 Liwei Wu1,2
Liwei Wu1,2 Chunnian He
Chunnian He Jingyuan Song
Jingyuan Song Hui Yao
Hui Yao- 1Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2Engineering Research Center of Chinese Medicine Resources, Ministry of Education, Beijing, China
Analysis of the relationships among wild species of section Moutan in the plant genus Paeonia has traditionally been problematic. Interspecies relationships cannot be effectively determined using phenotypic traits alone or through analysis of nuclear or chloroplast DNA fragments. Elucidation of complete chloroplast genome sequences will aid the identification and phylogeny of these species. In this study, the complete chloroplast genomes of three sect. Moutan plants were sequenced and analyzed. Comparative and phylogenetic analyses of the complete chloroplast genomes of all eight species of sect. Moutan were then conducted. The three complete chloroplast genomes gained in this study showed four-part annular structures, and the genome length, structure, GC content, codon usage, and gene distribution were highly similar. There was greater variation in the noncoding regions of the sequences than in the conserved protein-coding regions. Sequence variations in the small single copy (SSC) regions and large single copy (LSC) regions were considerably greater than those in the inverted repeat (IR) regions. Phylogenetic analysis revealed that the species of sect. Moutan clustered in one branch and then subdivided into smaller branches. As for the three complete chloroplast genome sequences obtained in this study, Paeonia jishanensis clustered with another P. jishanensis sequence from the GenBank database, Paeonia qiui clustered with Paeonia rockii, and Paeonia delavayi var. lutea clustered with Paeonia ludlowii. It was also found that the complete chloroplast genomes, LSC regions, and SSC regions all showed great abilities in identification and phylogenetic analysis of the species of sect. Moutan, while IRs regions and highly variable regions were not suitable for the species of sect. Moutan.
Introduction
Section Moutan belongs to the genus Paeonia, which was previously classified in the family Ranunculaceae but is now under the family Paeoniaceae. In 1946, Stern divided the genus Paeonia into three groups, namely sect. Moutan, sect. Paeonia and sect. Onaepia. Section Moutan can be further divided into subsect. Vaginatae and subsect. Delavayanae, the latter consisting of the Suffruticosa and Delavayi groups (Stern, 1946). In the latest classification, sect. Moutan comprises eight species (Hong and Pan, 1999). Section Moutan plants are economically important ornamental plants known for their attractive flowers. Moreover, these plants also have high medicinal value as they exhibit anti-oxidant, anti-tumor, anti-pathogenic, anti-inflammatory, antidiabetic, analgesic, and anti-osteoporotic effects, and have been shown to exert protective effects against cardiovascular disease (Qi and Hu, 1993; Okubo et al., 2000; Kim et al., 2004; Abdel-Aty, 2007; Tsai et al., 2008; Zhang X. J. et al., 2008). These medicinal effects are attributed to the presence of monoterpene glucosides, flavonoids, tannins, triterpenoids, steroids, paeonols, phenolic acids, and other compounds in the plants (Okubo et al., 2000). In addition, the pollen of sect. Moutan plants contains various nutrients that can be used in the development of health and beauty products (Zhao, 2007).
Species of sect. Moutan are all subshrubs (Guo, 2002). All eight wild species of sec. Moutan are endemic to China (Hao et al., 2008): the wild species of sect. Moutan originated and evolved in China, which is also the birthplace of cultivated species of sect. Moutan. The wild species of sect. Moutan that are endemic to China are regarded as a valuable germplasm resource worldwide (Ji et al., 2012). Elucidating the relationships between species of sect. Moutan is crucial for understanding and harnessing the medicinal and ornamental properties of the different species. Progress has been made in studies on the relationships of wild species of sect. Moutan. However, the origin, genetic background, evolution, relationships, and classification systems of these species are different (Hong et al., 1998; Lin et al., 2004). According to classical taxonomy, using phenotypic traits alone to infer phylogenetic relationships between taxa with different genotypes is problematic. Moreover, different interpretations for the morphological variations in sect. Moutan species have been reported, and the different classification treatments applied vary largely (Zhang J. M. et al., 2008). Therefore, the relationships between wild species of sect. Moutan require further study and discussion. Molecular markers are a reliable alternative that are independent of morphological features, enabling the taxonomic challenges arising from the differences in interpretation of the morphological variations to be addressed. Molecular systematics have previously been used to study the evolutionary relationships among sect. Moutan species (Sang et al., 1997a,b; Zou et al., 1999; Zhao et al., 2004). However, the complex network evolution and polyploidy evolution of sect. Moutan species result that limited nuclear or chloroplast DNA fragments provide insufficient phylogenetic information to effectively solve interspecies relationships. Furthermore, the results of previous studies are inconsistent with each other (Sang et al., 1995; Sun and Hong, 2012).
The chloroplast genome is independent of the nuclear genome and corresponds to matrilineal inheritance with a separate transcription and transport system. Chloroplast genomes are very conservative (Jansen and Ruhlman, 2012) in terms of genome structure, gene sequence and gene type. Most chloroplast genomes of angiosperms have a circular tetrad structure existing as multiple copies of covalent, closed, circular double-stranded DNA. The circular tetrad structure comprises two inverted repeats (IRs), a large single copy (LSC), and a small single copy (SSC; Wang et al., 2012). Chloroplast genomes are typically 120–160 kb in length and are characterized by their small molecular weight, multiple copies, and slow molecular evolution (Shimada and Sugiura, 1991; Wakasugi et al., 2001; Kahlau et al., 2006). Since the chloroplast genomes of Nicotiana tabacum and Marchantia polymorpha were first reported in 1986 (Ohyama et al., 1986; Sugiura et al., 1986), the complete chloroplast genomes of various plants have been sequenced, and the structure, function, and expression of their genes have been studied. Chloroplast genome sequencing is increasingly used in the identification and investigation of molecular markers and phylogeny of medicinal plants (Wu et al., 2010; Kuang et al., 2011; Nock et al., 2011; Takano and Okada, 2011). Over the past years, chloroplast genomes have been shown to be an efficient tool in revealing phylogenetic relationships (Jansen et al., 2007), identifying close species as a super barcode (Chen et al., 2018; Park et al., 2018a), and developing chloroplast genetic engineering (Daniell et al., 2016).
In this study, the complete chloroplast genomes of three wild species of sect. Moutan were sequenced. A comparative analysis of the complete chloroplast genomes was then conducted. Phylogenetic analysis was performed by constructing phylogenetic trees based on different datasets of the chloroplast genomes of 16 species of the genus Paeonia, including all eight sect. Moutan species. Data obtained in this study provided a basis for the identification and investigation of the phylogenetic relationships of species of sect. Moutan.
Materials and Methods
DNA Sources
Fresh leaves of Paeonia qiui, Paeonia jishanensis, and Paeonia delavayi var. lutea were collected from Shennongjia in Hubei Province, Jiyuan in Henan Province and Shangri-la in Yunnan Province, respectively. The three species were identified by Professor Peigen Xiao and Professor Chunnian He from the Institute of Medicinal Plant Development (IMPLAD), Chinese Academy of Medical Sciences and Peking Union Medical College. Voucher specimens were deposited in the herbarium at IMPLAD.
Total DNA Extraction and Sequencing
Total DNA was extracted using a DNeasy Plant Mini Kit (Qiagen, Germany). DNA concentration was determined using a microspectrophotometer (Nanodrop 2000, United States), and DNA quality was detected by 1% agarose gel electrophoresis. Illumina HiSeq X sequencing platform was used to construct a library with an insertion fragment of 500 bp. Paired-end sequencing was performed to obtain 150-bp sequences at both ends of each read.
Assembly of Chloroplast Genome Sequences
Low-quality regions in the original sequencing data were removed using Trimmomatic software (Bolger et al., 2014). A local sequence comparison retrieval (BLASTn) database was constructed from the chloroplast genome sequences published in the National Center for Biological Information (NCBI). Clean reads were compared with this database, and mapped reads were extracted based on coverage and similarity. SOAPdenovo 2 (Luo et al., 2012) was used to assemble extracted reads into contigs. A scaffold of the chloroplast genome was constructed using SSPACE software (Boetzer et al., 2011). Gaps were filled using GapFiller (Nadalin et al., 2012).
Chloroplast Genome Annotation and Structural Analysis
Dual Organellar GenoMe Annotator (Wyman et al., 2004) and chloroplast genome annotation, visualization, analysis, and genbank submission (CPGAVAS) (Liu et al., 2012) were used to initially annotate the sequences, and the annotations were then manually corrected. tRNAscan-SE software (Schattner et al., 2005) was used to annotate tRNA. Genes, introns and the boundaries of coding regions were compared with reference sequences. Chloroplast genome maps were generated using Organellar Genome DRAW v1.2 (Lohse et al., 2007) and then manually corrected. GC content was analyzed using MEGA 6.0 (Tamura et al., 2013). CodonW software (Sharp and Li, 1987) was adopted to analyze the relative synonymous codon usage (RSCU). The assembled complete chloroplast genome sequences of the three species of sect. Moutan were submitted to NCBI under the accession numbers MT210544 (P. qiui), MT210545 (P. jishanensis), and MT210546 (P. delavayi var. lutea).
Structural Analysis of Repeats
REPuter software (Kurtz et al., 2001) was used to identify long repeat sequences of the chloroplast genomes. Microsatellite identification tool (MISA) software (Beier et al., 2017) was used to determine the type and number of simple sequence repeats (SSRs), employing the parameters used by Li et al. (2013). Completely repetitive SSRs were searched, and cyclically arranged or inversely complementary SSRs were treated as the same type.
Analysis of Sequence Variations and Phylogenetic Relationships
Chloroplast genome sequences of all eight species of sect. Moutan were compared using the online genome comparison tool mVISTA (Frazer et al., 2004). Nucleic acid variation values of the three chloroplast genomes sequenced in this study were determined by DnaSP v5.10 (Librado and Rozas, 2009). The boundaries of four regions of chloroplast genomes were compared using IRscope (Ali et al., 2018). MAFFT version 5 software (Katoh and Standley, 2013) was used to compare the complete chloroplast genome sequences and LSC, SSC, and IRs regions of the chloroplast genomes of 16 species of the genus Paeonia (Supplementary Table S1). IQTREE software (Nguyen et al., 2015) and MrModeltest 2.3 (Nylander, 2004) were utilized to select tree models. Maximum likelihood (ML) and Bayesian inference (BI) phylogenetic trees were constructed based on the complete chloroplast genomes and LSC, SSC, and IRs regions using the program IQTREE and MrBayes v3.2.7 (Ronquist and Huelsenbeck, 2003), respectively. MEGA 6.0 (Tamura et al., 2013) was used to construct ML phylogenetic trees based on 19 highly variable regions, which were selected by the percent identity of the complete chloroplast genomes using mVISTA (Frazer et al., 2004). The positions of these highly variable regions were shown in Supplementary Table S2.
