-pathogen interaction making it a good candidate for downstream functional analysis.
Core Effectors of Pseudoperonospora humuli
Natural sources of DM resistance in hop are rare and for this reason, identifying new sources of resistance is a priority (Woods and Gent, 2016). However, breeding for resistance to DM is a laborious and time-consuming process, especially in a perennial crop sensitive to inbreeding depression (Henning et al., 2004). Pathogen effectors can be used to accelerate disease resistance through effector-assisted breeding (Vleeshouwers and Oliver, 2014) and loss of susceptibility breeding (Pavan et al., 2010). However, core effectors that are conserved across different isolates need to be validated in order to make an informed choice on the candidate effector that is selected. Effectors that are present in various pathogen isolates and also expressed during infection are considered core effectors, which are key to identifying broad spectrum and durable plant resistance genes (Jones et al., 2014; Vleeshouwers and Oliver, 2014). The P. humuli effectorome contained effectors that exhibited transcript evidence in all P. humuli isolates and showed enhanced expression during infection. We propose that these core effectors are ideal candidates for downstream functional analysis aiming toward the identification of robust sources of resistance to DM in hop.
Data Availability Statement
The datasets generated for this study can be found in the National Center for Biotechnology Information database under the Bioproject number PRJNA354153.
Author Contributions
LQ-O, SP, and LC conceived and designed the experiments and wrote the manuscript. SP, LC, MB, KC, and DG performed the experiments. SP, LC, MB, and KC analyzed the data. LQ-O, DG, and KC contributed reagents, materials, and analysis tools. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the United States Department of Agriculture (USDA) Animal and Plant Health Inspection Service (APHIS) Award 13-8130-0254-CA, USDA National Institute of Food and Agriculture Hatch project numbers NC02628, FLA-IRC-005637, and USDA-Agricultural Research Service CRIS 2072-21000-046-00.
Conflict of Interest
MB is employed by the company Ball Horticultural Company, West Chicago, IL, United States.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all the members of the Quesada lab and Nanci Adair for their valuable help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00910/full#supplementary-material
TABLE S1 | List of genes used for RT-qPCR and their primer sequences.
TABLE S2 | Secretome annotations and amino acid sequences Pseudoperonospora humuli.
TABLE S3 | Predicted apoplastic effectors in Pseudoperonospora humuli, their annotation, SignalP HMM score, signal peptide position, and amino acid sequences. The different classes are separated in different sheets. The degenerate amino acids in the catalytic triad of glucanase inhibitors are also presented.
TABLE S4 | List of Pseudoperonospora cubensis predicted contaminant coding genes (CDSs) identified using BLASTN search against NCBI nr database.
TABLE S5 | Secretome annotations and amino acid sequences Pseudoperonospora cubensis.
TABLE S6 | Predicted apoplastic effectors in Pseudoperonospora cubensis, their annotation, SignalP HMM score, signal peptide position, and amino acid sequences. The different classes are separated in different sheets.
TABLE S7 | RXLR effector annotations and amino acid sequences for Pseudoperonospora humuli.
TABLE S8 | Predicted sub-cellular localizations of RXLR effectors in Pseudoperonospora humuli. Predictions were made using DeepLoc-1.0 (Almagro Armenteros et al., 2017).
TABLE S9 | RXLR effector annotations and amino acid sequences for Pseudoperonospora cubensis.
TABLE S10 | Average number of motifs in permuted sequences and secretome in Pseudoperonospora humuli and Pseudoperonospora cubensis.
TABLE S11 | CRN effectors of Pseudoperonospora humuli. Motif sequences and positions of LFLAK and HVLVXXP motifs are presented. Single protein with predicted signal peptide are highlighted.
TABLE S12 | CRN effectors of Pseudoperonospora cubensis. Motif sequences and positions of LFLAK and HVLVXXP motifs are presented.
TABLE S13 | Reads per kilobase million (RPKM) values of RXLR effectors in the sporangia of different Pseudoperonospora humuli isolates.
