 Todd McLaughlin
Todd McLaughlin Narayan Dhimal1
Narayan Dhimal1 Sarah Xin Zhang
Sarah Xin Zhang- 1Departments of Ophthalmology and Biochemistry, Ross Eye Institute, University at Buffalo, State University of New York, Buffalo, NY, United States
- 2SUNY Eye Institute, State University of New York, Buffalo, NY, United States
p58IPK is an endoplasmic reticulum (ER)-resident chaperone playing a critical role in facilitating protein folding and protein homeostasis. Previously, we have demonstrated that p58IPK is expressed broadly in retinal neurons including retinal ganglion cells (RGCs) and loss of p58IPK results in age-related RGC degeneration. In the present study, we investigate the role of p58IPK in neuroprotection by in vitro and in vivo studies using primary RGC culture and two well-established disease-relevant RGC injury models: retinal ischemia/reperfusion (I/R) and microbead-induced ocular hypertension. Our results demonstrate that in both in vivo models, p58IPK −/− mice exhibit significantly increased RGC loss compared to wild type (WT) mice. In vitro, p58IPK-deficient RGCs show reduced viability and are more susceptible to cell death induced by the ER stress inducer tunicamycin (TM). Overexpression of p58IPK by adeno-associated virus (AAV) significantly diminishes TM-induced cell death in both WT and p58IPK −/− RGCs. Interestingly, we find that loss of p58IPK leads to reduced mRNA expression, but not the protein level, of mesencephalic astrocyte-derived neurotrophic factor (MANF), a neurotrophic factor that resides in the ER. Treatment with recombinant MANF protein protects R28 retinal neural cells and mouse retinal explants from TM-induced cell death. Taken together, our study suggests that p58IPK functions as an endogenous neuroprotectant for RGCs. The mechanisms underlying p58IPK’s neuroprotective action and the potential interactions between p58IPK and MANF warrant future investigation.
Introduction
Accumulating evidence suggests that disturbed protein homeostasis in the endoplasmic reticulum (ER), or ER stress, is a significant contributing factor to neurodegeneration in the central nervous system including the retina (Zhang et al., 2014). Prolonged ER stress activates pro-apoptotic genes such as C/EBP homologous protein (CHOP) resulting in apoptosis, while reducing ER stress alleviates cell death of retinal neurons, such as retinal ganglion cells (RGCs), induced by a variety of disease-provoking insults (Hu et al., 2012; Jing et al., 2012; Chen et al., 2014; Huang et al., 2015). In addition, ER stress reduces the production of neurotrophic factors but increases the secretion of pro-inflammatory cytokines, which further exacerbate neuronal death (Zhong et al., 2012; Kim et al., 2014; Cai et al., 2016). Thus, restoring ER protein homeostasis may provide a new strategy to prevent and treat retinal neurodegeneration.
The ER chaperone p58IPK is an important component of the ER protein folding system and is upregulated by the highly conserved unfolded protein response (UPR) genes XBP1 and ATF6 (Lee et al., 2003; van Huizen et al., 2003). Activation of p58IPK facilitates protein folding, reduces ER stress, and promotes cell survival (Rutkowski et al., 2007). In pancreatic β cells, deletion of p58IPK results in CHOP-mediated apoptosis and reduced insulin production (Huber et al., 2013). In a previous study, we demonstrated that p58IPK is present throughout the retina and mice lacking p58IPK display increased susceptibility to retinal injury caused by glutamate toxicity and age-related cell death (Boriushkin et al., 2015). In R28 retinal neural cells, p58IPK overexpression suppresses ER stress and improves cell survival under conditions of oxidative stress (Boriushkin et al., 2015). Furthermore, p58IPK acts as a potent inhibitor of NLRP3 inflammasome and reduces IL-1β secretion in bone marrow macrophages (Boriushkin et al., 2016). These findings suggest that p58IPK may function as a neuroprotectant and play an important role in maintaining the viability of retinal neurons such as RGCs in pathogenic conditions related to ischemic retinopathy and glaucoma.
In the present study, we tested this hypothesis in two commonly used RGC injury models induced by retinal ischemia/reperfusion (I/R) or ocular hypertension and explored the potential mechanism of p58IPK neuroprotection and its interaction with mesencephalic astrocyte-derived neurotrophic factor (MANF), an ER stress responsive neurotrophic factor.