Results
Molecular Features of the Chloroplast Genomes
The complete chloroplast genomes of three species of sect. Moutan show a common tetrad structure comprising two IRs (25,645–25,648 bp), an LSC (84,242–84,462 bp), and an SSC (17,032–17,046 bp; Figure 1). The total lengths of the chloroplast genomes were 152,578 bp (P. qiui), 152,631 bp (P. jishanensis) and 152,790 bp (P. delavayi var. lutea), while the total GC content ranged from 38.35 to 38.42%. Moreover, GC content in different regions was unbalanced; the IR regions had the highest GC content (43.06–43.10%) among the four sections, followed by the LSC (36.64–36.73%) and SSC regions (32.59–32.73%) (Table 1).
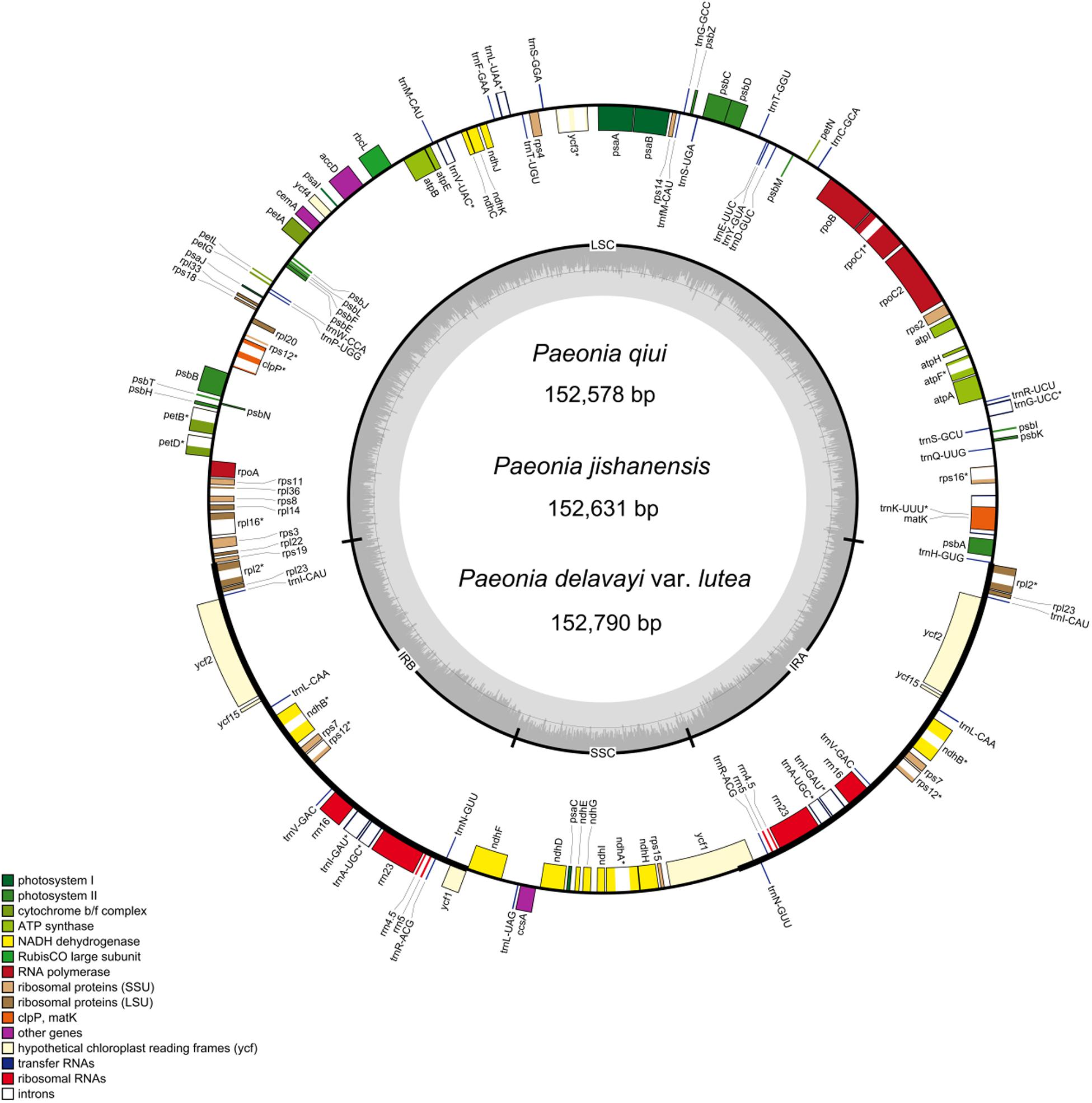
Figure 1. Gene maps of the complete chloroplast genomes of the three sect. Moutan species. Genes on the inside of the circle are transcribed clockwise, while those outside are transcribed counter clockwise. The darker gray in the inner circle corresponds to GC content, whereas the lighter gray corresponds to AT content. matK in P. delavayi var. lutea is a pseudo gene.
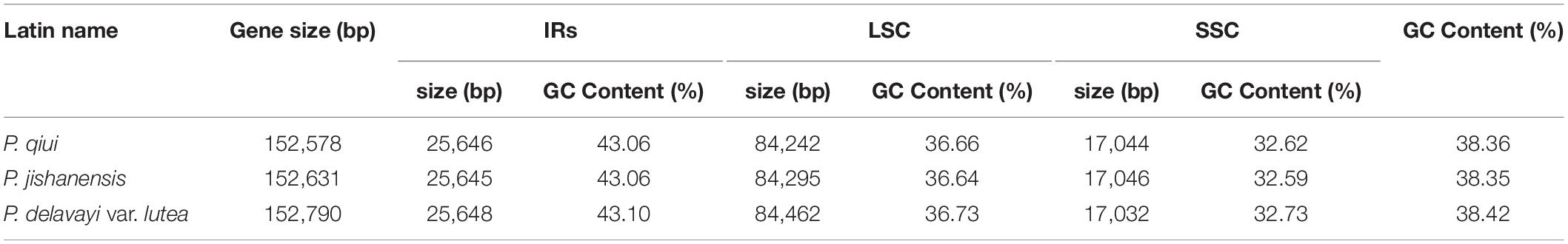
Table 1. Statistics for assembly of the three chloroplast genomes.
The chloroplast genome structure and gene composition of sect. Moutan species could be divided into four categories: genes related to photosynthesis, genes related to self-replication, protein-coding genes with unknown functions, and other genes (e.g., mature enzyme gene matK and cystic protein gene cemA). A total of 84 protein-coding genes were annotated in P. qiui and P. jishanensis, while 83 protein-coding genes were annotated in P. delavayi var. lutea. Compared with other species, the polyT repeat region of matK gene in P. delavayi var. lutea lost one T base, and the frame shift caused stop codon to appear prematurely, so the gene was annotated as a pseudo gene. Thirty-seven tRNA genes and 8 rRNA genes were annotated in the three species. Seven protein-coding genes (rpl2, rpl23, ycf2, ycf15, ndhB, rps7, and rps12), seven tRNAs (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, and trnN-GUU) and four rRNAs (rrn16, rrn23, rrn4.5, and rrn5) were located in the IR regions. Among the protein-coding genes, 18 genes contained introns; three of these genes (clpP, rps12, and ycf3) had two introns, and 15 genes had only one intron. The gene rps12, which contained two introns, is a trans-splicing gene with the 5’ exon in the LSC region and the 3’ exon in the IR region (Table 2).
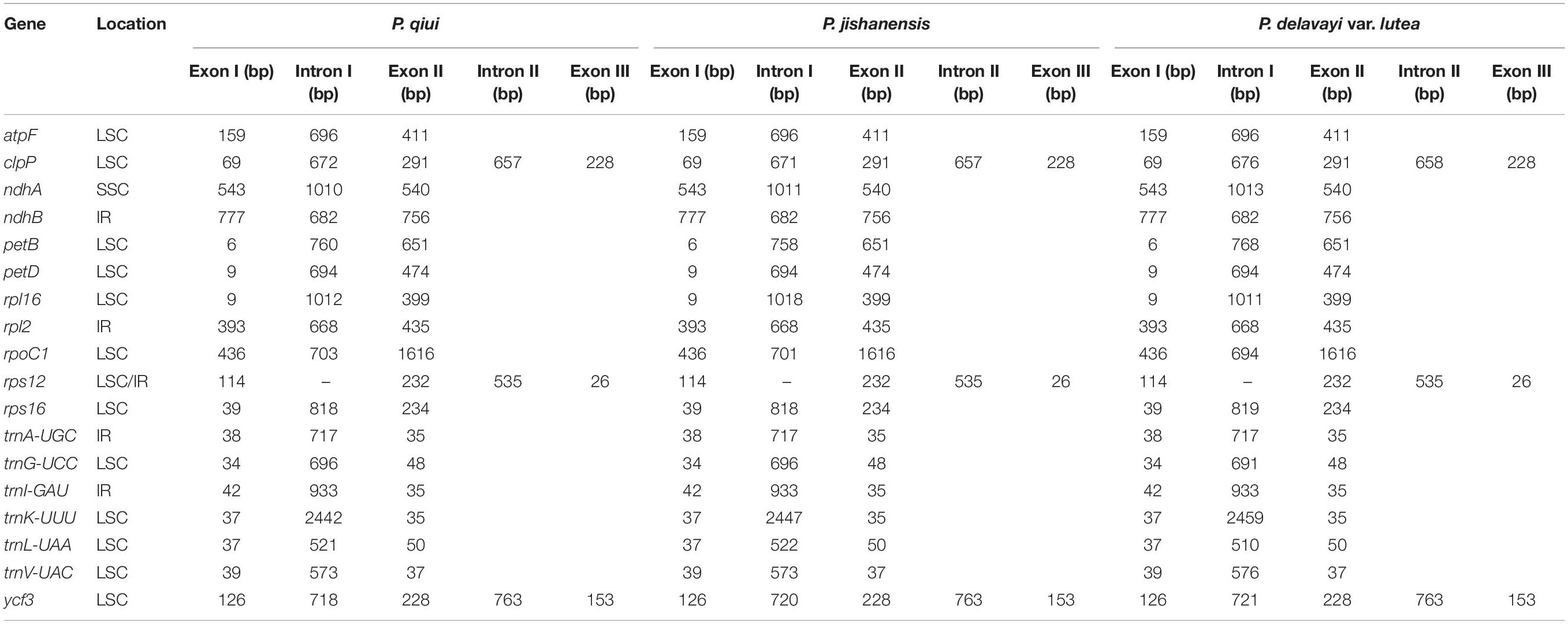
Table 2. Genes with introns in the chloroplast genomes of P. qiui, P. jishanensis and P. delavayi var. lutea as well as the lengths of the exons and introns.
