 Brooke A. Clemmons1
Brooke A. Clemmons1 Sydney T. Reese1
Sydney T. Reese1 Felipe G. Dantas1
Felipe G. Dantas1 Gessica A. Franco1
Gessica A. Franco1 Timothy P. L. Smith2Olusoji I. Adeyosoye3
Timothy P. L. Smith2Olusoji I. Adeyosoye3 Ky G. Pohler1*
Ky G. Pohler1* Phillip R. Myer1*
Phillip R. Myer1*- 1University of Tennessee Institute of Agriculture, University of Tennessee, Knoxville, TN, United States
- 2U.S. Meat Animal Research Center, Agricultural Research Service, United States Department of Agriculture, Clay Center, NE, United States
- 3Department of Animal Sciences, Obafemi Awolowo University, Ile-Ife, Nigeria
Reproductive inefficiency in cattle has major impacts on overall productivity of cattle operations, increasing cost of production, and impacting the sustainability of the cattle enterprise. Decreased reproductive success and associated disease states have been correlated with the presence of specific microbes and microbial community profiles, yet details of the relationship between microbial communities and host physiology are not well known. The present study profiles and compares the microbial communities in the bovine uterus and vagina using 16S rRNA sequencing of the V1–V3 hypervariable region at the time of artificial insemination. Significant differences (p < 0.05) between the vaginal and uterine communities were observed at the level of α-diversity metrics, including Chao1, Shannon’s Diversity Index, and observed OTU. Greater clustering of vaginal OTU was apparent in principal coordinate analysis compared to uterine OTU, despite greater diversity in the vaginal community in both weighted and unweighted UniFrac distance matrices (p < 0.05). There was a significantly greater relative abundance of unassigned taxa in the uterus (p = 0.008), otherwise there were few differences between the overall community profiles. Both vaginal and uterine communities were dominated by Firmicutes, although the relative abundance of rRNA sequences corresponding to species in this phylum was significantly (p = 0.007) lower in the uterine community. Additional differences were observed at the genus level, specifically in abundances within Clostridium (p = 0.009), Anaerofustis (p = 0.018), Atopobium (p = 0.035), Oscillospira (p = 0.035), 5-7N15 (p = 0.035), Mycoplasma (p = 0.035), Odoribacter (p = 0.042), and within the families Clostridiaceae (p = 0.006), Alcaligenaceae (p = 0.021), and Ruminococcaceae (p = 0.021). Overall, the comparison revealed differences and commonalities among bovine reproductive organs, which may be influenced by host physiology. The increased abundance of unassigned taxa found in the uterus may play a significant biological role in the reproductive status of the animal. The study represents an initial dataset for comparing bacterial communities prior to establishment of pregnancy.
Introduction
Reproductive efficiency is necessary for the survival of any species. Failure to reproduce or maintain pregnancy represents a significant cost to many species, including humans, exotic or endangered species, and domestic livestock. One in vitro fertilization cycle in humans averages ∼$12,000 (Thompson, 2016). In endangered or exotic species, breeding can often be a limiting factor, particularly in captivity, leading to possible extinction (Snyder et al., 1996). In domestic livestock, specifically cattle, reproductive loss and inefficiency results in ∼$600 million to $1.4 billion lost annually (Bellows et al., 2002; De Vries, 2006). While these losses are multifaceted and influenced by many factors such as disease states (Michi et al., 2016), male and female gametes (Druet et al., 2009; Valour et al., 2015), and genetics related to male and female fertility (Berry et al., 2014; Carthy et al., 2014), these factors cannot account for all reproductive related losses.
Human studies using next generation sequencing have revealed correlations between various reproductive issues in women, such as higher risk of preterm labor or fertility issues, and microbial factors, including presence of specific populations of microbes. One study found that lower abundances of Lactobacillus and increased bacterial diversity in the vaginas of pregnant women were associated with shorter gestational length (DiGiulio et al., 2015). The vaginal environment is an important aspect of the reproductive system because it represents the first obstacle to sperm, as well as the first barrier to protect the upper reproductive tract. Vaginal pH has been associated with different bacterial populations in humans, namely Lactobacillus, and is thought to aid in protection against infection (Boris and Barbés, 2000) providing evidence that the microbiota of a healthy woman may contribute significantly to the reproductive success and prevention of a diseased state. The study conducted by DiGiulio et al. (2015) concludes that such findings contribute toward a better understanding of the human body, possibly leading to novel methods for resolving or preventing reproductive issues or idiopathic infertility. At minimum, the presence or absence of specific microbial species might be useful indicators of pregnancy complications. Moreover, microbial biotechnology holds promise for combating some of these aforementioned reproductive issues (Huang et al., 2014; DiGiulio et al., 2015; Prince et al., 2016).
Culture-dependent methods examine only a small proportion of the overall microbial population (Amann et al., 1995; Wintzingerode et al., 1997), because many species of microbes, including those found in sensitive and unique microenvironments such as the uterus and vagina, cannot be cultured in a lab, possibly due to difficulty replicating their normal environment (Amann et al., 1995; Hugenholtz and Pace, 1996; Zoetendal et al., 2004). However, recent projects, based on next generation sequencing, such as the Human Microbiome Project, have propelled microbiome–host interaction research forward within the past decade, and that knowledge base continues to increase (Schuster, 2007; Human Microbiome Project Consortium, 2012).
Reproductive inefficiency in cattle can be extremely costly to producers. In dairy herds, economic based models have suggested that an average single pregnancy at conception is valued at US $278, and pregnancies lost after day 30 will cost a producer upward of US $550 (De Vries, 2006). While researchers continue to scrutinize mechanisms and approaches to reduce these losses and advance reproductive health, little has progressed regarding the microbiota within the reproductive tract of female bovines (Santos and Bicalho, 2012; Swartz et al., 2014; Stevenson et al., 2015). In addition to few studies conducted characterizing the microbiota of vaginal communities, fewer yet have interrogated uterine microbial communities in cattle. This limited research has predominantly been conducted in dairy cattle, which can vary significantly from beef cattle physiologically (Hart et al., 1975; Hayes et al., 2009; Knudsen et al., 2012; Santos and Bicalho, 2012). Determining the host-microbe relationship within the reproductive tract will provide stakeholders with better tools for improving reproductive success.
A few studies have examined both the uterine and vaginal microbiomes in humans, but little data exists about these microbiomes in livestock species or their potential role in reproductive success of animals. The influence of estrous cycles and their effects on these microbiomes are of particular interest in bovine reproductive physiology. For this study, the use of standard synchronization protocols for artificial insemination (AI), involving injections of hormones to bring a group of animals into similar hormonal status, reduces possible confounding effects due to varying hormonal levels and physiological states. The present study explores the potential of this approach, by focusing on vaginal and uterine microbiomes of synchronized animals 2 days prior to AI. These data are used to assess the approach of bacterial community profiling in the context of similar hormonal statuses and the same period in the bovine reproductive cycle. Non-pregnant cows were chosen for this initial screening to remove any influence that pregnancy, including hormonal and physiological factors, might have on the vaginal and uterine bacterial communities present. Ultimately, this foundational information could form part of a more comprehensive approach profiling reproductive tract microbiomes across the full reproductive cycle, and potentially create novel diagnostic criteria to elucidate likelihood of reproductive success.