TABLE S14 | Log2 transformed reads per kilobase million (RPKM) values of Pseudoperonospora humuli genes during infection on hop.
TABLE S15 | List of core effectors in Pseudoperonospora humuli.
Footnotes
References
Ai, G., Yang, K., Ye, W., Tian, Y., Du, Y., Zhu, H., et al. (2020). Prediction and characterization of RXLR effectors in Pythium species. Mol. Plant Microbe Interact. [Epub ahead of print].
Almagro Armenteros, J. J., Sønderby, C. K., Sønderby, S. K., Nielsen, H., and Winther, O. (2017). DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 33, 3387–3395. doi: 10.1093/bioinformatics/btx431
Anders, S., Pyl, P. T., and Huber, W. (2014). HTSeq–A Python framework to work with high-throughput sequencing data. Bioinformatics 38, 166–169. doi: 10.1093/bioinformatics/btu638
Asai, S., Rallapalli, G., Piquerez, S. J. M., Caillaud, M. C., Furzer, O. J., Ishaque, N., et al. (2014). Expression profiling during Arabidopsis/downy mildew interaction reveals a highly-expressed effector that attenuates responses to salicylic acid. PLoS Pathog. 10:e1004443. doi: 10.1371/journal.ppat.1004443
Bailey, K., Cevik, V., Holton, N., Byrne-Richardson, J., Sohn, K. H., Coates, M., et al. (2011). Molecular cloning of ATR5(Emoy2) from Hyaloperonospora arabidopsidis, an avirulence determinant that triggers RPP5-mediated defense in Arabidopsis. Mol. Plant Microbe Interact. 24, 827–838. doi: 10.1094/mpmi-12-10-0278
Baxter, L., Tripathy, S., Ishaque, N., Boot, N., Cabral, A., Kemen, E., et al. (2010). Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330, 1549–1551. doi: 10.1126/science.1195203
Bos, J. I. B., Armstrong, M. R., Gilroy, E. M., Boevink, P. C., Hein, I., Taylor, R. M., et al. (2010). Phytophthora infestans effector AVR3a is essential for virulence and manipulates plant immunity by stabilizing host E3 ligase CMPG1. Proc. Natl. Acad. Sci. U.S.A. 107, 9909–9914. doi: 10.1073/pnas.0914408107
Boutemy, L. S., King, S. R. F., Win, J., Hughes, R. K., Clarke, T. A., Blumenschein, T. M. A., et al. (2011). Structures of Phytophthora RXLR effector proteins: a conserved but adaptable fold underpins functional diversity. J. Biol. Chem. 286, 35834–35842. doi: 10.1074/jbc.m111.262303
Bozkurt, T. O., Schornack, S., Banfield, M. J., and Kamoun, S. (2012). Oomycetes, effectors, and all that jazz. Curr. Opin. Plant Biol. 15, 1–10.
Brouwer, H., Coutinho, P. M., Henrissat, B., and de Vries, R. P. (2014). Carbohydrate-related enzymes of important Phytophthora plant pathogens. Fungal Genet. Biol. 72, 192–200. doi: 10.1016/j.fgb.2014.08.011
Cabral, A., Oome, S., Sander, N., Küfner, I., Nürnberger, T., and Van den Ackerveken, G. (2012). Nontoxic Nep1-like proteins of the DM pathogen Hyaloperonospora arabidopsidis: repression of necrosis-inducing activity by a surface-exposed region. Mol. Plant Microbe Interact. 25, 697–708. doi: 10.1094/mpmi-10-11-0269
Chou, S., Krasileva, K. V., Holton, J. M., Steinbrenner, A. D., Alber, T., and Staskawicz, B. J. (2011). Hyaloperonospora arabidopsidis ATR1 effector is a repeat protein with distributed recognition surfaces. Proc. Natl. Acad. Sci. U.S.A. 108, 13323–13328. doi: 10.1073/pnas.1109791108
Coffey, M. J., Sleebs, B. E., Uboldi, A. D., Garnham, A., Franco, M., Marino, N. D., et al. (2015). An aspartyl protease defines a novel pathway for export of Toxoplasma proteins into the host cell. elife 4, 1–34.