Materials and Methods
Animals
The generation and maintenance of p58IPK knockout mice were previously described elsewhere (Ladiges et al., 2005; Boriushkin et al., 2015). All experimental procedures were performed in compliance with protocols approved by the Institutional Animal Care and Use Committees at the State University of New York at Buffalo.
Isolation and Culture of Mouse RGCs
RGCs were isolated from retinas of postnatal day 6 neonatal mice using the Miltenyi Biotec magnetic cell sorting (MACS) system following a published protocol (Huang et al., 2003; Jiao et al., 2005). Immediately after isolation, cells were incubated with adeno-associated virus (AAV)-GFP or AAV-p58IPK (Vector Biolabs, Malvern, PA, USA) at 10∧12 GC/ml following standard procedures approved by the Institutional Biosafety Committee at the University at Buffalo. After 24 h, transduced cells were treated with 1 μg/ml tunicamycin (TM) or a vehicle control (0.05% DMSO) for an additional 16 h. Cell viability was examined using the live/dead cytotoxicity assay (Molecular Probes) following the manufacturer’s protocol. Images were analyzed for live cells (green) and dead cells (red), blind to treatment or genotype (see Supplementary Material).
Retinal Ischemia/Reperfusion (I/R) Mouse Model
The anterior chamber of anesthetized mice was canulated with a 30g needle attached to a reservoir of sterile PBS. Retinal ischemia was induced by elevating the reservoir to generate a hydrostatic pressure of 90 mmHg for 60 min. After 7 days, mice were sacrificed and retinal whole-mounts were prepared for Tuj1 immunostaining to visualize RGCs (see Supplementary Material).
Microbead-Induced Ocular Hypertension Mouse Model
Microbead injection was performed as described previously (Chen et al., 2011). Briefly, the anterior chamber of anesthetized mice was canulated and 2 μl PBS containing 10 μm diameter polystyrene, fluorescent microspheres at 1.5 × 107 beads/ml (FluoSpheres, Invitrogen) was injected. Intraocular pressure (IOP) was recorded by bounce tonometer (Icare). Two weeks after microbead injection mice were sacrificed and retina whole-mounts were prepared for Tuj1 immunostaining (see Supplementary Material).
R28 Cell Treatment and Analysis
R28 cells were cultured as described previously (Boriushkin et al., 2015). At approximately 60%–70% confluence, cells were pretreated with 50 ng/ml recombinant human MANF (hMANF; PeproTech) or vehicle for 16 h and then treated with 5 μg/ml TM or vehicle for 24 h. Cells were labeled using the live/dead cytotoxicity assay, photographed, and analyzed (see Supplementary Material).
Immunohistochemistry and Morphological Analysis
Mouse eye cryosections were prepared for immunohistochemistry as described previously (McLaughlin et al., 2018). The primary antibodies used were: Ribeye (Synaptic Systems 192003, 1:800); PKCα (Santa Cruz sc-8393, 1:400); Pax6 (DSHB Pax6-s, 1:25); and calretinin (Millipore Mab1658, 1:800). The secondary antibodies used were: Texas-red conjugated goat-anti-rabbit (ThermoFisher T6391, 1:800); Alexa594 conjugated goat-anti-mouse (ThermoFisher A11005, 1:800). Photomontages were assembled with Adobe Photoshop.
Quantitative RT-PCR
Total RNA was isolated with Trizol (Invitrogen) per manufacturer instructions. cDNA was made from 500 ng of total RNA using Bio-Rad cDNA kit followed by quantitative RT-PCR (qPCR) with Bio-Rad iQ Sybr Green Supermix. The following primers were used: MANF f1: 5’-CAC CAG CCA CTA TTG AAG AAG A-3’ with MANF r1: 5’-AGC ATC ATC GTG GTC CAA-3’; MANF f2: 5’-AAA GAG AAT CGG TTG TGC TAC T-3’ with MANF r2: 5’-CCA GGA TCT TCT TCA GCT CTT T-3’; 18sF 5’-GTA ACC CGT TGA ACC CCA TT-3’ with 18sR 5’-CCA TCC AAT CGG TAG TAG CG-3’.
