 Cristina Zalfa
Cristina Zalfa Silke Paust
Silke Paust- Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA, United States
The tumor microenvironment (TME) is a complex and heterogeneous environment composed of cancer cells, tumor stroma, a mixture of tissue-resident and infiltrating immune cells, secreted factors, and extracellular matrix proteins. Natural killer (NK) cells play a vital role in fighting tumors, but chronic stimulation and immunosuppression in the TME lead to NK cell exhaustion and limited antitumor functions. Myeloid-derived suppressor cells (MDSCs) are a heterogeneous group of myeloid cells with potent immunosuppressive activity that gradually accumulate in tumor tissues. MDSCs interact with innate and adaptive immune cells and play a crucial role in negatively regulating the immune response to tumors. This review discusses MDSC-mediated NK cell regulation within the TME, focusing on critical cellular and molecular interactions. We review current strategies that target MDSC-mediated immunosuppression to enhance NK cell cytotoxic antitumor activity. We also speculate on how NK cell-based antitumor immunotherapy could be improved.
Introduction
Tumorigenesis is a complex and dynamic process involving three stages: initiation, progression, and metastasis (1). Besides blood and lymphatic vessels, composed in part of vascular endothelial cells, the major constituents of the tumor microenvironment (TME) are a heterogeneous population of cancer cells, fibroblasts, immune and inflammatory infiltrated cells, and secreted protein elements of the extracellular matrix (ECM) (2). The functional and physical interactions of these tumor elements determine clinical outcomes. At the beginning of the nineteenth century, Virchow described cancer as originating from chronic unresolved inflammation (3). Many studies have demonstrated that cancer-associated inflammation plays a critical role in tumor formation, contributing to genomic instability and epigenetic modification, and regulating the creation of a protected TME to promote cancer proliferation and metastasis (4–6). Tumor-associated inflammatory cells are observed in human cancers from the earliest phases of carcinogenesis (7). The first line of defense, represented by natural killer (NK) cells and cytotoxic CD8+ T cells, usually recognizes and kills malignant cells; however, a few immunogenic cancer cell variants can escape immune recognition (8).
The TME orchestrates multiple mechanisms to impair the antitumor functions of immune cells. These mechanisms include destabilizing the innate cell compartment, composed of NK cells, macrophages, neutrophils, and dendritic cells (DCs), and suppressing adaptive immune cell (T and B)-mediated antitumor activity. The presence and recruitment of atypical innate and adaptative immune cells in the tumor site are thought to occur during both the early and later stages of tumor development (6, 9). Tumor immune surveillance also fails because immunosuppression and its associated chronic inflammation further destabilize tumor-fighting immune cells, defending rather than eradicating tumors (10). The TME supports the growth of cancer-associated fibroblasts, stromal cells, and endothelial cells, contributing to tumor-associated capillary and lymphatic vessels that support tumor growth (11). These mechanisms select potentially aggressive tumor clones early during tumor development (9).
This review will discuss the crosstalk between NK cells, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and the critical cellular and molecular mechanisms within the TME that impact tumor development, progression, and angiogenesis, as well as how current therapies target these immunosuppressive cells in the TME. We will also review different NK cell exhaustion mechanisms and novel approaches for enhancing NK cell therapeutic potential against tumors.
Cytotoxic T Lymphocytes and NK Cells in Cancer
One of the adaptive and innate immune systems' essential activities is to kill infected and tumor cells. Mounting epidemiological and experimental evidence points to a critical role for cytotoxic T lymphocyte (CTL) and NK cell-mediated effector functions in host resistance to cancer. The collaboration between innate and adaptative effector cells can lead to tumor rejection (12, 13). CTL and NK cell activity has been linked to tumor immune surveillance and protection from cancer. Both cell types can form cytotoxic immunological synapses (14), which are specialized antigen-specific cell–cell junctions with a synaptic cleft to directly communicate and transduce highly controlled secretory signals between immune cells and their target cells (14). This mechanism, also present in NK cells, improves the efficiency of cytotoxic cell-mediated killing (15). Higher numbers of tumor-infiltrating CTL and/or NK cells are a favorable prognostic indicator for many cancer types (16, 17). T cell activation occurs when a clonal T cell receptor (TCR) is triggered by a tumor-derived antigen presented on class I human leukocyte antigen (HLA-I), in combination with co-receptor ligation and co-stimulation, leading to CTL activation, proliferation, cytokine and chemokine secretion, and tumor cell killing (18). Similar to CTL cells, NK cells are also cytotoxic lymphocytes and important tumor fighters.
NK cells do not express a TCR but instead have many activating receptors and also inhibitory receptors, which bind major histocompatibility complex class I (MHC-I) (19, 20). MHC-I is often downregulated by infected and malignant cells to avoid CTL killing. NK cells sense this “lack of inhibition” and are further activated by tumor cell-expressed stress ligands that ligate NK cell-expressed activating receptors (20–22). NK cells secrete cytokines, chemokines, pore-forming proteins (perforin), and cytotoxic mediators (granzymes) that trigger target cell apoptosis upon activation. A potent NK cell-activating receptor is NK group 2 member D (NKG2D), and NKG2D ligands are commonly upregulated on tumors (23).
Substantial evidence supports the conclusion that NK cells play a crucial role in eliminating tumors and tumor metastases (24). First, low cytotoxicity in peripheral blood (PB) NK cells correlates with a higher risk of developing cancer (25). Second, in many types of cancer, NK cells exhibit an altered phenotype and hypo-functionality (26, 27). Third, in mice, resistance to NK cell killing favors polyclonal metastasis (28). Several mechanisms contribute to NK cell exhaustion, including modulating adhesion and epithelial genes, decreasing the expression of NK cell-activating ligands (28), and the suppressive effects of regulatory immune cells and soluble factors within the TME (29).
NK Origin, Development, and Tissue Distribution
NK cells are bone marrow (BM)-derived granular lymphocytes that lyse target cells rapidly and continuously upon activation (30). In the BM, NK cells develop from CD34+ hematopoietic stem cells through a common lymphoid progenitor (CLP) intermediate that can seed and develop further in lymphoid and non-lymphoid organs (31). NK cell maturation requires several cytokines, among which interleukin (IL)-15, released by BM stromal cells, is crucial for the differentiation of CLPs toward the NK cell lineage (32). NK cells are well-represented in the PB, spleen, and BM and are found in most organs, including the liver, lungs, skin, gut, lymph nodes, tonsils, uterus, thymus, kidney, pancreas, and adipose tissue. (33, 34). Their recruitment to different tissues depends on the expression of several chemokine receptors and is reviewed elsewhere (35, 36).
Human NK cells express the hematopoietic cell marker CD45, the glycoprotein CD56 and mature NK cells, and the cluster of differentiation molecule CD16 also known as Fc receptor FcγRIIIa, but they do not express any T or B cell receptors. NK cell maturation is generally assessed by the amount of NK cell-expressed CD56 and CD16 expression. Specifically, CD56dim and CD56bright subsets show profound differences in cytokine secretion, response to cytokines, and killing efficiency. CD56dim CD16bright NK cells in the PB are about 90% of the total NK cell population and include the alloreactive NK cells described as “mature” with a higher cytotoxic potential. The remaining 10% of NK cells are CD56bright CD16dim and reside in the lymphoid tissue; they are considered “immature or unlicensed.” These NK cells are more sensitive to cytokine stimulation, which will activate the secretion of a variety of cytokines, including interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), IL-5, IL-10, and IL-13 (37–39).
A sophisticated array of germline-encoded activating and inhibiting receptors regulates NK cell development and, subsequently, their activation (40). Upon activation, NK cells employ several mechanisms of target cell killing and secrete chemokines and cytokines to interact with other immune cells (41, 42). NK cells are traditionally defined as innate immune response cells because they lack recombinase-dependent clonal antigen receptors (RAG) (43). Nonetheless, recent findings have revealed that NK cells display adaptive immunity features (44–47), including several developmental and functional characteristics of the adaptive immune system (43, 48–51). These similarities include vaccination or sensitization-dependent antigen-specific immunological memory (45, 46, 52–55). How NK memory impacts the tumor-specific NK cell response is currently unknown.
Antitumor Responses of NK Cells
Many studies have demonstrated that NK cells can kill tumor cells (56). NK cells survey their environment with a distinct receptor repertoire, including activating and inhibitory receptors, adhesion molecules, and cytokine and chemokine receptors (57). NK cells recognize the expression of HLA-I, also called MHC-I, on autologous cells. This interaction is generally inhibitory and prevents NK cells from attacking healthy host tissue. Specifically, HLA-I or MHC-I binds NK cell inhibitory receptors, including killer cell immunoglobulin-like receptors (KIRs) in humans, Ly49 in mice, and CD94/NKG2A (58, 59). In contrast to infected or malignant cells, healthy nucleated cells express robust levels of HLA-I/MHC-I molecules and escape NK cell immune attack. However, during malignant transformation or viral infection, the expression of MHC-I antigens on the cell surface can be downmodulated. This variation in the expression of MHC-I molecules on target cells (missing self) reduces the strength of inhibitory signals delivered to NK cells, thus promoting NK cell activation. NK cells survey tissues for low MHC-I molecule expression (60, 61) and for the expression of activating ligands, such as the NKG2D ligands (62, 63), and ligands for the natural cytotoxicity receptors NKp30 and NKp44 in humans and NKp46 in humans and mice (64).
NK cells also have the unique ability to exert antibody-dependent cell-mediated cytotoxicity (ADCC) upon engagement of CD16 with the Fc portion of the antibodies (65). Activation through these receptors elicits rapid target cell killing by several mechanisms. NK cells form an immunological synapse with target cells and kill them rapidly by secretion of lytic granules that contain an arsenal of effector molecules (perforin, granzymes) and cytokines (IFN-γ, TNF-α) that induce cell death in targeted cells (66–68). Perforin and granzyme are proteins that play a significant role in cell-mediated cytotoxicity. These molecules are expressed in NK cells, and several cytokines regulate their level of expression in CTLs. The role of perforin, which is involved in T cell- and NK cell-mediated target cell lysis, was demonstrated in mice lacking perforin with respect to their capability to eradicate a syngeneic lymphoid tumor mammary adenocarcinoma (69). Smyth and coworkers demonstrated that mice with lymphoma and deficient in the pore-forming protein perforin [(pfp)-deficient] showed an increased number of premalignant cells than their immunocompetent counterparts. In fact, pfp-deficient mice were 1,000-fold more susceptible to tumor (70), demonstrating that lymphocyte-mediated cytotoxicity plays an essential role in promoting host resistance to spontaneous tumor formation. NK cells also express several TNF superfamily proteins and death-inducing ligands, such as TNF-related apoptosis-inducing ligand (TRAIL) and FAS ligand (FASL), which induce target cell apoptosis via binding to their corresponding receptors (TRAIL-R and FAS). Activated NK cells also produce growth factors, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and chemokines (XCL1, CCL3, CCL4, and CCL5) l (26, 33, 71–73).
NK Cells in Solid Tumors
Genetic alterations in oncogenic pathways associated with an aberrant inflammatory milieu (74, 75), abnormal activations of transcription factors (nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κB] and signal transducer and activator of transcription-3 [STAT3]) (76, 77), and hypoxia (78) may contribute to development and maintenance of the TME. The TME is responsible for tumor onset and progression by orchestrating cell growth, proliferation, malignancy, and immune escape processes. TME can impair and “polarize” the innate, adaptive, stromal, and endothelial cell compartments by several mechanisms (2, 79–82). Tumor cells, MDSCs, and tumor-associated cells, such as tumor-associated fibroblasts (TAFs) and endothelial cells (83), can contribute to tissue modifications of the ECM by matrix metalloproteases (MMPs) and fibroblast activation proteins and the release of soluble factors (basic fibroblast growth factor [FGF], platelet-derived growth factor, hepatocyte growth factor, insulin-like growth factor), chemokines (CCL2, SDF1a/CXCL12, CXCL8), and immunosuppressive cytokines, such as transforming growth factor beta (TGF-β), IL-10, and IL-6 (84, 85). Due to the adverse TME, immune cells lose their ability to reach, recognize, and target tumor cells (2, 79, 86, 87). The TME impairs immune cell homing to the lymphoid organs promoting tumor immune cell escape, invasiveness, and angiogenesis (88–90). Thus, chronic inflammation orchestrated by immunosuppressive mediators of the TME supports tumor progression by exhausting immune cells, such as NK cells (74, 78).
NK cells also express inhibitory receptors targeting non-MHC molecules on healthy cells. One of these inhibitory receptors expressed on NK cells is killer cell lectin-like receptor G1 (KLRG1), a well-conserved member of the C-type lectin receptor superfamily. KLRG1 is known for its role in NK cell maturation, development, and homeostasis (91). Recently, a new role for KLRG1 has emerged as an inhibitory receptor impacting NK cell function in tumor surveillance. Impairment in the migration and/or retention of NK cells in the BM has been observed in multiple myeloma (MM). BM localization of the more functional NK KLRG1− subtypes is impaired in MM by altering the chemokine microenvironment (increasing chemokine [CXC] ligand 9 [CXCL9] and CXCL10 and reducing CXCL12 expression in BM) in a mouse tumor model of an early cancer growth stage (88). This is due to significant dysregulation of the CXCR3 and CXCR4 chemokine receptor/ligand axes, influencing NK cell responses (88, 92). Using murine models of chronic NK cell stimulation, Alvarez and colleagues have identified a “phenotypic signature of NK cell exhaustion,” characterized by upregulation of KLRG1 and downregulation of the activating receptor NKG2D (93). KLRG1 ligands, such as E- and N-cadherin, were upregulated in tumor specimens from patients with melanoma, breast, prostate, and colorectal cancer (94). The same authors showed that anti-KLRG1 antibody monotherapy in a 4T1 breast cancer mouse model enhanced tumor control compared to controls (94). Tata and colleagues also demonstrated that KLRG1-deficient mice had significantly fewer lung tissue tumors than wild-type controls (95).
Several studies have demonstrated the prognostic significance of tumor-infiltrating lymphocytes and their antitumor actions in cancer (16, 17, 96–99). It has recently become clear that CTL and NK cell cooperation are essential in many types of tumors (100–110). Using mouse models of mastocytoma (mice heterozygous for the H-2Ld/P1A35–43-specific TCR transgene: TCRP1A on the DBA/2, B10.D2; TCRP1A tg B10.D2[×DBA/2] F1; RAG-1°/°B10.D2), a study demonstrated that the frequency of cancer antigen-specific T cell precursors and the rate of antigen variants can contribute to the efficacy of adaptive T cell responses to cancer (107). Moreover, the efficiency of an effective antitumor antigen-specific T-cell response can depend on the complementary interaction between effector T cells and NK cells (107). Another study also demonstrated that the NK cell's antitumor effect requires interaction with specific activated tumor antigen-CD8+ T cells (12). However, further studies are necessary to clarify the mechanism of interaction between NK cells and specific effector T cells.
In clinical trials studying solid tumors, impaired NK cell function correlated with a poor prognosis in patients with advanced disease (111, 112). The TME plays a critical role in reducing NK cell persistence and trafficking in the tumor site by inhibiting NK cell activation, leading to tumor invasion and metastasis (26). Moreover, several studies showed anergic and hypo-functional NK cells (26, 27, 113–120), including tumor-associated NK cells in the PB and tumor-infiltrating NK cells within the tumor tissue. These CD56bright CD16low/− Perforinlow NK cells even exhibited pro-tumorigenic functions and pro-angiogenic activities (80, 121–124) and have been identified preferentially in many solid tumors (80, 87, 115–121, 123, 125–129). Some of these NK cells also downregulated their expression of NKG2D, impairing their antitumor functions further.
MDSCs and Tregs in Cancer
Development and Phenotypes of MDSCs
Common myeloid progenitors differentiate from hematopoietic stem cells in the BM. Later, they migrate to the peripheral lymphoid organs and differentiate into myeloid cells. This pathway involves granulocyte-macrophage progenitor and various myeloblast intermediate precursors, including common monocyte progenitors (130) (Figure 1). Immunological stress, as well as cancer, prolonged inflammation, trauma, and autoimmune disorders, can impair the differentiation of these immature myeloid cells (131). Tumor-associated myeloid cells are mainly represented by tumor-associated macrophages (TAMs) and MDSCs, which are one of the crucial players within the TME (Figure 1). The TME often subverts immunosurveillance by generating MDSCs with strong immunosuppressive activity and functional plasticity (130). MDSCs are a heterogeneous population of myeloid-derived cells represented by myeloid progenitors, immature granulocytes, DCs, and macrophages. Therefore, the characteristics that separate MDSCs from other myeloid cells are still under investigation. It is widely accepted that MDSCs are divided into two main subsets: granulocytic or polymorphonuclear (PMN)-MDSCs and monocytic (M)-MDSCs, cells showing a phenotype and morphology similar to neutrophils and macrophages, respectively (132–136) (Figure 1). However, MDSC subtypes can be distinguished from neutrophils and TAMs that are present in the TME (137, 138). Studies have shown additional mechanisms to describe the evolution and roles of these polarized neutrophils in the TME, and some evidence supports the idea that these cells are similar to MDSCs and could be described as PMN-MDSCs (139–141), which represent the most prevalent cells in several types of tumors (142, 143). MDSCs with granulocyte and monocyte hallmarks have genomic profiles, biochemistry, and in vitro properties that differ from neutrophils, monocytes, and DCs (144). Recently, whole-transcriptomic and proteomic analyses (134, 145–147) provided specific gene expression patterns for the characterization of these different cell types. Cell-surface markers have been identified to distinguish MDSC-specific phenotypes from TAMs, tumor-associated neutrophils, and neutrophils. M-MDSCs can be separated from TAMs by their differential expression of F4/80, M-CSF and CD115high, Ly6Clowtointermediate, IRF8low, and S100A9verylow (148). In contrast, PMN-MDSCs are CD11b+Ly6G+Ly6CloLOX−1 in mice and CD11b+CD14−CD15+ or CD11b+ CD14−CD66b in humans, while M-MDSCs are CD11b+Ly6G−Ly6Chi in mice and CD11b+CD14+HLA-DR−/loCD15− in humans (Figure 1). A low percentage of MDCSs (around 3%) consists of a mixture of more immature progenitors and precursors with myeloid-colony-forming activity termed “early-stage MDSCs” (e-MDSC) and are described as Lin−HLA-DR−CD33+ (132, 144). These and other novel marker combinations are currently under further investigation (133, 134, 149).