Combier, M., Evangelisti, E., Piron, M., Rengel, D., Legrand, L., Shenhav, L., et al. (2019). A secreted WY-domain-containing protein present in European isolates of the oomycete Plasmopara viticola induces cell death in grapevine and tobacco species. PLoS ONE 14:e0220184. doi: 10.1371/journal.pone.0220184
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Correll, J. C., Bluhm, B. H., Feng, C., Lamour, K., du Toit, L. J., and Koike, S. T. (2011). Spinach: better management of downy mildew and white rust through genomics. Eur. J. Plant Pathol. 129, 193–205. doi: 10.1007/s10658-010-9713-y
Damasceno, C. M., Bishop, J. G., Ripoll, D. R., Win, J., Kamoun, S., and Rose, J. K. (2008). Structure of the glucanase inhibitor protein (GIP) family from Phytophthora species suggests coevolution with plant endo-beta-1,3-glucanases. Mol. Plant Microbe Interact. 21, 820–830. doi: 10.1094/mpmi-21-6-0820
Delaunois, B., Jeandet, P., Clement, C., Baillieul, F., Dorey, S., and Cordelier, S. (2014). Uncovering plant-pathogen crosstalk through apoplastic proteomic studies. Front. Plant Sci. 5:249. doi: 10.3389/fpls.2014.00249
Derevnina, L., Chin-Wo-Reyes, S., Martin, F., Wood, K., Froenicke, L., Spring, O., et al. (2015). Genome sequence and architecture of the tobacco downy mildew pathogen Peronospora tabacina. Mol. Plant Microbe Interact. 28, 1198–1215. doi: 10.1094/mpmi-05-15-0112-r
Doehlemann, G., and Hemetsberger, C. (2013). Apoplastic immunity and its suppression by filamentous plant pathogens. New Phytol. 198, 1001–1016. doi: 10.1111/nph.12277
Dou, D., Kale, S. D., Wang, X., Chen, Y., Wang, Q., Wang, X., et al. (2008). Conserved C-terminal motifs required for avirulence and suppression of cell death by Phytophthora sojae effector Avr1b. Plant Cell 20, 1118–1133. doi: 10.1105/tpc.107.057067
Du, J., Verzaux, E., Chaparro-Garcia, A., Bijsterbosch, G., Keizer, L. C. P., Zhou, J., et al. (2015). Elicitin recognition confers enhanced resistance to Phytophthora infestans in potato. Nat. Plants 1:15034.
Dussert, Y., Gouzy, J., Richart-Cervera, S., Mazet, I. D., DelieÌre, L., Couture, C., et al. (2016). Draft genome sequence of Plasmopara viticola, the grapevine downy mildew pathogen. GenomeAnnounc. 4:e987-16.
Dussert, Y., Mazet, I. D., Couture, C., Gouzy, J., Piron, M.-C., Kuchly, C., et al. (2019). A high-quality grapevine downy mildew genome assembly reveals rapidly evolving and lineage-specific putative host adaptation genes. Genome Biol. Evol. 11, 954–969. doi: 10.1093/gbe/evz048
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ellis, J. G., and Dodds, P. N. (2011). Showdown at the RXLR motif: serious differences of opinion in how effector proteins from filamentous eukaryotic pathogens enter plant cells. Proc. Natl. Acad. Sci. U.S.A. 108, 14381–14382. doi: 10.1073/pnas.1111668108
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238.
Fawke, S., Doumane, M., and Schornack, S. (2015). Oomycete interactions with plants: infection strategies and resistance principles. Microbiol. Mol. Biol. Rev. 79, 263–280. doi: 10.1128/mmbr.00010-15
Fletcher, K., Gil, J., Bertier, L. D., Kenefick, A., Wood, K. J., Zhang, L., et al. (2019). Genomic signatures of heterokaryosis in the oomycete pathogen Bremia lactucae. Nature Comm. 10:2645.