Statistical Analysis
The quantitative data were expressed as mean ± SD. Statistical analyses were performed using Student’s t-test for two-group comparisons and one-way ANOVA with Bonferroni post hoc test for three groups or more. Statistical differences were considered significant at a p value of less than 0.05.
Results
p58IPK Deletion Does Not Affect Retinal Morphology in Young Adult Mice
We first examined overall retinal morphology of young adult p58IPK −/− and wild type (WT) mice using a variety of retinal cell type specific markers. We found that there was no significant difference in retinal expression of PKCα, Pax6, ribeye, or calretinin in p58IPK −/− and WT mice (Figure 1A). The retinal layer thickness was identical in these mice (not shown). We conclude that the retinas of p58IPK −/− mice were morphologically indistinguishable from WT at the ages we performed the retinal injury interventions described below.
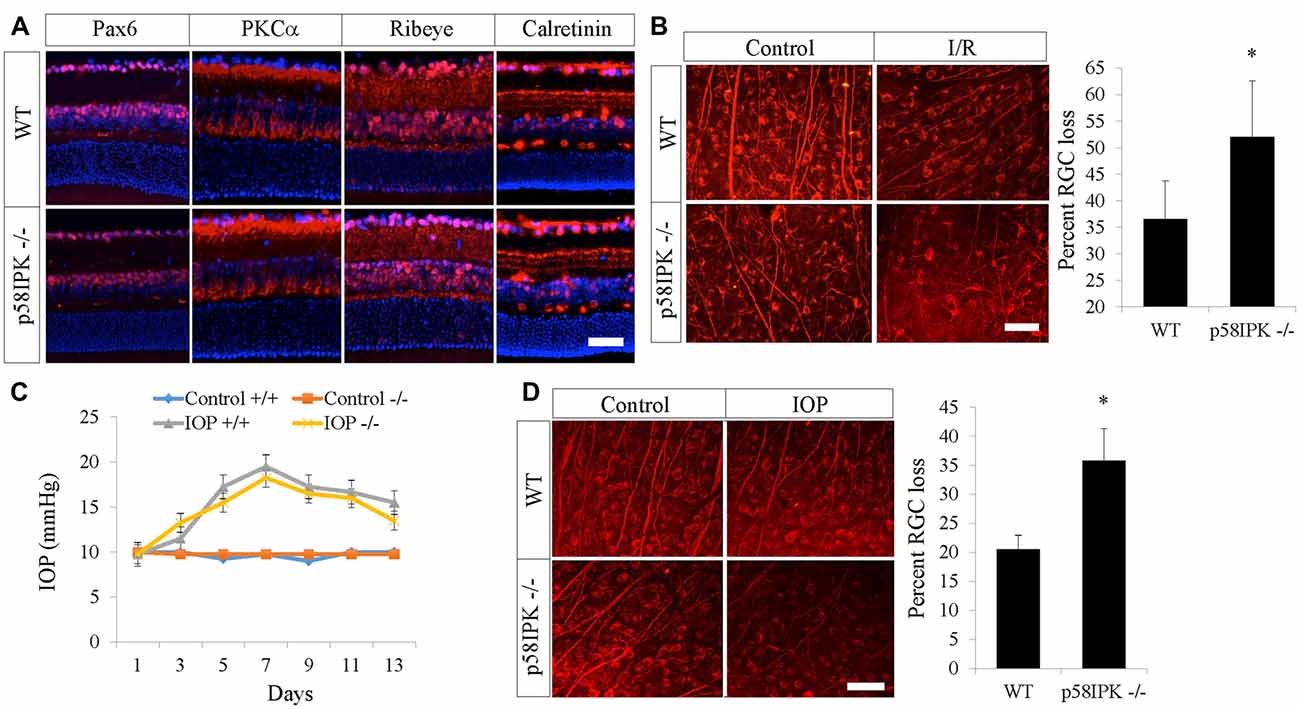
Figure 1. Loss of p58IPK exacerbates retinal ganglion cell (RGC) cell death in retinal ischemia/reperfusion (I/R) and microbead-induced ocular hypertension. (A) Retinal cryosections (20 μM) from 2-month-old wild type (WT) and p58IPK −/− mice labeled with primary antibodies against various retinal markers and a fluorescent secondary antibody (red). Anti-Pax6 labels amacrine cells and some RGCs; anti-PKCα labels rod bipolar cells and their dendrites as well as a subset of amacrine cells; anti-Ribeye labels ribbon synapses between bipolar cells and photoreceptors in the OPL, as well as synapses between amacrine cells and RGCs in the IPL; anti-calretinin labels three synaptic lamina within the IPL. There is no clear difference between WT and p58IPK −/− retina in labeling for any of these markers. All sections are counterstained with DAPI (blue) to label nuclei. n = 3 mice per group, scale bar = 50 μm. (B) Retinal whole mounts from WT and p58IPK −/− mice at 7 days after retinal I/R were stained with RGC marker, Tuj1. Representative images are shown on the left. Graph shows that the percentage of RGC loss, as determined from wholemount retinal Tuj1 staining, is significantly higher in p58IPK −/− mice compared to WT. Mean ± SD, n = 5 mice. *p < 0.05 (Student’s t-test). Scale bar = 100 μm. (C) Graph depicts the average