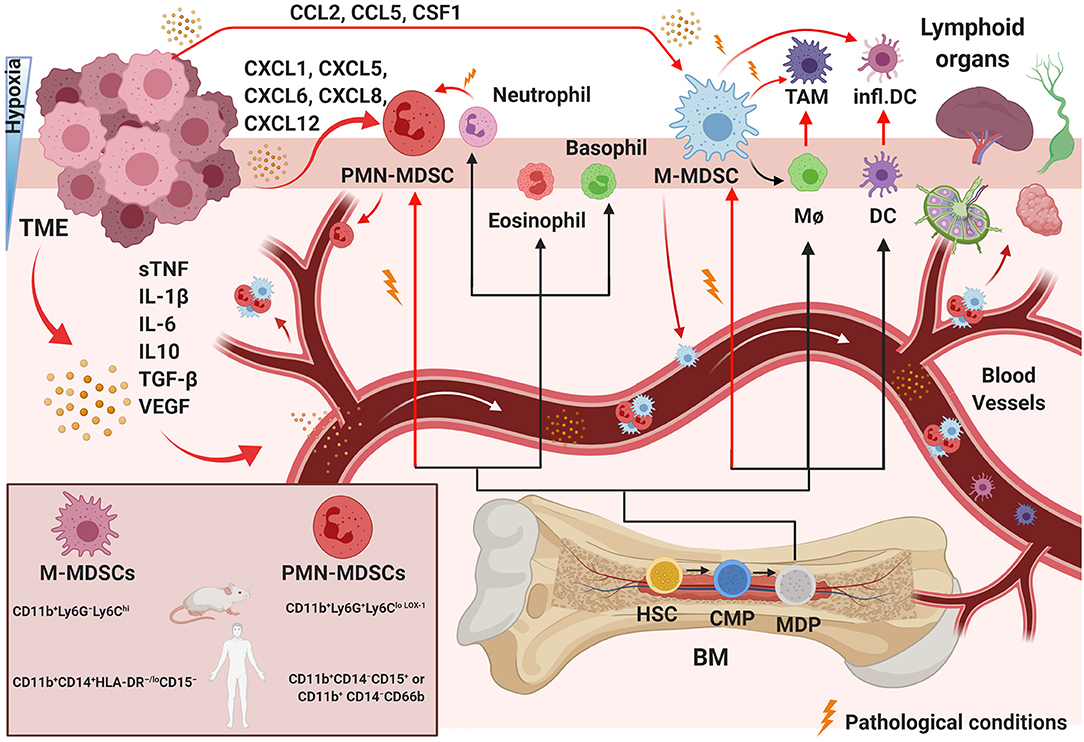
Figure 1. Pathological myelopoiesis in cancer. Under physiological conditions and chronic inflammation, hematopoietic progenitor cells (HPCs) differentiate via common myeloid progenitor cells (CMPs) into monocytic/dendritic progenitor cells (MDPs). Myelopoiesis is altered under pathological conditions, such as in a tumor. Tumor-immunosuppressive factors produced in the TME alter myelopoiesis, leading to aberrant differentiation and accumulation of myeloid lineage cells. The black lines show the normal pathways of myeloid cell differentiation from immature myeloid precursor cells to dendritic cells (DCs), macrophages (MΦ), and granulocytes, as depicted. The red lines indicate the aberrant pathways of myeloid cell differentiation that arise in cancer, in which the TME promotes the development of immunosuppressive populations, including monocytic myeloid-derived suppressor cells (M-MDSCs), polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), inflammatory DCs, and tumor-associated macrophages (TAMs). The MDSC mouse and human phenotypes are shown in the left square on the bottom. BM, bone marrow; sTNF, soluble tumor necrosis factors. Created with BioRender.com.
The number of MDSCs is associated with the clinical cancer stage and metastatic disease (150). Thus, MDSCs could be a potential biomarker of disease in several types of cancer. For example, the frequencies of MDSCs change during tumor progression in glioma (151) and cervical cancer (152). The frequency of peripheral PMN-MDSCs has been correlated with cancer prognosis, while the percentage of M-MDSCs has been shown to be higher in patients with advanced cervical cancer (152). Moreover, PMN-MDSCs' rate is negatively correlated with CD8+ T cells' rate (151, 152). MDSCs have been reported as prognostic markers in non-small cell lung cancer (NSCLC) (153); breast and colorectal cancer (154); gastric, esophageal, and pancreatic cancer (155); and melanoma (156). Therefore, MDSCs can also be used as a predictive marker for immunotherapy. Additionally, a diminished number of MDSCs helped to eradicate metastatic disease after the removal of primary tumors in a mammary carcinoma model (157).
Recruitment and Expansion of MDSCs in Tumors
While absent in normal physiological conditions (144), MDSCs can be detected in the BM, blood, spleen, tumor, and lymph nodes in pathological conditions (Figure 1). MDSCs have been shown to increase significantly in early- and late-stage cancer in preclinical animal models and human tumors (6, 145, 151). Upon their recruitment, MDSCs gradually expand in the TME and support the development of an immunosuppressive tumor environment by interacting with several components of the innate and adaptive immune systems (158, 159) and by stimulation of neo-angiogenesis (160, 161). Vetsika et al. described all phases of this process (162); here, we will summarize. The network of transcriptional regulators that directs MDSC development can be combined into two partially overlapping groups: (i) factors promoting myelopoiesis and avoiding differentiation of mature myeloid cells and (ii) factors contributing to pathologic activation of MDSCs. In different types of mouse and human cancers (132, 144, 163–166), MDSCs are gradually recruited and increase in the TME. They support tumor progression through non-immune activities by stimulating pre-metastatic niche formation, invasion (167, 168), and inducing pro-tumor angiogenesis (169). Some authors have proposed a “two-signal model” for describing how MDSCs can acquire the modifications guiding their pathological activation, immunosuppressive activity, and expansion in the TME under tumor pathologic signals (170, 171). Myeloid cells exposed to pathological conditions (autoimmunity, cancer, trauma, graft vs. host disease, and infections) can be activated in response to damage-associated molecular pattern molecules, pathogen-associated molecular pattern molecules, or pro-inflammatory cytokines (144). Because patient blood has been observed to have increased tumor-released macro- and micro-vehicles during tumor progression, tumor niches could potentially gather MDSCs from the BM by releasing exosomes. Their contents have been demonstrated to reprogram target cells in different types of cancer, increase the mobility of the progenitor myeloid population to the tumor site, and increase tumor immunosuppression (162).
MDSCs migrate in response to several chemo-attractant molecules released from cells in the TME using two main pathways: PMN-MDSC migration includes the secretion of CXCL1, CXCL5, CXCL6, CXCL8, and CXCL12, and M-MDSCs respond to CCL2, CCL5, and CSF1 (172) (Figure 1). The TME can guide the differentiation of incoming MDSCs in several different directions. The commitment of myeloid progenitor and precursor cells into MDSCs can be triggered by immunosuppressive cytokines released in the TME, such as soluble tumor necrosis factors (sTNF), IL-1β, IL-6, IL10, TGF-β, and vascular endothelial growth factor (VEGF) (173, 174) (Figure 1). MDSC phenotypes develop under conditions of acute or chronic inflammation, stress and hypoxia, high concentrations of oxidative molecules, and reduced nutrients (172, 175) (Figure 1). Hypoxia, specifically hypoxia-inducible factor 1 alpha (HIF-1α), appears to be one of the most critical stresses (35, 56) and was shown to be essential in M2-type TAM generation from Ly6Chi monocytes inside a tumor (176). During hypoxia, immunomodulatory proteins and chemokines also mediate the differentiation of TAMs or M2 macrophages (177) from M-MDSCs or/and guide later events in tumor progression (177, 178).
Because M-MDSCs have a longer lifespan (179), their differentiation has been studied more extensively than that of PMN-MDSCs. TAMs can be derived from tissue-resident macrophages proliferating in situ in pancreatic and mammary tumors (180, 181). Several studies have shown the ability of M-MDSCs to differentiate into TAMs after migrating from the spleen to a tumor (178, 180, 182, 183), and TAMs can be “regenerated” by the arrival of new M-MDSCs from other organs during tumor progression (184–186) (Figure 1). MDSCs can also differentiate into DCs and fibrocytes during cancer progression (187). Moreover, Ly6Chi, Ly6CX3CR1, and Ly6C+CCR2+ monocytes can differentiate into TAM subsets (Figure 1) in mammary adenocarcinoma, lung adenocarcinoma, and lung carcinoma models (188, 189). In the chronic inflammation present in tumor tissues, IL-18 can support the function of TGF-β1 that is produced and activated by M2-polarized TAMs (190, 191). IL-18 promotes the differentiation of CD11b− BM progenitor cells into M-MDSCs and increases their suppressive functions, including arginase expression (ARG1) and NO secretion (192). MDSCs induced by IL-18 can inhibit CD4+ T cell proliferation and IFN-γ production (192), contributing to the negative regulation of immune responses in tumors through immunosuppressive functions.
Treg Origin and Development in the TME
CD4+CD25+ Tregs are a subpopulation of suppressor T cells that mediate immune homeostasis, maintain peripheral tolerance, and prevent immune and auto-immune disease by suppressing autoreactive T cells (193). These cells regulate immune responses in the context of immunity and infections differently (194, 195). In cancers, Tregs are linked to the development of an immunosuppressive TME, promoting immune evasion and cancer progression and preventing antitumor immunity (196–198). Tregs represent about 1–3% of CD4+ T cells in human tumors and about 10% in rodents. These cells express cell surface molecules associated with activated/memory T cells, CD25, FoxP3, CD45RBlow, CD62L, CD103, cytotoxic T-lymphocyte associated protein 4 (CTLA-4), and glucocorticoid-induced TNF receptor (199). An increase in Tregs prevalence has been shown in several tumor malignances (7), and they are recruited and expand within the TME via several mechanisms (200). We will summarize these stages in the following steps. Tregs are recruited into tumors in response to chemokines secreted by tumor cells and innate immune cells. Tregs then expand and proliferate in response to tumor-derived factors (TGF-β, adenosine, VEGF, and IL-10) within the TME. TGF-β and adenosine, released from cancer cells and also MDSCs, seem to play a key role in generating suppressive CD25+ FoxP3+ Tregs from non-suppressive CD25− FoxP3− conventional Tregs. The recruitment of Tregs occurs at early tumor stages, as demonstrated by their presence in pre-malignant lesions, and their prevalence increases with pancreatic and breast tumor progression and worsening clinical outcomes (114, 201–203). Moreover, it has been shown that the depletion of Tregs cells in pancreatic ductal adenocarcinoma slows tumor growth and prolongs survival (204–207).
Crosstalk Between MDSCs and Tregs
MDSC expansion in PB is directly correlated with poor clinical outcomes (208–210). MDSCs can support the conversion of naive CD4+ T cells into Tregs by secreting retinoic acid and TGF-β (Figure 2), promoting the trans-differentiation of Th17 cells into Foxp3+ Tregs (211). Moreover, MDSCs can also induce Tregs immunosuppressive functions by mediating the release of IL-10 and IFN-γ (162). Tumor-infiltrating M-MDSCs express high levels of C-C chemokine receptor type 5 (CCR5) ligands and recruit high numbers of Tregs into the TME (212), establishing an additional cooperative network between MDSCs and Tregs (24, 35). Therefore, Tregs are accumulated in the TME and produce VEGF, which promotes angiogenesis (161, 213). By using light-sheet fluorescent microscopy, Siret and colleagues demonstrated direct interactions between MDSCs and Tregs in pancreatic ductal adenocarcinoma (214), and in vivo depletion of MDSCs significantly reduced the Tregs population in pancreatic tumors (215). Furthermore, video-microscopy and ex vivo functional assays have demonstrated that MDSCs can induce Treg cells by cell–cell-dependent contact at different stages of human cancer, and Tregs can also affect the survival and/or the proliferation of MDSCs (214). The molecular mechanisms guiding MDSC/Tregs interplay are not fully understood. The role of co-stimulatory molecules, protein membranes, and receptor candidates, respectively, such as PD-L1 (216), CD80 (217), and CD40 (218), is currently under investigation. Together, Tregs and MDSCs contribute to establishing an immunosuppressive TME in multiple solid neoplasms (114, 201, 202, 214).
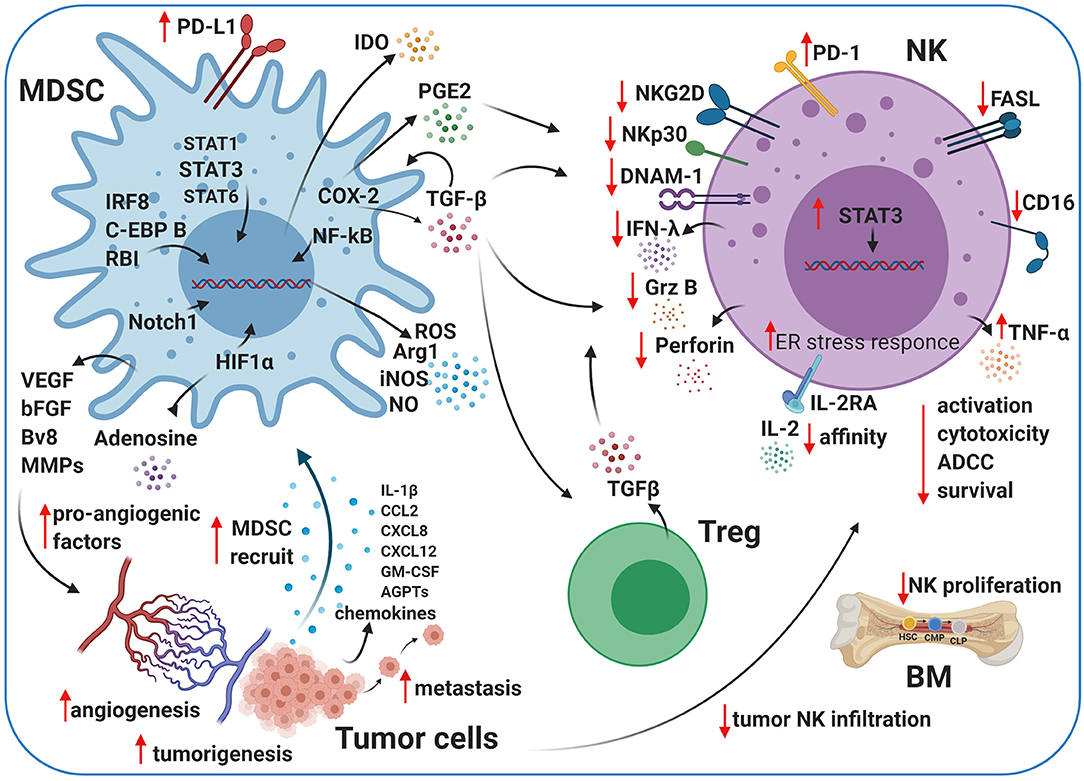
Figure 2. Crosstalk between MDSCs, NK cells, and Tregs in the TME. Myeloid-derived suppressor cells (MDSCs) and natural killer (NK) cells crosstalk within the TME. The cartoon shows the immunosuppressive activity of MDSCs and regulatory T cells (Tregs) on NK cells using different pathways. MDSCs impair NK cell activation, cytotoxicity, survival, and maturation by several secreted factors, including transforming growth factor beta (TGF-β), indoleamine 2,3-dioxygenase (IDO), nitrogen oxide (NO), reactive oxygen species (ROS), and prostaglandin-E2 (PGE2). MDSCs contribute to tumor angiogenesis by releasing pro-angiogenic factors, such as adenosine and vascular endothelial growth factor (VEGF), under pathological conditions. Created with BioRender.com.
Cytokines and Other Mediators in MDSC-Mediated NK Cell Regulation
In addition to cell-intrinsic defects caused by chronic stimulation (219), an immunosuppressive TME represses CTL and NK killing (29, 220) via the recruitment of other cells types, such as MDSCs, M2 macrophages (221), and Tregs (222), which contribute to immune exhaustion via the expression of inhibitory ligands, suppressive cytokines, and tumor-promoting factors (221, 223). MDSCs display potent immunosuppressive activity and play a critical role in regulating tumors and metastasis development (144). MDSCs can impair CD8+ CTLs and NK cells directly by influencing the pro-tumor TME (224). Their contribution to regulating T lymphocytes is well-described, while their interactions with other immune cells, such as NK cells, DCs, or macrophages, in the TME are less understood (158). The suppressive effects of MDSCs are mediated through cell–cell contact, as well as the secretion of soluble factors, and result in antigen-specific or antigen-non-specific suppression of T-cell responses (225). The presence of MDSCs in tumors is associated with chronic inflammation and antigen-specific tolerance by T cells (226). MDSCs can also regulate the innate immune response by inhibiting NK cell functions (227–230) and/or by modulating macrophages' cytokine production (159, 225). The immunosuppressive activity of MDSCs plays an essential role in the regulation of the NK cell response to the tumor. In vitro co-culture of MDSCs and NK cells showed a reduction in NK cell-mediated cytotoxicity and higher tumor cell tolerance. MDSCs inhibit antitumor responses in part through immune checkpoint inhibition (ICI), including programmed death (PD)-1/PD-L1, galectin-9/T-cell immunoglobulin domain and mucin domain 3, and CTLA-4/B7 interactions (231).
STAT3 regulates NK cell biology at several levels, including activation, cytokine/cytolytic-mediated functions, and interactions with other immune system components (232). Many growth factors and cytokine receptors signal through STAT family transcription factors. STATs are impaired in several types of cancer and play a crucial role in innate and adaptive immunity (225, 233–235). In particular, STAT3 regulates several pathways involved in NK cell development, cytotoxic activities, and killing (232). Similarly, several cytokines, transcription factors/regulators, and signaling pathways are involved in the expansion and differentiation of MDSCs. These include interferon regulatory factor 8 (IRF8), CCAAT-enhancer-binding protein, and retinoblastoma protein; signaling pathways STAT3, STAT1, STAT6, Notch, NF-κB, and cyclooxygenase 2 (COX-2); and endoplasmic reticulum. Stress pathways are involved in MDSCs' expansion (170) (Figure 2) and include STAT3 activation in e-MDSC subtypes, which is critical for NF-κB activation and increasing indoleamine 2,3-dioxygenase (IDO), the release of which inhibits NK cell activation, proliferation, and effector functions (236). The molecular mechanisms regulating PMN-MDSC and M-MDSC populations differ from the afore-described pathways and are currently under investigation. Danvatirsen, a STAT3 antisense oligonucleotide, reversed the immunosuppressive TME and enhanced immune activity, as well as checkpoint blockades, in patient tumor samples from two phase I clinical trials and murine models (237).
Tumor-derived IL-1β release into the TME has been shown to enhance the recruitment of specific MDSCs during chronic inflammation (173). The augmented suppressive potential of IL-1β-induced MDSCs in mice was due to the activity of a novel subset of MDSCs lacking Ly6C expression (173). When these cells are prevalent under inflammatory conditions, they can impair NK cell development and function in vitro and in vivo by reducing the expression of the NK cell activating receptor NKG2D (173). Ly6C− MDSCs may be a valuable therapeutic target.