Fletcher, K., Klosterman, S. J., Derevnina, L., Martin, F., Bertier, L. D., Koike, S., et al. (2018). Comparative genomics of downy mildews reveals potential adaptations to biotrophy. BMC Genomics 19:851. doi: 10.1186/s12864-018-5214-8
Gent, D. H., Barbour, J. D., Dreves, A. J., James, D. G., Parker, R., and Walsh, D. B. (eds) (2009). Field guide for Integrated Field Management in Hops. Pullman, WA: U.S. Hop Industry Plant Protection Committee.
Gent, D. H., Block, M., and Claassen, B. J. (2020). High levels of insensitivity of phosphonate fungicides in Pseudoperonospora humuli. Plant Dis. 104, 1400–1406. doi: 10.1094/pdis-10-19-2067-re
Gent, D. H., Nelson, M. E., and Grove, G. G. (2008). Persistence of phenylamide insensitivity in Pseudoperonospora humuli. Plant Dis. 92, 463–468. doi: 10.1094/pdis-92-3-0463
Gent, D. H., Nelson, M. E., Grove, G. G., Mahaffee, W. F., Turechek, W. W., and Woods, J. L. (2012). Association of spring pruning practices with severity of powdery mildew and downy mildew on hop. Plant Dis. 96, 1343–1351. doi: 10.1094/pdis-01-12-0084-re
Gent, D. H., and Ocamb, C. M. (2009). Predicting infection risk of hop by Pseudoperonospora humuli. Phytopathology 99, 1190–1198. doi: 10.1094/phyto-99-10-1190
Gent, D. H., Ocamb, C. M., and Farnsworth, J. L. (2010). Forecasting and management of hop downy mildew. Plant Dis. 94, 425–431. doi: 10.1094/pdis-94-4-0425
Gessler, C., Pertot, I., and Perazzolli, M. (2011). Plasmopara viticola: a review of knowledge on downy mildew of grapevine and effective disease management. Phytopathol. Mediterr. 50, 3–44.
Goss, E. M., Press, C. M., and Grünwald, N. J. (2013). Evolution of RXLR-class effectors in the oomycete plant pathogen Phytophthora ramorum. PLoS ONE 8:e79347. doi: 10.1371/journal.pone.0079347
Guo, B., Wang, H., Yang, B., Jiang, W., Jing, M., Li, H., et al. (2019). Phytophthora sojae effector PsAvh240 inhibits host aspartic protease secretion to promote infection. Mol. Plant Pathol. 12, 552–564. doi: 10.1016/j.molp.2019.01.017
Haas, B. J., Kamoun, S., Zody, M. C., Jiang, R. H., Handsaker, R. E., Cano, L. M., et al. (2009). Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461, 393–398.
Henning, J. A., Townsend, M. S., and Kenny, S. T. (2004). Potential heterotic crosses in hops as estimated by AFLP-based genetic diversity and coefficient of coancestry. J. Am. Soc. Brew. Chem. 62, 63–70. doi: 10.1094/asbcj-62-0063
Hiller, N. L., Bhattacharjee, S., Ooij, C., Liolios, K., Harrison, T., Lopez-Estraño, C., et al. (2012). A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science 10, 1934–1938.