intraocular pressure (IOP) of sham control eyes and microbead-injected eyes in WT (+/+) and p58IPK −/− mice at the time points as indicated. (D) Retinal whole mounts from WT and p58IPK −/− mice at 14 days after microbead injection were stained with RGC marker, Tuj1. Representative images are shown on the left. Bar graph shows the percentage of RGC loss is significantly greater in p58IPK −/− mice compared to WT. Mean ± SD, n = 4 mice. *p < 0.05 (Student’s t-test).
p58IPK Deficiency Exacerbates RGC Loss Induced by Retinal I/R
Retinal I/R is a widely used model for investigating the mechanisms and treatment of inner retinal neuron injury caused by acute ischemia. Seven days after I/R, RGC numbers were examined by immunostaining for Tuj1, a putative RGC marker, in retinal whole mounts. We found a 36.6% RGC loss in WT mice and a 52.0% RGC loss in p58IPK −/− mice (Figure 1B). This indicates that I/R induced a significantly higher rate of RGC loss (a 42% increase) in p58IPK −/− mice than in WT, suggesting a protective role of p58IPK in RGCs.
Loss of p58IPK Increases RGC Damage in Microbead-Induced Ocular Hypertension
We used a second model that is highly relevant to glaucoma to validate the role of p58IPK in RGC protection. We found no significant difference in IOP elevation after microbead injection in p58IPK −/− and WT mice (Figure 1C). Two weeks after induction, WT retinas with increased IOP exhibited a 20% loss of RGCs; the percentage of RGC loss increased significantly to over 35% in p58ipk −/− mice (Figure 1D). This again indicates that a loss of p58IPK results in an increase in RGC death.
Overexpression of p58IPK Protects RGCs From ER Stress-Induced Cell Death
Next, we examined whether overexpressing p58IPK by AAV is sufficient to protect primary mouse RGCs isolated from p58IPK −/− and WT mice. Under control conditions, we found that the percentage of RGCs survival in culture was significantly decreased in p58IPK −/− RGCs compared to WT (Figure 2A), although this decrease was not rescued by acute overexpression of p58IPK. TM exposure caused a significant loss of cells in both p58IPK −/− and WT RGCs infected with control virus (Figure 2A). Overexpressing p58IPK led to a significant recovery to near control levels of survivability in both WT and p58IPK −/− RGCs.