Materials and Methods
Experimental Design and Sampling
This study was carried out in accordance with the recommendations of the Institutional Animal Care and Use Committee at the University of Tennessee, Knoxville. The protocol was approved by the University of Tennessee, Knoxville Institutional Animal Care and Use Committee.
All cows involved in this study were part of the East Tennessee Research and Education Center (ETREC) registered Angus herd. Thirty cows were selected from the fall calving herd ranging from 3 to 11 years of age with an average of 5 years at initiation of the study. Cows were an average of 82 days postpartum at AI. Cows included in the study had daughters retained at ETREC for use in future longitudinal studies of the microbiome. All cows were kept on cool-season grass pastures supplemented with corn silage for the duration of the study.
All 30 cows were inseminated following an industry-standard 7-Day Co-Synch Protocol, with the addition of a pre-synchronization step to help establish baseline cyclicity in the herd. Controlled internal release device (CIDR) were not used due to collection methods. The pre-synchronization protocol was performed 21 days prior to AI (D-21), by intramuscular injection of 5 mL of 25 mg/mL prostaglandin. A standard Co-Synch Protocol was then begun at D-9 by injection of 2 mL of 100 mcg/mL gonadotropin releasing hormone, to initiate a follicular wave. The animals were given an administration of 5 mL of 25 mg/mL prostaglandin on D-2 to regress the corpus luteum, at the same time that flushes for microbiome profiling were collected. The uterine and vaginal flush with saline solution in all cows were performed as follows: syringe injection of 60 mL into the vagina, followed by rectal massage to collect the fluid (vaginal flush); inflation of a Foley catheter in the uterus after passing through the vagina, to sequester the uterus from the vagina, followed by introduction of 180 mL saline and rectal massage to collect the fluid (uterine wash). All wash samples were stored at -80°C until used for microbiome profiling. The animals then had injection of 2 mL of 100 mcg/mL gonadotropin releasing hormone on D0 to induce ovulation and AI was performed using semen from the same sire for all cows. Animals were subjected to pregnancy check at D30, and 10 non-pregnant (open) cows were identified. Non-pregnant animals were used for this study to eliminate any possibility of the flushing technique having an effect on the pregnancy establishment, thus biasing the results. The flushes from these 10 open animals were used to obtain total DNA for 16S rRNA profiling. Immediately after flush, pH of each sample was measured using UltraBasic pH meter (Denver Instruments, Arvada, CO, United States).
DNA Extraction and Sequencing
Each flush sample was vortexed to homogenize, then a 5 mL aliquot was taken and centrifuged in 15 mL tubes at 4,696 ×g and 4°C, and pelleted material was resuspended in 180 μL of sterile saline solution to concentrate microbial cells. DNA was then extracted from the cell concentrate using the Qiagen DNEasy Blood and Tissue kit (Qiagen, Hilden, Germany) according to manufacturer instruction. Extractions were stored at -20°C until amplification. DNA was amplified using polymerase chain reaction (PCR) for 30 cycles at an annealing temperature of 58°C, targeting the V1–V3 hypervariable regions of the bacterial 16S rRNA gene. Modified universal primers 27F (5′-Adapter/Index/AGAGTTTGATCCTGGCTCAG) and 519R (5′-Adapter/Index/GTATTACCGCGGCTGCTG) including TruSeq indices and adapters were used with AccuPrime Taq high fidelity DNA Polymerase (Life Technologies, Carlsbad, CA, United States) to produce the sequencing libraries. Products were quality checked with gel electrophoresis. Libraries were then purified using AmPure beads (Agencourt, Beverly, MA, United States) and quantified using a Nanodrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, United States) and by real-time PCR on the LightCycler 480 system (Roche Diagnostics, Mannheim, Germany). The PCR amplicon libraries were sequenced using the 2 × 300, v3 600-cycle kit and the Illumina MiSeq sequencing platform (Illumina, Inc., San Diego, CA, United States) at the United States Meat Animal Research Center (US MARC; Clay Center, NE, United States).
Sequence Read Processing and Analysis
Sequence data is available from the NCBI Sequence Read Archive (SRA Accession SRP103314). Additional descriptive information is associated with NCBI BioProject PRJNA382146. Amplicon sequence reads were processed using the Quantitative Insights Into Microbial Ecology (QIIME) bioinformatics pipeline, version 1.9.1 (Caporaso et al., 2010) and Mothur version 1.36.1 (Schloss et al., 2009). Sequences were quality trimmed using the Galaxy server (Afgan et al., 2016) and those with a score ≥ Q25 were retained. Sequences that contained read lengths shorter than 300 bp were removed and adapters/index sequences were trimmed. Chimeric sequences were identified and filtered using usearch61 (Edgar, 2010). Sequences classified as chloroplasts and mitochondria were removed from the analysis. To avoid biases generated by differences in sequencing depth, each sample was subsampled to an even depth of 50,000 sequences. Operational taxonomic unit (OTU) picking was completed utilizing the cleaned subsamples and were clustered with a pairwise identity threshold of 97% using the UCLUST module from QIIME and further assigned to taxonomy using UCLUST and the Greengenes v13_8 16S rRNA database as a reference (Caporaso et al., 2010). Phylogenic trees were built with FastTree (Price et al., 2010) to determine α- and β-diversity metrics. Alpha-diversity was analyzed using observed species, Faith’s phylogenetic diversity, Shannon diversity, and Chao1 richness indices. Beta-diversity analyses were performed using weighted and unweighted UniFrac distance matrices, as implemented in QIIME. Principal coordinates analysis (PCoA) was performed using weighted and unweighted UniFrac analyses (Lozupone and Knight, 2005). Heatmaps were constructed utilizing OTU found within ≥50% of samples and at a relative abundance of ≥0.01%.
Statistical Analysis
All variables were tested for normality using the PROC UNIVARIATE procedure in SAS 9.4 (SAS Institute, Cary, NC, United States). Differences in pH and bacterial community characteristics by normally distributed variables (number of observed OTU, and Chao1) were statistically analyzed by a one-way analysis of variance (ANOVA) for multiple independent groups (Weiss et al., 2017). Differences which do not follow a normal distribution and all multiple-group comparisons (Shannon’s index, Faith’s phylogenetic diversity and relative abundances of taxonomic profiles) were completed using the Kruskal–Wallis H test with Benjamini–Hochberg FDR multiple test correction (Weiss et al., 2017). Where appropriate, reported p-values are those corrected for multiple testing. Correlations were performed in SAS 9.4 using the PROC CORR procedure. Spearman’s correlation was used for all non-parametric data. For all analyses, the significance level was set at 0.05. Abundance values reported in the results and in the additional files are reported as mean relative abundances for the environment groups along with the standard error. Statistical differences in bacterial community composition between environments were calculated using analysis of similarity (ANOSIM) with 9999 permutations and principal coordinate analysis (PCoA) of weighted and unweighted UniFrac distances within QIIME.
Results
Uterine and Vaginal pH
All uterine and vaginal samples were measured for pH. Vaginal pH ranged from 6.15 to 7.44 with a mean of 6.69 ± 0.14 for individual vaginal samples. Uterine pH ranged from 5.62 to 6.52 with a mean of 6.06 ± 0.09. Individual uterine and vaginal pH data is available in Supplementary Material S1. Differences in pH did exist between reproductive environments (p = 0.0016), with a greater pH in the vagina than in the uterus. There was a weak negative correlation between the uterine and vaginal pH values (r = -0.2242; p = 0.5334).