Holmes, G. J., Ojiambo, P. S., Hausbeck, M. K., Quesada-Ocampo, L. M., and Keinath, A. P. (2015). Resurgence of cucurbit downy mildew in the United States: a watershed event for research and extension. Plant Dis. 99, 428–441. doi: 10.1094/pdis-09-14-0990-fe
Jiang, R. H., De Bruijn, I., Haas, B. J., Belmonte, R., Lobach, L., Christie, J., et al. (2013). Distinctive expansion of potential virulence genes in the genome of the oomycete fish pathogen Saprolegnia parasitica. PLoS Genet. 9:e1003272. doi: 10.1371/journal.pgen.1003272
Jiang, R. H., Tripathy, S., Govers, F., and Tyler, B. M. (2008). RXLR effector reservoir in two Phytophthora species is dominated by a single rapidly evolving superfamily with more than 700 members. Proc. Natl. Acad. Sci. U.S.A. 105, 4874–4879. doi: 10.1073/pnas.0709303105
Jones, J. D. G., Witek, K., Verweij, W., Jupe, F., Cooke, D., Dorling, S., et al. (2014). Elevating crop disease resistance with cloned genes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130087. doi: 10.1098/rstb.2013.0087
Kale, S. D., Gu, B., Capelluto, D. G. S., Dou, D. L., Feldman, E., Rumore, A., et al. (2010). External lipid PI-3-P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell 142, 284–295. doi: 10.1016/j.cell.2010.06.008
Kamoun, S. (2006). A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 44, 41–60. doi: 10.1146/annurev.phyto.44.070505.143436
Kemen, E., Gardiner, A., Schultz-Larsen, T., Kemen, A. C., Balmuth, A. L., Robert-Seilaniantz, A., et al. (2011). Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biol. 9:e1001094. doi: 10.1371/journal.pbio.1001094
King, S. R. F., McLellan, H., Boevink, P. C., Armstrong, M. R., Bukharova, T., Sukarta, O., et al. (2014). Phytophthora infestans RXLR effector PexRD2 interacts with host MAPKKK to suppress plant immune signaling. Plant Cell 26, 1345–1359. doi: 10.1105/tpc.113.120055
Klein, J., Neilen, M., van Verk, M., Dutilh, B. E., and Van den Ackerveken, G. (2019). Genome reconstruction of the non-culturable spinach downy mildew Peronospora effusa by metagenome filtering. bioRxiv [Preprint] doi: 10.1101/842658
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Laurence, M., Hatzis, C., and Brash, D. E. (2014). Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS ONE 9:e97876. doi: 10.1371/journal.pone.0097876
Levasseur, A., Drula, E., Lombard, V., Coutinho, P. M., and Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6:41. doi: 10.1186/1754-6834-6-41
Levesque, C. A., Brouwer, H., Cano, L., Hamilton, J. P., Holt, C., Huitema, E., et al. (2010). Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 11:R73.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lyu, X., Shen, C., Fu, Y., Xie, J., Jiang, D., Li, G., et al. (2015). Comparative genomic and transcriptional analyses of the carbohydrate-active enzymes and secretomes of phytopathogenic fungi reveal their significant roles during infection and development. Sci. Rep. 5:15565. doi: 10.1038/srep15565
Meijer, H. J., Mancuso, F. M., Espadas, G., Seidl, M. F., Chiva, C., Govers, F., et al. (2014). Profiling the secretome and extracellular proteome of the potato late blight pathogen Phytophthora infestans. Mol. Cell Proteomics 13, 2101–2113. doi: 10.1074/mcp.m113.035873
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Natsume, S., Takagi, H., Shiraishi, A., Murata, J., Toyonaga, H., Patzak, J., et al. (2014). The draft genome of hop (Humulus lupulus), an essence for brewing. Plant Cell Physiol. 56, 428–441. doi: 10.1093/pcp/pcu169