Relative Synonymous Codon Usage Analysis of the Chloroplast Genomes
The RSCU of the chloroplast genomes of species of sect. Moutan was calculated using all protein-coding genes. The RSCU value is the ratio of the frequency of use of a particular codon to the expected frequency. It enables the detection of synonymous codons that do not uniformly occur in the coding sequence. Codons with no preference value are set to 1.00. The actual usage of codons with an RSCU value >1.00 is higher than expected, and that of codons with an RSCU value <1.00 is lower than expected. Among all amino acids, leucine (Leu) had the highest number of codons in all protein-coding gene sequences in the chloroplast genomes. The amino acids with a total codon number <1000 were methionine (Met), tyrosine (Tyr), histidine (His), glutamine (Gln), cysteine (Cys), tryptophan (Trp), serine (Ser), and terminator (TER); Cys had the lowest number codons (Supplementary Table S3). Figure 2 shows the codon contents of 20 amino acids and stop codons of all protein-coding genes in the chloroplast genomes of the three species of sect. Moutan sequenced in this study.
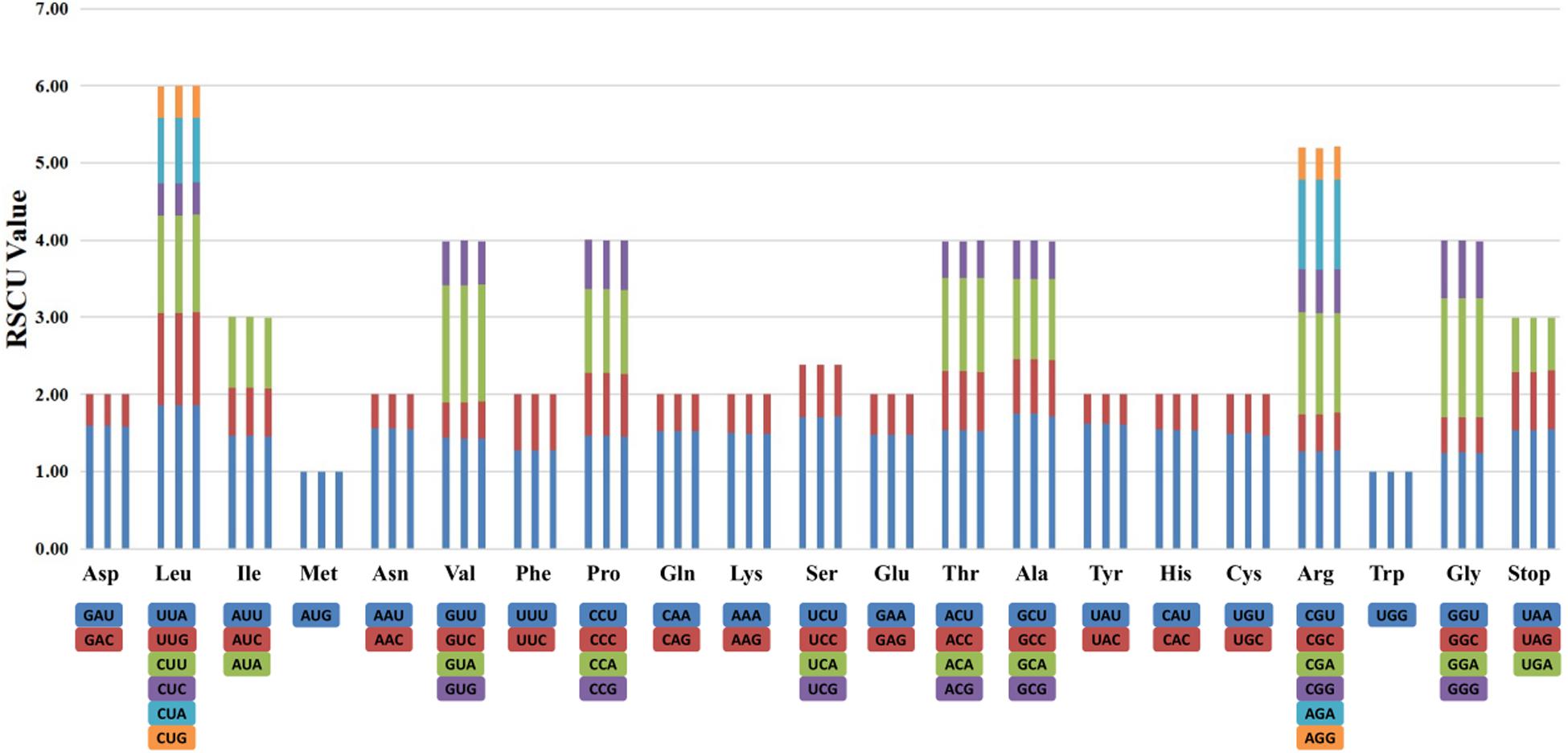
Figure 2. Codon distribution of 20 amino acid and stop codons in all protein-coding genes of the chloroplast genomes of three sect. Moutan species. The order of every three columns is P. qiui, P. jishanensis, and P. delavayi var. lutea, respectively.
Long Repeat Sequence and SSR Analyses
Long repeat sequences are classified as forward (F), palindrome (P), reverse (R), or complement (C). For all repeat types, the repeat length is ≥30 bp and sequence similarity is ≥90%. In the chloroplast genome of P. qiui, there were 21 F repeats, 23 P repeats, and 4 R repeats. Furthermore, 22 F repeats and 24 P repeats were present in the chloroplast genome of P. jishanensis, while the chloroplast genome of P. delavayi var. lutea contained 22 F repeats and 23 P repeats. No C repeats were found in the chloroplast genomes of the three sect. Moutan species, and no R repeats were identified in the chloroplast genomes of P. jishanensis and P. delavayi var. lutea. The length of the repeat sequences that were found predominantly ranged from 30 to 39 bp. R repeat sequences of the P. qiui chloroplast genome only contained 30–39 bp (Figure 3).
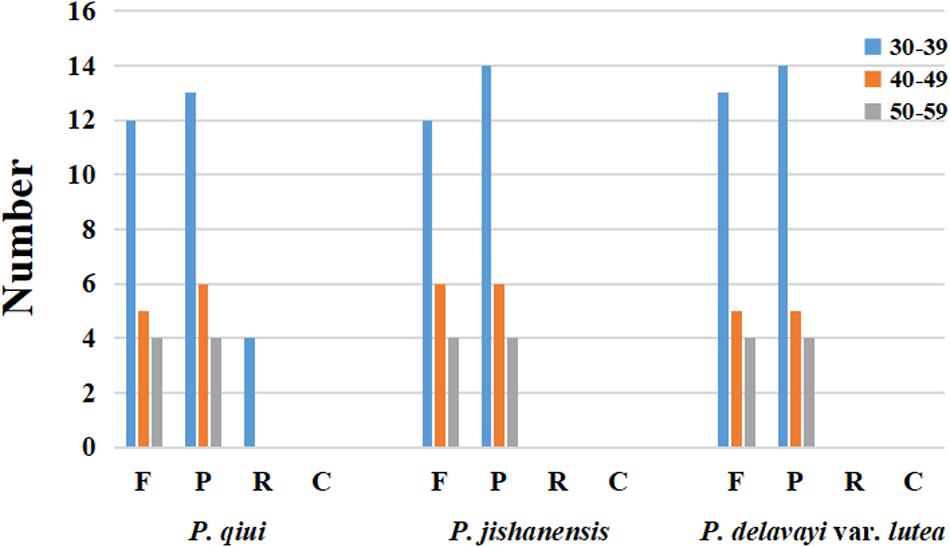
Figure 3. Repeat sequences in three chloroplast genomes. REPuter was used to identify repeat sequences with length ≥30 bp and sequences identified ≥90% in the chloroplast genomes. F, P, R, and C indicate the repeat types F (forward), P (palindrome), R (reverse), and C (complement). Repeats with different lengths are indicated in different colors.
Simple sequence repeats in the chloroplast genomes have abundant polymorphisms, and they are an efficient molecular marker (Tang et al., 2010). In this study, 73, 72, and 61 SSRs were identified in the chloroplast genomes of P. qiui, P. jishanensis, and P. delavayi var. lutea, respectively. In addition, the base composition of the repeating motifs from mononucleotide repeats to trinucleotide repeats had a certain base preference, mainly repeating motifs rich in A–T. In these SSRs, mononucleotide repeats were the most abundant, being found 49, 47, and 39 times in the chloroplast genomes of P. qiui, P. jishanensis, and P. delavayi var. lutea, respectively. A/T repeats were the most common mononucleotide repeats (93.9, 97.9, and 100% for P. qiui, P. jishanensis, and P. delavayi var. lutea, respectively). Dinucleotide repeat sequences predominantly comprised AT/AT repeats (91.7, 92.3, and 92.3% for P. qiui, P. jishanensis, and P. delavayi var. lutea, respectively), and all trinucleotide repeats were AAT/ATT. This was consistent with A–T enrichment in the complete chloroplast genomes of sect. Moutan species (61.6, 61.7, and 61.6% for P. qiui, P. jishanensis, and P. delavayi var. lutea, respectively) (Table 3).
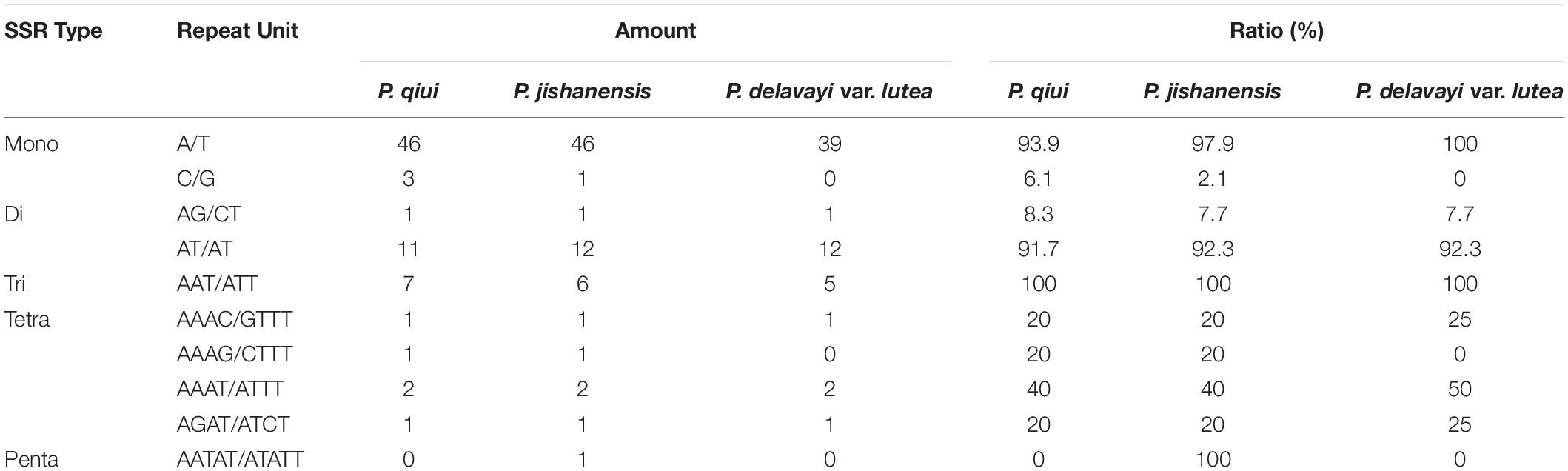
Table 3. Types and amounts of SSRs in the three chloroplast genomes.