Tumor-derived prostaglandin-E2 (PGE2) may also play a crucial pro-tumor role in inducing MDSCs, mainly via COX-2 (238) (Figure 2). Monocytes exposed to PGE2 acquire MDSC-like functions, gaining the ability to inhibit NK cells via TGF-β (239, 240). High levels of TGF-β in the plasma were observed in patients with advanced tumors and correlated with worse outcomes (241, 242). TGF-β is released from cancer cells and can increase the expansion of M-MDSCs (243), recruit suppressive cells to the TME (MDSCs, Tregs, DCs, and stromal cells), and compromise the function of NK cells (230, 244–246), cytotoxic CD8+ cells, DCs, Tregs, and macrophages (247). NK cells exposed to MDSCs secrete less IFN-γ and downregulate NKG2D and CD247 in vitro and in vivo (229, 230, 248, 249). TGF-β not only impairs NK cell functions by downregulating the expression of activating receptors (NKG2D and NKp30) and inhibiting their transcription but also downregulates tumor cell-expressed NKG2D ligands (250). Thereby, NK cells lose their capacity to recognize and kill tumor cells via NKG2D (250). Furthermore, TGF-β inhibits CD16-mediated IFN-γ production and ADCC in human NK cells through mothers against decapentaplegic homolog 3 (SMAD3) activation (251) and affects CD34+ hematopoietic progenitors by inhibiting the maturation of CD56brightCD16+ NK cells fraction in the PB (246). The incubation of PB-NK cells with stromal cells, isolated from decidual tissue conditioned media, mimicked the suppressive effects of TGF-β1. NK cell interactions with CXC chemokine ligands and progesterone at the maternal–fetal interface after TGF-β1 exposure resulted in the reduction of CD56brightCD16+ NK cells and induced decidual-like NK cells that showed an exhausted phenotype (246). As such, STAT3 blockade and TGF-β inhibition improve tumor immune surveillance by NK cells (252). Specifically, tumor-infiltrating and tumor-associated NK cells from STAT3-deficient tumor-bearing mice express enhanced levels of NKG2D, CD69, FASL, granzyme B, perforin, and IFN-γ, reducing tumor growth and improving survival (252, 253). TGF-β signaling is deregulated in many diseases, including cancer. In early-stage tumor cells, this pathway has tumor-suppressor functions, including cell-cycle arrest and apoptosis (254). TGF-β signaling in late-stage cancer can promote tumorigenesis, angiogenesis, metastasis, and immunosuppression (254–256). MDSCs release TGF-β in the TME (144, 163, 175), and NK cell anergy (Figure 2) correlates with the marked increase of MDSCs in the liver and spleen in orthotropic liver cancer-bearing mice (230). Also, MDSCs prevent cytotoxicity, NKG2D expression, and IFN-γ production by NK cells (Figure 2) in vitro and in vivo through membrane-bound TGF-β (230).
IDO is an intracellular enzyme, and it regulates tryptophan catabolism into kynurenine (257, 258), which inhibits the proliferation and function of NK and T cells (259–261). IDO synthesized by MDSCs impairs NK cell activation, development, and expansion, resulting in dramatically decreased expression of NKG2D and DNAM-1 and limiting IFN-γ secretion (262, 263) (Figure 2).
ARG1 and reactive oxygen species (ROS), soluble factors secreted by MDSCs, also impair NK cell functions (Figure 2) in cancer models in vivo (248, 264). Moreover, several pro-inflammatory cytokines have been identified to mediate MDSC/NK crosstalk in the TME (264). These phenotypic and functional TME alterations due to MDSC/NK cell and/or T cell interactions contribute to pro-tumor, pro-angiogenic, and pre-metastatic activities in the tumor. Further studies are necessary to elucidate the mechanisms involved in MDSC/NK cell interactions to identify potential therapeutic candidates or pathways to limit NK cell MDSC-mediated suppression in the TME. MDSCs secrete high levels of soluble factors, such as ROS, inducible nitrogen-oxygen synthase (iNOS), nitrogen oxide (NO), peroxynitrate, and ARG1 (144), and show an elevated endoplasmic reticulum stress response (132, 265) (Figure 2).
NO is a ubiquitous, water-soluble, gaseous transmitter, which plays an essential role in various physiological conditions, inflammation, and cancer (266, 267). NO can play different roles in regulating immunity depending on the exact circumstances of its secretion. The autocrine production of NO by NK cells can improve NK cell function, but when MDSCs produce NO, it plays a vital role in mediating immunosuppression (266, 267). Co-cultures of autologous NK cells and MDSCs from patients with cancer showed that MDSCs suppress FcR-mediated function and signal transduction, leading to reduced responses to monoclonal antibody (mAb) therapies, and inhibit the secretion of IFN-γ and TNF-α by NK cells. Elimination of MDSCs or abrogation of NO production can improve responses to mAb immunotherapy (228).
MDSCs also support tumor progression by inducing tumor angiogenesis through the release of VEGF, basic FGF (bFGF), prokineticin 2 (Bv8), and some MMPs (169, 172, 268–270) (Figure 2). Some studies showed that MDSCs in the TME produce high levels of MMPs, including MMP2, MMP8, MMP9, MMP13, and MMP14 (167, 268, 271, 272). MDSCs in the presence of high levels of MMP9 can promote VEGF function by raising its bioavailability (273). VEGF stimulates MDSCs via STAT3 in the TME and potentiates their immunosuppressive activity by expanding other immune cell populations (213, 274, 275) and stimulating the secretion of numerous additional angiogenic factors and chemokines, which further enhance MDSCs accumulation within tumors. IL-1β, C-C motif chemokine ligand 2 (CCL2), CXCL8, CXCL2, angiopoietin 1 and 2 (AGPTs), and GM-CSF (Figure 2) have been shown to contribute to MDSC-mediated angiogenesis and involve STAT3 for their expression (80, 276–278).
The major components of the TME are the endothelial cells of the blood and lymphatic vessels, fibroblasts, immune cells, and the ECM (2). During tumor development and progression, cancer and stromal cells often have restricted access to nutrients and oxygen. Most solid tumors have hypoxic regions due to abnormal vascularization and inadequate blood supply (279). The changes to cancer and stromal cells that are necessary for tumor progression in a hypoxic environment are attributed to HIF-dependent signaling. The HIF family of transcription factors includes HIF-1, HIF-2, and HIF-3. Signaling by HIF-1 and HIF-2 induces the expression of multiple pro-angiogenic factors (VEGF, angiopoietin-2 [ANG-2], phosphatidylinositol-glycan biosynthesis class F protein [PlGF], bFGF, and semaphorin 4D), and angiogenesis was promoted in MDSCs by HIF-1 through VEGF and S100 calcium-binding protein A8 (S100A8) (280).
The TME has high levels of adenosine during hypoxia and inflammation. Adenosine/adenosine receptor interactions increase immunosuppression and angiogenesis through immune cells (281, 282). MDSCs express increased levels of CD39 and CD73 under hypoxic conditions or TGF-β stimulation (283, 284). These enzymes can convert adenosine triphosphate (ATP) and adenosine monophosphate (AMP) to adenosine, resulting in increased adenosine levels in tumor lesions (Figure 2). Adenosine accumulation impairs IL-2 and Ly49D activation, NKp46-receptor crosslinking, and maturation in NK cells (285). It has also been shown that adenosine signaling is involved in reducing the engagement of A2A adenosine receptor (A2AR) as a checkpoint in NK cell maturation (286).
Tregs interact with different components of the TME (287) and exert their suppressive function via contact-dependent and -independent mechanisms, which have been previously reviewed (288, 289). NK cell–Treg crosstalk in the human tumor has not been studied extensively (290). However, it has been shown that peripheral Tregs isolated from healthy donors and patients with gastrointestinal stromal tumors impaired NK cells by downregulating the expression of NKG2D activated receptors and also inhibited NK cell functions via membrane-bound TGF-β (291) (Figure 2). A similar effect was observed in vitro with cervical carcinomas (292).
Through these mechanisms, MDSCs and Tregs inhibit CTL and NK cell activity, promote tumor progression, and hinder antitumor immunity (169, 172, 287, 293–296). Therefore, it is not surprising that high MDSC infiltration of tumors correlates with poor patient prognosis and resistance to immunotherapy and chemotherapy (150, 175, 297–300). MDSC and Treg numbers also positively correlate with disease stage and tumor burden (114, 132, 144, 163–166, 201–203) and are predictors of poor outcome in patients with solid tumors (301–304). Both MDSCs and Tregs increase suppressive activity via signaling pathways, and their interactions in tumors have recently been reviewed (287, 305). Consistent with these findings, pharmacological targeting of MDSCs and Tregs in animal models and cancer patients significantly improves antitumor immunity, enabling tumor control (306, 307).
Targeting MDSCs and Their Ligands—Crosstalk With NK Cells
The complexity of the TME impairs immune cell functions and affects their phenotype. Several strategies have been developed, investigated, and applied in clinical trials to target MDSC immunosuppression and enhance NK cells' cytotoxic activity in the TME.
Studies have indicated that the types of mediators responsible for the differentiation, inhibition, and recruitment of MDSCs into the tumor were dependent on the different MDSCs subsets and tumor models (175). Targeting MDSCs is an approach designed to limit their immunosuppression within the TME and reduce neo-angiogenesis. Many therapies are focused on blocking intratumoral recruitment and expansion of MDSCs (Figure 3). Other approaches are dedicated to increasing MDSC differentiation or inhibiting their immunosuppression activity (264) (Figure 3). MDSCs are robust immunosuppressors in the TME and are an impediment for many cancer immune therapies (161).
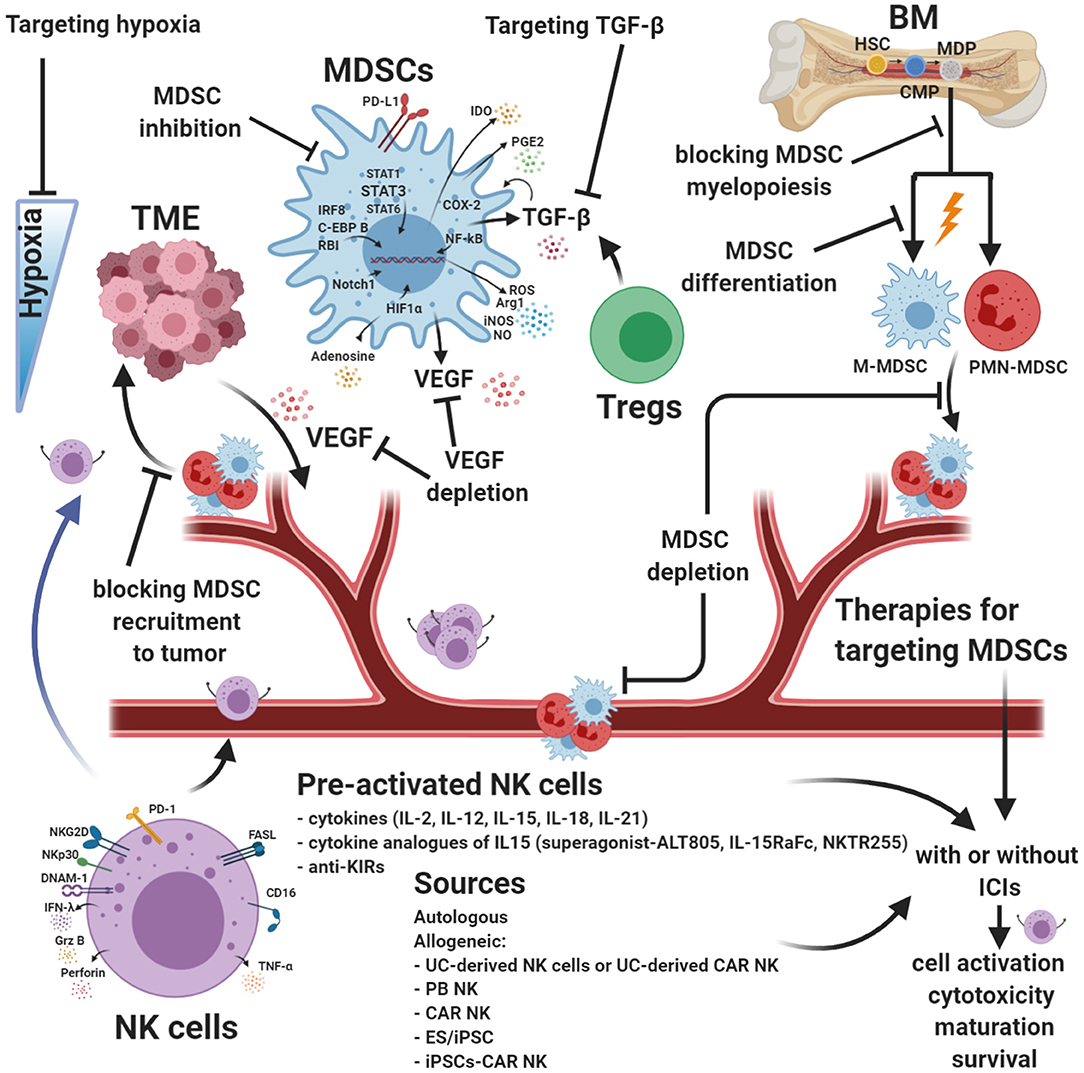
Figure 3. Strategies for targeting MDSCs and augmenting NK cell functions. Myeloid-derived suppressor cells (MDSCs) are robust immunosuppressors in the tumor microenvironment (TME) and represent an obstacle for many cancer immune therapies. Targeting MDSCs is a different approach designed to limit their immunosuppression within the TME and reduce neo-angiogenesis. Many combinatorial therapies are focused on blocking intratumoral recruitment and expansion of MDSCs. Other approaches are dedicated to increasing MDSC differentiation or inhibiting their immunosuppression activity. Natural killer (NK) cell-based immunotherapy strategies have been studied in an increasing number of clinical trials. Although NK cells can be useful in some types of cancer, it is necessary to improve the efficacy of currently available NK cell products to avoid cell exhaustion. Innovative combinatorial approaches intended to improve NK cell function and block MDSCs are in development. MDSC targeting, enhanced NK cell cytotoxic activity, and drugs targeting hypoxia can be used with current cancer therapies, including immunotherapy, and may improve antitumor response efficacy. ICIs, immune checkpoint molecules. Created with BioRender.com.
Low doses of chemotherapy induce MDSC exhaustion (308, 309). Chemotherapy drugs such as gemcitabine (310), 5-Fluorouracil (311), and doxorubicin (312) reduce MDSC frequency, which enhances NK-mediated antitumor cytotoxicity (313–315). Phenformin and metformin, two antidiabetic drugs, impair MDSC functions mainly by blocking 5' AMP-activated protein kinase (AMPK). These drugs also upregulate the expression of MHC class I polypeptide-related sequence A (MICA) and heat shock protein 70 (HSP70) on cancer cells through the phosphatidylinositide 3-kinase/protein kinase B pathway leading to NK cell activation (316). In addition to chemotherapy and radiotherapy, many studies have combined MDSC targeting methods with immune-based therapies to increase the antitumor effects (317). Encouraging progress has been made combining MDSC targeting with immunotherapy strategies.
Blocking TGF-β pathways is a promising strategy in some preclinical and clinical trials. Some of the approaches currently under investigation in preclinical models and clinical trials consist of decreasing circulating TGF-β, blocking ligand–receptor interactions using neutralizing antibodies, and inhibiting TGF-β signaling pathways (NCT00356460 and NCT01722825) (318–323). Blocking TGF-β is a viable strategy to prevent myeloid precursors from differentiating into M-MDSCs and impair the migration and expansion of MDSCs and Tregs in the TME (Figure 3). Alternatively, manipulating NK cells to block TGF-β signaling pathways is an attractive and promising strategy for solid tumors. In SMAD3-silenced NK cells (NK-92-S3KD), TGFβ1-mediated immunosuppression was blocked, inhibiting cancer progression in mouse models with human hepatoma and melanoma (324). Similarly, NK-92 cells genetically modified to express a chimeric TGF-β type II receptor (extracellular and transmembrane domains) and the intracellular domain of NK cell-activating receptor NKG2D were resistant to TGF-β-induced suppressive signaling and did not downmodulate expression of NKG2D (325). The same authors demonstrated that NK-92-TN cells inhibited the differentiation of human naïve CD4+ T cells to Tregs and decreased tumor volumes in vivo in a hepatocellular carcinoma xenograft cancer model (325).
A recent study demonstrated that NK cell sensitivity to TGF-β can be reduced by stimulation of NK cells with IL-2 (Figure 3), which increases IFN-γ and TNF-α production by NK cells in the tumor, compared to NK cells that encountered acute TGF-β exposure or were not TGF-β imprinted (326). A new approach to overcoming tumor resistance mechanisms to ICIs has been tested by combining TGF-β, CXCR1/2 signaling, and PD-L1 (327). This simultaneous inhibition reduced mesenchymal tumor features and infiltration of suppressive PMN-MDSCs into the TME, improving antitumor activity by promoting immune cell infiltration and activation in tumors (327).
Cytotoxic agents and tyrosine kinase inhibitors (TKIs) deplete MDSCs and regulate myelopoiesis (264). TKIs, such as axitinib, sorafenib, and sunitinib, induce DNA damage by histone γ-H2AX phosphorylation and checkpoint kinase 1 activation, leading to senescence of human renal carcinoma cells (328). The presence of DNA damage in cancer cells also improved the identification of tumor cells by NK cells (328). TKIs can directly target VEGF and/or involve c-KIT signaling and interact with other factors, such as CSF or STAT3. These mechanisms impaired MDSC function and inhibited tumor angiogenesis (329, 330). These processes upregulate NKG2DLs and, consequently, stimulate NK cell antitumor cytotoxicity (329, 330). Several in vivo studies combined TKI and ICI therapies; the combination therapy improved antitumor mechanisms and simultaneously reduced immune cell exhaustion (330–333).
STATs are activated in tumor cells by multiple soluble factors. This activation causes impaired cytolytic functions mediated by perforin and granzyme B in NK cells and alters the expression of NK cell receptors NKG2D and DNAX accessory molecule-1 (DNAM-1) (232). STAT3 activation in tumor cells also represses the expression of NK cell-chemotactic factors, which reduce the migration of NK cells in the TME. TGF-β and IDO produced by tumor cells and MDSCs impair NK cell development, proliferation, and activation (232), leading to reduced NK-mediated cytotoxicity. Exhausted NK cells and T cells have a diminished secretion of IFN-γ (334). MDSCs secrete type I interferons to maintain a high level of PD-L1 expression and preserve their immunosuppressive activity in the TME as a compensatory mechanism (335); therefore, the autocrine IFNα/IFNβ-pSTAT1-PD-L1 circuit represents a pivotal pathway to targeted MDSCs (335).
STAT3 also increases the expression of PD-L1, which engages PD-1 expressed on NK cells, reducing their antitumor response (232). Janus kinase (JAK)/STAT3 inhibitors decrease MDSC trafficking in the TME, diminishing angiogenesis by inhibiting VEGFA and casein kinase 2 (336) and driving NK cell activation (336, 337). Targeting STAT3 in tumor-bearing mice leads to tumor reduction, better survival, and a significantly higher number of activated NK cells following treatment, as compared to control mice (252, 253).
Other approaches using STAT3 inhibitors combined with ICIs, such as nivolumab (NCT03647839), STAT3 small interfering RNA (siRNA), or decoy STAT3 oligonucleotide inhibitors alone or combined with ICIs (AZD9190), are in phase I/II clinical trials (NCT03421353). STAT3 siRNAs coupled to CpG oligonucleotides reduced the immunosuppression of toll-like receptor 9 (TLR9)-expressing PMN-MDSCs in preclinical data (338). Mouse and human PMN-MDSCs overexpress fatty acid transporter protein 2 (FATP2) due to stimulation by GM-CSF through activation of the STAT5 transcription factor (339). FATP2 inhibitors alone, or in combination with ICIs, delayed tumor progression in tumor-bearing mice (339). CSF-1R inhibitors, combined with anti-PD-1, improved the immune response in a mouse model of neuroblastoma (340).
MDSC immune functions are also impaired using class I deacetylase, entinostat, or ARG1 small-molecule inhibitors by decreasing iNOS and COX-2 levels (341). Some of these treatments enhanced NK killing and blocked MDSC-mediated suppression of T cells in vitro and in vivo in tumor models (342–344). One study showed that an ARG1 small peptide inhibitor combined with anti-PD-L1 slowed tumor growth (345). ARG1 inhibitors (such as CB-1158) showed encouraging preclinical results (346), increasing tumor-infiltrating NK cells and CD8+ T cells (345, 347), reducing tumor burden (348, 349), and decreasing MDSCs recruitment into the TME (345).