Nielsen, H., Engelbrecht, J., Brunak, S., and Von Heijne, G. (1997). A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8, 581–599. doi: 10.1142/s0129065797000537
Ojiambo, P. S., Gent, D. H., Quesada-Ocampo, L. M., Hausbeck, M. K., and Holmes, G. J. (2015). Epidemiology and population biology of Pseudoperonospora cubensis: a model system for management of downy mildews. Annu. Rev. Phytopathol. 53, 223–246. doi: 10.1146/annurev-phyto-080614-120048
Oome, S., Raaymakers, T. M., Cabral, A., Samwel, S., Böhm, H., Albert, I., et al. (2014). Nep1-like proteins from three kingdoms of life act as a microbe-associated molecular pattern in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111, 16955–16960. doi: 10.1073/pnas.1410031111
Ospina-Giraldo, M. D., Griffith, J. G., Laird, E. W., and Mingora, C. (2010). The CAZyome of Phytophthora spp: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genomics 28:525. doi: 10.1186/1471-2164-11-525
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Parra, L., Maisonneuve, B., Lebeda, A., Schut, J., Christopoulou, M., Jeuken, M., et al. (2016). Rationalization of genes for resistance to Bremia lactucae in lettuce. Euphytica 210, 309–326. doi: 10.1007/s10681-016-1687-1
Pavan, S., Jacobsen, E., Visser, R. G. F., and Bai, Y. (2010). Loss of susceptibility as a novel breeding strategy for durable and broad-spectrum resistance. Mol. Breed. 25, 1–12. doi: 10.1007/s11032-009-9323-6
Pecrix, Y., Buendia, L., Penouilh-Suzette, C., Marechaux, M., Legrand, L., Bouchez, O., et al. (2019). Sunflower resistance to multiple DM pathotypes revealed by recognition of conserved effectors of the oomycete Plasmopara halstedii. Plant J. 97, 730–748. doi: 10.1111/tpj.14157
Purayannur, S., Miles, T. D., Gent, D. H., Pigg, S., and Quesada-Ocampo, L. M. (2020). Hop downy mildew caused by Pseudoperonospora humuli: a diagnostic guide. Plant Health Prog. 21, 173–179. doi: 10.1094/php-10-19-0072-dg
Qutob, D., Kamoun, S., and Gijzen, M. (2002). Expression of a Phytophthora sojae necrosis inducing protein occurs during transition from biotrophy to necrotrophy. Plant J. 32, 361–373. doi: 10.1046/j.1365-313x.2002.01439.x
Raaymakers, T. M., and den Ackerveken, G. V. (2016). Extracellular recognition of oomycetes during biotrophic infection of plants. Front. Plant Sci. 7:906. doi: 10.3389/fpls.2016.00906
Raffaele, S., and Kamoun, S. (2012). Genome evolution in filamentous plant pathogens: why bigger can be better. Nat. Rev. Microbiol. 10, 417–430. doi: 10.1038/nrmicro2790
Rahman, A., Góngora-Castillo, E., Bowman, M. J., Childs, K. L., Gent, D. H., Martin, F. N., et al. (2019). Genome sequencing and transcriptome analysis of the hop downy mildew pathogen Pseudoperonospora humuli reveal species-specific genes for molecular detection. Phytopathology 109, 1354–1366. doi: 10.1094/phyto-11-18-0431-r
Rose, J. K., Ham, K. S., Darvill, A. G., and Albersheim, P. (2002). Molecular cloning and characterization of glucanase inhibitor proteins: coevolution of a counter defense mechanism by plant pathogens. Plant Cell 14, 1329–1345. doi: 10.1105/tpc.002253
Runge, F., and Thines, M. (2012). Reevaluation of host specificity of the closely related species Pseudoperonospora humuli and P. cubensis. Plant Dis. 96, 55–61. doi: 10.1094/pdis-01-11-0035
Savory, E. A., Adhikari, B. N., Hamilton, J. P., Vaillancourt, B., Buell, C. R., and Day, B. (2012a). mRNA-Seq analysis of the Pseudoperonospora cubensis transcriptome during cucumber (Cucumis sativus L.) infection. PLoS ONE 7:e35796. doi: 10.1371/journal.pone.0035796