Comparative Analysis of the Chloroplast Genomes
In this study, the complete chloroplast genomes of all species of sect. Moutan were compared using mVISTA (Frazer et al., 2004) with the P. qiui genome as the reference genome (Figure 4). There were more variations in the noncoding regions of the sequences than in the conserved protein-coding regions. Variations in the SSC and LSC regions were considerably greater than those in the IR regions, while the rRNA genes were highly conserved with almost no variation. As shown in Figure 4, the genes with large variations included trnK, trnR, psbZ, ycf3, rps3, and rps19, whereas the other genes had a very high degree of conservation (most had >90% similarity). Variations in intergenic regions were notably greater than those in gene regions; such intergenic regions included trnK-rps16, rps16-trnQ, rpoC1-rpoB, rpoB-trnC, psbM-trnD, psbZ-trnG, ndhC-trnV, atpB-rbcL, petA-psbJ, rpl20-rps12, rpl16-rps3, ndhG-ndhI, ndhA-ndhH, and ndhB-trnL.
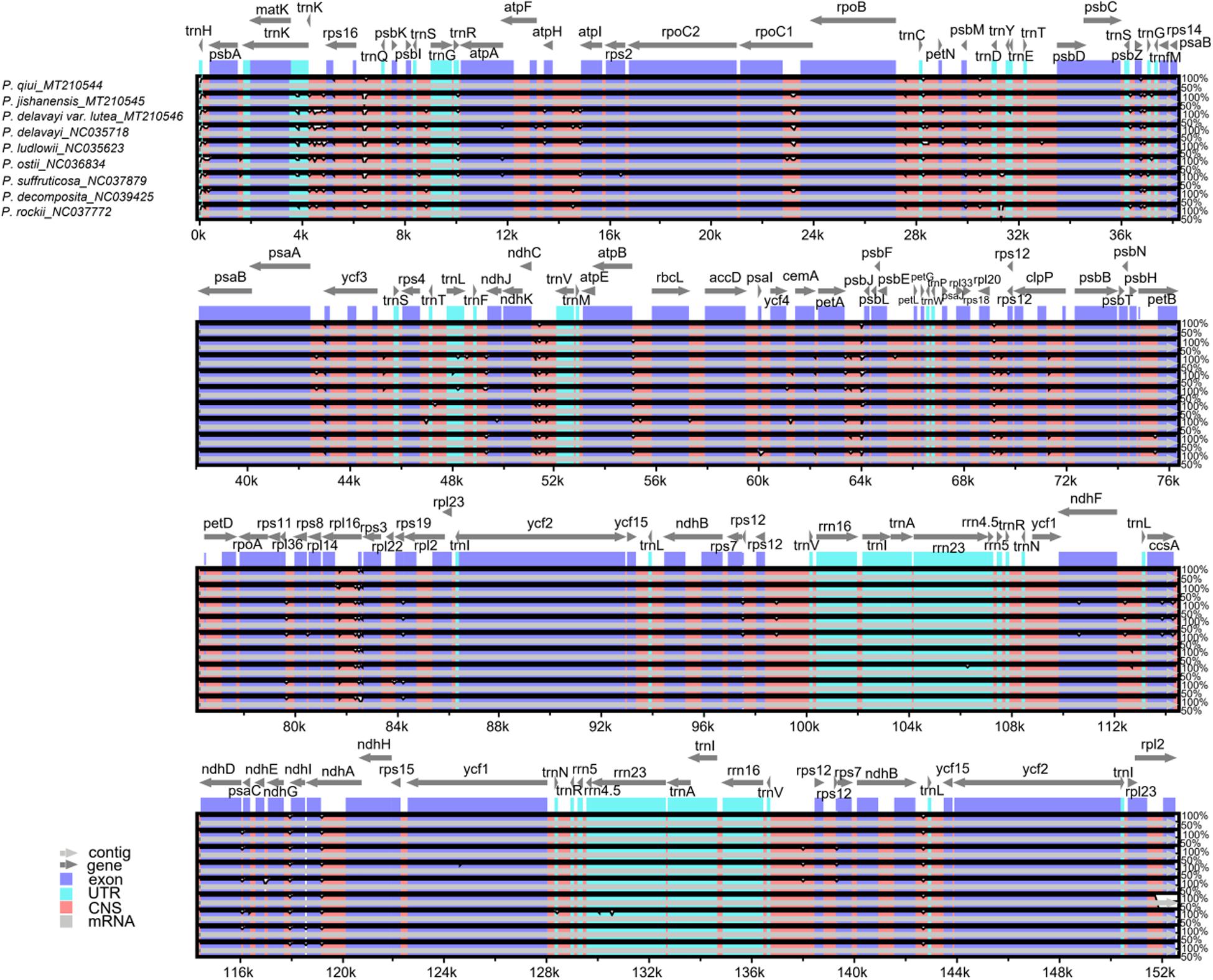
Figure 4. Global alignment of chloroplast genomes of all sect. Moutan species. Gray arrows and thick black lines above the alignment indicate genes with their orientation and the position of the IRs, respectively. A cutoff of 70% identity was used for the plots, and the Y-scale represents the percent identity ranging from 50 to 100%.
DnaSP (Librado and Rozas, 2009) was used to analyze and detect highly variable regions in the chloroplast genomes sequenced in this study. The K value was calculated by pairwise comparisons to determine variations at the sequence level (Figure 5). Variation in the IR regions of the chloroplast genomes was markedly lower than that in the LSC and SSC regions, consistent with the mVISTA results. Furthermore, the K value was generally below 0.005. In the LSC regions, two pairwise-comparison peaks with a K value >0.005 were particularly prominent. As shown in the mVISTA map and specific sites, the two peaks were petA-psbJ and rpl16-rps3. The average K value was 0.00372 between P. delavayi var. lutea and P. jishanensis, 0.00378 between P. delavayi var. lutea and P. qiui, and 0.00100 between P. jishanensis and P. qiui. Thus, the largest nucleic acid variation was observed between P. delavayi var. lutea and P. qiui, followed by that between P. delavayi var. lutea and P. jishanensis, and that between P. jishanensis and P. qiui.
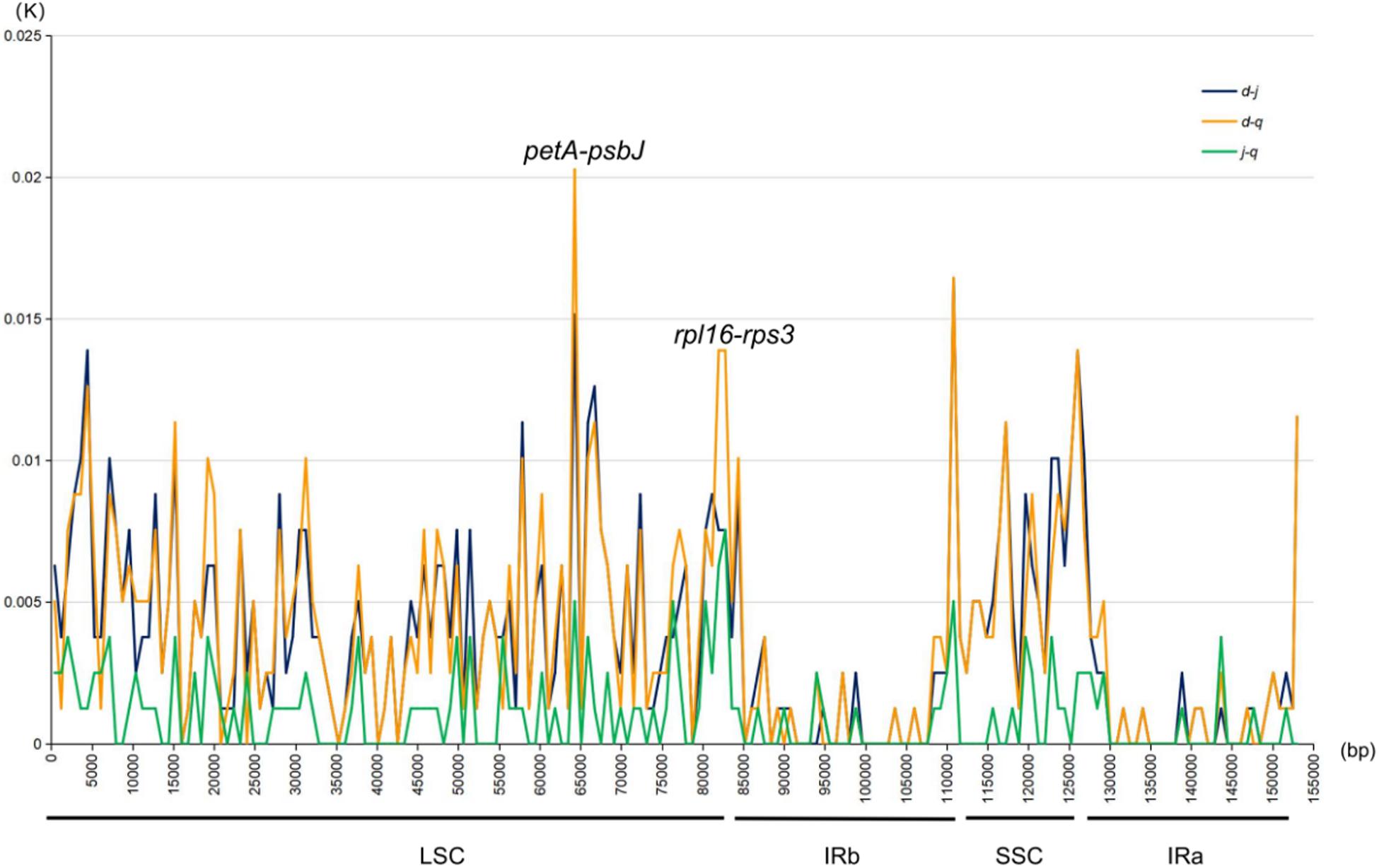
Figure 5. Nucleic acid variation information of chloroplast genomes of sect. Moutan species. d: P. delavayi var. lutea; j: P. jishanensis; q: P. qiui.