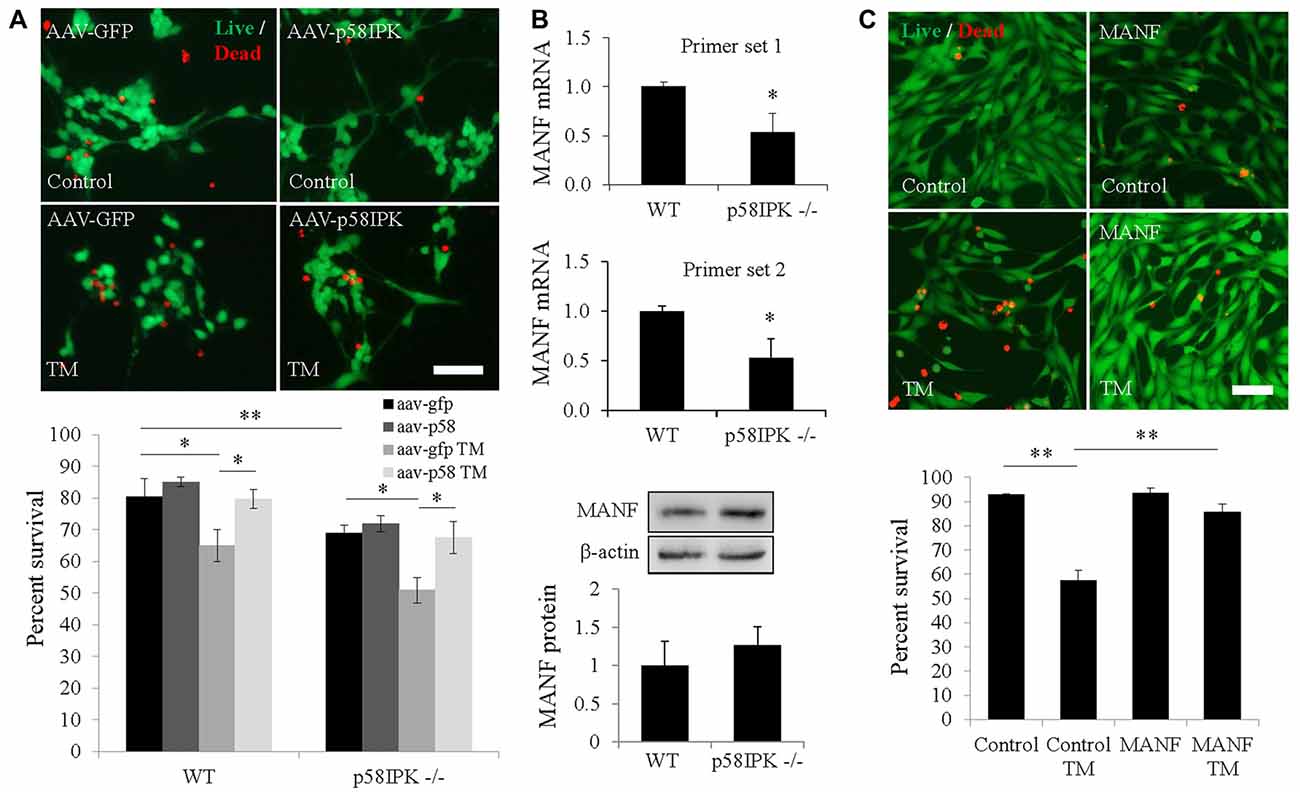
Figure 2. Overexpressing p58IPK by adeno-associated virus (AAV) protects RGCs from endoplasmic reticulum (ER) stress-induced cell death. (A) Cultured primary mouse RGCs isolated from p58IPK −/− and WT retinas were infected with AAV-GFP or AAV-p58IPK 24 h prior to a 16 h-treatment with tunicamycin (TM; 1 μg/ml) or control. Cells are labeled with the live/dead cytotoxicity assay (calcein-AM to label live cells (green) and ethidium homodimer-1 to label dead cells (red)). Representative images of live/dead assay are shown above. Graph depicts the survival percentage of RGCs from WT and p58IPK −/− mice. Under control conditions, p58IPK −/− RGCs infected with AAV-GFP have a significantly lower survival percentage than the matched WT group. RGCs from both WT and p58IPK −/− mice infected with AAV-GFP show a significant decrease in survival percentage after TM treatment compared to control treatment. The survival percentage is significantly increased for RGCs infected with AAV-p58IPK, compared to infection with AAV-GFP for both WT and p58IPK −/− mice. Mean ± SD, n = 3 independent experiments. *p < 0.05 (one-way ANOVA with Bonferroni post hoc test). Scale bar = 80 μm. (B) Quantitative RT-PCR (qPCR; top) using total RNA from whole retina from 2-month-old WT and p58IPK −/− mice. Two sets of independent primers specific for mesencephalic astrocyte-derived neurotrophic factor (MANF) reveal a mean significant 50% reduction in mRNA level in p58IPK −/− retina compared to WT. All qPCR data are normalized to 18 s. Mean ± SD, n = 3 mice per genotype. *p < 0.05 (Student’s t-test). Western blot analysis (bottom) of whole retina lysate from WT or p58IPK −/− mice reveal that the level of MANF protein is not statistically different for whole retina (n = 6 mice per group). (C) R28 cells in culture exposed to 16-h pre-treatment with 50 ng/ml MANF or vehicle, followed by 5 μg/ml TM or control treatment for 24 h. Cells are labeled with the live/dead cytotoxicity assay as in (A). Quantification of the survival of R28 cells reveals that approximately 60% of R28 cells survive TM treatment in the absence of MANF. With MANF pre-treatment the survival percentage is significantly increased to over 85%. Mean ± SD, n = 4 independent experiments; *p < 0.05, **p < 0.01 (one-way ANOVA with Bonferroni post hoc test). Scale bar = 100 μm.