Conventionally, phosphodiesterase-5 (PDE5) inhibitors (sildenafil and tadalafil) are used as therapies for non-malignant conditions (350). Recent evidence suggests that PDE5 inhibitors could improve antitumor cell responses by inhibiting the suppressive functions of MDSCs in the TME (306, 351, 352). Preclinical and clinical data show that the PDE5 inhibitor tadalafil enhanced the immune response in head and neck squamous cell carcinoma (HNSCC) patients through inhibition of MDSCs (NCT01697800) (352). However, another PDE5 inhibitor, Sildenafil, reduced PMN-MDSC function through downregulation of ARG1, IL4Ra, and ROS expression (248). PDE5 inhibitors also enabled NK cell antitumor cytotoxicity and reduced postoperative disease recurrence. These studies have also shown Treg reduction and enhanced CD4/CD8 T cell function in the TME (248, 306, 352). Phase I/II Clinical Trial NCT02544880 used tadalafil as a therapy for decreasing MDSCs and Tregs, improving the antitumor response. The treatment might also enhance antitumor mucin 1 (MUC1) vaccine efficacy in patients with resectable and recurrent HNSCC by promoting a permissive environment (NCT02544880).
All-trans retinoic acid (ATRA) and vitamin A metabolites (retinol) have been used to treat acute myelogenous leukemia (353–355). Several studies reported that ATRA inhibited cell migration, metastasis, and proliferation, and promoted apoptosis of tumor cells (356–358). ATRA alone (359–361) or in combination with a DC vaccine against p53 (360) or IL-2 administration (361) showed lower MDSC frequencies and enhanced differentiation of MDSCs (359–361) into mature DCs, macrophages, and neutrophils (362). ATRA decreased the expression of immunosuppressive genes through the downregulation of TGF-β, PD-L1, IL-10, and IDO in MDSCs and upregulated MHC class I homologs MICA and MICB on tumor cells, enhancing NK cell activity (363, 364) and cytotoxicity of T cells (365) in the CD8− and CD4− immune response (362). In clinical studies, ATRA alone or combined with ipilimumab significantly reduced the level of circulating MDSCs in advanced-stage melanoma patients (366). In a preclinical breast cancer model, ATRA therapy improved the efficacy of anti-angiogenic treatments (367).
VEGF is produced by tumor cells in the TME and supports neo-angiogenesis, metastasis dissemination, and also acts as a chemoattractant for MDSCs in the tumor site (302, 368, 369). This has been shown in both NSCLC and renal cell carcinoma patients, especially under hypoxic conditions (370, 371). In a NSCLC model, VEGF attracts MDSCs from the BM to the periphery, expanding their presence throughout the individual (372). The binding of VEGF to its receptor is correlated with increased production ROS via the JAK2/STAT3 activation pathway. MDSCs can also secrete VEGF (Figure 2), creating, together with the tumor cells, a positive autocrine feedback loop in the TME (373, 374). Therefore, anti-VEGFR2 reduced the accumulation of intratumoral MDSCs, decreased hypoxia, and interfered with the formation of tumor microvessels through S100A8 (367). Genetic inactivation of VEGFA in MDSCs improved clearance of senescent tumor cells by NK cells, inhibited tumor regrowth after chemotherapy and, prevented cachexia in tumor-bearing mice (375). A phase II clinical trial evaluated the efficacy and pharmacokinetics of bevacizumab, an anti-VEGF recombinant human mAb, combined with capecitabine and paclitaxel chemotherapy in subjects with triple-negative, metastatic, or locally advanced breast cancer. In 77% of patients, the therapy showed an objective response rate, with complete response in 19%, and the median progression-free survival was 7.6 months (376). Several studies also demonstrated that MDSCs possess secondary pro-angiogenic mechanisms involving MMPs, as discussed in Vetsika et al.' review (162).
Because MDSCs are recruited to the TME by tumor cells, blocking their migration using a CCR5 antagonist and CCL2 inhibitors seems to be a promising therapeutic approach. Accumulation of CCR5+ MDSCs with high suppressive activity, associated with increased concentrations of CCR5 ligands and tumor progression, has been shown in a tumor-bearing melanoma study model (377). The upregulation of CCR5+ on CD11b+Gr1+ myeloid cells was induced in vitro by CCR5 ligands and other inflammatory factors. Blocking the CCR5/CCR5 ligand interaction improved survival by reducing the migration and the immunosuppressive functions of MDSCs in melanoma lesions of tumor-bearing mice (377). This strategy can also enhance the suppression of MDSCs by NK cells, as shown in a premetastatic lung animal model (378). CCL2 expression in MDSCs has been elucidated in a lung tumor model, and anti-CCL2 treatment can decrease peripheral and intratumoral PMN-MDSCs and M-MDSCs by inhibiting the ARG1 expression and iNOS (379). The same study showed enhancement in CD4+ and CD8+ T cell infiltration and production of IFN-γ in the TME. As a result, anti-CCL2 therapy increased the survival time of tumor-bearing mice. Anti-CCL2 therapy could be a potential approach to improve NK cell activity and the efficacy of ICI immunotherapy.
Some studies have shown that COX-2 inhibitors (celecoxib or nimesulide) reduced the expansion of MDSC subtypes and decreased cancer progression (380–382). COX-2 inhibitors also stimulate NKG2D ligand expression on tumor cells, enhancing NK cell-mediated cytotoxicity (383) and reducing angiogenic pathways via VEGF (384). A phase II clinical trial showed that perioperative treatment with a COX-2 inhibitor (etodolac) in combination with a β-adrenergic antagonist (propranolol) reduced circulating CD14+ monocytes and improved NK cell activation (385). Thus, this study supports the rationale for targeting MDSCs in the perioperative period to enhance clinical outcomes. A decrease in MDSC COX-2 and PEG2 activity and NO and ROS production has been shown in animals and humans after Vitamin D3 or E treatments (386). MDSCs impair NK cell function through NO production, and clinical trials are evaluating novel therapies to block this mechanism (228). A clinical trial using celecoxib, in combination with nivolumab and ipilimumab, is currently recruiting for the treatment of colon carcinoma (NCT03026140). A colorectal cancer phase III trial combining vitamin D3 with standard chemotherapy and bevacizumab is also ongoing (NCT04094688). Vitamin E supplementation decreased PGE2 production by inhibiting COX-2 activity, resulting from decreased NO production, in mice and humans (386). It also improved the activation of T and NK cells, increased lymphocyte proliferation, and modulated DC function (386).
ROS molecules are involved in many pathways, thereby controlling a wide range of biological events, such as immunosuppression in the TME (387). Following pathogenic and inflammatory immune responses, MDSCs release ROS (387). Although ROS has toxic effects on most cells, MDSCs survive despite the elevated levels and constant production of ROS in the TME (388). The production of ROS by MDSCs is upregulated in many murine tumor models and human cancers (389, 390) and plays a crucial role in maintaining MDSCs in an undifferentiated state (387). ROS production is regulated by the nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Synthetic triterpenoid C-28 methyl ester of 2-cyano-3,12-dioxooleana-1,9,-dien-28-oic acid (CDDO-Me; bardoxolone methyl) in an MC38 colon carcinoma model has been shown to reduce MDSC ROS production via Nrf2 activation (307) and IL-17D production (391). A phase I clinical trial showed encouraging results using this therapy (392). Forcing IL-17D production using Nrf2 agonists can enhance NK cell activation and recruitment, leading to tumor regression (391, 393).
The pathological intratumoral accumulation of CXCR2+ PMN-MDSCs impairs the functions of NK cells in mice bearing oral carcinoma tumors by secreting TGF-β, NO, and ROS (394). CXCR1/2 inhibitors (SX-682) significantly abrogated MDSCs trafficking within a tumor and improved tumor infiltration, activation, and therapeutic efficacy of adoptively transferred murine NK cells in the HNSCC preclinical model (394), and in combination with anti-PD-1, they improved ICI therapy (395).
HIF-1α, released in the TME from tumor cells, is another protein that has a crucial role in MDSC differentiation toward TAMs (178, 396). Some progress has been achieved targeting hypoxia using hypoxia-activated prodrugs (TH-302) (Figure 3), hypoxia-modulator drugs (modulating expression, DNA-binding, transcriptional activity, and degradation of HIF proteins), drugs directly modulating HIF mRNA (antisense oligonucleotides), and inhibiting pathways involved in the control of HIF-1α mRNA (397). Combination therapy of TH-302 with gemcitabine in a phase II clinical trial for advanced or metastatic pancreatic cancer (NCT01144455) (398) or with doxorubicin in a phase III trial for advanced soft tissue sarcoma (NCT01440088) (399) showed encouraging results. The combination of TH-302 therapy with anti-CTLA-4 and PD-1 cured more than 80% of tumors in a mouse prostate cancer model by reducing MDSCs and granulocytic subsets and driving T cell migration into the hypoxic tumor sites (400). Innovative combinatorial approaches, such as drugs targeting hypoxia used with current cancer therapies, including immunotherapy and MDSCs targeting, may improve antitumor response efficacy.
IL-18 and IL-33 are involved in CD11b− BM progenitor cell differentiation to M-MDSCs (192) and PMN-MDSCs (401), respectively. Targeting of CD11b− BM treated with anti-IL-18 or IL-33 has been shown to enhance T cell proliferation and IFN-γ secretion (192, 401). Other MDSC molecules targeting aminoacyl tRNA synthetase complex interacting multifunctional protein 1, TLR agonists, tumor-derived exosome-associated HSP72, and inflammasome component NLR family pyrin domain containing 3 have also been shown to contribute to MDSC differentiation and have been reviewed previously (402). MDSCs can be blocked by combining ICIs (anti-PD-1, PD-L1, and CTLA-4) with anti-IL-18 (192), inhibitors of FATP2 (339), long non-coding RNA Pvt1 (403), downregulation of the pseudogene Olfr29-ps1 (404), or deletion of the nuclear factor 1 A gene (405).
Pre-Activated NK Cell Products for Cancer Immunotherapy
Cancer immunotherapy strategies have focused on T-cell-based immunotherapy using expanded tumor-specific CD8+ CTLs from tumor-infiltrating lymphocytes (TILs) (406). Adoptive transfer of TILs following lymph-depleting strategies have shown promising effectiveness in metastatic melanoma studies (407–409), but short-lived responses (410) and side effects, such as vitiligo, uveitis, and retinitis, have been reported (407, 411).
Pre-clinically and clinically, studies have identified several cytokines and other novel soluble factors that increase NK cell numbers, function, and persistence. The two most common strategies are pretreating NK cells with cytokines (before the adoptive transfer) or in vivo cytokine administration. IL-2 (412), IL-15 (413), IL-12 (414, 415), IL-18 (416, 417), IL-21, their combinations, and administrations with other immunotherapeutic agents have all been described in the context of regulating NK cell function, maturation, survival, and improving activation and cytotoxicity (Figure 3). Results are expected from clinical trials evaluating the safety and efficacy of combining IL-21 with other immunotherapeutic mediators: IL-21/anti-PD-1 against solid tumors (NCT01629758) and IL-21/ipilimumab against melanoma (NCT01489059). Cytokine analogs of IL-15, such as IL-15 superagonist ALT-803 (NCT03228667, NCT03127098, NCT03022825, NCT02384954, NCT02138734, NCT02890758, NCT02559674, and NCT03520686) and NKTR-255, are under investigation. Furthermore, multiple ongoing clinical trials are evaluating the safety and efficacy of several other immune cytokines alone or in combination with other therapeutic strategies, such as immune checkpoint inhibitors (NCT03209869, NCT03386721, NCT02627274, and NCT02350673) (Figure 3).
Combination therapies of PD-1 blockade and IL-15 stimulation and also IL-15 and IL-15RaFc have been reported as safe in mouse models (418) and patients with NSCLC (419). IL-15 stimulation increases the expression of the activating receptors CD16 and NKG2D on NK cells and increases the activation, proliferation, cytotoxic activity, and survival of NK cells and CTLs (420). One of our preclinical studies found that the combination of PD-1 blockade and IL-15 signaling resulted in eradication of transplanted lung adenocarcinoma (LUAD) cells in about one-half of treated LUAD-SIS-PDX mice, while the other half presented with a partial response. Notably, IL-15 alone, without PD-1 blockade, significantly reduced tumor burden in all treated LUAD-SIS-PDX animals (418). PD-1 blockade alone transiently prevented tumor growth, but tumors grew at a similar rate to untreated control tumors after 2 weeks. The addition of IL-15 to PD-1 blockade completely abrogated tumor escape from ICI, resulting in a powerful additive therapeutic effect capable of tumor eradication. These findings support a key role for adjuvant IL-15 treatment to induce an immune cell-mediated tumor attack, which can prevent tumor escape from checkpoint blockade therapy, as shown using our novel LUAD-SIS-PDX model (418).
In addition to cytokine stimulation, mAb can be used to block the activity of NK cell inhibitory receptors. The ligation of inhibitory KIRs by HLA molecules triggers NK cell inhibition (64), and anti-KIR antibodies are under investigation to improve NK cell cytotoxicity in cancer (421). Because the expression of NK cell-expressed inhibitory KIRs and PD-1 correlate in patients with solid tumors (e.g., NSCLC), combining anti-KIR antibodies with anti-PD-1 treatments to avoid immune escape of tumors in these patients may be an effective treatment strategy (422). Several clinical trials are currently evaluating anti-KIR antibodies against solid tumors combined with other immune treatments (NCT03341936, NCT03203876, and NCT03347123).
NK Cell Sources
NK cells do not need prior activation to target tumor cells. The fine-tuning of NK cell functions occurs during their maturation and instills a form of tolerance. NK cells are “educated” to recognize healthy MHC-I-expressing cells with KIR receptors, resulting in inhibitory signals and preventing NK cell activation. In addition to KIRs, NK cells also express two other types of inhibitory receptors (423): leukocyte immunoglobulin-like receptors (LILRs) (424) and C-type lectin receptors (NKG2A/CD94). The balance of inhibitory and activating signals expressed on target cells mediate NK cell activation and response (425–427).
NK cells can kill tumor target cells through a variety of mechanisms, as discussed previously in this review. Since NK cells represent an important defense against tumors, NK cell infusion products have been evaluated as a possible cancer immunotherapy. Different NK cell sources have been tested in patients with tumors combined with chemotherapy (428) and, allogeneic NK cells have been selected in many studies for their increased alloreactivity, achieved by mismatching of inhibitory KIRs and tumor HLA (429) (Figure 3). This is referred to as a haploidentical or half-matched setting. Haploidentical NK cells are more reactive against recipient tumor cells because of reduced KIR-mediated inhibition (429). NK cells for adoptive transfers can be obtained from five different sources: autologous NK cells, allogeneic NK cells (430), umbilical cord-derived NK cells, NK cell lines, and embryonic stem cell-derived/induced pluripotent stem cell (ES/iPSC)-derived NK cells (431) (Figure 3), allowing for a multitude of choices to match or mismatch KIR-HLA receptor pairings.
Chimeric Antigen Receptor–NK Cells for Cancer Immunotherapy
One currently promising strategy is redirecting NK cells with chimeric antigen receptors (CARs) (Figure 3). Autologous CAR-T cell successes in patients with leukemia and lymphoma (432) have raised considerable interest in using immune cells as a cancer treatment. The advantage of a CAR strategy is that one CAR can be applied for many tumor types expressing the matching ligands. Modification with CAR is also proposed for reprogramming NK cells to improve their cytotoxicity. CAR-NK cells represent an exciting approach for cancer immunotherapy. NK cells can be targeted with CARs against surface molecules expressed by tumor cells and might avoid some of limitations or side effects of CAR-T cells. While conventional T cells are HLA-restricted, CAR-T cells are designed to recognize their target antigens independent of HLA expression and deliver their costimulatory signal. However, CAR-T cells are expensive and labor-intensive to generate (433), and the application of CAR-T cell therapy is often limited by intrinsic risks, such as graft-vs.-host disease (GvHD) (434). Also, off-target effects, cytokine release syndrome, and other side effects restrict their clinical applications. CAR-T cell therapy has been successful in treating blood cancers. However, a significant obstacle for the treatment of solid tumors is an extremely immunosuppressive TME that decreases the ability of immune cells to infiltrate tumors (435). CAR-T cell functions are often suppressed in solid tumors due to T cell expression of PD-1 and PD-L1 expression in the TME. These data may explain why the use of CAR-T cells has not been as useful for treating solid tumors as it has been to treat hematological malignancies (436). However, PD-1 levels expressed by NK cells are substantially lower, making NK cells good candidates for eradicating solid tumors (437).
Allogeneic NK cell transplantation rarely induces GvHD (438) and has the potential to become “off-the-shelf” products, making CAR-NK cell therapies a possible widespread product (433). Large-scale culture and genetic modification of allogenic human NK cells are feasible and could readily be used to treat a broad range of cancer patients. NK cell-expressed CARs typically include a single-chain variable fragment from a mAb, a transmembrane hinge region, and a costimulatory signaling domain, such as CD28, CD3-zeta, 4-1BB (CD137), or 2B4 (CD244) heterodimers (439, 440). These main signaling domains frequently have been derived from TCR moieties (441). Four generations of CARs are currently under development (442). The first generation of CARs usually contains only the CD3ζ activation signaling domain (443). In the second and the third generations of CARs, costimulatory molecules like CD28, 4-1BB, and CD134 are also included to increase NK cell activation (444). The fourth generation of CARs is engineered to secrete transgenic cytokine-like IL-12, which should help to remodel the tumor environment to promote therapeutic success (442, 445). Once CAR-modified NK cells recognize their specific targets, such as CD19−, CD20−, or CD138− cells, the CAR receptors trigger an intracellular signaling cascade that activates CAR-NK cells to kill the antigen-expressing target cell.
Methods to generate CAR-NK cells that target solid tumors include lentiviral transduction or electroporation of the NK-92 NK-like cell line (438), primary NK cells, and the differentiation of NK cells from modified pluripotent stem cells (431). NK cells express NKG2D, an activating receptor triggered by MICA/B and UL binding protein (ULBPs) expressed on the surface of stressed cells upon DNA damage, hypoxia, or viral infection (446). NKG2D ligands are often overexpressed on solid tumors and tumor-infiltrating cells like MDSCs (447). However, the NKG2D cytotoxic adapter molecule, DNAX-activation protein 10 (DAP10), is downregulated by suppressive molecules like TGF-β, which is abundantly expressed in the TME (448). NK cells and CAR-NK cells expressing the native NKG2D receptors are thus downmodulated in the TME due to the reduction in DAP10 expression. To overcome the repressive effects of the solid TME on NKG2D functions, one group (449) established a gene-modified NK cell bearing a chimeric receptor in which the activating receptor NKG2D is fused to the cytotoxic ζ-chain of the T-cell receptor (NKG2D.ζ) (450). This specific CAR has been designed to target MDSCs in the TME of solid tumors, which are refractory to other types of immunotherapy. The NKG2D.ζ-NK cells are cytotoxic against MDSCs, but unmodified NK cells are not. They also showed that NKG2D.ζ-NK cells generated from patients with neuroblastoma successfully killed autologous MDSCs in the TME, which were capable of suppressing CAR-T functions (449). CAR-NK cells have been established and engineered against several antigens for solid tumors, including epidermal growth factor receptor, human epidermal growth factor receptor-2 (HER2), egeria, disialoganglioside, epithelial cell adhesion molecule, mesothelin, and tyrosine-protein kinase transmembrane receptor ROR1, with promising results in preclinical or clinical studies (39). In a recently published phase I/II clinical trial, HLA-mismatched anti-CD19 CAR-NK cells were administered to 11 patients with relapsed or refractory CD19-positive cancers. The majority of treated patients showed a response to treatment with anti-CD19 CAR-NK cells without developing significant toxic side effects (451). The phase I/II trial was approved for B-cell lymphoma in 2017 (NCT03056339). In another study, NK cells derived from umbilical cord blood were transfected with a CAR containing inducible caspase 9/IL-15 (iC9/CAR.19/IL-15) (452). They produce IL-15, which supports CAR-NK cell survival, and are engineered to express the inducible suicide gene caspase 9 for their pharmacologically induced elimination. Liu and coworkers showed efficient iC9/CAR.19/IL-15 cell killing of CD19-expressing tumor cell lines in vitro and improved clinical outcome in a xenograft Raji lymphoma murine model (452). In the clinical trial, iC9/CAR.19/IL-15 cells were used to treat patients with relapsed/refractory CD19+ B lymphoid malignancies. This cell therapy was applied with high-dose chemotherapy (NCT03579927).