Savory, E. A., Zou, C., Adhikari, B. N., Hamilton, J. P., Buell, C. R., Shiu, S. H., et al. (2012b). Alternative splicing of a multi-drug transporter from Pseudoperonospora cubensis generates an RXLR effector protein that elicits a rapid cell death. PLoS ONE 7:e34701. doi: 10.1371/journal.pone.0034701
Schornack, S., Huitema, E., Cano, L. M., Bozkurt, T. O., Oliva, R., Van Damme, M., et al. (2009). Ten things to know about oomycete effectors. Mol. Plant Pathol. 10, 795–803. doi: 10.1111/j.1364-3703.2009.00593.x
Schornack, S., van Damme, M., Bozkurt, T. O., Cano, L. M., Smoker, M., Thines, M., et al. (2010). Ancient class of translocated oomycete effectors targets the host nucleus. Proc. Natl. Acad. Sci. U.S.A. 107, 17421–17426. doi: 10.1073/pnas.1008491107
Sharma, R., Mishra, B., Runge, F., and Thines, M. (2014). Gene loss rather than gene gain is associated with a host jump from monocots to dicots in the smut fungus Melanopsichium pennsylvanicum. Genome Biol. Evol. 6, 2034–2049. doi: 10.1093/gbe/evu148
Sharma, R., Xia, X., Cano, L. M., Evangelisti, E., Kemen, E., Judelson, H., et al. (2015). Genome analyses of the sunflower pathogen Plasmopara halstedii provide insights into effector evolution in DMs and Phytophthora. BMC Genomics 16:741. doi: 10.1186/s12864-015-1904-7
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Manners, J. M., Singh, K. B., et al. (2015a). Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 11:e1004806. doi: 10.1371/journal.ppat.1004806
Sperschneider, J., Williams, A. H., Hane, J. K., Singh, K. B., and Taylor, J. M. (2015b). Evaluation of secretion prediction highlights differing approaches needed for oomycete and fungal effectors. Front. Plant Sci. 6:1168. doi: 10.3389/fpls.2015.01168
Sperschneider, J., Dodds, P. N., Singh, K. B., and Taylor, J. M. (2017). ApoplastP: prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 217, 1764–1778. doi: 10.1111/nph.14946
Stamatakis, A., Ludwig, T., and Meier, H. (2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463. doi: 10.1093/bioinformatics/bti191
Stassen, J. H. M., den Boer, E., Vergeer, P. W. J., Andel, A., Ellendorff, U., Pelgrom, K., et al. (2013). Specific in planta recognition of two GKLR proteins of the downy mildew Bremia lactucae revealed in a large effector screen in lettuce. Mol. Plant Microbe Interact. 26, 1259–1270. doi: 10.1094/mpmi-05-13-0142-r
Thines. (2014). Phylogeny and evolution of plant pathogenic oomycetes—a global overview. Eur. J. Plant Pathol. 138, 431–447. doi: 10.1007/s10658-013-0366-5
Thines, M. (2019). An evolutionary framework for host shifts –jumping ships for survival. New Phytol. 224, 605–617. doi: 10.1111/nph.16092
Thines, M., and Kamoun, S. (2010). Oomycete–plant coevolution: recent advances and future prospects. Curr. Opin. PlantBiol. 13, 427–433. doi: 10.1016/j.pbi.2010.04.001
Tian, M., Huitema, E., Da Cunha, L., Torto-Alalibo, T., and Kamoun, S. (2004). A Kazal-like extracellular serine protease inhibitor from Phytophthora infestans targets the tomato pathogenesis-related protease P69B. J. Biol. Chem. 279, 26370–26377. doi: 10.1074/jbc.m400941200
Tian, M., Win, J., Savory, E., Burkhardt, A., Held, M., Brandizzi, F., et al. (2011). 454 genome sequencing of Pseudoperonospora cubensis reveals effector proteins with a QXLR translocation motif. Mol. Plant Microbe Interact. 24, 543–553. doi: 10.1094/mpmi-08-10-0185