The boundaries of the four regions of chloroplast genomes of all species of sect. Moutan were comprehensively compared. At the junctions, the gene positions in the boundary regions of the three chloroplast genomes sequenced in this study and that of P. suffruticosa were very similar, whereas the chloroplast genomes of the five other species of sect. Moutan were different (Figure 6). In addition, it could be seen from the figure that the genome length of P. delavayi was much longer than that of its variety P. delavayi var. lutea, mainly because the length of the LSC region of P. delavayi was about 1600 bp longer than that of P. delavayi var. lutea. There were one more infA, trnP-GGG, trnT-GGU and trnM-CAU genes in the LSC region of P. delavayi than those in P. delavayi var. lutea.

Figure 6. Comparison of the borders of LSC, SSC, and IR regions among all sect. Moutan species chloroplast genomes. Number above the gene features means the distance between the ends of genes and the border sites. These features are not to scale. JLB, junction of LSC/IRb; JSB, junction of IRb/SSC; JSA, junction of SSC/IRa; JLA, junction of IRa/LSC.
Phylogenetic Analysis
Chloroplast genomes play an important role in phylogenetic studies (Zhang et al., 2011; Hu et al., 2016). In the current study, the complete chloroplast genome sequences and LSC, SSC, and IRs regions of the chloroplast genomes of 16 species of the genus Paeonia, including all eight wild species of sect. Moutan, were used to construct ML and BI trees, with Bergenia scopulosa and Coptis chinensis as the outgroups. The two phylogenetic analyses (ML and BI) revealed congruent topologies based on the complete chloroplast genomes, LSC regions and SSC regions, and all of the nodes in the phylogenetic trees had high bootstrap support values (Figures 7–9). The resulting phylogenetic trees demonstrated that species of sect. Moutan were located on one branch, whereas species of the sect. Onaepia and sect. Paeonia were located on another branch. Species of subsect. Vaginatae and subsect. Delavayanae in sect. Moutan clustered in different branches, and the bootstrap support values for these were 100%. For the three chloroplast genomes sequenced in this study, P. jishanensis (GenBank accession no. MT210545) clustered with another P. jishanensis sequence obtained from the GenBank database, P. qiui (MT210544) clustered with P. rockii, and P. delavayi var. lutea (MT210546) clustered with P. ludlowii. Bootstrap support rates were all >95%. However, the two phylogenetic analyses (ML and BI) revealed incongruent topologies based on the IRs regions (Supplementary Figure S1). In the phylogenetic trees based on IRs regions, the two sequences of P. suffruticosa did not cluster together, and in subsect. Delavayanae, P. delavayi clustered with P. ludlowii and then with P. delavayi var. lutea, which was different from that based on complete chloroplast genomes. In addition, some of the nodes had very low bootstrap support values. It showed that IRs regions were not suitable for the identification and phylogenetic analysis of the species of sect. Moutan.
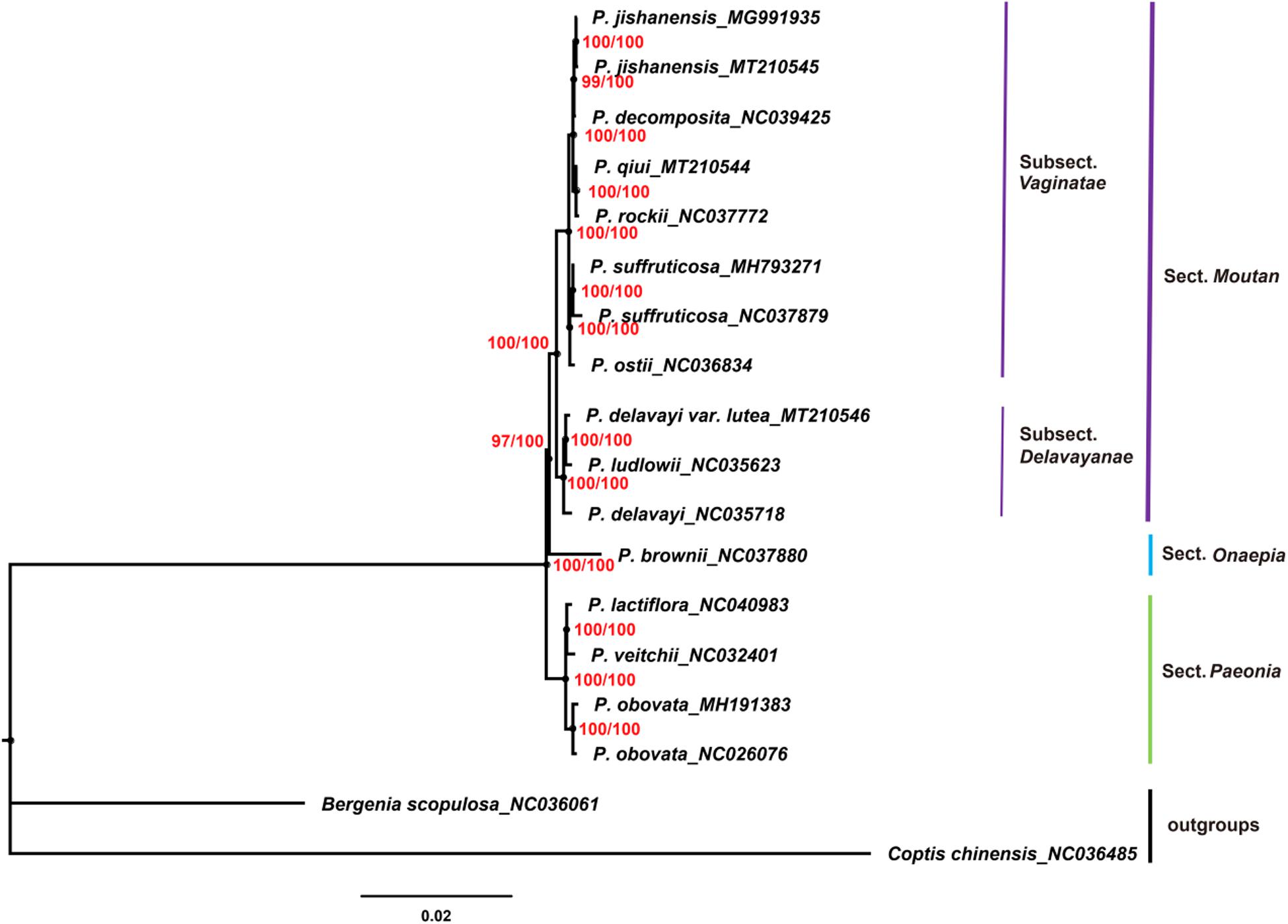
Figure 7. Phylogenetic trees constructed using Maximum Likelihood (ML) and Bayesian Inference (BI) methods based on the complete chloroplast genome sequences of 16 Paeonia species, including all eight species of sect. Moutan. Red numbers at nodes are values for bootstrap support (ML/BI).
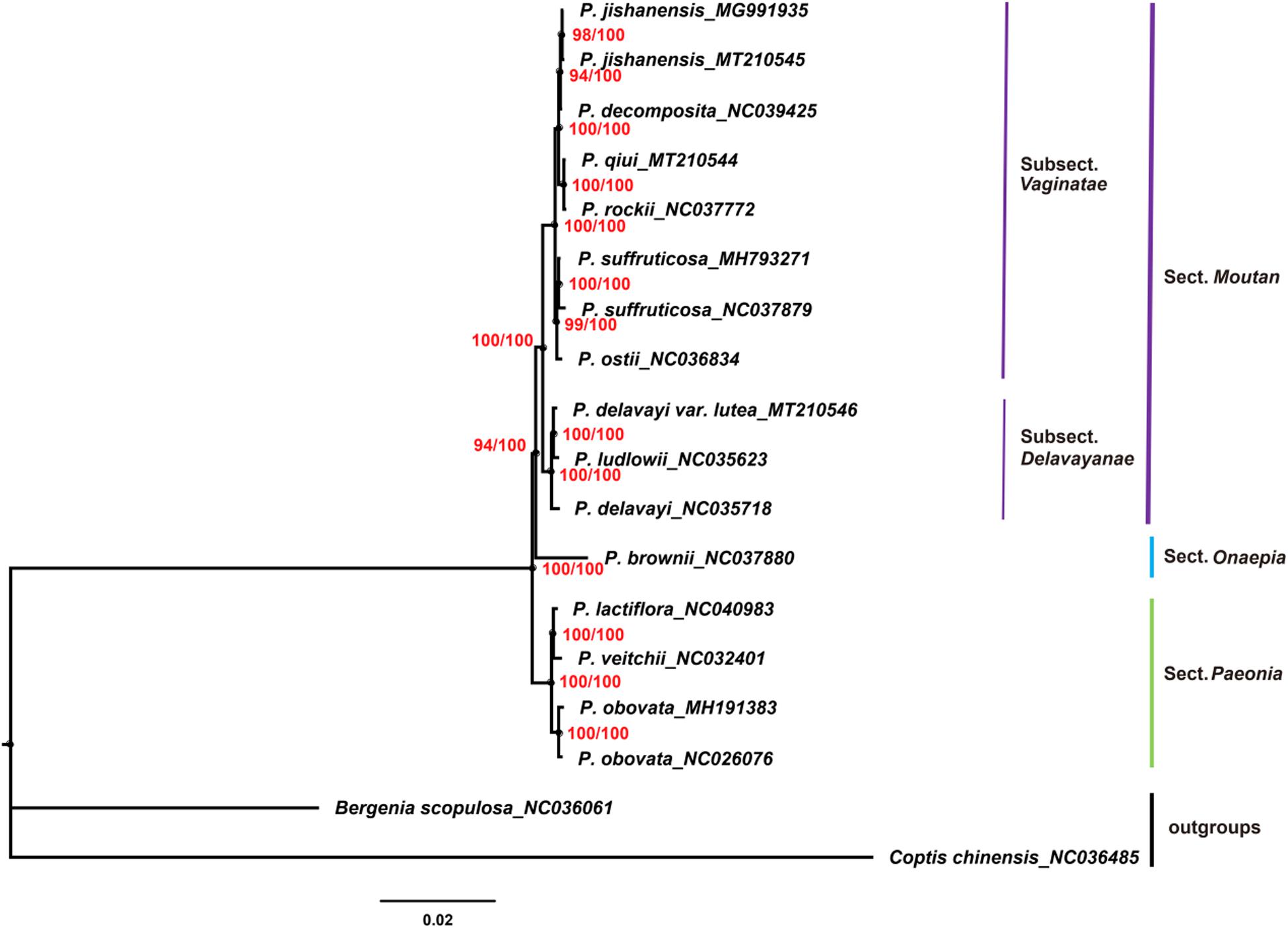
Figure 8. Phylogenetic trees constructed using ML and BI methods based on the LSC regions of the chloroplast genomes of 16 Paeonia species, including all eight species of sect. Moutan. Red numbers at nodes are values for bootstrap support (ML/BI).