An alternative approach for the generation of CAR-NK cells is the use of iPSC as a platform to generate CAR-NK cells (453) (Figure 3). In 2018, a NK cell line was established from human iPSCs that expresses a CAR containing the transmembrane domain of NKG2D, the 2B4 costimulatory domain, and the CD3ζ signaling domain able to mediate strong antigen-specific NK cell signaling (iPSC-CAR-NK cells) (453). The combination of NKG2D-2B4ζ in this CAR construct conferred strong upregulation and activation of phospholipase C gamma, Syk-vav1-Erk, and NF-κB pathways, improving iPSC-CAR-NK cell activation, proliferation, and antitumor activity. This cell line maintains the same NK cell phenotype as wild-type cells but enhanced antitumor activity compared to CAR-T cells, iPSC-NK cells, or non-CAR PB-NK cells. In an ovarian cancer xenograft model, iPSC-CAR-NK cells significantly inhibited tumor growth and improved survival in mice and showed less toxicity than CAR-T cells (453).
A pilot study investigated the use of NKG2D ligand targeted CAR-NK cells in patients with metastatic colorectal cancer and evaluated the safety and feasibility of CAR-NK cell treatment against solid metastatic tumors (454). For these studies, autologous or allogeneic NK cells were transfected by mRNA electroporation to generate CAR-NK cells with transiently enhanced specificity and activity against NKG2D ligand-expressing cancer cells. This approach has also been used in a clinical trial involving solid metastatic tumors (NCT03415100).
About 200 clinical trials are currently using CAR-T cells, and only 20 clinical trials so far are utilizing CAR-NK cells (http://www.clinicaltrials.gov). For these trials, CAR-NK cell products are derived from either primary NK cells or NK cell lines. Only five clinical trials were conducted to evaluate the safety of CAR-NK-92 infusion products, while 15 trials evaluated CAR-NK cells from other sources. CD7 (NCT02742727), CD19 (NCT02892695), and CD33 (NCT02944162) targeted blood cancer (455); HER2 targeted glioblastoma (NCT03383978); co-stimulating conversion receptors targeted NSCLC (NCT03656705); and MUC1 targeted multiple refractory solid tumors, including hepatocellular carcinoma, NSCLC, pancreatic tumors, and triple-negative metastatic breast tumors (NCT02839954) (455).
Additional preclinical or clinical trials targeting CD19− (NCT03690310) or CD22− expressing (NCT03692767) cells are ongoing, and bivalent CD19/22 (NCT03824964) CAR-NK cell products are also being examined in patients with relapsed and refractory B cell lymphoma (456). One clinical trial targeting CD19-positive cells is testing CD19-CAR-NK cell infusions in a pediatric setting (NCT00995137), using irradiated K562-mb15-41BBL expanded PB-derived NK cells transfected with CD19-41BBz-CAR (455).
Roundabout homolog 1 (ROBO1), a member of the axon guidance receptor family (Robo1–4), is a potential target for immunotherapy (457). ROBO1 modulates the chemotaxis of T cells and tumor angiogenesis to counteract the tumor growth (458–460). Some solid tumors, such as pancreatic cancer, have increased expression of ROBO1 (461). Three clinical trials are currently studying CAR-NK cells directed against ROBO1, using CAR-NK (NCT03940820), bi-chimeric antigen receptor-NK cell BiCAR-NK (NCT03941457), and bi-chimeric antigen receptor-NK cell or T cell BiCAR-NK/T (NCT03931720) cells on patients with solid tumors (456).
Additional strategies for optimizing NK cell-mediated cytotoxicity have been employed. ADCC is a potent mechanism of cytotoxicity used by NK cells. An increasing number of mAbs are currently being examined in preclinical and clinical studies for their ability to improve antitumor ADCC (462). Limiting shedding of CD16 from NK cell membranes also enhances ADCC, and iPSC-NK cells modified to overexpress a non-cleavable version of CD16 showed improved ADCC in vitro and in vivo in a human B-cell lymphoma model (463). iPSC can also be modified to improve in vivo persistence of iPSC-NK cells. Furthermore, the deletion of cytokine-inducible SH2-containing protein (CISH) in iPSC differentiated NK cells improved persistence and enhanced antitumor activity in a leukemia xenograft model (464). Other relevant strategies to strengthen NK killing potential use antibody therapy to target NK cell checkpoints that inhibit NK cell activity in the TME, such as PD-1 (465), T cell immunoreceptor with Ig and ITIM domains (466), and single Ig IL-1-related receptor (467). These findings provide some flexibility in the combining of therapeutic approaches. NK cells can be modified to express CARs and/or be used in combination with ICI or additional antibody immunotherapy. While significantly more work is needed to optimize these approaches, these data nevertheless demonstrate the potential for “off-the-shelf” NK cell platforms for treating solid tumors and hematological malignancies. Further research needs to be done to identify and understand possible CAR-NK exhaustion mechanisms after transplantation in preclinical and clinical studies.
Conclusion
The TME plays a critical role in regulating NK cell antitumor functions and regulating NK cell trafficking, persistence, proliferation, and activation. MDSCs are one of the critical players inducing and regulating the immunosuppressive environment of the TME. MDSCs interact with numerous innate immune cells, modulating their functions and suppressing strong tumor-specific immunity. As such, MDSCs represent an essential target in oncology. While NK cell-MDSC interactions have been investigated, few studies have evaluated how NK cell cytotoxicity can be exploited to attack both the immunosuppressive TME as well as the tumor. We have provided an updated review of several current prospective therapies for targeting the MDSC–Treg axis and improving antitumor NK cell functions (several mechanisms are summarized in Figure 3). These pathways have substantial implications for the tumor and are currently under investigation. The combinatorial treatments that target these pathways have been showing promising results. More work is needed to fully exploit NK cell functions to eradicate hematological malignancies and solid tumors.
Author Contributions
CZ and SP conceptualized the content and wrote the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by unrestricted funds from The Scripps Research Institute, La Jolla, CA (SP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. (2017) 8:761–73. doi: 10.7150/jca.17648
2. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. (2012) 125:5591–6. doi: 10.1242/jcs.116392
3. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. (2001) 357:539–45. doi: 10.1016/S0140-6736(00)04046-0
5. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
6. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
7. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
8. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. (2015) 125:3338–46. doi: 10.1172/JCI80004
9. Palucka AK, Coussens LM. The basis of oncoimmunology. Cell. (2016) 164:1233–47. doi: 10.1016/j.cell.2016.01.049
10. Wang D, DuBois RN. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis. (2015) 36:1085–93. doi: 10.1093/carcin/bgv123
11. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. (2018) 9:115. doi: 10.1038/s41419-017-0061-0
12. Shanker A, Verdeil G, Buferne M, Inderberg-Suso EM, Puthier D, Joly F, et al. CD8 T cell help for innate antitumor immunity. J Immunol. (2007) 179:6651–62. doi: 10.4049/jimmunol.179.10.6651
13. Uzhachenko RV, Shanker A. CD8(+) T lymphocyte and NK cell network: circuitry in the cytotoxic domain of immunity. Front Immunol. (2019) 10:1906. doi: 10.3389/fimmu.2019.01906
14. Dustin ML, Colman DR. Neural and immunological synaptic relations. Science. (2002) 298:785–9. doi: 10.1126/science.1076386
15. Dustin ML, Long EO. Cytotoxic immunological synapses. Immunol Rev. (2010) 235:24–34. doi: 10.1111/j.0105-2896.2010.00904.x
16. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. (2011) 105:93–103. doi: 10.1038/bjc.2011.189
17. Lee WS, Kang M, Baek JH, Lee JI, Ha SY. Clinical impact of tumor-infiltrating lymphocytes for survival in curatively resected stage IV colon cancer with isolated liver or lung metastasis. Ann Surg Oncol. (2013) 20:697–702. doi: 10.1245/s10434-012-2752-1
18. Halle S, Halle O, Forster R. Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol. (2017) 38:432–43. doi: 10.1016/j.it.2017.04.002
19. Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol. (2008) 8:259–68. doi: 10.1038/nri2276
20. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
21. El-Gazzar A, Cai X, Reeves RS, Dai Z, Caballero-Benitez A, McDonald DL, et al. Effects on tumor development and metastatic dissemination by the NKG2D lymphocyte receptor expressed on cancer cells. Oncogene. (2014) 33:4932–40. doi: 10.1038/onc.2013.435
22. Spear P, Wu MR, Sentman ML, Sentman CL. NKG2D ligands as therapeutic targets. Cancer Immunity. (2013) 13:8.
23. Trembath AP, Sharma N, Raju S, Polic B, Markiewicz MA. A protective role for NKG2D-H60a interaction via homotypic T cell contact in nonobese diabetic autoimmune diabetes pathogenesis. Immunohorizons. (2017) 1:198–212. doi: 10.4049/immunohorizons.1700011
24. Tanaka J, Miller JS. Recent progress in and challenges in cellular therapy using NK cells for hematological malignancies. Blood Rev. (2020) 44:100678. doi: 10.1016/j.blre.2020.100678
25. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. (2012) 12:239–52. doi: 10.1038/nri3174
26. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. (2016) 17:1025–36. doi: 10.1038/ni.3518
27. Baginska J, Viry E, Paggetti J, Medves S, Berchem G, Moussay E, et al. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front Immunol. (2013) 4:490. doi: 10.3389/fimmu.2013.00490
28. Lo HC, Xu Z, Kim IS, Pingel B, Aguirre S, Kodali S, et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nature Cancer. (2020) 1:709–22. doi: 10.1038/s43018-020-0068-9
30. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer. (1975) 16:216–29. doi: 10.1002/ijc.2910160204
31. Cichocki F, Grzywacz B, Miller JS. Human NK cell development: one road or many? Front Immunol. (2019) 10:2078. doi: 10.3389/fimmu.2019.02078
32. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, clinical utilization. Front Immunol. (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
33. Crinier A, Narni-Mancinelli E, Ugolini S, Vivier E. SnapShot: natural killer cells. Cell. (2020) 180:1280.e1. doi: 10.1016/j.cell.2020.02.029
34. Bjorkstrom NK, Ljunggren HG, Michaelsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol. (2016) 16:310–20. doi: 10.1038/nri.2016.34
35. Shi FD, Ljunggren HG, La Cava AL. Van Kaer, Organ-specific features of natural killer cells. Nat Rev Immunol. (2011) 11:658–71. doi: 10.1038/nri3065
36. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
37. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. (2001) 22:633–40. doi: 10.1016/S1471-4906(01)02060-9
38. Gras Navarro, Bjorklund AT, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol. (2015) 6:202. doi: 10.3389/fimmu.2015.00202
39. Nayyar G, Chu Y, Cairo MS. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front Oncol. (2019) 9:51. doi: 10.3389/fonc.2019.00051
40. Vivier E, Ugolini S. Natural killer cells: from basic research to treatments. Front Immunol. (2011) 2:18. doi: 10.3389/fimmu.2011.00018
41. Lanier LL. NK cell recognition. Annu Rev Immunol. (2005) 23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526
42. Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l'union fait la force”. Blood. (2005) 106:2252–8. doi: 10.1182/blood-2005-03-1154
43. Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. (2014) 159:94–107. doi: 10.1016/j.cell.2014.08.026
44. Nikzad R, Angelo LS, Aviles-Padilla K, Le DT, Singh VK, Bimler L, et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci Immunol. (2019) 4:eaat8116. doi: 10.1126/sciimmunol.aat8116
45. Stary V, Pandey RV, Strobl J, Kleissl L, Starlinger P, Pereyra D, et al. A discrete subset of epigenetically primed human NK cells mediates antigen-specific immune responses. Sci Immunol. (2020) 5:eaba6232. doi: 10.1126/sciimmunol.aba6232
46. Paust S, Blish CA, Reeves RK. Redefining memory: building the case for adaptive NK cells. J Virol. (2017) 91:e00169-17. doi: 10.1128/JVI.00169-17
47. Paust S, Senman B, von Andrian UH. Adaptive immune responses mediated by natural killer cells. Immunol Rev. (2010) 235:286–96. doi: 10.1111/j.0105-2896.2010.00906.x
48. Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. (1997) 91:661–72. doi: 10.1016/S0092-8674(00)80453-5
49. Di Santo JP. Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol. (2006) 24:257–86. doi: 10.1146/annurev.immunol.24.021605.090700
50. Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. (2010) 142:847–56. doi: 10.1016/j.cell.2010.08.031
51. Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat Rev Immunol. (2011) 11:645–57. doi: 10.1038/nri3044
52. Reeves RK, Li H, Jost S, Blass E, Li H, Schafer JL, et al. Antigen-specific NK cell memory in rhesus macaques. Nat Immunol. (2015) 16:927–32. doi: 10.1038/ni.3227
53. Paust S, von Andrian UH. Natural killer cell memory. Nat Immunol. (2011) 12:500–8. doi: 10.1038/ni.2032
54. Sun JC, Ugolini S, Vivier E. Immunological memory within the innate immune system. Embo J. (2014) 33:1295–303. doi: 10.1002/embj.201387651
55. O'Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol. (2006) 7:507–16. doi: 10.1038/ni1332
56. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
57. Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med. (2015) 7:297ra115. doi: 10.1126/scitranslmed.aac5722
58. Ljunggren HG, Karre K. Experimental strategies and interpretations in the analysis of changes in MHC gene expression during tumour progression. Opposing influences of T cell and natural killer mediated resistance? J Immunogenet. (1986) 13:141–51. doi: 10.1111/j.1744-313X.1986.tb01095.x
59. Diefenbach A, Hsia JK, Hsiung MY, Raulet DH. A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur J Immunol. (2003) 33:381–91. doi: 10.1002/immu.200310012
60. Ljunggren HG, Karre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol Today. (1990) 11:237–44. doi: 10.1016/0167-5699(90)90097-S
61. Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. (2015) 6:578. doi: 10.3389/fimmu.2015.00578
62. Trembath AP, Markiewicz MA. More than decoration: roles for natural killer group 2 member d ligand expression by immune cells. Front Immunol. (2018) 9:231. doi: 10.3389/fimmu.2018.00231
63. Sivori S, Parolini S, Marcenaro E, Millo R, Bottino C, Moretta A. Triggering receptors involved in natural killer cell-mediated cytotoxicity against choriocarcinoma cell lines. Hum Immunol. (2000) 61:1055–8. doi: 10.1016/S0198-8859(00)00201-9
64. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol. (2013) 132:536–44. doi: 10.1016/j.jaci.2013.07.006
65. Nimmerjahn F, Ravetch JV. FcgammaRs in health and disease. Curr Top Microbiol Immunol. (2011) 350:105–25. doi: 10.1007/82_2010_86
66. Bradley M, Zeytun A, Rafi-Janajreh A, Nagarkatti PS, Nagarkatti M. Role of spontaneous and interleukin-2-induced natural killer cell activity in the cytotoxicity and rejection of Fas+ and Fas- tumor cells. Blood. (1998) 92:4248–55. doi: 10.1182/blood.V92.11.4248.423k20_4248_4255
67. Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H, et al. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J Immunol. (1999) 163:1906–13.
68. Habif G, Crinier A, Andre P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
69. Street SE, Zerafa N, Iezzi M, Westwood JA, Stagg J, Musiani P, et al. Host perforin reduces tumor number but does not increase survival in oncogene-driven mammary adenocarcinoma. Cancer Res. (2007) 67:5454–60. doi: 10.1158/0008-5472.CAN-06-4084
70. Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. (2000) 192:755–60. doi: 10.1084/jem.192.5.755
71. Robertson MJ. Role of chemokines in the biology of natural killer cells. J Leukoc Biol. (2002) 71:173–83.
72. Mace EM, Orange JS. Genetic causes of human NK cell deficiency and their effect on NK cell subsets. Front Immunol. (2016) 7:545. doi: 10.3389/fimmu.2016.00545
73. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
74. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. (2008) 454:436–44. doi: 10.1038/nature07205
75. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. (2009) 30:1073–81. doi: 10.1093/carcin/bgp127
76. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. (2007) 7:41–51. doi: 10.1038/nri1995
77. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. (2006) 441:431–6. doi: 10.1038/nature04870
78. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. (2014) 44:1582–92. doi: 10.1002/eji.201344272
79. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
80. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. (2018) 9:527. doi: 10.3389/fimmu.2018.00527
81. Granot Z, Fridlender ZG. Plasticity beyond cancer cells and the “immunosuppressive switch”. Cancer Res. (2015) 75:4441–5. doi: 10.1158/0008-5472.CAN-15-1502
82. Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer. (2013) 13:365–76. doi: 10.1038/nrc3498
83. Servais C, Erez N. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. J Pathol. (2013) 229:198–207. doi: 10.1002/path.4103
84. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. (2004) 172:7335–40. doi: 10.4049/jimmunol.172.12.7335
85. Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. (2015) 67:3037–46. doi: 10.1002/art.39295
86. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. (2014) 122:91–128. doi: 10.1016/B978-0-12-800267-4.00003-1
87. Cantoni C, Huergo-Zapico L, Parodi M, Pedrazzi M, Mingari MC, Moretta A, et al. NK cells, tumor cell transition, and tumor progression in solid malignancies: new hints for NK-based immunotherapy? J Immunol Res. (2016) 2016:4684268. doi: 10.1155/2016/4684268
88. Ponzetta A, Benigni G, Antonangeli F, Sciume G, Sanseviero E, Zingoni A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. (2015) 75:4766–77. doi: 10.1158/0008-5472.CAN-15-1320
89. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat Immunol. (2017) 18:995–1003. doi: 10.1038/ni.3809
90. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
91. Huntington ND, Tabarias H, Fairfax K, Brady J, Hayakawa Y, Degli-Esposti MA, et al. NK cell maturation and peripheral homeostasis is associated with KLRG1 up-regulation. J Immunol. (2007) 178:4764–70. doi: 10.4049/jimmunol.178.8.4764
92. Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. (2008) 68:8437–45. doi: 10.1158/0008-5472.CAN-08-1440
93. Alvarez M, Simonetta F, Baker J, Pierini A, Wenokur AS, Morrison AR, et al. Regulation of murine NK cell exhaustion through the activation of the DNA damage repair pathway. JCI Insight. (2019) 5:e127729. doi: 10.1172/jci.insight.127729
94. Greenberg SA, Kong SW, Thompson E, Gulla SV. Co-inhibitory T cell receptor KLRG1: human cancer expression and efficacy of neutralization in murine cancer models. Oncotarget. (2019) 10:1399–406. doi: 10.18632/oncotarget.26659
95. Tata A, Vivier E, Brossay L. Role of the KLRG1 pathway during the immune response to cancer. J Immunol. (2020) 204.
96. Ibrahim EM, Al-Foheidi ME, Al-Mansour MM, Kazkaz GA. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: a meta-analysis. Breast Cancer Res Treat. (2014) 148:467–76. doi: 10.1007/s10549-014-3185-2
97. Deschoolmeester V, Baay M, Van Marck E, Weyler J, Vermeulen P, Lardon F, et al. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol. (2010) 11:19. doi: 10.1186/1471-2172-11-19
98. Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. (1997) 79:2320–8.