Reduced Retinal mRNA Expression of MANF in p58IPK −/− Mice
MANF is a member of a newly identified ER-localized neurotrophic factor family and is upregulated during ER stress (Glembotski et al., 2012). We investigated whether MANF expression is altered in p58IPK −/− retinas. Using qPCR, we found that MANF mRNA expression was significantly reduced in the retina of p58IPK −/− mice (Figure 2B). However, the protein level appeared to be insignificantly different (Figure 2B). To further explore whether there is a reciprocal regulation between p58IPK and MANF, we transduced R28 cells with adenovirus to overexpress p58IPK and then treated the cells with TM to induce ER stress. We found that both MANF and p58IPK levels were significantly increased after TM treatment (Supplementary Figure S1A). Overexpression of p58IPK did not alter MANF protein expression in unstimulated cells, but significantly reduced the induction of MANF by TM (Supplementary Figure S1A).
Recombinant MANF Protects R28 Cells or Retinal Explants From ER Stress Induced Cell Death and Caspase-3 Activation
To examine whether MANF is important for neuronal survival under ER stress, we pretreated R28 cells with 50 ng/ml recombinant hMANF protein or vehicle (0.1% PBS) followed by treatment with 5 μg/ml TM, or vehicle, for 24 h. We find TM treatment induces cell death in 43% of R28 cells in the absence of MANF treatment (Figure 2C). With MANF pretreatment, this number was reduced to 16%, which represents a recovery to 91% of the survival rate of control R28 cells treated with vehicle (Figure 2C). In addition, MANF treatment significantly reduced TM-elicited caspase-3 activation in ex vivo cultured retinal explants (Supplementary Figure S1B).
Discussion
The findings from the present study provide strong evidence for an endogenous role of p58IPK in protection of RGCs in conditions of ER stress, acute ischemia and ocular hypertension. Previous studies, including our own, have implicated p58IPK in a broad range of neurodegenerative diseases and diabetes (Ladiges et al., 2005; Boriushkin et al., 2014, 2015, 2016; Han et al., 2015; McLaughlin and Zhang, 2015). Clinically, patients bearing loss-of-function mutations in the DNAJC3 gene, which encodes p58IPK, suffer from diabetes and multisystemic neurodegeneration (Synofzik et al., 2014). In animal models, loss of p58IPK results in increased ER stress and oxidative stress, decreased β cell function, progressive cell death and diabetes in male p58IPK KO mice (Ladiges et al., 2005; Han et al., 2015). In contrast to the relatively thoroughly studied role of p58IPK in diabetes, how p58IPK regulates neuronal function remains unexplored. Herein, we examined the impact of p58IPK deficiency on RGC survival in disease-relevant models. Our data clearly indicate an important role of p58IPK in RGC survival during acute retinal ischemia and in ocular hypertension. Notably, we used young adult p58IPK KO mice that demonstrate no significant morphological defects in the retina compared to their age-matched littermate controls. In addition, most of our experiments were carried out in female mice at 2 months of age, which do not experience increased blood glucose levels as some male p58IPK KO mice do with age.
Our in vitro results suggest a therapeutic potential of overexpressing p58IPK or enhancing its function for neuroprotection of RGCs, which warrants an in vivo study in disease models. Another interesting finding is that there is likely a reciprocal regulation between p58IPK and MANF in retinal cells undergoing ER stress. Recent studies show that MANF protects CNS neurons (Voutilainen et al., 2017) and markedly improves RGC survival in a rat glaucoma model (Gao et al., 2017). Our data support these findings and demonstrate that MANF inhibits ER stress-induced cell death and caspase-3 activation in retinal cells and explants. In R28 cells, both p58IPK and MANF are upregulated during ER stress; interestingly, overexpressing p58IPK reduces ER stress-stimulated MANF upregulation. This suggests a potential interaction between the two chaperones, which both bind to GRP78 and regulate the UPR, although it remains unclear whether MANF secretion is affected by p58IPK manipulation. Future studies will investigate how MANF and p58IPK are regulated in RGCs and whether overexpression of p58IPK and MANF would synergistically promote RGC survival and function.