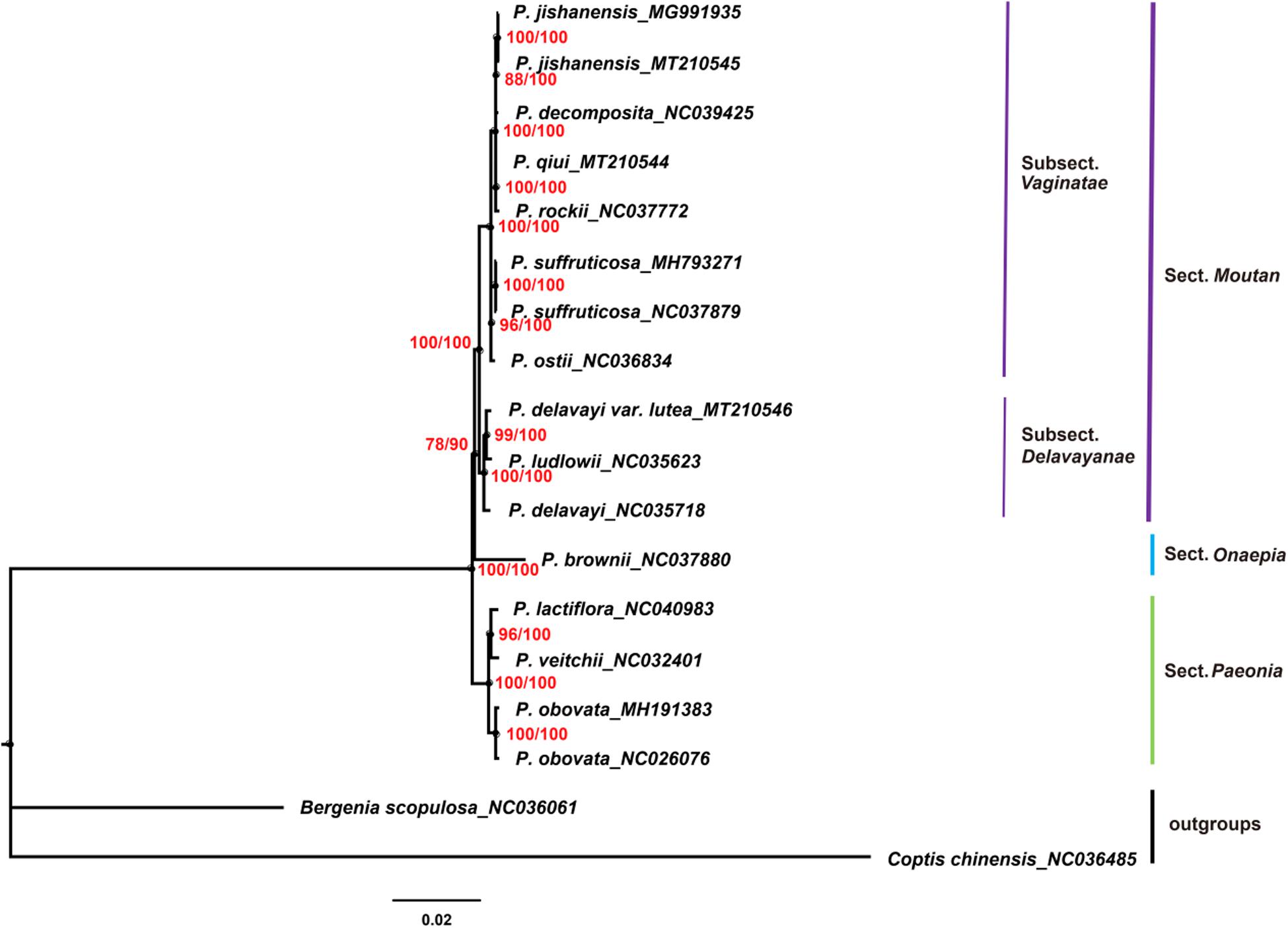
Figure 9. Phylogenetic trees constructed using ML and BI methods based on the SSC regions of the chloroplast genomes of 16 Paeonia species, including all eight species of sect. Moutan. Red numbers at nodes are values for bootstrap support (ML/BI).
Then 19 highly variable regions of chloroplast genomes were used to construct ML trees (Supplementary Figure S2). Most of the bootstrap support values of the nodes in the phylogenetic trees were much lower than that based on complete chloroplast genomes. Species of sect. Moutan did not cluster in one branch in some phylogenetic trees, such as the trees based on psbZ, rps19, trnR, ndhG-ndhI, psbM-trnD, rpl16-rps3, rpoC1-rpoB, and rps16-trnQ. In the phylogenetic trees based on ycf3, ndhB-trnL, psbZ-trnG, rpl20-rps12, and trnK-rps16, species of subsect. Vaginatae and subsect. Delavayanae did not cluster in one branch, respectively. The other highly variable regions were also not suitable for the identification and phylogenetic analysis of the species of sect. Moutan.
Discussion
Analysis of the Chloroplast Genomes of Sect. Moutan Species
In the current study, the GC content distribution in the chloroplast genomes of the three species of sect. Moutan was the same as that reported for most other angiosperms (Xiang et al., 2016; Zhou et al., 2017); the IR regions had the highest GC content among the four regions, followed by the LSC and SSC regions. The high GC content in the IR regions may be attributed to the fact that these regions contain rRNAs with low A/T content in the chloroplast genome, including rrna4.5, rrna5, rrna23, and rrna16. In all types of SSR in this study, A and T were the most often-used bases. Chloroplast genome SSRs are typically composed of polyA or polyT repeats, as a result of the A/T base preference of chloroplast genomes (Qian et al., 2013). These SSRs not only have the advantages of an abundant number of markers, codominant inheritance and high repeatability, but also have the characteristics of a simple structure, the single-parent inheritance of chloroplast genomes and are relatively conservative (Yang et al., 2014). Chloroplast genome SSRs have been widely used in species identification, phylogenetic analysis, population genetic structure and system geography of several species (Park et al., 2018b).
In the current study, a common feature of the chloroplast genomes of sect. Moutan species was that the IR regions were substantially more conserved than the LSC and SSC regions. rrn4.5, rrn5, rrn16, and rrn23 were the most conserved sequence regions in the IR regions. In addition, the degree of variation in noncoding regions was considerably greater than that in coding regions. The evolution rate of coding regions is slow and thus these regions are suitable for phylogenetic analysis at high levels of taxonomic hierarchy, such as families and orders (Li et al., 2012). In contrast, the sequence of noncoding regions rapidly evolves, cannot encode proteins, and contains an abundance of variation information. Noncoding regions are therefore suitable for phylogenetic analysis at low levels of taxonomic hierarchy, such as genera (Shaw et al., 2007). Noncoding regions can be subdivided into introns and intergenic regions and can be used for molecular identification of subspecies (Shaw et al., 2007). In the current study, 18 genes contained introns; three of these genes (clpP, rps12, and ycf3) contained two introns, and 15 genes contained only one intron. Furthermore, the intergenic regions of the sect. Moutan chloroplast genomes were variable.
The study of chloroplast genomes is highly relevant for revealing the structure and origin of chloroplast DNA, plant molecular markers and species relationships (Tang et al., 2011). Chloroplast genome sequencing and phylogenetic analysis of species of sect. Moutan can enrich the number of chloroplast genome sequences and lay the foundation for species identification, phylogenetic relationship, breeding of improved varieties and sustainable exploitation of plant resources. Furthermore, this sequencing and analysis also provides a theoretical basis for studying chloroplast genetic engineering in species of sect. Moutan.
Phylogenetic Analysis of Sect. Moutan Species and Chloroplast Genome Super Barcode
Species of sect. Moutan are economically important ornamental plants that are also commonly used as medicinal plants. However, the phylogenetic relationships and taxonomic systems of wild species of sect. Moutan were different in previous studies (Lin et al., 2004), and this could affect their applications. Plant classification and identification have been based on morphological evidence for a long time. However, morphological traits are easily affected by the environment, and convergence and parallel evolution often occur (Xing et al., 2013). DNA studies can provide reliable molecular evidence for phylogenetic evolution of species and identification of similar species within a genus. Numerous genes, such as the nuclear genes ITS, Adh and GPAT and the chloroplast genes matK, psbA-trnH, and trnL(UAA)-trnf(GAA), have been applied to phylogenetic relationship studies of species of sect. Moutan (Sang et al., 1995, 1997a,b; Ferguson and Sang, 2001; Tank and Sang, 2001; Sang, 2002; Sun and Hong, 2012). Although these studies have advanced our understanding of the relationships among species of sect. Moutan, the results of different studies are not completely consistent.
In the current study, phylogenetic trees were constructed based on the complete chloroplast genomes and LSC, SSC, and IRs regions of the chloroplast genomes of 16 species of the genus Paeonia, including all eight wild species of sect. Moutan. Among them, complete chloroplast genomes, LSC regions and SSC regions showed good abilities in the phylogenetic analysis of Sect. Moutan species. Species of sect. Moutan, sect. Onaepia and sect. Paeonia clustered in large distinct branches of the phylogenetic tree, and the two subgroups of sect. Moutan were further subdivided into different branches. In subsect. Vaginatae, P. jishanensis, P. decomposita, P. qiui, and P. rockii clustered in one small branch, while P. ostii and P. suffruticosa clustered in a different small branch, consistent with previous studies (Zhao et al., 2004; Zhao, 2007). Previous studies (Zou et al., 1999; Tank and Sang, 2001; Feng et al., 2015) showed that P. decomposita was related to P. rockii, and P. qiui was related to P. jishanensis; however, the current study indicated that P. decomposita was related to P. jishanensis, while P. rockii was related to P. qiui. Wang (1996) and Zhang J. M. et al. (2008) used gene fragments of the chloroplast genomes of sect. Moutan species to analyze their relationships and found that P. decomposita and P. jishanensis were closely related and clustered together in the phylogenetic tree. This is consistent with the results of the present study. Furthermore, a pathway proposed by Zhou and Yao (2002) for the phylogenetic evolution of subsect. Vaginatae based on morphological traits produced the same conclusion as the current study. In subsect. Delavayanae, P. delavayi var. lutea clustered with P. ludlowii and then with P. delavayi. This demonstrated that P. delavayi var. lutea and P. ludlowii are more closely related to each other than to P. delavayi. This is consistent with the finding that P. delavayi var. lutea used to be an independent species named P. lutea and that P. ludlowii was a variety of this species known as P. lutea var. ludlowii (Stern and Taylor, 1951). Feng et al. (2015) came to the same conclusion using chloroplast genes psbA-trnH. However, IRs regions were not suitable for the identification and phylogenetic analysis of the species of sect. Moutan. It was mainly because IRs regions were more conserved and had less variations, just as described above.