99. Villegas FR, Coca S, Villarrubia VG, Jimenez R, Chillon MJ, Jareno J, et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer. (2002) 35:23–8. doi: 10.1016/S0169-5002(01)00292-6
100. Barkin J, Rodriguez-Suarez R, Betito K. Association between natural killer cell activity and prostate cancer: a pilot study. Can J Urol. (2017) 24:8708–713.
101. Gutkin DW, Shurin MR. Clinical evaluation of systemic and local immune responses in cancer: time for integration. Cancer Immunol Immunother. (2014) 63:45–57. doi: 10.1007/s00262-013-1480-0
102. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. (2000) 356:1795–9. doi: 10.1016/S0140-6736(00)03231-1
103. Jobin G, Rodriguez-Suarez R, Betito K. Association between natural killer cell activity and colorectal cancer in high-risk subjects undergoing colonoscopy. Gastroenterology. (2017) 153:980–7. doi: 10.1053/j.gastro.2017.06.009
104. Kastelan M, Kovacic K, Tarle R, Kraljic I, Tarle M. Analysis of NK cell activity, lymphocyte reactivity to mitogens and serotest PSA and TPS values in patients with primary and disseminated prostate cancer, PIN and BPH. Anticancer Res. (1997) 17:1671–5.
105. Koo KC, Shim DH, Yang CM, Lee SB, Kim SM, Shin TY, et al. Reduction of the CD16(-)CD56bright NK cell subset precedes NK cell dysfunction in prostate cancer. PLoS ONE. (2013) 8:e78049. doi: 10.1371/journal.pone.0078049
106. Lin CF, Lin CM, Lee KY, Wu SY, Feng PH, Chen KY, et al. Escape from IFN-gamma-dependent immunosurveillance in tumorigenesis. J Biomed Sci. (2017) 24:10. doi: 10.1186/s12929-017-0317-0
107. Shanker A, Buferne M, Schmitt-Verhulst AM. Cooperative action of CD8 T lymphocytes and natural killer cells controls tumour growth under conditions of restricted T-cell receptor diversity. Immunology. (2010) 129:41–54. doi: 10.1111/j.1365-2567.2009.03150.x
108. Knudson KM, Hicks KC, Alter S, Schlom J, Gameiro SR. Mechanisms involved in IL-15 superagonist enhancement of anti-PD-L1 therapy. J Immunother Cancer. (2019) 7:82. doi: 10.1186/s40425-019-0551-y
109. Ferlazzo G, Morandi B. Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front Immunol. (2014) 5:159. doi: 10.3389/fimmu.2014.00159
110. Stojanovic A, Cerwenka A. Natural killer cells and solid tumors. J Innate Immun. (2011) 3:355–64. doi: 10.1159/000325465
111. Konjevic G, Jurisic V, Jovic V, Vuletic A, Mirjacic Martinovic K, Radenkovic S, et al. Investigation of NK cell function and their modulation in different malignancies. Immunol Res. (2012) 52:139–56. doi: 10.1007/s12026-012-8285-7
112. Tarle M, Kraljic I, Kastelan M. Comparison between NK cell activity and prostate cancer stage and grade in untreated patients: correlation with tumor markers and hormonal serotest data. Urol Res. (1993) 21:17–21. doi: 10.1007/BF00295186
113. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. (2008) 27:5932–43. doi: 10.1038/onc.2008.267
114. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
115. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia. (2013) 15:133–42. doi: 10.1593/neo.121758
116. Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer. (2008) 112:863–75. doi: 10.1002/cncr.23239
117. Bruno A, Bassani B, D'Urso DG, Pitaku I, Cassinotti E, Pelosi G, et al. Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J. (2018) 32:5365–77. doi: 10.1096/fj.201701103R
118. Rocca YS, Roberti MP, Arriaga JM, Amat M, Bruno L, Pampena MB, et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun. (2013) 19:76–85. doi: 10.1177/1753425912453187
119. Schleypen JS, Baur N, Kammerer R, Nelson PJ, Rohrmann K, Grone EF, et al. Cytotoxic markers and frequency predict functional capacity of natural killer cells infiltrating renal cell carcinoma. Clin Cancer Res. (2006) 12:718–25. doi: 10.1158/1078-0432.CCR-05-0857
120. Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. (2011) 17:700–7. doi: 10.1038/nm.2366
121. Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, et al. Characterization of tumor infiltrating natural killer cell subset. Oncotarget. (2015) 6:13835–43. doi: 10.18632/oncotarget.3453
122. Bosi A, Zanellato S, Bassani B, Albini A, Musco A, Cattoni M, et al. Natural killer cells from malignant pleural effusion are endowed with a decidual-like proangiogenic polarization. J Immunol Res. (2018) 2018:2438598. doi: 10.1155/2018/2438598
123. Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst. (2014) 106:dju200. doi: 10.1093/jnci/dju200
124. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov. (2016) 6:414–29. doi: 10.1158/2159-8290.CD-15-0732
125. Bruno A, Pagani A, Pulze L, Albini A, Dallaglio K, Noonan DM, et al. Orchestration of angiogenesis by immune cells. Front Oncol. (2014) 4:131. doi: 10.3389/fonc.2014.00131
126. Messaoudene M, Fregni G, Fourmentraux-Neves E, Chanal J, Maubec E, Mazouz-Dorval S, et al. Mature cytotoxic CD56(bright)/CD16(+) natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res. (2014) 74:81–92. doi: 10.1158/0008-5472.CAN-13-1303
127. Parisi L, Bassani B, Tremolati M, Gini E, Farronato G, Bruno A. Natural killer cells in the orchestration of chronic inflammatory diseases. J Immunol Res. (2017) 2017:4218254. doi: 10.1155/2017/4218254
128. Pasero C, Gravis G, Granjeaud S, Guerin M, Thomassin-Piana J, Rocchi P, et al. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget. (2015) 6:14360–73. doi: 10.18632/oncotarget.3965
129. Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V, et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J Immunol. (2014) 192:3805–15. doi: 10.4049/jimmunol.1301889
130. Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol. (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
131. Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol. (2010) 199:273–81. doi: 10.1007/s00430-010-0151-4
132. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
133. Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. (2012) 61:1155–67. doi: 10.1007/s00262-012-1294-5
134. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. (2016) 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
135. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. (2014) 124:5466–80. doi: 10.1172/JCI77053
136. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
137. Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. (2009) 70:325–30. doi: 10.1016/j.humimm.2009.02.008
138. Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci USA. (1996) 93:13119–24. doi: 10.1073/pnas.93.23.13119
139. Cimen Bozkus C, Elzey BD, Crist SA, Ellies LG, Ratliff TL. Expression of cationic amino acid transporter 2 is required for myeloid-derived suppressor cell-mediated control of t cell immunity. J Immunol. (2015) 195:5237–50. doi: 10.4049/jimmunol.1500959
140. Mairhofer DG, Ortner D, Tripp CH, Schaffenrath S, Fleming V, Heger L, et al. Impaired gp100-specific CD8(+) T-cell responses in the presence of myeloid-derived suppressor cells in a spontaneous mouse melanoma model. J Invest Dermatol. (2015) 135:2785–93. doi: 10.1038/jid.2015.241
141. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. (2014) 134:2853–64. doi: 10.1002/ijc.28622
142. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425. doi: 10.1158/0008-5472.CAN-06-3037
143. Nagaraj S, Youn JI, Gabrilovich DI. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol. (2013) 191:17–23. doi: 10.4049/jimmunol.1300654
144. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
145. Choksawangkarn W, Graham LM, Burke M, Lee SB, Ostrand-Rosenberg S, Fenselau C, et al. Peptide-based systems analysis of inflammation induced myeloid-derived suppressor cells reveals diverse signaling pathways. Proteomics. (2016) 16:1881–8. doi: 10.1002/pmic.201500102
146. Gato M, Blanco-Luquin I, Zudaire M, de Morentin XM, Perez-Valderrama E, Zabaleta A, et al. Drafting the proteome landscape of myeloid-derived suppressor cells. Proteomics. (2016) 16:367–78. doi: 10.1002/pmic.201500229
147. Gato-Cañas M, Martinez de Morentin X, Blanco-Luquin I, Fernandez-Irigoyen J, Zudaire I, Liechtenstein T, et al. A core of kinase-regulated interactomes defines the neoplastic MDSC lineage. Oncotarget. (2015) 6:27160–75. doi: 10.18632/oncotarget.4746
148. Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, et al. RORC1 regulates tumor-promoting “Emergency” granulo-monocytopoiesis. Cancer Cell. (2015) 28:253–69. doi: 10.1016/j.ccell.2015.07.006
149. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother. (2016) 65:161–9. doi: 10.1007/s00262-015-1782-5
150. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. (2009) 58:49–59. doi: 10.1007/s00262-008-0523-4
151. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M, et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. (2016) 18:1253–64. doi: 10.1093/neuonc/now034
152. Liang Y, Lü B, Zhao P, Lü W. Increased circulating GrMyeloid-derived suppressor cells correlated with tumor burden and survival in locally advanced cervical cancer patient. J Cancer. (2019) 10:1341–8. doi: 10.7150/jca.29647
153. Vetsika EK, Koinis F, Gioulbasani M, Aggouraki D, Koutoulaki A, Skalidaki E, et al. A circulating subpopulation of monocytic myeloid-derived suppressor cells as an independent prognostic/predictive factor in untreated non-small lung cancer patients. J Immunol Res. (2014) 2014:659294. doi: 10.1155/2014/659294
154. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. (2011) 118:2254–65. doi: 10.1182/blood-2010-12-325753
155. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. (2011) 60:1419–30. doi: 10.1007/s00262-011-1028-0
156. Kitano S, Postow MA, Ziegler CG, Kuk D, Panageas KS, Cortez C, et al. Computational algorithm-driven evaluation of monocytic myeloid-derived suppressor cell frequency for prediction of clinical outcomes. Cancer Immunol Res. (2014) 2:812–21. doi: 10.1158/2326-6066.CIR-14-0013
157. Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. (2005) 174:636–45. doi: 10.4049/jimmunol.174.2.636
158. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
159. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. (2007) 179:977–83. doi: 10.4049/jimmunol.179.2.977
160. Ben-Meir K, Twaik N, Baniyash M. Plasticity and biological diversity of myeloid derived suppressor cells. Curr Opin Immunol. (2018) 51:154–61. doi: 10.1016/j.coi.2018.03.015
161. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
162. Vetsika EK, Koukos A, Kotsakis A. Myeloid-derived suppressor cells: major figures that shape the immunosuppressive and angiogenic network in cancer. Cells. (2019) 8:1647. doi: 10.3390/cells8121647
163. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. (2001) 166:5398–406. doi: 10.4049/jimmunol.166.9.5398
164. Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. (2003) 102:2138–45. doi: 10.1182/blood-2003-01-0190
165. Budhwar S, Verma P, Verma R, Rai S, Singh K. The Yin and Yang of myeloid derived suppressor cells. Front Immunol. (2018) 9:2776. doi: 10.3389/fimmu.2018.02776
166. Sica A, Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J Autoimmun. (2017) 85:117–25. doi: 10.1016/j.jaut.2017.07.010
167. Safarzadeh E, Orangi M, Mohammadi H, Babaie F, Baradaran B. Myeloid-derived suppressor cells: important contributors to tumor progression and metastasis. J Cell Physiol. (2018) 233:3024–36. doi: 10.1002/jcp.26075
168. Baniyash M. Myeloid-derived suppressor cells as intruders and targets: clinical implications in cancer therapy. Cancer Immunol Immunother. (2016) 65:857–67. doi: 10.1007/s00262-016-1849-y
169. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
170. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. (2015) 98:913–22. doi: 10.1189/jlb.4RI0515-204R
171. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. (2011) 32:19–25. doi: 10.1016/j.it.2010.10.002
172. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
173. Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, et al. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. (2010) 40:3347–57. doi: 10.1002/eji.201041037
174. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. (2012) 122:4094–104. doi: 10.1172/JCI64115
175. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
176. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. (2016) 126:3672–9. doi: 10.1172/JCI84427
177. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. (2019) 38:5158–73. doi: 10.1038/s41388-019-0782-x
178. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
179. Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. (2014) 124:2626–39. doi: 10.1172/JCI74056
180. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–38.e6. doi: 10.1016/j.immuni.2017.07.014
181. Tymoszuk P, Evens H, Marzola V, Wachowicz K, Wasmer MH, Datta S, et al. In situ proliferation contributes to accumulation of tumor-associated macrophages in spontaneous mammary tumors. Eur J Immunol. (2014) 44:2247–62. doi: 10.1002/eji.201344304
182. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. (2015) 17:170–82. doi: 10.1038/ncb3090
183. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
184. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci USA. (2012) 109:2491–6. doi: 10.1073/pnas.1113744109
185. Movahedi K, Van Ginderachter JA. The ontogeny and microenvironmental regulation of tumor-associated macrophages. Antioxid Redox Signal. (2016) 25:775–91. doi: 10.1089/ars.2016.6704
186. Shand FH, Ueha S, Otsuji M, Koid SS, Shichino S, Tsukui T, et al. Tracking of intertissue migration reveals the origins of tumor-infiltrating monocytes. Proc Natl Acad Sci USA. (2014) 111:7771–6. doi: 10.1073/pnas.1402914111
187. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. (2015) 125:3356–64. doi: 10.1172/JCI80005
188. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
189. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
190. Bellora F, Castriconi R, Dondero A, Pessino A, Nencioni A, Liggieri G, et al. TLR activation of tumor-associated macrophages from ovarian cancer patients triggers cytolytic activity of NK cells. Eur J Immunol. (2014) 44:1814–22. doi: 10.1002/eji.201344130
191. Bellora F, Castriconi R, Doni A, Cantoni C, Moretta L, Mantovani A, et al. M-CSF induces the expression of a membrane-bound form of IL-18 in a subset of human monocytes differentiating in vitro toward macrophages. Eur J Immunol. (2012) 42:1618–26. doi: 10.1002/eji.201142173
192. Lim HX, Hong HJ, Cho D, Kim TS. IL-18 enhances immunosuppressive responses by promoting differentiation into monocytic myeloid-derived suppressor cells. J Immunol. (2014) 193:5453–60. doi: 10.4049/jimmunol.1401282
193. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
194. Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. (2000) 101:455–8. doi: 10.1016/S0092-8674(00)80856-9
195. Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. (2002) 2:389–400. doi: 10.1038/nri821
196. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. (2019) 110:2080–9. doi: 10.1111/cas.14069
197. Elkord E, Alcantar-Orozco EM, Dovedi SJ, Tran DQ, Hawkins RE, Gilham DE. T regulatory cells in cancer: recent advances and therapeutic potential. Expert Opin Biol Ther. (2010) 10:1573–86. doi: 10.1517/14712598.2010.529126
198. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. (2014) 27:1–7. doi: 10.1016/j.coi.2013.12.005
199. Ghiringhelli F, Ménard C, Martin F, Zitvogel L. The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression. Immunol Rev. (2006) 214:229–38. doi: 10.1111/j.1600-065X.2006.00445.x
200. Ondondo B, Jones E, Godkin A, Gallimore A. Home sweet home: the tumor microenvironment as a haven for regulatory T cells. Front Immunol. (2013) 4:197. doi: 10.3389/fimmu.2013.00197
201. Magnuson AM, Kiner E, Ergun A, Park JS, Asinovski N, Ortiz-Lopez A, et al. Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc Natl Acad Sci USA. (2018) 115:E10672–e681. doi: 10.1073/pnas.1810580115
202. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. (2007) 67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175
203. Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene. (2017) 36:3048–58. doi: 10.1038/onc.2016.458
204. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. (2009) 182:1746–55. doi: 10.4049/jimmunol.182.3.1746
205. Wachsmann MB, Pop LM, Vitetta ES. Pancreatic ductal adenocarcinoma: a review of immunologic aspects. J Investig Med. (2012) 60:643–63. doi: 10.2310/JIM.0b013e31824a4d79
206. Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest. (2017) 127:3472–83. doi: 10.1172/JCI90499
207. Siddiqui SA, Frigola X, Bonne-Annee S, Mercader M, Kuntz SM, Krambeck AE, et al. Tumor-infiltrating Foxp3-CD4+CD25+ T cells predict poor survival in renal cell carcinoma. Clin Cancer Res. (2007) 13:2075–81. doi: 10.1158/1078-0432.CCR-06-2139
208. Messmer MN, Netherby CS, Banik D, Abrams SI. Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol Immunother. (2015) 64:1–13. doi: 10.1007/s00262-014-1639-3
209. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci. (2014) 1319:47–65. doi: 10.1111/nyas.12469
210. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PLoS ONE. (2016) 11:e0164514. doi: 10.1371/journal.pone.0164514
211. Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood. (2011) 117:6532–41. doi: 10.1182/blood-2010-11-317321
212. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. (2012) 189:5602–11. doi: 10.4049/jimmunol.1201018
213. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. (2018) 9:978. doi: 10.3389/fimmu.2018.00978
214. Siret C, Collignon A, Silvy F, Robert S, Cheyrol T, André P, et al. Deciphering the crosstalk between myeloid-derived suppressor cells and regulatory T cells in pancreatic ductal adenocarcinoma. Front Immunol. (2019) 10:3070. doi: 10.3389/fimmu.2019.03070
215. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. (2014) 63:1769–81. doi: 10.1136/gutjnl-2013-306271
216. Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol. (2012) 132:1239–46. doi: 10.1038/jid.2011.416
217. Yang R, Cai Z, Zhang Y, W.Yutzy Ht, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res. (2006) 66:6807–15. doi: 10.1158/0008-5472.CAN-05-3755
218. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. (2010) 70:99–108. doi: 10.1158/0008-5472.CAN-09-1882
219. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
220. Bianchi G, Borgonovo G, Pistoia V, Raffaghello L. Immunosuppressive cells and tumour microenvironment: focus on mesenchymal stem cells and myeloid derived suppressor cells. Histol Histopathol. (2011) 26:941–51.
221. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front Immunol. (2020) 11:940. doi: 10.3389/fimmu.2020.00940
222. M. De Simone, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating t regulatory cells. Immunity. (2016) 45:1135–47. doi: 10.1016/j.immuni.2016.10.021
223. Yuan Y, Jiang YC, Sun CK, Chen QM. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol Rep. (2016) 35:2499–515. doi: 10.3892/or.2016.4660
224. Monu NR, Frey AB. Myeloid-derived suppressor cells and anti-tumor T cells: a complex relationship. Immunol Invest. (2012) 41:595–613. doi: 10.3109/08820139.2012.673191
225. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
226. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. (2010) 32:790–802. doi: 10.1016/j.immuni.2010.05.010
227. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. (2009) 50:799–807. doi: 10.1002/hep.23054
228. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin Cancer Res. (2018) 24:1891–904. doi: 10.1158/1078-0432.CCR-17-0691
229. Vaknin I, Blinder L, Wang L, Gazit R, Shapira E, Genina O, et al. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood. (2008) 111:1437–47. doi: 10.1182/blood-2007-07-100404
230. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
231. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
232. Cacalano NA. Regulation of natural killer cell function by STAT3. Front Immunol. (2016) 7:128. doi: 10.3389/fimmu.2016.00128
233. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. (2015) 66:97–110. doi: 10.1146/annurev-med-051013-052304
234. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. (2004) 172:464–74. doi: 10.4049/jimmunol.172.1.464
235. Rebe C, Vegran F, Berger H, Ghiringhelli F. STAT3 activation: a key factor in tumor immunoescape. Jakstat. (2013) 2:e23010. doi: 10.4161/jkst.23010
236. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. (2014) 193:2574–86. doi: 10.4049/jimmunol.1400833
237. Proia TA, Singh M, Woessner R, Carnevalli L, Bommakanti G, Magiera L, et al. STAT3 Antisense oligonucleotide remodels the suppressive tumor microenvironment to enhance immune activation in combination with anti-PD-L1. Clin Cancer Res. (2020) 26:6335–49. doi: 10.1158/1078-0432.CCR-20-1066
238. Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. (2002) 23:144–50. doi: 10.1016/S1471-4906(01)02154-8
239. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. (2014) 20:4096–106. doi: 10.1158/1078-0432.CCR-14-0635
240. Newson J, Motwani MP, Kendall AC, Nicolaou A, Muccioli GG, Alhouayek M, et al. Inflammatory resolution triggers a prolonged phase of immune suppression through COX-1/mPGES-1-derived prostaglandin E2. Cell Rep. (2017) 20:3162–75. doi: 10.1016/j.celrep.2017.08.098
241. Santin AD, Bellone S, Ravaggi A, Roman J, Smith CV, Pecorelli S, et al. Increased levels of interleukin-10 and transforming growth factor-beta in the plasma and ascitic fluid of patients with advanced ovarian cancer. Bjog. (2001) 108:804–8. doi: 10.1016/S0306-5456(00)00206-0
242. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
243. Lee CR, Lee W, Cho SK, Park SG. Characterization of multiple cytokine combinations and TGF-β on differentiation and functions of myeloid-derived suppressor cells. Int J Mol Sci. (2018) 19:869. doi: 10.3390/ijms19030869
244. Allan DS, Rybalov B, Awong G, Zúñiga-Pflücker JC, Kopcow HD, Carlyle JR, et al. TGF-β affects development and differentiation of human natural killer cell subsets. Eur J Immunol. (2010) 40:2289–95. doi: 10.1002/eji.200939910
245. Cerdeira AS, Rajakumar A, Royle CM, Lo A, Husain Z, Thadhani RI, et al. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. J Immunol. (2013) 190:3939–48. doi: 10.4049/jimmunol.1202582
246. Keskin DB, Allan DS, Rybalov B, Andzelm MM, Stern JN, Kopcow HD, et al. TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc Natl Acad Sci USA. (2007) 104:3378–83. doi: 10.1073/pnas.0611098104
247. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. (2001) 29:117–29. doi: 10.1038/ng1001-117
248. Tai LH, Alkayyal AA, Leslie AL, Sahi S, Bennett S, Tanese de Souza C, et al. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of Natural Killer cell cytotoxicity. Oncoimmunology. (2018) 7:e1431082. doi: 10.1080/2162402X.2018.1431082
249. Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood. (2008) 112:4080–9. doi: 10.1182/blood-2008-03-143776
250. Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. (2004) 64:7596–603. doi: 10.1158/0008-5472.CAN-04-1627
251. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. (2008) 181:3784–92. doi: 10.4049/jimmunol.181.6.3784
252. Sun X, Sui Q, Zhang C, Tian Z, Zhang J. Targeting blockage of STAT3 in hepatocellular carcinoma cells augments NK cell functions via reverse hepatocellular carcinoma-induced immune suppression. Mol Cancer Ther. (2013) 12:2885–96. doi: 10.1158/1535-7163.MCT-12-1087
253. Sui Q, Zhang J, Sun X, Zhang C, Han Q, Tian Z. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J Immunol. (2014) 193:2016–23. doi: 10.4049/jimmunol.1302389
254. Lebrun JJ. The dual role of TGFbeta in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. (2012) 2012:381428. doi: 10.5402/2012/381428
255. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. (2006) 6:506–20. doi: 10.1038/nrc1926
257. Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer. (2009) 9:445–52. doi: 10.1038/nrc2639
258. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
259. Sucher R, Kurz K, Weiss G, Margreiter R, Fuchs D, Brandacher G. IDO-mediated tryptophan degradation in the pathogenesis of malignant tumor disease. Int J Tryptophan Res. (2010) 3:113–20. doi: 10.4137/IJTR.S4157
260. Gostner JM, Becker K, Uberall F, Fuchs D. The potential of targeting indoleamine 2,3-dioxygenase for cancer treatment. Expert Opin Ther Targets. (2015) 19:605–15. doi: 10.1517/14728222.2014.995092
261. Hornyak L, Dobos N, Koncz G, Karanyi Z, Pall D, Szabo Z, et al. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. (2018) 9:151. doi: 10.3389/fimmu.2018.00151
262. Zhang J, Han X, Hu X, Jin F, Gao Z, Yin L, et al. IDO1 impairs NK cell cytotoxicity by decreasing NKG2D/NKG2DLs via promoting miR-18a. Mol Immunol. (2018) 103:144–55. doi: 10.1016/j.molimm.2018.09.011
263. M. Della Chiesa, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. (2006) 108:4118–25. doi: 10.1182/blood-2006-03-006700
264. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front Immunol. (2019) 10:771. doi: 10.3389/fimmu.2019.00771
265. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
266. Choudhari SK, Chaudhary M, Bagde S, Gadbail AR, Joshi V. Nitric oxide and cancer: a review. World J Surg Oncol. (2013) 11:118. doi: 10.1186/1477-7819-11-118
267. Ying L, Hofseth LJ. An emerging role for endothelial nitric oxide synthase in chronic inflammation and cancer. Cancer Res. (2007) 67:1407–10. doi: 10.1158/0008-5472.CAN-06-2149
268. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
269. Karakhanova S, Link J, Heinrich M, Shevchenko I, Yang Y, Hassenpflug M, et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: importance of myeloid-derived suppressor cells. Oncoimmunology. (2015) 4:e998519. doi: 10.1080/2162402X.2014.998519
270. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. (2007) 450:825–31. doi: 10.1038/nature06348
271. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. (2008) 13:23–35. doi: 10.1016/j.ccr.2007.12.004
272. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. (2008) 8:618–31. doi: 10.1038/nrc2444
273. Jacob A, Prekeris R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front Cell Dev Biol. (2015) 3:4. doi: 10.3389/fcell.2015.00004
274. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res. (2017) 23:587–99. doi: 10.1158/1078-0432.CCR-16-0387
275. Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, et al. Control of the immune response by pro-angiogenic factors. Front Oncol. (2014) 4:70. doi: 10.3389/fonc.2014.00070
276. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. (2015) 12:244–57. doi: 10.1016/j.celrep.2015.06.024
277. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. (2011) 71:7463–70. doi: 10.1158/0008-5472.CAN-11-2449
278. Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. (2016) 22:3924–36. doi: 10.1158/1078-0432.CCR-15-2463
279. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. (2006) 441:437–43. doi: 10.1038/nature04871
280. Ahn GO, Seita J, Hong BJ, Kim YE, Bok S, Lee CJ, et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc Natl Acad Sci USA. (2014) 111:2698–703. doi: 10.1073/pnas.1320243111
281. Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. (2016) 29:7–16. doi: 10.1016/j.coph.2016.04.001
282. Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. (2013) 13:842–57. doi: 10.1038/nrc3613
283. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. (2017) 8:517. doi: 10.1038/s41467-017-00530-7
284. Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol. (2014) 193:3155–64. doi: 10.4049/jimmunol.1400578
285. Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I). Immunol Res. (2006) 36:91–9. doi: 10.1385/IR:36:1:91
286. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. (2014) 74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583
287. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. (2013) 138:105–15. doi: 10.1111/imm.12036
288. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
289. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
290. Pedroza-Pacheco I, Madrigal A, Saudemont A. Interaction between natural killer cells and regulatory T cells: perspectives for immunotherapy. Cell Mol Immunol. (2013) 10:222–9. doi: 10.1038/cmi.2013.2
291. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
292. Chang WC, Li CH, Chu LH, Huang PS, Sheu BC, Huang SC. Regulatory T cells suppress natural killer cell immunity in patients with human cervical carcinoma. Int J Gynecol Cancer. (2016) 26:156–62. doi: 10.1097/IGC.0000000000000578
293. Hansen GL, Gaudernack G, Brunsvig PF, Cvancarova M, Kyte JA. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol Immunother. (2015) 64:1609–21. doi: 10.1007/s00262-015-1766-5
294. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
295. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol. (2013) 91:493–502. doi: 10.1038/icb.2013.29
296. Dannenmann SR, Thielicke J, Stöckli M, Matter C, von Boehmer L, Cecconi V, et al. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunology. (2013) 2:e23562. doi: 10.4161/onci.23562
297. Mantovani A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol. (2010) 40:3317–20. doi: 10.1002/eji.201041170
298. Katoh H, Watanabe M. Myeloid-derived suppressor cells and therapeutic strategies in cancer. Mediators Inflamm. (2015) 2015:159269. doi: 10.1155/2015/159269
299. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. (2009) 182:6562–8. doi: 10.4049/jimmunol.0803831
300. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. (2009) 182:4499–506. doi: 10.4049/jimmunol.0802740
301. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
302. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
303. Yamauchi Y, Safi S, Blattner C, Rathinasamy A, Umansky L, Juenger S, et al. Circulating and tumor myeloid-derived suppressor cells in resectable non-small-cell lung cancer. Am J Resp Crit Care Med. (2018) 198:777–87. doi: 10.1164/rccm.201708-1707OC
304. Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH, et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14(-)/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. (2010) 136:35–45. doi: 10.1007/s00432-009-0634-0
305. Pyzer AR, Cole L, Rosenblatt J, Avigan DE. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int J Cancer. (2016) 139:1915–26. doi: 10.1002/ijc.30232
306. Weed DT, Vella JL, Reis IM, De la Fuente AC, Gomez C, Sargi Z, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. (2015) 21:39–48. doi: 10.1158/1078-0432.CCR-14-1711
307. Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. (2010) 16:1812–23. doi: 10.1158/1078-0432.CCR-09-3272
308. Alizadeh D, Larmonier N. Chemotherapeutic targeting of cancer-induced immunosuppressive cells. Cancer Res. (2014) 74:2663–8. doi: 10.1158/0008-5472.CAN-14-0301
309. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology. (2015) 4:e954829. doi: 10.4161/21624011.2014.954829
310. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. (2005) 11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883
311. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
312. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. (2014) 74:104–18. doi: 10.1158/0008-5472.CAN-13-1545
313. Gujar SA, Clements D, Dielschneider R, Helson E, Marcato P, Lee PW. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br J Cancer. (2014) 110:83–93. doi: 10.1038/bjc.2013.695
314. Cao G, Wang J, Zheng X, Wei H, Tian Z, Sun R. Tumor therapeutics work as stress inducers to enhance tumor sensitivity to natural killer (NK) cell cytolysis by up-regulating NKp30 Ligand B7-H6. J Biol Chem. (2015) 290:29964–73. doi: 10.1074/jbc.M115.674010
315. Shi L, Lin H, Li G, Sun Y, Shen J, Xu J, et al. Cisplatin enhances NK cells immunotherapy efficacy to suppress HCC progression via altering the androgen receptor (AR)-ULBP2 signals. Cancer Lett. (2016) 373:45–56. doi: 10.1016/j.canlet.2016.01.017
316. Xia C, Liu C, He Z, Cai Y, Chen J. Metformin inhibits cervical cancer cell proliferation by modulating PI3K/Akt-induced major histocompatibility complex class I-related chain A gene expression. J Exp Clin Cancer Res. (2020) 39:127. doi: 10.1186/s13046-020-01627-6
317. Wang Y, Jia A, Bi Y, Wang Y, Yang Q, Cao Y, et al. Targeting myeloid-derived suppressor cells in cancer immunotherapy. Cancers. (2020) 12:2626. doi: 10.3390/cancers12092626
318. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFβR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res. (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
319. Shaim H, Basar R, Wang F, Daher M, Zamler D, Gumin J, et al. Inhibition of the αv integrin-TGF-β axis improves natural killer cell function against glioblastoma stem cells. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.03.30.016667
320. Yingling JM, McMillen WT, Yan L, Huang H, Sawyer JS, Graff J, et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget. (2018) 9:6659–77. doi: 10.18632/oncotarget.23795
321. Fujiwara Y, Nokihara H, Yamada Y, Yamamoto N, Sunami K, Utsumi H, et al. Phase 1 study of galunisertib, a TGF-beta receptor I kinase inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. (2015) 76:1143–52. doi: 10.1007/s00280-015-2895-4
322. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE. (2018) 13:e0191358. doi: 10.1371/journal.pone.0191358
323. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE. (2014) 9:e90353. doi: 10.1371/journal.pone.0090353
324. Wang QM, Tang PM, Lian GY, Li C, Li J, Huang XR, et al. Enhanced cancer immunotherapy with Smad3-silenced NK-92 cells. Cancer Immunol Res. (2018) 6:965–77. doi: 10.1158/2326-6066.CIR-17-0491
325. Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-βR II and NKG2D. Cancer Immunol Immunother. (2017) 66:537–48. doi: 10.1007/s00262-017-1959-1
326. Foltz JA, Moseman JE, Thakkar A, Chakravarti N, Lee DA. TGFβ imprinting during activation promotes natural killer cell cytokine hypersecretion. Cancers. (2018) 10:423. doi: 10.3390/cancers10110423
327. Horn LA, Riskin J, Hempel HA, Fousek K, Lind H, Hamilton DH, et al. Simultaneous inhibition of CXCR1/2, TGF-β, and PD-L1 remodels the tumor and its microenvironment to drive antitumor immunity. J Immunother Cancer. (2020) 8:e000326. doi: 10.1136/jitc-2019-000326
328. Morelli MB, Amantini C, Santoni M, Soriani A, Nabissi M, Cardinali C, et al. Axitinib induces DNA damage response leading to senescence, mitotic catastrophe, and increased NK cell recognition in human renal carcinoma cells. Oncotarget. (2015) 6:36245–59. doi: 10.18632/oncotarget.5768
329. Huang Y, Wang Y, Li Y, Guo K, He Y. Role of sorafenib and sunitinib in the induction of expressions of NKG2D ligands in nasopharyngeal carcinoma with high expression of ABCG2. J Cancer Res Clin Oncol. (2011) 137:829–37. doi: 10.1007/s00432-010-0944-2
330. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. (2009) 69:2506–13. doi: 10.1158/0008-5472.CAN-08-4323
331. Guislain A, Gadiot J, Kaiser A, Jordanova ES, Broeks A, Sanders J, et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol Immunother. (2015) 64:1241–50. doi: 10.1007/s00262-015-1735-z
332. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. (2009) 69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709
333. Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, et al. Myeloid-derived suppressor cells express Bruton's tyrosine kinase and can be depleted in tumor-bearing hosts by ibrutinib treatment. Cancer Res. (2016) 76:2125–36. doi: 10.1158/0008-5472.CAN-15-1490
334. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. (2018) 9:847. doi: 10.3389/fimmu.2018.00847
335. Xiao W, Klement JD, Lu C, Ibrahim ML, Liu K. IFNAR1 Controls autocrine type I IFN regulation of PD-L1 expression in myeloid-derived suppressor cells. J Immunol. (2018) 201:264–77. doi: 10.4049/jimmunol.1800129
336. Liu JF, Deng WW, Chen L, Li YC, Wu L, Ma SR, et al. Inhibition of JAK2/STAT3 reduces tumor-induced angiogenesis and myeloid-derived suppressor cells in head and neck cancer. Mol Carcinog. (2018) 57:429–39. doi: 10.1002/mc.22767
337. Bill MA, Fuchs JR, Li C, Yui J, Bakan C, Benson DM, et al. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol Cancer. (2010) 9:165. doi: 10.1186/1476-4598-9-165
338. Kortylewski M, Moreira D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol Immunother. (2017) 66:979–88. doi: 10.1007/s00262-017-1966-2
339. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. (2019) 569:73–8. doi: 10.1038/s41586-019-1118-2
340. Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin Cancer Res. (2016) 22:3849–59. doi: 10.1158/1078-0432.CCR-15-1912
341. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. (2017) 23:5187–201. doi: 10.1158/1078-0432.CCR-17-0741
342. Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C, et al. The histone deacetylase inhibitor valproic acid inhibits NKG2D expression in natural killer cells through suppression of STAT3 and HDAC3. Sci Rep. (2017) 7:45266. doi: 10.1038/srep45266
343. Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J Jr. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett. (2007) 581:1317–22. doi: 10.1016/j.febslet.2007.02.045
344. Kiany S, Huang G, Kleinerman ES. Effect of entinostat on NK cell-mediated cytotoxicity against osteosarcoma cells and osteosarcoma lung metastasis. Oncoimmunology. (2017) 6:e1333214. doi: 10.1080/2162402X.2017.1333214
345. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. A. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. (2017) 5:101. doi: 10.1186/s40425-017-0308-4
346. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. (2017) 8:93. doi: 10.3389/fimmu.2017.00093
347. Heys SD, Segar A, Payne S, Bruce DM, Kernohan N, Eremin O. Dietary supplementation with L-arginine: modulation of tumour-infiltrating lymphocytes in patients with colorectal cancer. Br J Surg. (1997) 84:238–41. doi: 10.1046/j.1365-2168.1997.02528.x
348. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
349. Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
350. Sandner P, Hütter J, Tinel H, Ziegelbauer K, Bischoff E. PDE5 inhibitors beyond erectile dysfunction. Int J Impot Res. (2007) 19:533–43. doi: 10.1038/sj.ijir.3901577
351. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. (2006) 203:2691–702. doi: 10.1084/jem.20061104
352. Califano JA, Khan Z, Noonan KA, Rudraraju L, Zhang Z, Wang H, et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. (2015) 21:30–8. doi: 10.1158/1078-0432.CCR-14-1716
353. Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. (1990) 76:1704–9. doi: 10.1182/blood.V76.9.1704.bloodjournal7691704
354. Warrell RP Jr., Frankel SR, Miller WH Jr., Scheinberg DA, Itri LM, Hittelman WM, et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med. (1991) 324:1385–93. doi: 10.1056/NEJM199105163242002
355. Breitman TR, Collins SJ, Keene BR. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood. (1981) 57:1000–4. doi: 10.1182/blood.V57.6.1000.1000
356. Liang C, Yang L, Guo S. All-trans retinoic acid inhibits migration, invasion and proliferation, and promotes apoptosis in glioma cells in vitro. Oncol Lett. (2015) 9:2833–8. doi: 10.3892/ol.2015.3120
357. Hsu SL, Hsu JW, Liu MC, Chen LY, Chang CD. Retinoic acid-mediated G1 arrest is associated with induction of p27(Kip1) and inhibition of cyclin-dependent kinase 3 in human lung squamous carcinoma CH27 cells. Exp Cell Res. (2000) 258:322–31. doi: 10.1006/excr.2000.4933
358. Cui J, Gong M, He Y, Li Q, He T, Bi Y. All-trans retinoic acid inhibits proliferation, migration, invasion and induces differentiation of hepa1-6 cells through reversing EMT in vitro. Int J Oncol. (2016) 48:349–57. doi: 10.3892/ijo.2015.3235
359. Li Y, Wongsiriroj N, Blaner WS. The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surg Nutr. (2014) 3:126–39. doi: 10.3978/j.issn.2304-3881.2014.05.04
360. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. (2013) 62:909–18. doi: 10.1007/s00262-013-1396-8
361. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. (2006) 66:9299–307. doi: 10.1158/0008-5472.CAN-06-1690
362. Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. (2003) 63:4441–9.