Sequencing Information
A total of 20 samples were processed for DNA, including vaginal and uterine samples from 10 non-pregnant animals at D-2 prior to AI. Open animals were chosen to establish native uterine and vaginal bacteriomes of cows undergoing timed AI. Bacterial communities were analyzed by amplifying and sequencing the V1–V3 hypervariable regions of the 16S rRNA gene. After quality control and chimera removal, a total of 2,080,553 remained among all 20 samples, for an average of 100,000 per sample (Table 1). Total number of cleaned sequences from uterine samples were 1,149,373 sequences and ranged from 97,855 sequences to 149,286 sequences for individual uterine samples. Total number of cleaned sequences from the vaginal samples were 931,180 sequences and ranged from 63,302 sequences to 125,486 sequences for individual vaginal samples. Average number of cleaned sequences with standard error are presented in Table 1.
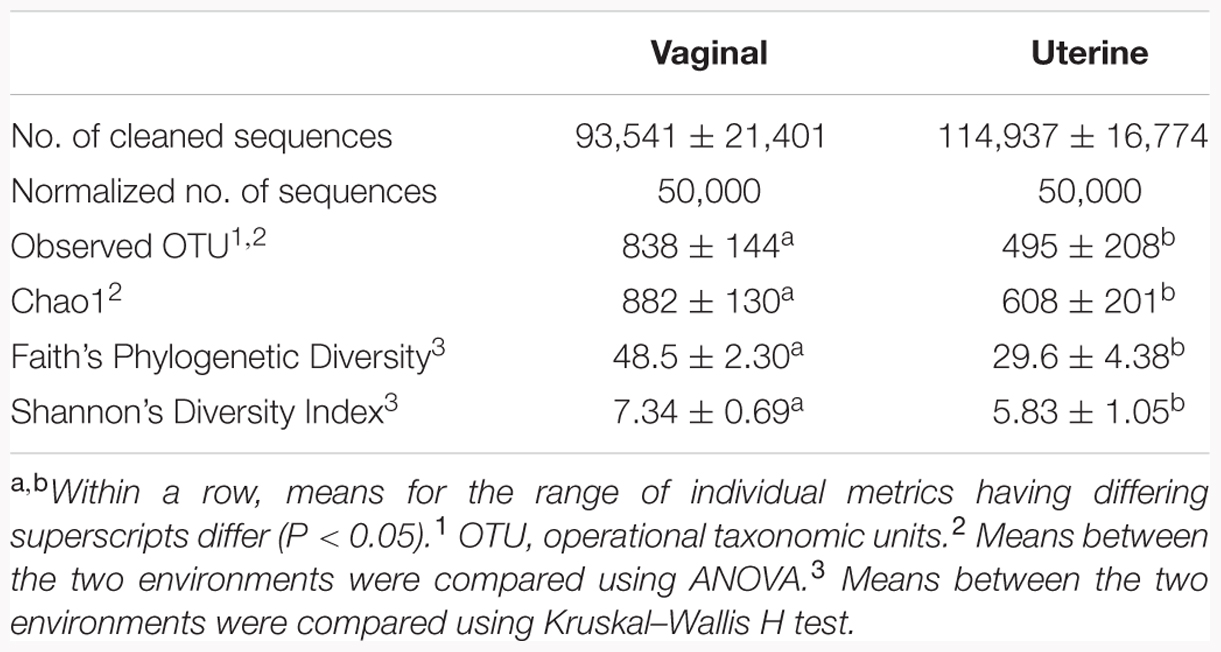
TABLE 1. Sequence and alpha-diversity statistics of the 16S rRNA gene sequences for bacterial populations in the vaginal and uterine environments.
Alpha- and Beta-Diversity
After binning reads at 97% similarity, a total of 13,328 OTU were detected, with an average of 838 and 495 OTU for the vaginal and uterine samples, respectively. Alpha-diversity was measured using observed OTU, Chao1, Faith’s Phylogenetic Diversity and Shannon’s Diversity Index, and is presented in Table 1. Significant differences existed in alpha-diversity between the uterine and vaginal bacterial observed OTU, Chao1, Faith’s Phylogenetic Diversity and Shannon’s Diversity Index (p < 0.05). The vagina had a significantly greater number of OTU than did the uterus, increased richness as measured by Chao1, and greater diversity as measured by Shannon’s Diversity Index and Faith’s Phylogenetic Diversity all of which are presented in Table 1. Core OTU are found in 100% of samples from each environment, and the shared core phylotypes are those core OTU present in both the uterus and vagina. The uterus contained 76 core OTU, the vagina had 279 core OTU, and of those core OTU, 36 OTU were shared between the environments.
Beta-diversity was also analyzed to examine differences in microbial communities between samples. Using an OTU-centric approach, PCoA matrices were employed using weighted and unweighted UniFrac distance matrices to compare the phylogenetic divergence among the OTU between vaginal and uterine samples (Figure 1). While vaginal samples contained a greater phylogenetic diversity and greater number of bacterial OTU, their spatial heterogeneity was reduced and thus tended to cluster more closely than the uterine samples in both the weighted and unweighted UniFrac distance matrices, where uterine samples had a greater spatial heterogeneity. Specifically, ANOSIM testing of the samples revealed that the compositions of the bacterial communities differed significantly (p = 0.005) between the uterus and vagina. The positive value of the R statistic (R = 0.1538) indicates clustering of samples based on the region interrogated, however, proximity of the value to zero supports the greater spatial heterogeneity of the samples observed in the PCoA. Overall, although the ANOSIM indicates significance, the R statistic dictates that the clustering is only moderate.