Author Contributions
TM designed and performed the experiments, analyzed the data and drafted the manuscript. ND and JL performed the experiments, analyzed the data and revised the manuscript. JW and SZ conceived and designed the study, reviewed all the data, drafted and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by National Institutes of Health/National Eye Institute (NIH/NEI) grants EY019949 and EY025061, and an Unrestricted Grant to the Department of Ophthalmology, SUNY-Buffalo, from Research to Prevent Blindness.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Michael Katze, Ph.D. (University of Washington, Seattle, WA, USA) for p58IPK knockout mice and Gail M. Seigel, Ph.D. (University at Buffalo, Buffalo, NY, USA) for R28 cells.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2018.00267/full#supplementary-material
References
Boriushkin, E., Wang, J. J., Li, J., Bhatta, M., and Zhang, S. X. (2016). p58(IPK) suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages. Sci. Rep. 6:25013. doi: 10.1038/srep25013
Boriushkin, E., Wang, J. J., Li, J., Jing, G., Seigel, G. M., and Zhang, S. X. (2015). Identification of p58IPK as a novel neuroprotective factor for retinal neurons. Invest. Ophthalmol. Vis. Sci. 56, 1374–1386. doi: 10.1167/iovs.14-15196
Boriushkin, E., Wang, J. J., and Zhang, S. X. (2014). Role of p58IPK in endoplasmic reticulum stress-associated apoptosis and inflammation. J. Ophthalmic. Vis. Res. 9, 134–143.
Cai, M., Wang, H., Li, J. J., Zhang, Y. L., Xin, L., Li, F., et al. (2016). The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neuronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain Behav. Immun. 57, 347–359. doi: 10.1016/j.bbi.2016.05.010
Chen, C., Cano, M., Wang, J. J., Li, J., Huang, C., Yu, Q., et al. (2014). Role of unfolded protein response dysregulation in oxidative injury of retinal pigment epithelial cells. Antioxid. Redox Signal. 20, 2091–2106. doi: 10.1089/ars.2013.5240
Chen, H., Wei, X., Cho, K.-S., Chen, G., Sappington, R., Calkins, D. J., et al. (2011). Optic neuropathy due to microbead-induced elevated intraocular pressure in the mouse. Invest. Ophthalmol. Vis. Sci. 52, 36–44. doi: 10.1167/iovs.09-5115
Gao, F.-J., Wu, J.-H., Li, T.-T., Du, S.-S., and Wu, Q. (2017). Identification of mesencephalic astrocyte-derived neurotrophic factor as a novel neuroprotective factor for retinal ganglion cells. Front. Mol. Neurosci. 10:76. doi: 10.3389/fnmol.2017.00076
Glembotski, C. C., Thuerauf, D. J., Huang, C., Vekich, J. A., Gottlieb, R. A., and Doroudgar, S. (2012). Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J. Biol. Chem. 287, 25893–25904. doi: 10.1074/jbc.m112.356345
Han, J., Song, B., Kim, J., Kodali, V. K., Pottekat, A., Wang, M., et al. (2015). Antioxidants complement the requirement for protein chaperone function to maintain β-cell function and glucose homeostasis. Diabetes 64, 2892–2904. doi: 10.2337/db14-1357
Hu, Y., Park, K. K., Yang, L., Wei, X., Yang, Q., Cho, K.-S., et al. (2012). Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 73, 445–452. doi: 10.1016/j.neuron.2011.11.026
Huang, C., Wang, J. J., Ma, J. H., Jin, C., Yu, Q., and Zhang, S. X. (2015). Activation of the UPR protects against cigarette smoke-induced RPE apoptosis through up-regulation of Nrf2. J. Biol. Chem. 290, 5367–5380. doi: 10.1074/jbc.m114.603738
Huang, X., Wu, D. Y., Chen, G., Manji, H., and Chen, D. F. (2003). Support of retinal ganglion cell survival and axon regeneration by lithium through a Bcl-2-dependent mechanism. Invest. Ophthalmol. Vis. Sci. 44, 347–354. doi: 10.1167/iovs.02-0198