In addition, highly variable regions of chloroplast genomes were used to analyze their phylogenetic relationships, but most of the support values in the phylogenetic trees were very low, and none of them effectively resolved relationships among the sect. Moutan species. The relationships among sect. Moutan species were resolved with very high support values in the phylogenetic trees based on complete chloroplast genomes. The complete chloroplast genomes were better than the highly variable regions in the analysis of phylogenetic relationships of the sect. Moutan species. It was mainly due to the inadequate variations provided by a limited number of DNA loci (Xu et al., 2015), while the complete chloroplast genome could provide sufficient informative sites, which can help to clarify the relationships of intractable groups at low taxonomic levels (Yang et al., 2013; Li et al., 2017; Yu et al., 2017). In fact, chloroplast genomes have been proposed as super barcodes for species identification (Li et al., 2015). Super barcodes overcome many limitations of traditional barcodes (Parks et al., 2009; Steele and Pires, 2011; Coissac et al., 2016). Zhu et al. (2018) experimentally demonstrated that complete chloroplast genome sequences have higher resolution than DNA barcodes or highly variable regions of chloroplast genomes and can be used to identify related species, consistent with the current study. Super barcodes have been successfully used to identify species and individuals (Doorduin et al., 2011; Kane et al., 2012; Chen et al., 2018; Ma et al., 2018). The phylogenetic trees constructed in this study demonstrated that complete chloroplast genome sequences can also be used as a reference for the identification of species of sect. Moutan. With the rapid development of sequencing technology and analytical methods, chloroplast genome assembly sequencing is predicted to become widely used as a super barcode.
Conclusion
The chloroplast genome structure and gene content of species of section Moutan in the genus Paeonia were relatively conserved, while the GC content and variations in LSC, SSC, and IRs regions of the sequences were different. In addition, abundant repetitive sequences were identified, and their nucleotide composition was analyzed. The phylogenetic analysis illustrated that the two subgroups of sect. Moutan clustered in different branches. In subsect. Vaginatae, P. jishanensis, P. decomposita, P. qiui, and P. rockii clustered in a small branch, while P. ostii and P. suffruticosa clustered in a different branch. P. decomposita was found to be related to P. jishanensis, and P. rockii was related to P. qiui. In subsect. Delavayanae, P. delavayi var. lutea and P. ludlowii were more closely related to each other than to P. delavayi. Furthermore, it was found that the complete chloroplast genomes, LSC regions and SSC regions had higher discrimination than IRs regions and highly variable regions for the species of sect. Moutan, and the complete chloroplast genomes could also be used as a super barcode for the identification of species of sect. Moutan.
Data Availability Statement
The assembled chloroplast genomes of P. qiui, P. jishanensis, and P. delavayi var. lutea were deposited in GenBank with the accession numbers MT210544, MT210545, and MT210546. The sequences are available on NCBI now https://www.ncbi.nlm.nih.gov/nuccore/MT210544.1/, https://www.ncbi.nlm.nih.gov/nuccore/MT210545, and https://www.ncbi.nlm.nih.gov/nuccore/MT210546.
Author Contributions
LW, LN, and PL performed the experiments. ZX and JS assembled the sequences. LW and LN analyzed the data. LW wrote the manuscript. LN, YW, and CH collected the plant material. HY conceived the research and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Major Scientific and Technological Special Project for “Significant New Drugs Creation” (No. 2018ZX09711001-008-007) and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-3-016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00980/full#supplementary-material
FIGURE S1 | Phylogenetic trees constructed using Maximum Likelihood (ML) and Bayesian Inference (BI) methods based on the IRs regions of the chloroplast genomes of 16 Paeonia species.
FIGURE S2 | Phylogenetic trees constructed using the ML method based on 19 highly variable regions of chloroplast genomes of 16 Paeonia species.
TABLE S1 | Information on the 16 Paeonia species.
TABLE S2 | The positions of the 19 highly variable regions.
TABLE S3-1 | Codons in chloroplast genome of P. qiui.
TABLE S3-2 | Codons in chloroplast genome of P. jishanensis.
TABLE S3-3 | Codons in chloroplast genome of P. delavayi var. lutea.
References
Abdel-Aty, A. S. (2007). Nematicidal effect of Paeonia suffruticosa constituents. Alexandria Sci. Exch. 28, 1–8. doi: 10.21608/asejaiqjsae.2007.1837
Ali, A., Jaakko, H., and Peter, P. (2018). IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chen, X. L., Zhou, J. G., Cui, Y. X., Wang, Y., Duan, B. Z., and Yao, H. (2018). Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 9:695. doi: 10.3389/fphar.2018.00695
Coissac, E., Hollingsworth, P. M., Lavergne, S., and Taberlet, P. (2016). From barcodes to genomes: extending the concept of DNA barcoding. Mol. Ecol. 25, 1423–1428. doi: 10.1111/mec.13549
Daniell, H., Lin, C. S., Ming, Y., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Doorduin, L., Gravendeel, B., Lammers, Y., Ariyurek, Y., Chin-A-Woeng, T., and Vrieling, K. (2011). The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 18, 93–105. doi: 10.1093/dnares/dsr002
Feng, Y. L., Cheng, J., Zang, R. X., Wang, M. M., Chen, H., and He, L. X. (2015). Phylogenetic relationships of Paeonia Sect. Moutan based on chloroplast psbA-trnH and trnL-F sequences. J. Biol. 32, 55–58. doi: 10.3969/j.issn.2095-1736.2015.01.055
Ferguson, D., and Sang, T. (2001). Speciation through homoploid hybridization between allotetraploids in peonies (Paeonia). Proc. Natl. Acad. Sci. U. S. A. 98, 3915–3919. doi: 10.1073/pnas.061288698
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Guo, X. F. (2002). Advances in classification of Chinese herbaceous peony. J. Beij. Forest. Univer. 24, 99–102.
Hao, Q., Liu, Z. A., Shu, Q. Y., Zhang, R., De Rick, J., and Wang, L. S. (2008). Studies on Paeonia cultivars and hybrids identification based on SRAP analysis. Hereditas 145, 38–47. doi: 10.1111/j.0018-0661.2008.2013.x
Hong, D. Y., and Pan, K. Y. (1999). Taxonomical history and revision of Paeonia sect. Moutan (Paeoniaceae). Acta Phytotaxon. Sin. 37, 351–368.
Hong, D. Y., Pan, K. Y., and Yu, H. (1998). Taxonomy of the Paeonia delavayi complex (Paeoniaceae). Ann. Mo. Bot. Gard. 85, 554–564. doi: 10.2307/2992016
Hu, Y., Woeste, K. E., and Zhao, P. (2016). Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7, 1955–1970. doi: 10.3389/fpls.2016.01955
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebensmack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 104, 19369–19374. doi: 10.1073/pnas.0709121104
Ji, L. J., Wang, Q., Teixeira, da Silva, J. A., and Yu, X. N. (2012). The genetic diversity of Paeonia L. Sci. Hortic. 143, 62–74. doi: 10.1016/j.scienta.2012.06.011
Kahlau, S., Aspinall, S., Gray, J. C., and Bock, R. (2006). Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J. Mol. Evol. 63, 194–207. doi: 10.1007/s00239-005-0254-5
Kane, N., Sveinsson, S., Dempewolf, H., Yang, J. Y., Zhang, D. P., Engels, J. M. M., et al. (2012). Ultra-barcoding in cacao (Theobroma spp., Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 99, 320–329. doi: 10.3732/ajb.1100570
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, S. H., Kim, S. A., Park, M. K., Kim, S. H., Park, Y. D., Na, H. J., et al. (2004). Paeonol inhibits anaphylactic reaction by regulating histamine and TNF-a. Int. Immunopharmacol. 4, 279–287. doi: 10.1016/j.intimp.2003.12.013
Kuang, D. Y., Wu, H., Wang, Y. L., Gao, L. M., Zhang, S. Z., and Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae) : implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/g11-026
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, P., Zhang, S., Li, F., Zhang, S., Zhang, H., Wang, X., et al. (2017). A phylogenetic analysis of chloroplast genomes elucidates the relationships of the six economically important Brassica species comprising the triangle of U. Front. Plant Sci. 8:111. doi: 10.3389/fpls.2017.00111
Li, X. W., Gao, H. H., Wang, Y. T., Song, J. Y., Henry, R., Wu, H. Z., et al. (2013). Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. China-Life Sci. 56, 189–198. doi: 10.1007/s11427-012-4430-8
Li, X. W., Hu, Z. G., Lin, X. H., Li, Q., Gao, H. H., Luo, G. A., et al. (2012). High-throughput pyrosequencing of the complete chloroplast genome of Magnolia officinalis and its application in species identification. Acta Pharm. Sin. 47, 124–130.
Li, X. W., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., and Chen, S. L. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. 90, 157–166. doi: 10.1111/brv.12104
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lin, Q. B., Zhou, Z. Q., Zhao, X., Pan, K. Y., and Hong, D. Y. (2004). Interspecific relationships among the wild species of Paeonia Sect. Moutan DC. based on DNA sequence of Adh gene family. Acta Hortic. Sin. 31, 627–632. doi: 10.1300/J064v24n01_09
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13:715. doi: 10.1186/1471-2164-13-715
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18. doi: 10.1186/2047-217X-1-18
Ma, S. J., Zhou, J. G., Li, Y., Chen, X. L., Wu, M. L., Sun, W., et al. (2018). Complete chloroplast genomes of Dioscorea opposite and D. collettii and screening specific DNA barcodes. Scientia Sinica Vitae 48, 571–582. doi: 10.1360/N052017-00160
Nadalin, F., Vezzi, F., and Policriti, A. J. B. B. (2012). GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinf. 13:s8. doi: 10.1186/1471-2105-13-S14-S8
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nock, C. J., Waters, D. L. E., Edwards, M. A., Bowen, S. G., Rice, N., Cordeiro, G. M., et al. (2011). Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9, 328–333. doi: 10.1111/j.1467-7652.2010.00558.x
Nylander, J. A. A. (2004). MrModeltest v.2. Program distributed by the author. Uppsala: Uppsala University.