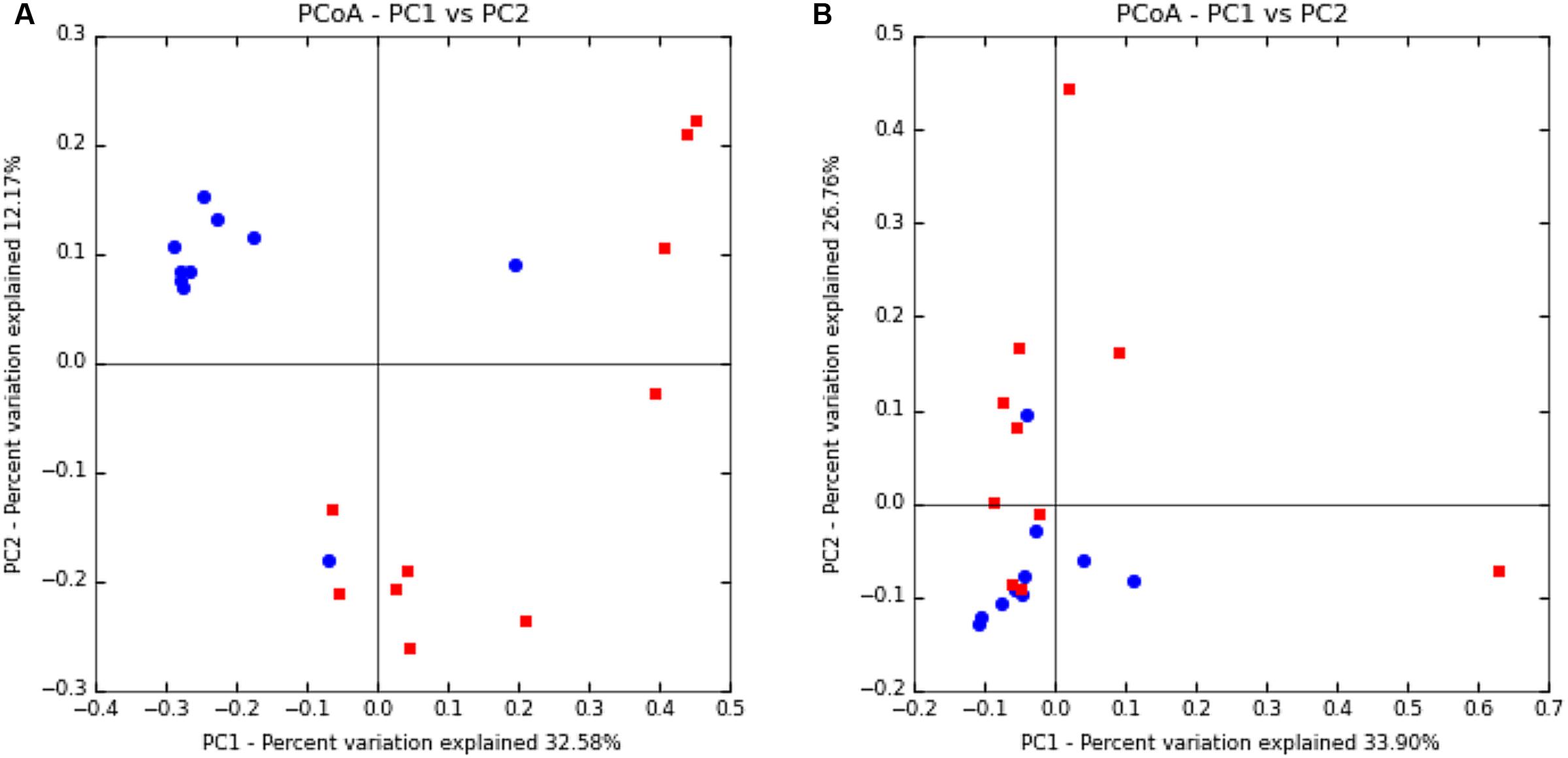
FIGURE 1. Principal coordinate analysis of uterine and vaginal samples using UniFrac unweighted (A) and weighted (B) metrics. Samples were analyzed from rarefied subsets of 50,000 sequences from each sample. Uterine samples (n = 10) are represented by red squares and vaginal samples (n = 10) are represented by blue circles.
Taxonomic Composition
In both the uterus and vagina, Firmicutes was the most dominant phyla with an average relative abundance of 31.3 ± 5.6% and 65.9 ± 4.1%, respectively, across all samples. In the uterus, the next most prevalent bacterial phyla based on relative abundance were Proteobacteria (22.9 ± 7.7%), Actinobacteria (13.2 ± 6.7%), and Bacteroidetes (8.5 ± 1.9%), with all other phyla present at <1% and unassigned taxa at 16.1 ± 4.6%. In the vagina, the other dominant phyla besides Firmicutes consisted of Bacteroidetes (16.8 ± 2.7%), Proteobacteria (7.4 ± 3.8%), Tenericutes (2.8 ± 1.5%), and Actinobacteria (2.3 ± 0.8%), with all other phyla representing <1% relative abundance and unassigned taxa at 3.4 ± 1.1%. The only significantly different phyla between the uterus and vagina were Firmicutes, which were greater in abundance in vaginal samples (p = 0.007), and unassigned taxa, which were reduced in abundance in vaginal samples (p = 0.008). A visual distribution of the phyla across cows and environment are available in Figure 2. Additional information containing phylum- and genus-level relative abundances as well as statistical information can be found in Supplementary Material S2.
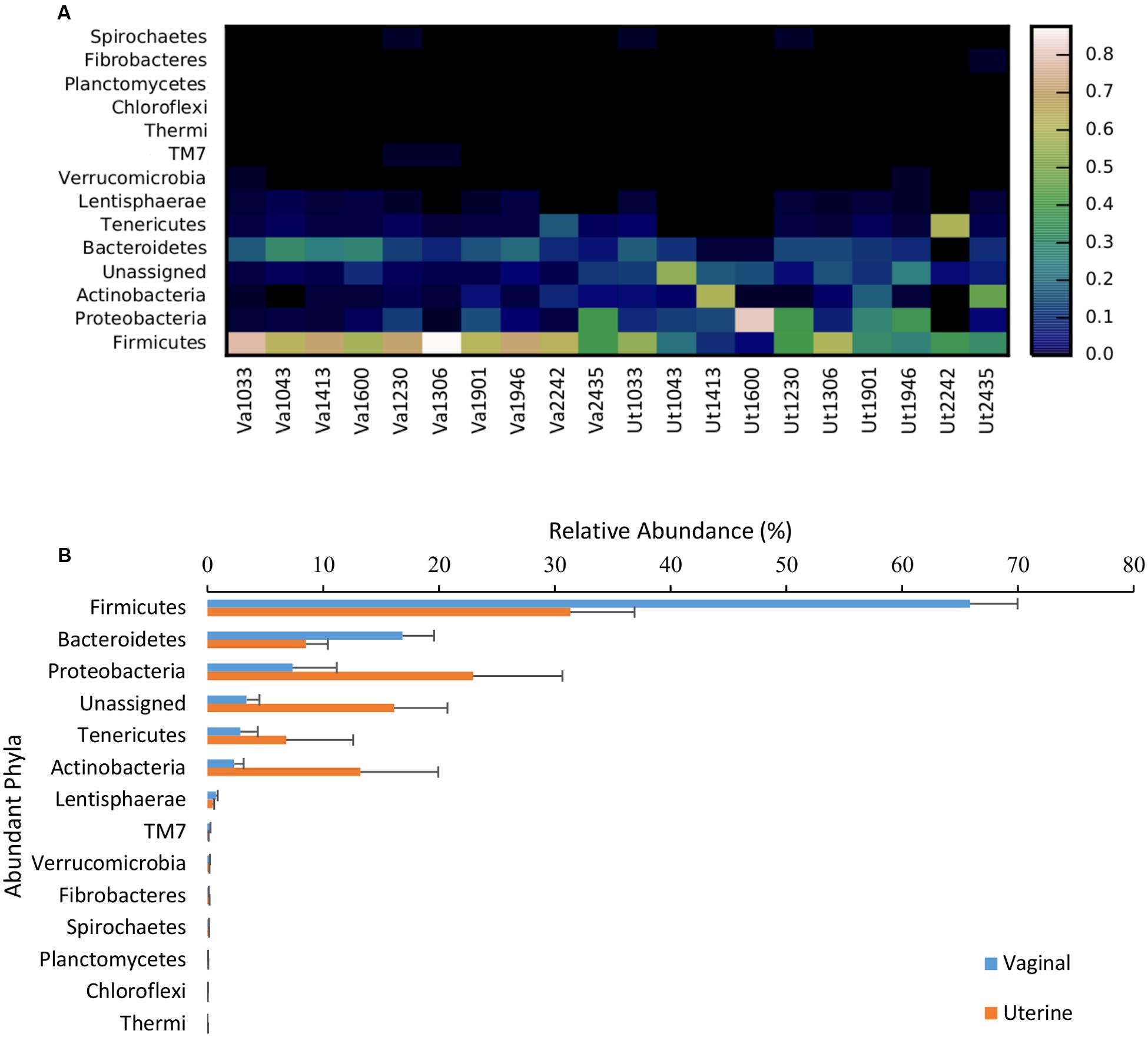
FIGURE 2. Relative abundance of bacterial phyla present in the uterus and vagina among samples (heatmap; A) and as a function of region (bar graph; B). Data consisted of phyla present in ≥0.01% relative abundance in ≥50% of the vaginal and uterine samples. Uterine (Ut) and Vaginal (Va) samples are denoted by animal ID.