Huber, A.-L., Lebeau, J., Guillaumot, P., Pétrilli, V., Malek, M., Chilloux, J., et al. (2013). p58IPK-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Mol. Cell 49, 1049–1059. doi: 10.1016/j.molcel.2013.01.009
Jiao, J., Huang, X., Feit-Leithman, R. A., Neve, R. L., Snider, W., Dartt, D. A., et al. (2005). Bcl-2 enhances Ca2+ signaling to support the intrinsic regenerative capacity of CNS axons. EMBO J. 24, 1068–1078. doi: 10.1038/sj.emboj.7600589
Jing, G., Wang, J. J., and Zhang, S. X. (2012). ER stress and apoptosis: a new mechanism for retinal cell death. Exp. Diabetes Res. 2012:589589. doi: 10.1155/2012/589589
Kim, S., Joe, Y., Jeong, S. O., Zheng, M., Back, S. H., Park, S. W., et al. (2014). Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun. 20, 799–815. doi: 10.1177/1753425913508593
Ladiges, W. C., Knoblaugh, S. E., Morton, J. F., Korth, M. J., Sopher, B. L., Baskin, C. R., et al. (2005). Pancreatic β-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 54, 1074–1081. doi: 10.2337/diabetes.54.4.1074
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/mcb.23.21.7448-7459.2003
McLaughlin, T., Falkowski, M., Park, J. W., Keegan, S., Elliott, M., Wang, J. J., et al. (2018). Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 13:16. doi: 10.1186/s13024-018-0250-z
McLaughlin, T., and Zhang, S. X. (2015). The neuroprotective potential of endoplasmic reticulum chaperones. Neural Regen. Res. 10, 1211–1213. doi: 10.4103/1673-5374.162696
Rutkowski, D. T., Kang, S.-W., Goodman, A. G., Garrison, J. L., Taunton, J., Katze, M. G., et al. (2007). The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 18, 3681–3691. doi: 10.1091/mbc.e07-03-0272
Synofzik, M., Haack, T. B., Kopajtich, R., Gorza, M., Rapaport, D., Greiner, M., et al. (2014). Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am. J. Hum. Genet. 95, 689–697. doi: 10.1016/j.ajhg.2014.10.013
van Huizen, R., Martindale, J. L., Gorospe, M., and Holbrook, N. J. (2003). P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2α signaling. J. Biol. Chem. 278, 15558–15564. doi: 10.1074/jbc.m212074200
Voutilainen, M. H., De Lorenzo, F., Stepanova, P., Bäck, S., Yu, L.-Y., Lindholm, P., et al. (2017). Evidence for an additive neurorestorative effect of simultaneously administered CDNF and GDNF in hemiparkinsonian rats: implications for different mechanism of action. eNeuro 4:ENEURO.0117-16.2017. doi: 10.1523/eneuro.0117-16.2017
Zhang, S. X., Sanders, E., Fliesler, S. J., and Wang, J. J. (2014). Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 125, 30–40. doi: 10.1016/j.exer.2014.04.015
Keywords: p58IPK, retinal ganglion cells, retinal ischemia/reperfusion, ocular hypertension, X-box binding protein 1
Citation: McLaughlin T, Dhimal N, Li J, Wang JJ and Zhang SX (2018) p58IPK Is an Endogenous Neuroprotectant for Retinal Ganglion Cells. Front. Aging Neurosci. 10:267. doi: 10.3389/fnagi.2018.00267
Received: 03 June 2018; Accepted: 20 August 2018;
Published: 07 September 2018.
Edited by:
Daniel Ortuño-Sahagún, Universidad de Guadalajara, MexicoReviewed by:
Rebecca M. Sappington, Vanderbilt University Medical Center, United StatesPatrick William Keeley, University of California, Santa Barbara, United States
Denise M. Inman, Northeast Ohio Medical University, United States
Copyright © 2018 McLaughlin, Dhimal, Li, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah Xin Zhang, eHpoYW5nMzhAYnVmZmFsby5lZHU=