Ohyama, K., Fukuzawa, H., Kohchi, T., Shirai, H., Sano, T., Sano, S., et al. (1986). Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha, chloroplast DNA. Nature 322, 572–574. doi: 10.1038/322572a0
Okubo, T., Nagai, F., Seto, T., Satoh, K., Ushiyama, K., and Kano, I. (2000). The inhibition of phenylhydroquinone-induced oxidative DNA cleavage by constituents of Moutan Cortex and Paeoniae radix. Biol. Pharm. Bull. 23, 199–203. doi: 10.1248/bpb.23.199
Park, I., Yang, S. Y., Kim, W. J., Noh, P., Lee, H. O., and Moon, B. C. (2018a). Authentication of herbal medicines dipsacus asper and phlomoides umbrosa using DNA barcodes, chloroplast genome, and sequence characterized amplified region (SCAR) marker. Molecules 23:1748. doi: 10.3390/molecules23071748
Park, I., Yang, S. Y., Kim, W. J., Noh, P., Lee, H. O., and Moon, B. C. (2018b). The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic pharbitidis semen (Seeds of I. nil or I. Purpurea). Front. Plant Sci. 9, 965–978. doi: 10.3389/fpls.2018.00965
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Qi, S. P., and Hu, P. R. (1993). Effect of the esters of gallic acid on model and human blood platelet membranes studied by Fourier-transform infrared spectroscopy. Sci. China Ser. B 36, 702–709.
Qian, J., Song, J. Y., Gao, H. H., Zhu, Y. J., Xu, J., Pang, X. H., et al. (2013). The Complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One 6:e57607. doi: 10.1371/journal.pone.0057607
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Sang, T. (2002). Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit. Rev. Biochem. Mol. Biol. 37, 121–147. doi: 10.1080/10409230290771474
Sang, T., Crawford, D. J., and Stuessy, T. F. (1995). Documentation of reticulate evolution in peonies (Paeonia) using internal transcribed spacer sequences of nuclear ribosomal DNA: implications for biogeography and concerted evolution. Proc. Natl. Acad. Sci. U. S. A. 92, 6813–6817. doi: 10.1073/pnas.92.15.6813
Sang, T., Crawford, D. J., and Stuessy, T. F. (1997a). Chloroplast DNA phylogeny, reticulate evolution and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 84, 1120–1136. doi: 10.2307/2446155
Sang, T., Donoghue, M. J., and Zhang, D. (1997b). Evolution of alcohol dehydrogenase genes in peonies (Paeonia): phylogenetic relationships of putative nonhybrid species. Mol. Biol. Evol. 14, 994–1007. doi: 10.1093/oxfordjournals.molbev.a025716
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sharp, P. M., and Li, W. H. (1987). The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1002/ajpa.21279
Shaw, J., Lickey, E. B., Schilling, E. E., and Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94, 275–288. doi: 10.3732/ajb.94.3.275
Shimada, H., and Sugiura, M. (1991). Fine structural features of the chloroplast genome: Comparison of the sequenced chloroplast genome. Nucleic Acids Res. 19, 983–995. doi: 10.1093/nar/19.5.983
Steele, P. R., and Pires, J. C. (2011). Biodiversity assessment: State of the art techniques in phylogenomics and species identification. Am. J. Bot. 98, 415–425. doi: 10.3732/ajb.1000296
Stern, F. C., and Taylor, G. (1951). A new peony from S. E. Tibet. Journ. Roy. Hort. Soc. 76, 216–217.
Sugiura, M., Shinozaki, K., Zaita, N., Kusuda, M., and Kumano, M. (1986). Clone bank of the tobacco (Nicotiana tabacum) chloroplast genome as a set of overlapping restriction endonuclease fragments: mapping of eleven ribosomal protein genes. Plant Sci. 44, 211–217. doi: 10.1016/0168-9452(86)90093-2
Sun, Y. L., and Hong, S. K. (2012). Phylogenetic relationship and evolution analysis of the peony Paeonia species using multi-locus deoxyribonucleic acid (DNA) barcodes. J. Med. Plants Res. 6, 5048–5058. doi: 10.5897/JMPR11.1733
Takano, A., and Okada, H. (2011). Phylogenetic relationships among subgenera, species, and varieties of Japanese Salvia L. (Lamiaceae). J. Plant Res. 124, 245–252. doi: 10.1007/s10265-010-0367-9
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tang, D. Q., Lu, J. J., Fang, W., Zhang, S., and Zhou, M. B. (2010). Development, characterization and utilization of GenBank microsatellite markers in Phyllostachys pubescens and related species. Mol. Breed. 25, 299–311. doi: 10.1007/s11032-009-9333-4
Tang, P., Ruan, Q. Y., and Peng, C. (2011). Phylogeny in structure alterations of poaceae cpDNA. Chin. Agricul. Sci. Bull. 27, 171–176.
Tank, D. C., and Sang, T. (2001). Phylogenetic utility of the glycerol-3-phosphate acyltransferase gene: evolution and implications in Paeonia (Paeoniaceae). Mol. Phylogenet. Evol. 19, 421–429. doi: 10.1006/mpev.2001.0931
Tsai, H. Y., Lin, H. Y., Fong, Y. C., Wu, J. B., Chen, Y. F., Tsuzuki, M., et al. (2008). Paeonol inhibits RANKL-induced osteoclastogenesis by inhibiting ERK, p38 and NF-κB pathway. Eur. J. Pharmacol. 588, 124–133. doi: 10.1016/j.ejphar.2008.04.024
Wakasugi, T., Tsudzuki, T., and Sugiura, M. (2001). The genomics of land plant chloroplasts: Gene content and alteration of genomic information by RNA editing. Photosynth. Res. 70, 107–118. doi: 10.1023/a:1013892009589
Wang, L., Dong, W. P., and Zhou, S. L. (2012). Structural mutations and reorganizations in chloroplast genomes of flowering plants. Acta Bot. Boreali Occident. Sin. 32, 1282–1288. doi: 10.3969/j.issn.1000-4025.2012.06.031
Wang, Z. P. (1996). Molecular systematics and evolution of Paeonia suffruticosa andr. complex: evidence from nuclear ribosomal DNA (nrDNA) variation. Ph. D dissertation, Beijing: Institute of Botany.
Wu, F. H., Chan, M. T., Liao, D. C., Hsu, C. T., Lee, Y. W., Daniell, H., et al. (2010). Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 10, 68–79. doi: 10.1186/1471-2229-10-68
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xiang, B., Li, X., Qian, J., Wang, L., Ma, L., Tian, X., et al. (2016). The complete chloroplast genome sequence of the medicinal plant Swertia mussotii using the PacBio RS II Platform. Molecules 21:1029. doi: 10.3390/molecules21081029
Xing, W. R., Hou, B. W., Guan, J. J., Luo, J., and Ding, X. Y. (2013). Sequence analysis of LEAFY homologous gene from Dendrobium moniliforme and application for identification of medicinal Dendrobium. Acta Pharm. Sin. 48, 597–603.
Xu, S. Z., Li, D. Z., Li, J. W., Xiang, X. G., Jin, W. T., Huang, W. C., et al. (2015). Evaluation of the DNA Barcodes in Dendrobium (Orchidaceae) from Mainland Asia. PLoS One 10:e0115168. doi: 10.1371/journal.pone.0115168
Yang, J. B., Tang, M., Li, H. T., Zhang, Z. R., and Li, D. Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Yang, L., Zhao, H. S., Peng, Z. H., Dong, L. L., and Gao, Z. M. (2014). Development and application of SSR molecular markers from the chloroplast genome of bamboo. J. Trop. Subtrop. Bot. 22, 263–269. doi: 10.3969/j.issn.1005-3395.2014.03.008
Yu, X. Q., Drew, B. T., Yang, J. B., Gao, L. M., and Li, D. Z. (2017). Comparative chloroplast genomes of eleven Schima (Theaceae) species: Insights into DNA barcoding and phylogeny. PLoS One 12:e0178026. doi: 10.1371/journal.pone.0178026
Zhang, J. M., Wang, J. X., Xia, T., and Zhou, S. L. (2008). Application of DNA barcoding technique based on phylogenetic analysis in clarifying the species of Paeonia Section Moutan. Scientia Sinica Vitae 38, 1166–1176.
Zhang, X. J., Li, Z., Leung, W. M., Liu, L., Xu, H. X., and Bian, Z. X. (2008). The Analgesic Effect of Paeoniflorin on Neonatal Maternal Separation-Induced Visceral Hyperalgesia in Rats. J. Pain 9, 497–505. doi: 10.1016/j.jpain.2007.12.009
Zhang, Y. J., Ma, P. F., and Li, D. Z. (2011). High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS One 6:e20596. doi: 10.1371/journal.pone.0020596
Zhao, X. (2007). Phylogenetic relationship among the speices of sect. Moutan and origin of cultivated tree peony. Ph. D dissertation, Chongqing: Southwest University.
Zhao, X., Zhou, Z. Q., Lin, Q. B., Pan, K. Y., and Hong, D. Y. (2004). Molecular evidence for the interspecific relationships in Paeonia sect. Moutan: PCR-RFLP and sequence analysis of glycerol-3-phosphate acyltransferase (GPAT) gene. J. Syst. Evol. 42, 236–244. doi: 10.1088/1009-0630/6/5/011
Zhou, J. G., Chen, X. L., Cui, Y. X., Sun, W., Li, Y. H., Wang, Y., et al. (2017). Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species. Int. J. Mol. Sci. 18, 1839. doi: 10.3390/ijms18091839
Zhou, R. C., and Yao, C. H. (2002). Studies on phylogeny of Paeonia Section Moutan subsection Vaginatae. Bull. Botan. Res. 22, 72–75. doi: 10.1088/1009-1963/11/5/313
Zhu, S. Y., Niu, Z. T., and Xue, Q. Y. (2018). Accurate authentication of Dendrobium officinale and its closely related species by comparative analysis of complete plastomes. Acta Pharm. Sin. B 8, 969–980. doi: 10.1016/j.apsb.2018.05.009
Keywords: section Moutan, chloroplast genome, comparative analysis, phylogeny, species relationship
Citation: Wu L, Nie L, Xu Z, Li P, Wang Y, He C, Song J and Yao H (2020) Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Three Paeonia Section Moutan Species (Paeoniaceae). Front. Genet. 11:980. doi: 10.3389/fgene.2020.00980
Received: 02 May 2020; Accepted: 03 August 2020;
Published: 18 September 2020.
Edited by:
Denis Baurain, University of Liège, BelgiumReviewed by:
Yun Sun Lee, Seoul National University, South KoreaJinming Chen, Chinese Academy of Sciences, China
Copyright © 2020 Wu, Nie, Xu, Li, Wang, He, Song and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Yao, c2NhdXlhb2hAc2luYS5jb20=