At the genus level, the most prevalent genera in the uterus representing ≥1% relative abundance of the total sequences included Corynebacterium (10.6% ± 5.6), Ureaplasma (6.1% ± 5.9), Staphylococcus (3.7% ± 3.5), Microbacterium (1.6% ± 1.6), Butyrivibrio (1.4% ± 0.5), and Helcococcus (1.3% ± 1.0), which are represented in the heatmap in Figure 3A. The predominant genera in the vagina were an undetermined genus and family of the order Bacteroidales (3.4% ± 0.5), followed by genus 5-7N15 of family Bacteroidaceae (3.4% ± 0.6), and genera Oscillospira (2.0% ± 0.2), Butyrivibrio (1.8% ± 0.3), Ureaplasma (1.8% ± 1.6), Campylobacter (1.7% ± 1.4), Dorea (1.6% ± 0.2), CF231 of family Paraprevotellaceae (1.5% ± 0.3), Clostridium (1.3% ± 0.1), Helcococcus (1.2% ± 0.5), and Corynebacterium (1.0% ± 0.6), as presented in Figure 3B. In contrast to analysis at the phylum level, where only Firmicutes were significantly different between vagina and uterus, several significantly different genera were identified between the two environments. At the genus level, the vagina contained significantly greater levels of undetermined genera of the family Clostridiaceae (p = 0.006), Clostridium (p = 0.009), Anaerofustis (p = 0.018), undetermined genera of family Ruminococcaceae (p = 0.021), Atopobium (p = 0.035), Oscillospira (p = 0.035), 5-7N15 of family Bacteroidaceae (p = 0.035), Mycoplasma (p = 0.035), and Odoribacter (p = 0.042). The uterus contained a greater relative abundance of undetermined genera of family Alcaligenaceae (p = 0.021).
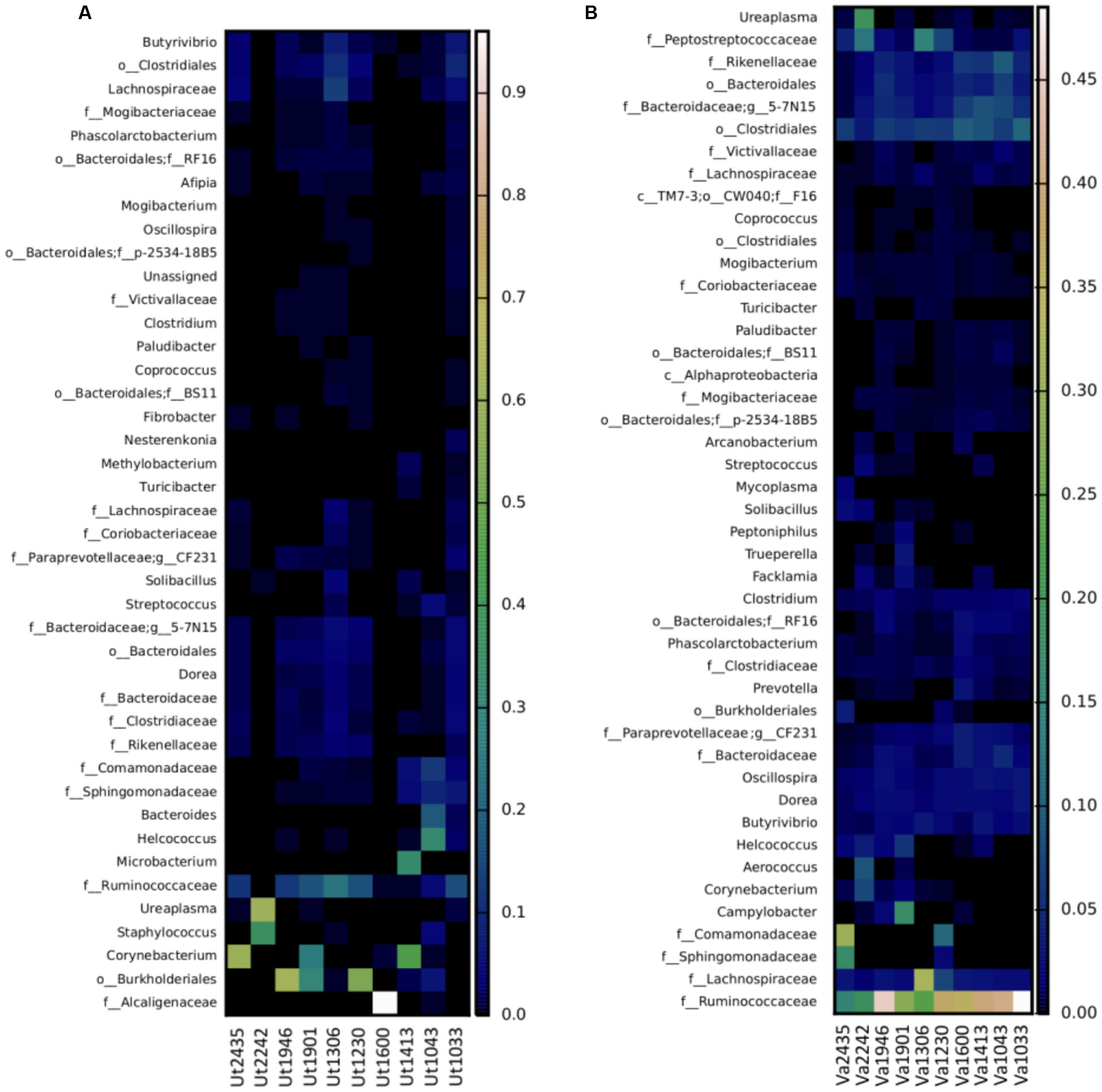
FIGURE 3. Heatmap of the taxonomic profile for bacterial genus-level relative abundance in uterine (A) and vaginal (B) samples. Data consisted of genera present in ≥0.01% relative abundance in ≥50% of the vaginal and uterine samples. Uterine (Ut) and Vaginal (Va) samples are denoted by animal ID.
The 16S rRNA sequences that could not be classified to the genus level across the uterine samples, included those in families Ruminococcaceae (10.89% ± 2.7), Alcaligenaceae (8.1% ± 8.0), Lachnospiraceae (2.3% ± 0.8), Sphingomonadaceae (1.3% ± 0.5), Comamonadaceae (1.2% ± 0.6), Bacteroidaceae (1.2% ± 0.3), and Rikenellaceae (1.1% ± 0.3). In some samples, genera and families could not be identified, including orders Burkholderiales (9.6% ± 4.8), Clostridiales (3.5% ± 1.0), and Bacteroidales (2.0% ± 0.4). Vaginal samples that contained genera that could not be identified or placed into the undefined category included family Ruminococcaceae (32.6% ± 3.5), undetermined genera and family member of the order Clostridiales (7.1% ± 0.6), family Lachnospiraceae (5.7% ± 2.4), family Peptostreptococcaceae (3.7% ± 1.3), family Rikenellaceae (3.3% ± 0.7), family Comamonadaceae (2.8% ± 2.1), family Sphingomonadaceae (1.3% ± 1.1), family Clostridiaceae (1.2% ± 0.2), and family Bacteroidaceae (1.8% ± 0.4).