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Tyler, B. M., Tripathy, S., Zhang, X. M., Dehal, P., Jiang, R. H. Y., Aerts, A., et al. (2006). Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 313, 1261–1266. doi: 10.1126/science.1128796
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Vleeshouwers, V. G., and Oliver, R. P. (2014). Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and necrotrophic plant pathogens. Mol. Plant Microbe Interact. 27, 196–206. doi: 10.1094/mpmi-10-13-0313-ia
Wagner, G. P., Kin, K., and Lynch, V. J. (2012). Measurement of mRNA abundance using RNA-seq. 131, 281–285. doi: 10.1007/s12064-012-0162-3
Wawra, S., Trusch, F., Matena, A., Apostolakis, K., Linne, U., Zhukov, I., et al. (2017). The RxLR motif of the host targeting effector AVR3a of Phytophthora infestans is cleaved before secretion. Plant Cell 29, 1184–1195. doi: 10.1105/tpc.16.00552
Whisson, S. C., Boevink, P. C., Moleleki, L., Avrova, A. O., Morales, J. G., Gilroy, E. M., et al. (2007). A translocation signal for delivery of oomycete effector proteins into host plant cells. Nature 450, 115–118. doi: 10.1038/nature06203
Win, J., Krasileva, K. V., Kamoun, S., Shirasu, K., Staskawicz, B. J., and Banfield, M. J. (2012). Sequence divergent RXLR effectors share a structural fold conserved across plant pathogenic oomycete species. PLoS Pathog. 8:e1002400. doi: 10.1371/journal.ppat.1002400
Win, J., Morgan, W., Bos, J., Krasileva, K. V., Cano, L. M., Chaparro-Garcia, A., et al. (2007). Adaptive evolution has targeted the C-terminal domain of the RXLR effectors of plant pathogenic oomycetes. Plant Cell 19, 2349–2369. doi: 10.1105/tpc.107.051037
Withers, S., Gongora-Castillo, E., Gent, D., Thomas, A., Ojiambo, P. S., and Quesada-Ocampo, L. M. (2016). Using next-generation sequencing to develop molecular diagnostics for Pseudoperonospora cubensis, the cucurbit downy mildwe pathogen. Phytopathology 106, 1105–1116. doi: 10.1094/phyto-10-15-0260-fi
Wood, K., Nur, M., Gil, J., Fletcher, K., Lakeman, K., Gothberg, A., et al. (2019). Effector prediction and characterization in the oomycete pathogen Bremia lactucae reveal host-recognized WY domain proteins that lack the canonical RXLR motif. bioRXiv [Preprint] doi: 10.1101/679787
Woods, J. L., and Gent, D. H. (2016). Susceptibility of hop cultivars to downy mildew: associations with chemical characteristics and region of origin. Plant Health Prog. 17, 42–48. doi: 10.1094/php-rs-15-0044
Zerillo, M. M., Adhikari, B. N., Hamilton, J. P., Buell, C. R., Lévesque, C. A., and Tisserat, N. (2013). Carbohydrate-active enzymes in Pythium and their role in plant cell wall and storage polysaccharide degradation. PLoS ONE 8:e72572. doi: 10.1371/journal.pone.0072572
Keywords: downy mildew, Pseudoperonospora, secretome, effectors, RXLR
Citation: Purayannur S, Cano LM, Bowman MJ, Childs KL, Gent DH and Quesada-Ocampo LM (2020) The Effector Repertoire of the Hop Downy Mildew Pathogen Pseudoperonospora humuli. Front. Genet. 11:910. doi: 10.3389/fgene.2020.00910
Received: 28 February 2020; Accepted: 22 July 2020;
Published: 11 August 2020.
Edited by:
Sucheta Tripathy, Indian Institute of Chemical Biology (CSIR), IndiaReviewed by:
Brett Merrick Tyler, Oregon State University, United StatesFranck Panabieres, Centre INRAE Provence-Alpes-Côte d’Azur, France
Copyright © 2020 Purayannur, Cano, Bowman, Childs, Gent and Quesada-Ocampo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liliana M. Cano, bG1jYW5vQHVmbC5lZHU=; Lina M. Quesada-Ocampo, bG1xdWVzYWRAbmNzdS5lZHU=