363. Ahmad SM, Haskell MJ, Raqib R, Stephensen CB. Markers of innate immune function are associated with vitamin a stores in men. J Nutr. (2009) 139:377–85. doi: 10.3945/jn.108.100198
364. Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int J Cancer. (2003) 104:354–61. doi: 10.1002/ijc.10966
365. Nwangwu CA, Weiher H, Schmidt-Wolf IGH. Increase of CIK cell efficacy by upregulating cell surface MICA and inhibition of NKG2D ligand shedding in multiple myeloma. Hematol Oncol. (2017) 35:719–25. doi: 10.1002/hon.2326
366. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol. (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
367. Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. (2018) 78:3220–32. doi: 10.1158/0008-5472.CAN-17-3415
368. Umansky V, Blattner C, Gebhardt C, Utikal J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines. (2016) 4:36. doi: 10.3390/vaccines4040036
369. Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. (2019) 10:172. doi: 10.3389/fimmu.2019.00172
370. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. (1996) 2:1096–103. doi: 10.1038/nm1096-1096
371. Kusmartsev S, Eruslanov E, Kübler H, Tseng T, Sakai Y, Su Z, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. (2008) 181:346–53. doi: 10.4049/jimmunol.181.1.346
372. Koinis F, Vetsika EK, Aggouraki D, Skalidaki E, Koutoulaki A, Gkioulmpasani M, et al. Effect of first-line treatment on myeloid-derived suppressor cells' subpopulations in the peripheral blood of patients with non-small cell lung cancer. J Thorac Oncol. (2016) 11:1263–72. doi: 10.1016/j.jtho.2016.04.026
373. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
374. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
375. Klose R, Krzywinska E, Castells M, Gotthardt D, Putz EM, Kantari-Mimoun C, et al. Targeting VEGF-A in myeloid cells enhances natural killer cell responses to chemotherapy and ameliorates cachexia. Nat Commun. (2016) 7:12528. doi: 10.1038/ncomms12528
376. Ferrero JM, Hardy-Bessard AC, Capitain O, Lortholary A, Salles B, Follana P, et al. Weekly paclitaxel, capecitabine, and bevacizumab with maintenance capecitabine and bevacizumab as first-line therapy for triple-negative, metastatic, or locally advanced breast cancer: results from the GINECO A-TaXel phase 2 study. Cancer. (2016) 122:3119–26. doi: 10.1002/cncr.30170
377. Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5(+) Myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. (2018) 78:157–67. doi: 10.1158/0008-5472.CAN-17-0348
378. Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. (2012) 72:3906–11. doi: 10.1158/0008-5472.CAN-11-3873
379. Wang Y, Zhang X, Yang L, Xue J, Hu G. Blockade of CCL2 enhances immunotherapeutic effect of anti-PD1 in lung cancer. J Bone Oncol. (2018) 11:27–32. doi: 10.1016/j.jbo.2018.01.002
380. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. (2010) 10:464. doi: 10.1186/1471-2407-10-464
381. Kosaka A, Ohkuri T, Okada H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother. (2014) 63:847–57. doi: 10.1007/s00262-014-1561-8
382. Iachininoto MG, Nuzzolo ER, Bonanno G, Mariotti A, Procoli A, Locatelli F, et al. Cyclooxygenase-2 (COX-2) inhibition constrains indoleamine 2,3-dioxygenase 1 (IDO1) activity in acute myeloid leukaemia cells. Molecules. (2013) 18:10132–45. doi: 10.3390/molecules180910132
383. Kim SJ, Ha GH, Bae JH, Kim GR, Son CH, Park YS, et al. COX-2- and endoplasmic reticulum stress-independent induction of ULBP-1 and enhancement of sensitivity to NK cell-mediated cytotoxicity by celecoxib in colon cancer cells. Exp Cell Res. (2015) 330:451–9. doi: 10.1016/j.yexcr.2014.09.008
384. Majumder M, Nandi P, Omar A, Ugwuagbo KC, Lala PK. EP4 as a therapeutic target for aggressive human breast cancer. Int J Mol Sci. (2018) 19:1019. doi: 10.20944/preprints201802.0095.v1
385. Shaashua L, Shabat-Simon M, Haldar R, Matzner P, Zmora O, Shabtai M, et al. Perioperative COX-2 and β-adrenergic blockade improves metastatic biomarkers in breast cancer patients in a phase-II randomized trial. Clin Cancer Res. (2017) 23:4651–61. doi: 10.1158/1078-0432.CCR-17-0152
386. Lee GY, Han SN. The role of vitamin e in immunity. Nutrients. (2018) 10:1614. doi: 10.3390/nu10111614
387. Ohl K, Tenbrock K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol. (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
388. Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, et al. Myeloid-derived suppressor cell survival and function are regulated by the transcription factor Nrf2. J Immunol. (2016) 196:3470–8. doi: 10.4049/jimmunol.1501785
389. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. (2009) 182:5693–701. doi: 10.4049/jimmunol.0900092
390. Dietlin TA, Hofman FM, Lund BT, Gilmore W, Stohlman SA, van der Veen RC. Mycobacteria-induced Gr-1+ subsets from distinct myeloid lineages have opposite effects on T cell expansion. J Leukoc Biol. (2007) 81:1205–12. doi: 10.1189/jlb.1006640
391. Saddawi-Konefka R, Seelige R, Gross ET, Levy E, Searles SC, Washington A, et al. Nrf2 induces IL-17D to mediate tumor and virus surveillance. Cell Rep. (2016) 16:2348–58. doi: 10.1016/j.celrep.2016.07.075
392. Wang YY, Yang YX, Zhe H, He ZX, Zhou SF. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Dev Ther. (2014) 8:2075–88. doi: 10.2147/DDDT.S68872
393. Seelige R, Saddawi-Konefka R, Adams NM, Picarda G, Sun JC, Benedict CA, et al. Interleukin-17D and Nrf2 mediate initial innate immune cell recruitment and restrict MCMV infection. Sci Rep. (2018) 8:13670. doi: 10.1038/s41598-018-32011-2
394. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin Cancer Res. (2020) 26:1420–31. doi: 10.1158/1078-0432.CCR-19-2625
395. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. (2014) 6:237ra67. doi: 10.1126/scitranslmed.3007974
396. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
397. Noman MZ, Hasmim M, Lequeux A, Xiao M, Duhem C, Chouaib S, et al. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: new opportunities and challenges. Cells. (2019) 8:1083. doi: 10.3390/cells8091083
398. Borad MJ, Reddy SG, Bahary N, Uronis HE, Sigal D, Cohn AL, et al. Randomized phase II trial of gemcitabine plus TH-302 versus gemcitabine in patients with advanced pancreatic cancer. J Clin Oncol. (2015) 33:1475–81. doi: 10.1200/JCO.2014.55.7504
399. Chawla SP, Cranmer LD, Van Tine BA, Reed DR, Okuno SH, Butrynski JE, et al. Phase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J Clin Oncol. (2014) 32:3299–306. doi: 10.1200/JCO.2013.54.3660
400. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. (2018) 128:5137–49. doi: 10.1172/JCI96268
401. Lim HX, Choi S, Cho D, Kim TS. IL-33 inhibits the differentiation and immunosuppressive activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Immunol Cell Biol. (2017) 95:99–107. doi: 10.1038/icb.2016.72
402. Lim HX, Kim TS, Poh CL. Understanding the differentiation, expansion, recruitment and suppressive activities of myeloid-derived suppressor cells in cancers. Int J Mol Sci. (2020) 21:3599. doi: 10.3390/ijms21103599
403. Zheng Y, Tian X, Wang T, Xia X, Cao F, Tian J, et al. Long noncoding RNA Pvt1 regulates the immunosuppression activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Mol Cancer. (2019) 18:61. doi: 10.1186/s12943-019-0978-2
404. Shang W, Gao Y, Tang Z, Zhang Y, Yang R. The pseudogene Olfr29-ps1 promotes the suppressive function and differentiation of monocytic MDSCs. Cancer Immunol Res. (2019) 7:813–27. doi: 10.1158/2326-6066.CIR-18-0443
405. Dai J, Kumbhare A, Williams DA, Youssef D, Yao ZQ, McCall CE, et al. El Gazzar, Nfia deletion in myeloid cells blocks expansion of myeloid-derived suppressor cells during sepsis. Innate Immun. (2018) 24:54–65. doi: 10.1177/1753425917742956
406. Geller MA, Miller JS. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy. (2011) 3:1445–59. doi: 10.2217/imt.11.131
407. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. (2002) 298:850–4. doi: 10.1126/science.1076514
408. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
409. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
410. Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. (2005) 175:6169–76. doi: 10.4049/jimmunol.175.9.6169
411. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. (2006) 24:e20–2. doi: 10.1200/JCO.2006.05.9964
412. Dhupkar P, Gordon N. Interleukin-2: old and new approaches to enhance immune-therapeutic efficacy. Adv Exp Med Biol. (2017) 995:33–51. doi: 10.1007/978-3-319-53156-4_2
413. Perez-Martinez A, Fernandez L, Valentin J, Martinez-Romera I, Corral MD, Ramirez M, et al. A phase I/II trial of interleukin-15–stimulated natural killer cell infusion after haplo-identical stem cell transplantation for pediatric refractory solid tumors. Cytotherapy. (2015) 17:1594–603. doi: 10.1016/j.jcyt.2015.07.011
414. Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK, et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol. (2008) 26:5896–903. doi: 10.1200/JCO.2007.15.6794
415. Little RF, Pluda JM, Wyvill KM, Rodriguez-Chavez IR, Tosato G, Catanzaro AT, et al. Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood. (2006) 107:4650–7. doi: 10.1182/blood-2005-11-4455
416. Robertson MJ, Kirkwood JM, Logan TF, Koch KM, Kathman S, Kirby LC, et al. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. (2008) 14:3462–9. doi: 10.1158/1078-0432.CCR-07-4740
417. Simpkins F, Flores A, Chu C, Berek JS, Lucci J, 3rd, Murray S, et al. Chemoimmunotherapy using pegylated liposomal Doxorubicin and interleukin-18 in recurrent ovarian cancer: a phase I dose-escalation study. Cancer Immunol Res. (2013) 1:168–78. doi: 10.1158/2326-6066.CIR-13-0098
418. Le DT, Burt B, Buren GV, Abeynaike S, Zalfa C, Nikzad R, et al. NK cells and CTLs are required to clear solid tumor in a novel model of patient-derived-xenograft. BioRxiv [Preprint]. (2020). doi: 10.1101/2020.05.24.112722
419. Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. (2018) 19:694–704. doi: 10.1016/S1470-2045(18)30148-7
420. Dubois S, Conlon KC, Müller JR, Hsu-Albert J, Beltran N, Bryant BR, et al. IL15 infusion of cancer patients expands the subpopulation of cytotoxic CD56(bright) NK cells and increases nk-cell cytokine release capabilities. Cancer Immunol Res. (2017) 5:929–38. doi: 10.1158/2326-6066.CIR-17-0279
421. Perez-Santos M, Guerrero-González T, Gómez-Conde E, Cebada J, Flores A, Villa-Ruano N. Treatment of cancer with an anti-KIR antibody: a patent evaluation of US9879082 and US2018208652. Expert Opin Ther Pat. (2020) 30:159–62. doi: 10.1080/13543776.2020.1717469
422. He Y, Liu S, Mattei J, Bunn PA Jr., Zhou C, et al. The combination of anti-KIR monoclonal antibodies with anti-PD-1/PD-L1 monoclonal antibodies could be a critical breakthrough in overcoming tumor immune escape in NSCLC. Drug Des Devel Ther. (2018) 12:981–6. doi: 10.2147/DDDT.S163304
423. Lanier LL, Follow the leader: NK cell receptors for classical nonclassical MHC class I. Cell. (1998) 92:705–7. doi: 10.1016/S0092-8674(00)81398-7
424. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. (2008) 9:495–502. doi: 10.1038/ni1581
425. Roder JC, Pross HF. The biology of the human natural killer cell. J Clin Immunol. (1982) 2:249–63. doi: 10.1007/BF00915064
426. Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. (2001) 1:41–9. doi: 10.1038/35095564
427. Miller JS. The biology of natural killer cells in cancer, infection, and pregnancy. Exp Hematol. (2001) 29:1157–68. doi: 10.1016/S0301-472X(01)00696-8
428. Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J. (2015) 21:486–91. doi: 10.1097/PPO.0000000000000156
429. Lee DA, Denman CJ, Rondon G, Woodworth G, Chen J, Fisher T, et al. Haploidentical natural killer cells infused before allogeneic stem cell transplantation for myeloid malignancies: a phase I trial. Biol Blood Marrow Transplant. (2016) 22:1290–8. doi: 10.1016/j.bbmt.2016.04.009
430. Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. (2019) 11:769. doi: 10.3390/cancers11060769
431. Bernareggi D, Pouyanfard S, Kaufman DS. Development of innate immune cells from human pluripotent stem cells. Exp Hematol. (2019) 71:13–23. doi: 10.1016/j.exphem.2018.12.005
432. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
433. Siegler EL, Zhu Y, Wang P, Yang L. Off-the-shelf CAR-NK cells for cancer immunotherapy. Cell Stem Cell. (2018) 23:160–1. doi: 10.1016/j.stem.2018.07.007
434. Chow VA, Gopal AK, Maloney DG, Turtle CJ, Smith SD, Ujjani CS, et al. Outcomes of patients with large B-cell lymphomas and progressive disease following CD19-specific CAR T-cell therapy. Am J Hematol. (2019) 94:E209–13. doi: 10.1182/blood-2018-99-110747
435. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. (2017) 168:724–40. doi: 10.1016/j.cell.2017.01.016
436. Wang W, Jiang J, Wu C. CAR-NK for tumor immunotherapy: clinical transformation and future prospects. Cancer Lett. (2020) 472:175–80. doi: 10.1016/j.canlet.2019.11.033
437. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
438. Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: recent innovations and future developments. Front Immunol. (2017) 8:631. doi: 10.3389/fimmu.2017.00631
439. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
440. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
441. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. (2017) 545:423–31. doi: 10.1038/nature22395
442. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
443. Sadelain M. Chimeric antigen receptors: driving immunology towards synthetic biology. Curr Opin Immunol. (2016) 41:68–76. doi: 10.1016/j.coi.2016.06.004
444. Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. (2007) 18:712–25. doi: 10.1089/hum.2007.028
445. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. (2014) 257:83–90. doi: 10.1111/imr.12125
446. González S, López-Soto A, Suarez-Alvarez B, López-Vázquez A, López-Larrea C. NKG2D ligands: key targets of the immune response. Trends Immunol. (2008) 29:397–403. doi: 10.1016/j.it.2008.04.007
447. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. (2004) 6:558–68. doi: 10.1593/neo.04316
448. Dasgupta S, Bhattacharya-Chatterjee M, O'Malley BW Jr., Chatterjee SK. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J Immunol. (2005) 175:5541–50. doi: 10.4049/jimmunol.175.8.5541
449. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res. (2019) 7:363–75. doi: 10.1158/2326-6066.CIR-18-0572
450. Barber A, Rynda A, Sentman CL. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J Immunol. (2009) 183:6939–47. doi: 10.4049/jimmunol.0902000
451. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
452. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. (2018) 32:520–31. doi: 10.1038/leu.2017.226
453. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92.e5. doi: 10.1016/j.stem.2018.06.002
454. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. (2019) 27:1114–25. doi: 10.1016/j.ymthe.2019.03.011
455. G. Del Zotto, Marcenaro E, Vacca P, Sivori S, Pende D, Della Chiesa M, et al. Markers and function of human NK cells in normal and pathological conditions. Cytometry B Clin Cytom. (2017) 92:100–14. doi: 10.1002/cyto.b.21508
456. Zhang C, Hu Y, Shi C. Targeting natural killer cells for tumor immunotherapy. Front Immunol. (2020) 11:60. doi: 10.3389/fimmu.2020.00060
457. Legg JA, Herbert JM, Clissold P, Bicknell R. Slits and Roundabouts in cancer, tumour angiogenesis and endothelial cell migration. Angiogenesis. (2008) 11:13–21. doi: 10.1007/s10456-008-9100-x
458. Kidd T, Brose K, Mitchell KJ, Fetter RD, Tessier-Lavigne M, Goodman CS, et al. Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell. (1998) 92:205–15. doi: 10.1016/S0092-8674(00)80915-0
459. Wang B, Xiao Y, Ding BB, Zhang N, Yuan X, Gui L, et al. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. (2003) 4:19–29. doi: 10.1016/S1535-6108(03)00164-8
460. Prasad A, Qamri Z, Wu J, Ganju RK. Slit-2/Robo-1 modulates the CXCL12/CXCR4-induced chemotaxis of T cells. J Leukoc Biol. (2007) 82:465–76. doi: 10.1189/jlb.1106678
461. He H, Di Y, Liang M, Yang F, Yao L, Hao S, et al. The microRNA-218 and ROBO-1 signaling axis correlates with the lymphatic metastasis of pancreatic cancer. Oncol Rep. (2013) 30:651–8. doi: 10.3892/or.2013.2516
462. Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med. (2019) 7:105. doi: 10.21037/atm.2019.01.42
463. Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. (2020) 135:399–410. doi: 10.1182/blood.2019000621
464. Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. (2020) 27:224–37. doi: 10.2139/ssrn.3544410
465. Pesce S, Greppi M, Grossi F, Del Zotto G, Moretta L, Sivori S, et al. PD/1-PD-Ls checkpoint: insight on the potential role of NK cells. Front Immunol. (2019) 10:1242. doi: 10.3389/fimmu.2019.01242
466. Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol. (2020) 11:167. doi: 10.3389/fimmu.2020.00167
Keywords: natural killer cells, myeloid derived suppressor cells, tumor mircroenvironment, natural killer cell immunotherapy, cancer
Citation: Zalfa C and Paust S (2021) Natural Killer Cell Interactions With Myeloid Derived Suppressor Cells in the Tumor Microenvironment and Implications for Cancer Immunotherapy. Front. Immunol. 12:633205. doi: 10.3389/fimmu.2021.633205
Received: 24 November 2020; Accepted: 12 February 2021;
Published: 05 May 2021.
Edited by:
Yumeng Mao, Uppsala University, SwedenReviewed by:
Hung-Chang Chen, University of Cambridge, United KingdomViktor Umansky, German Cancer Research Center (DKFZ), Germany
Copyright © 2021 Zalfa and Paust. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silke Paust, paust@scripps.edu