Discussion
In humans and other animals, healthy microbial ecosystems within the body contribute to overall health and wellness of the host organism (Human Microbiome Project Consortium, 2012; Shreiner et al., 2015). While many studies have been conducted to identify bacterial and other microbial communities in the human vagina and uterus, few have interrogated these communities in cattle. Knowledge of microbiome influences in beef cattle stems primarily from studies conducted in the gastrointestinal tract, with little emphasis on the microbiome of the reproductive tract. This study aimed to conduct foundational research to interrogate the uterine and vaginal bacteriomes of cows undergoing estrus synchronization. Understanding bacterial communities in the reproductive tracts of cows will provide foundational data in order to ultimately determine how these factors are correlated with reproductive success. An increased comprehension of these communities may lead to novel approaches to improving reproduction, such as treatment with probiotics, to introduce microbial communities that result in positive results with regards to reproduction.
The most abundant bacterial phyla identified in the uterus in this study were, in order of abundance, Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes, which corresponds with dominant phyla present in the vagina, with the exception of the presence of Tenericutes in vaginal communities. These phyla are representative of the most common phyla found in many environments, particularly in host-microbiome relationships. The uterine bacterial communities contained significantly greater unassigned or unidentified taxa than did the vaginal communities. Unassigned taxa accounted for 16.1% relative abundance in the uterus, compared to only 3.4% unassigned OTU in the vagina. The lack of existing studies of the uterine bacterial communities may indicate that many of these populations have yet to be determined, including their role in uterine physiology and possible correlations with reproductive health and success.
The common phyla found between the uterus and vagina are those found most commonly in many host-microbiome relationships in many species (Turnbaugh et al., 2009; Human Microbiome Project Consortium, 2012; Jami and Mizrahi, 2012; Myer et al., 2015); however, the ratios and relative abundances of these phyla are correlated with changes in host physiology. These common phyla have been shown to be independent of genetic lineage or sex (Ley et al., 2005). While the mechanisms explaining these relationships are not well characterized, there is a clear relationship between host phenotype and presence of different bacterial communities, abundances, and diversity in many species (Human Microbiome Project Consortium, 2012; Le Chatelier et al., 2013; Jami et al., 2014; Romero et al., 2014). Increased diversity in the microbiome has been associated with negative phenotypes in some species (Turnbaugh et al., 2009; Hyman et al., 2014; Durso et al., 2015). In contrast, stability of bacterial communities in the vagina have been associated with a healthy female reproductive tract (Gajer et al., 2012; Human Microbiome Project Consortium, 2012; DiGiulio et al., 2015).
Core OTU are those that are found across a habitat (Turnbaugh et al., 2007), and core OTU present a unique opportunity to identify those bacteria acting as keystone bacteria or functional groups that contribute to certain phenotypes in animals. While greater abundance of specific OTU does not necessarily indicate that those OTU perform necessary functions, it could provide insight into the health and maintenance of the animal. In mice and humans, core bacterial communities have been associated with phenotypic variation (Ley et al., 2005; Turnbaugh et al., 2009). Shared OTU between the uterus and vagina suggests that there is some interaction between the bacterial communities of the uterus and vagina. Yet, the core OTU differences between the uterus and vagina are likely a result of the functional differences associated with the tissues and microbial ecosystem niche.
Besides unassigned taxa, the uterus contained significantly greater abundances of unidentified genera in family Alcaligenaceae compared to the vagina. This family is predominantly aerobic, though some members of the family can use nitrate or nitrite as an electronic acceptor in lieu of oxygen (De Ley et al., 1986) since the uterus is predominantly an anaerobic environment. Heifers and cows can be grouped based on the level of aerobic bacterial communities in the vagina, regardless of age or pregnancy status (Laguardia-Nascimento et al., 2015). This suggests that variation among individuals, including anatomical and physiological differences, may influence the microbiome of the reproductive tract (Laguardia-Nascimento et al., 2015). Additional studies or analyses could be conducted to determine the impact of cow-specific differences on microbiome establishment.
Greater levels of diversity and increased richness in the vagina when compared with the uterus may be explained by its proximity to the external environment. The uterus is significantly more restricted from external exposure compared to the vagina (Sheldon et al., 2002). Protection of the uterine environment by the vagina and cervix may contribute to the decreased diversity present in the uterine cavity. The vaginal microbiome has been demonstrated to combat infectious bacteria, likely due to regulation of the vaginal pH (Boskey et al., 1999; Boris and Barbés, 2000; Brabin et al., 2005; van Oostrum et al., 2013; Huang et al., 2014). Maintaining a low vaginal pH prevents colonization of pathogenic microbes, which in turn can positively impact fertility (Boskey et al., 1999; Boris and Barbés, 2000; O’Hanlon et al., 2013).
Some bacteria may be responsible for maintaining the health of the vagina, which could in turn promote a healthy uterus. The relative abundances of OTU that were greater in the vagina than uterus are those that are abundant within the gastrointestinal tract or feces, particularly those in the genus Clostridium. The greater abundances of these bacteria in the vagina may be due to the anatomy of the cow’s reproductive tract but some mechanism likely reduces their presence in the uterus (Songer, 1996; Singh et al., 2008). Mechanisms may include a physical barrier provided by the cervix (Singer and Jordan, 2009; Nott et al., 2016); competition with other microbes more suited to the environment (Recine et al., 2016); antagonistic environment created by mutualistic microbes in coordination with the host (Klebanoff et al., 1991; Boskey et al., 2001; Kaewsrichan et al., 2006); or simply a constant introduction of these bacteria in the vagina from feces, providing constant opportunities for colonization.
Given the critical role that bacteria have in maintaining homeostasis within the uterine and vaginal environment (Mendez-Figueroa and Anderson, 2011), it is possible that microbiota associated with reproductive tracts may significantly affect the pregnancy rates and reproductive success in cows. Women with bacterial vaginosis (BV) have a greater incidence of infertility (van Oostrum et al., 2013). Cows and heifers that experience fertility or pregnancy maintenance issues may have different microbiota or proportions of core microbiota present in the reproductive tract. Additional research should aim to further analyze the microbiome of the uterus and vagina in cattle to determine relationships between presence (or absence) of certain microbiota and fertility rates. Such research would be utilized to document variation of the different abundances of microbiota in reproductive tracts of pregnant and non-pregnant cows, as well as identifying potentially unique microbes that affiliate with high versus low levels of fertility. It may be beneficial to also determine the microbiome changes associated with hormone shifts that occur throughout the estrous cycle, as these changes have been observed in humans (Human Microbiome Project Consortium, 2012). Determining the differences in bacterial and other microbial populations between cows and heifers with high and low fertility may allow researchers to manipulate the microbiota in cows with low fertility and increase their pregnancy rates, or use marker microbes as predictors of fertility in cows and heifers.
Determining methods to increase success of pregnancy and enhance fertility in domestic livestock would result in millions of dollars saved to producers annually. Providing foundational information and establishing baselines for native vaginal and uterine bacteriomes and complete microbiomes facilitates study of the impact of microbes on reproductive efficiency. In order to determine anomalies in the microbial communities that may lead to interference with successful pregnancies, healthy baselines should be established and shared throughout scientific and industrial communities. This study characterizes the native bacterial communities found in cows undergoing timed AI during a specific hormonal state. Future research may aim to determine the microbial communities during further stages of synchronization, or analyses to correlate the microbiome of the uterus and vagina with increased risk of disease, varying hormone levels throughout the reproductive cycle, changes introduced from sexual partners, or AI methods.
Author Contributions
BC was responsible for primary manuscript preparation, sampling, and data and statistical analyses. SR, FD, GF, and OA were responsible for sampling and manuscript preparation. TS was responsible for DNA sequencing, sequencing analyses, and manuscript preparation. KP and PM were responsible for experimental design development, sampling, data and statistical analyses, and manuscript preparation.
Funding
This study was supported by the Angus Foundation (Award A16-1356-001) and USDA-NIFA Hatch/Multistate Project W3112-TEN00506 – Reproductive performance in domestic ruminants. Mention of a trade name, proprietary product, or specific equipment does not constitute a guarantee or warranty by the USDA and does not imply approval to the exclusion of other products that may be suitable. USDA is an equal opportunity provider and employer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors would like to acknowledge and thank the technical support of Sue Hauver.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01047/full#supplementary-material
References
Afgan, E., Baker, D., van den Beek, M., Blankenberg, D., Bouvier, D., Čech, M., et al. (2016). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44, W3–W10. doi: 10.1093/nar/gkw343
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
Bellows, D., Ott, S., and Bellows, R. (2002). Review: cost of reproductive diseases and conditions in cattle1. Prof. Anim. Sci. 18, 26–32. doi: 10.15232/S1080-7446(15)31480-7
Berry, D. P., Wall, E., and Pryce, J. E. (2014). Genetics and genomics of reproductive performance in dairy and beef cattle. Animal 8, 105–121. doi: 10.1017/S1751731114000743
Boris, S., and Barbés, C. (2000). Role played by lactobacilli in controlling the population of vaginal pathogens. Microbes Infect. 2, 543–546. doi: 10.1016/S1286-4579(00)00313-0
Boskey, E. R., Cone, R. A., Whaley, K. J., and Moench, T. R. (2001). Origins of vaginal acidity: high D/L lactate ratio is consistent with bacteria being the primary source. Hum. Reprod. 16, 1809–1813. doi: 10.1093/humrep/16.9.1809
Boskey, E. R., Telsch, K. M., Whaley, K. J., Moench, T. R., and Cone, R. A. (1999). Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect. Immun. 67, 5170–5175.
Brabin, L., Roberts, S. A., Fairbrother, E., Mandal, D., Higgins, S. P., Chandiok, S., et al. (2005). Factors affecting vaginal pH levels among female adolescents attending genitourinary medicine clinics. Sex. Trans. Infect. 81, 483–487. doi: 10.1136/sti.2005.014621
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Method 7, 335–336. doi: 10.1038/nmeth.f.303
Carthy, T., Berry, D. P., Fitzgerald, A., McParland, S., Williams, E., Butler, S. T., et al. (2014). Risk factors associated with detailed reproductive phenotypes in dairy and beef cows. Animal 8, 695–703. doi: 10.1017/S1751731114000354
De Ley, J., Segers, P., Kersters, K., Mannheim, W., and Lievens, A. (1986). Intra- and intergeneric similarities of the bordetella ribosomal ribonucleic acid cistrons: proposal for a new family, alcaligenaceae. Int. J. Syst. Evol. Microbiol. 36, 405–414. doi: 10.1099/00207713-36-3-405
De Vries, A. (2006). Economic value of pregnancy in dairy cattle1. J. Dairy Sci. 89, 3876–3885. doi: 10.3168/jds.S0022-0302(06)72430-4
DiGiulio, D. B., Callahan, B. J., McMurdie, P. J., Costello, E. K., Lyell, D. J., Robaczewska, A., et al. (2015). Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. U.S.A. 112, 11060–11065. doi: 10.1073/pnas.1502875112
Druet, T., Fritz, S., Sellem, E., Basso, B., Gerard, O., Salas-Cortes, L., et al. (2009). Estimation of genetic parameters and genome scan for 15 semen characteristics traits of Holstein bulls. J. Anim. Breed. Genet. 126, 269–277. doi: 10.1111/j.1439-0388.2008.00788.x
Durso, L., Wells, J. E., and Kim, M. S. (2015). “Diversity of microbiomes in beef cattle,” in Encyclopedia of Metagenomics: Environmental Metagenomics, eds S. K. Highlander, F. Rodriguez-Valera, and B. A. White (Boston: Springer), 129–138.
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Gajer, P., Brotman, R. M., Bai, G., Sakamoto, J., Schutte, U. M., and Zhong, X. (2012). Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 4:132ra52. doi: 10.1126/scitranslmed.3003605
Hart, I. C., Bines, J. A., Balch, C. C., and Cowie, A. T. (1975). Hormone and metabolite differences between lactating beef and dairy cattle. Life Sci. 16, 1285–1291. doi: 10.1016/0024-3205(75)90313-6
Hayes, B. J., Chamberlain, A. J., Maceachern, S., Savin, K., McPartlan, H., MacLeod, I., et al. (2009). A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim. Genet. 40, 176–184. doi: 10.1111/j.1365-2052.2008.01815.x
Huang, B., Fettweis, J. M., Brooks, J. P., Jefferson, K. K., and Buck, G. A. (2014). The changing landscape of the vaginal microbiome. Clin. Lab. Med. 34, 747–761. doi: 10.1016/j.cll.2014.08.006
Hugenholtz, P., and Pace, N. R. (1996). Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends Biotechnol. 14, 190–197. doi: 10.1016/0167-7799(96)10025-1
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Hyman, R. W., Fukushima, M., Jiang, H., Fung, E., Rand, L., Johnson, B., et al. (2014). Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 21, 32–40. doi: 10.1177/1933719113488838
Jami, E., and Mizrahi, I. (2012). Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 7:e33306. doi: 10.1371/journal.pone.0033306
Jami, E., White, B. A., and Mizrahi, I. (2014). Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 9:e85423. doi: 10.1371/journal.pone.0085423
Kaewsrichan, J., Peeyananjarassri, K., and Kongprasertkit, J. (2006). Selection and identification of anaerobic lactobacilli producing inhibitory compounds against vaginal pathogens. FEMS Immunol. Med. Microbiol. 48, 75–83. doi: 10.1111/j.1574-695X.2006.00124.x
Klebanoff, S. J., Hillier, S. L., Eschenbach, D. A., and Waltersdorph, A. M. (1991). Control of the microbial flora of the vagina by H2O2-generating lactobacilli. J. Infect. Dis. 164, 94–100. doi: 10.1093/infdis/164.1.94
Knudsen, L. R. V., Rasmussen, E. L., Schou, K. K., Pedersen, H. G., Angen,Ø., Agerholm, J. S. et al. (2012). “Investigating the microbiome of the bovine uterus in relation to endometritis, a costly disease for dairy farmers,” in Proceedings of the 14th International Symposium on Microbial Ecology, Bella Center.
Laguardia-Nascimento, M., Branco, K. M., Gasparini, M. R., Giannattasio-Ferraz, S., Leite, L. R., Araujo, F. M., et al. (2015). Vaginal microbiome characterization of nellore cattle using metagenomic analysis. PLoS ONE 10:e0143294. doi: 10.1371/journal.pone.0143294
Le Chatelier, E., Nielsen, T., Qin, J., Prifti, E., Hildebrand, F., Falony, G., et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546. doi: 10.1038/nature12506
Ley, R. E., Bäckhed, F., Turnbaugh, P., Lozupone, C. A., Knight, R. D., and Gordon, J. I. (2005). Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U.S.A. 102, 11070–11075. doi: 10.1073/pnas.0504978102
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Mendez-Figueroa, H., and Anderson, B. (2011). Vaginal innate immunity: alteration during pregnancy and its impact on pregnancy outcomes. Exp. Rev. Obstet. Gynecol. 6, 629–641. doi: 10.1586/eog.11.63
Michi, A. N., Favetto, P. H., Kastelic, J., and Cobo, E. R. (2016). A review of sexually transmitted bovine trichomoniasis and campylobacteriosis affecting cattle reproductive health. Theriogenology 85, 781–791. doi: 10.1016/j.theriogenology.2015.10.037
Myer, P. R., Smith, T. P. L., Wells, J. E., Kuehn, L. A., and Freetly, H. C. (2015). Rumen microbiome from steers differing in feed efficiency. PLoS ONE 10:e0129174. doi: 10.1371/journal.pone.0129174
Nott, J. P., Bonney, E. A., Pickering, J. D., and Simpson, N. A. B. (2016). The structure and function of the cervix during pregnancy. Trans. Res. Anat. 2, 1–7. doi: 10.1016/j.tria.2016.02.001
O’Hanlon, D. E., Moench, T. R., and Cone, R. A. (2013). Vaginal pH and microbicidal lactic acid when lactobacilli dominate the microbiota. PLoS ONE 8:e80074. doi: 10.1371/journal.pone.0080074
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Prince, A. L., Ma, J., Kannan, P. S., Alvarez, M., Gisslen, T., Harris, R. A., et al. (2016). The placental microbiome is altered among subjects with spontaneous preterm birth with and without chorioamnionitis. Am. J. Obstet. Gynecol 214, 627.e1–627.e16. doi: 10.1016/j.ajog.2016.01.193
Recine, N., Palma, E., Domenici, L., Giorgini, M., Imperiale, L., Sassu, C., et al. (2016). Restoring vaginal microbiota: biological control of bacterial vaginosis. A prospective case–control study using Lactobacillus rhamnosus BMX 54 as adjuvant treatment against bacterial vaginosis. Arch. Gynecol. Obstet. 293, 101–107. doi: 10.1007/s00404-015-3810-2
Romero, R., Hassan, S. S., Gajer, P., Tarca, A. L., Fadrosh, D. W., and Nikita, L. (2014). The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2:4. doi: 10.1186/2049-2618-2-4
Santos, T. M., and Bicalho, R. C. (2012). Diversity and succession of bacterial communities in the uterine fluid of postpartum metritic, endometritic and healthy dairy cows. PLoS ONE 7:e53048. doi: 10.1371/journal.pone.0053048
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schuster, S. C. (2007). Next-generation sequencing transforms today’s biology. Nature 200, 16–18. doi: 10.1038/nmeth1156
Sheldon, I. M., Noakes, D. E., Rycroft, A. N., and Dobson, H. (2002). Effect of postpartum manual examination of the vagina on uterine bacterial contamination in cows. Vet. Rec. 151, 531–534. doi: 10.1136/vr.151.18.531
Shreiner, A. B., Kao, J. Y., and Young, V. B. (2015). The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 31, 69–75. doi: 10.1097/MOG.0000000000000139
Singer, A., and Jordan, J. A. (2009). The Functional Anatomy of the Cervix, the Cervical Epithelium and the Stroma. The Cervix. Hoboken, NJ: Blackwell Publishing Ltd, 13–37.
Singh, J., Murray, R. D., Mshelia, G., and Woldehiwet, Z. (2008). The immune status of the bovine uterus during the peripartum period. Vet. J. 175, 301–309. doi: 10.1016/j.tvjl.2007.02.003
Snyder, N. F., Derrickson, S. R., Beissinger, S. R., Wiley, J. W., Smith, T. B., Toone, W. D., et al. (1996). Limitations of captive breeding in endangered species recovery. Conserv. Biol. 10, 338–348. doi: 10.1046/j.1523-1739.1996.10020338.x
Songer, J. G. (1996). Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 9, 216–234.
Stevenson, J., Hill, S., Bridges, G., Larson, J., and Lamb, G. (2015). Progesterone status, parity, body condition, and days postpartum before estrus or ovulation synchronization in suckled beef cattle influence artificial insemination pregnancy outcomes. J. Anim. Sci. 93, 2111–2123. doi: 10.2527/jas.2014-8391
Swartz, J. D., Lachman, M., Westveer, K., O’Neill, T., Geary, T., Kott, R. W., et al. (2014). Characterization of the vaginal microbiota of ewes and cows reveals a unique microbiota with low levels of Lactobacilli and near-neutral pH. Front. Vet. Sci. 1:19. doi: 10.3389/fvets.2014.00019
Thompson, C. (2016). IVF global histories, USA: between rock and a marketplace. Reprod. Biomed. Soc. Online 2, 128–135. doi: 10.1016/j.rbms.2016.09.003
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C., Knight, R., and Gordon, J. I. (2007). The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature 449:804. doi: 10.1038/nature06244
Valour, D., Michot, P., Eozenou, C., Lefebvre, R., Bonnet, A., Capitan, A., et al. (2015). Dairy cattle reproduction is a tightly regulated genetic process: highlights on genes, pathways, and biological processes. Anim. Front. 5, 32–41. doi: 10.2527/af.2015-0006
van Oostrum, N., De Sutter, P., Meys, J., and Verstraelen, H. (2013). Risks associated with bacterial vaginosis in infertility patients: a systematic review and meta-analysis. Hum. Reprod. 28, 1809–1815. doi: 10.1093/humrep/det096
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5:27. doi: 10.1186/s40168-017-0237-y
Wintzingerode, F. V., Göbel, U. B., and Stackebrandt, E. (1997). Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 21, 213–229. doi: 10.1111/j.1574-6976.1997.tb00351.x
Keywords: uterine, vaginal, bacteriome, cow, reproductive success
Citation: Clemmons BA, Reese ST, Dantas FG, Franco GA, Smith TPL, Adeyosoye OI, Pohler KG and Myer PR (2017) Vaginal and Uterine Bacterial Communities in Postpartum Lactating Cows. Front. Microbiol. 8:1047. doi: 10.3389/fmicb.2017.01047
Received: 02 February 2017; Accepted: 24 May 2017;
Published: 08 June 2017.
Edited by:
David Berry, University of Vienna, AustriaReviewed by:
David William Waite, University of Queensland, AustraliaItzhak Mizrahi, Ben-Gurion University of the Negev, Beersheba, Israel
Copyright © 2017 Clemmons, Reese, Dantas, Franco, Smith, Adeyosoye, Pohler and Myer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ky G. Pohler, kpohler@utk.edu Phillip R. Myer, pmyer@utk.edu