 Md. Ezazul Haque
Md. Ezazul Haque In-Su Kim2
In-Su Kim2 Md. Jakaria
Md. Jakaria Mahbuba Akther
Mahbuba Akther Dong-Kug Choi
Dong-Kug Choi- 1Department of Applied Life Science, Graduate School, Konkuk University, Chungju, South Korea
- 2Department of Integrated Bioscience and Biotechnology, College of Biomedical and Health Science, Research Institute of Inflammatory Disease, Konkuk University, Chungju, South Korea
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder associated with impairment of cognition, memory deficits and behavioral abnormalities. Accumulation of amyloid beta (Aβ) is a characteristic hallmark of AD. Microglia express several GPCRs, which, upon activation by modulators, mediate microglial activation and polarization phenotype. This GPCR-mediated microglial activation has both protective and detrimental effects. Microglial GPCRs are involved in amyloid precursor protein (APP) cleavage and Aβ generation. In addition, microglial GPCRs are featured in the regulation of Aβ degradation and clearance through microglial phagocytosis and chemotaxis. Moreover, in response to Aβ binding on microglial Aβ receptors, they can trigger multiple inflammatory pathways. However, there is still a lack of insight into the mechanistic link between GPCR-mediated microglial activation and its pathological consequences in AD. Currently, the available drugs for the treatment of AD are mostly symptomatic and dominated by acetylcholinesterase inhibitors (AchEI). The selection of a specific microglial GPCR that is highly expressed in the AD brain and capable of modulating AD progression through Aβ generation, degradation and clearance will be a potential source of therapeutic intervention. Here, we have highlighted the expression and distribution of various GPCRs connected to microglial activation in the AD brain and their potential to serve as therapeutic targets of AD.
Introduction
Microglia or resident macrophages of the central nervous system (CNS) originate from the embryonic yolk sac and are incorporated into the CNS during the earlier stages of development (Lannes et al., 2017). Microglia contribute to protection and maintenance of the CNS (Datta et al., 2018). Their role in the pathophysiology of many neurodegenerative disorders has been studied over the previous decades (Gentleman, 2013; Giunti et al., 2014; Salter and Stevens, 2017; Dukhinova et al., 2018), suggesting that microglia can rapidly change their phenotype to express different receptors according to stimuli generated by CNS damage or infection. Moreover, they can respond with both pro- or anti-inflammatory activity through direct migration (McHugh et al., 2010).
The progressive irreversible neurodegeneration disorder, Alzheimer’s disease (AD), is characterized by visible abnormal microscopic structures, such as deposition of the extracellular insoluble amyloid-β peptide (Aβ) in neuritic plaques and hyperphosphorylated tau protein in the neurofibrillary tangles (NFT) of the brain (Amihăesei et al., 2013; Luna et al., 2013; Puzzo et al., 2015). Accumulation of characteristic plaques and tangles in the brain consequently result in memory deficits and cognitive impairment in AD (Bloom, 2014). Aβ proteins are 37–43 amino acid containing peptides that are produced from precursor transmembrane Aβ precursor protein (β-APP) through sequential cleavage by β-and γ-secretase (Thinakaran and Koo, 2008). In the AD brain, Aβ40 and Aβ42 are abundantly found, whereas Aβ42 are closely related to AD pathogenesis (Crouch et al., 2008). Depending on Aβ aggregation and conformational changes, Aβ proteins exist in two distinct forms—soluble and fibrillar. Senile plaque, a characteristic hallmark of AD, is comprised of compact and dense insoluble Aβ fibrils and more soluble toxic oligomeric Aβ species (Shankar et al., 2008; Koffie et al., 2009). On the other hand, tau is a microtubule-associated protein, its breakdown, phosphorylation and changes in conformation have been implicated in the pathological progression of AD (Mondragón-Rodríguez et al., 2014; Yang et al., 2014). However, these characteristic biomarkers of AD can be detected a decade before the first symptoms of AD appear (Craig-Schapiro et al., 2009).
The link between microglia and AD was reported 30 years ago by McGeer et al. (1988) where the accumulation of microglia near the senile plaques in the AD brain was discussed (McGeer et al., 1988). In the AD brain, accumulation of this misfolded Aβ has been demonstrated to induce neuroinflammation by binding with several microglial innate immune receptors, including G-Protein-Coupled Receptors (GPCRs), which were postulated to induce an inflammatory cascade known as the “amyloid cascade-inflammatory hypothesis of AD” (Hardy and Higgins, 1992; Karran et al., 2011; McGeer and McGeer, 2013). This hypothesis is one of the most influential hypotheses surrounding AD pathogenesis to date. According to this hypothesis, the accumulation of Aβ and deficiency in its clearance is considered to be the root cause of AD pathogenesis and leads to extracellular senile plaque and tau-immunoreactive NFT formation, neurodegeneration and eventually dementia in AD patients (Hardy and Higgins, 1992). However, this hypothesis is challenged by many experts and it is believed that Aβ and tau are not the reason underlying AD pathogenesis, but merely its byproducts (McGeer and McGeer, 2013). Furthermore, it has been suspected that Aβ production triggers abnormal tau processing and thereby demands further study to determine a link between Aβ and tau protein (Zempel and Mandelkow, 2014). The role of microglia in Aβ formation, maintenance and clearance is highly investigated in the literature.
Based on the chemotactic effect of Aβ, microglia can accumulate near the core dense amyloid plaques. It has been postulated that, the chemotactic effect of Aβ plaques is related to its size. Therefore, microglial response can be changed in proportion to the size of Aβ plaque (Serrano-Pozo et al., 2013). Moreover, microglial phenotypic switch or change in activation state is closely associated with Aβ and plaques (Figure 1). It has been reported that smaller oligomers are more likely associated with Aβ induced microglial activation. Incubation of microglia with Aβ1–40 which produces smaller oligomers than Aβ1–42 showed marked increased in IL-6, NO release and tumor necrosis factor-α (TNF-α) expression (Takata et al., 2003). In another study primary microglia cells treated with freshly dissolved Aβ1–40 resulted in release of IL-1β and treatment with Aβ1–42 increased release of IL-1α and IFN-γ (Lindberg et al., 2005). Furthermore, the shifting of the microglial activation state from alternative to classical activation is linked with degradation and clearance of Aβ peptides through phagocytosis (Bard et al., 2000; Wilcock et al., 2003). This microglial activity in Aβ clearance and phagocytosis has a connection with GPCR-mediated signaling. GPCRs can modulate microglial activity through altering their response to many external and internal stimuli and control microglial morphology, chemotaxis and phagocytosis (Nagele et al., 2004; Nasu-Tada et al., 2005; Koizumi et al., 2007; Kim et al., 2011; Peng et al., 2015). P2Y12, a purinergic GPCR, activating microglia upon binding with extracellular nucleotides. Generally, expression of P2Y12 is high in resting microglia but it decreases upon microglial activation by adenosine diphosphate (ADP) or adenosine triphosphate (ATP). Moreover, microglia from P2Y12 knockout mice are unable to polarize and their chemotaxis is suppressed (Haynes et al., 2006; Ohsawa et al., 2007). Similarly, the orphan GPCR, G-Protein-Coupled Receptor 34 (GPR34), which belongs to the P2Y12-like group, is abundantly expressed on microglial membrane plays a vital role in microglial morphology, function and phagocytosis. Its deficiency in microglia has been reported to reduce phagocytosis in Cryptococcus neoformans-infected mice (Preissler et al., 2015).
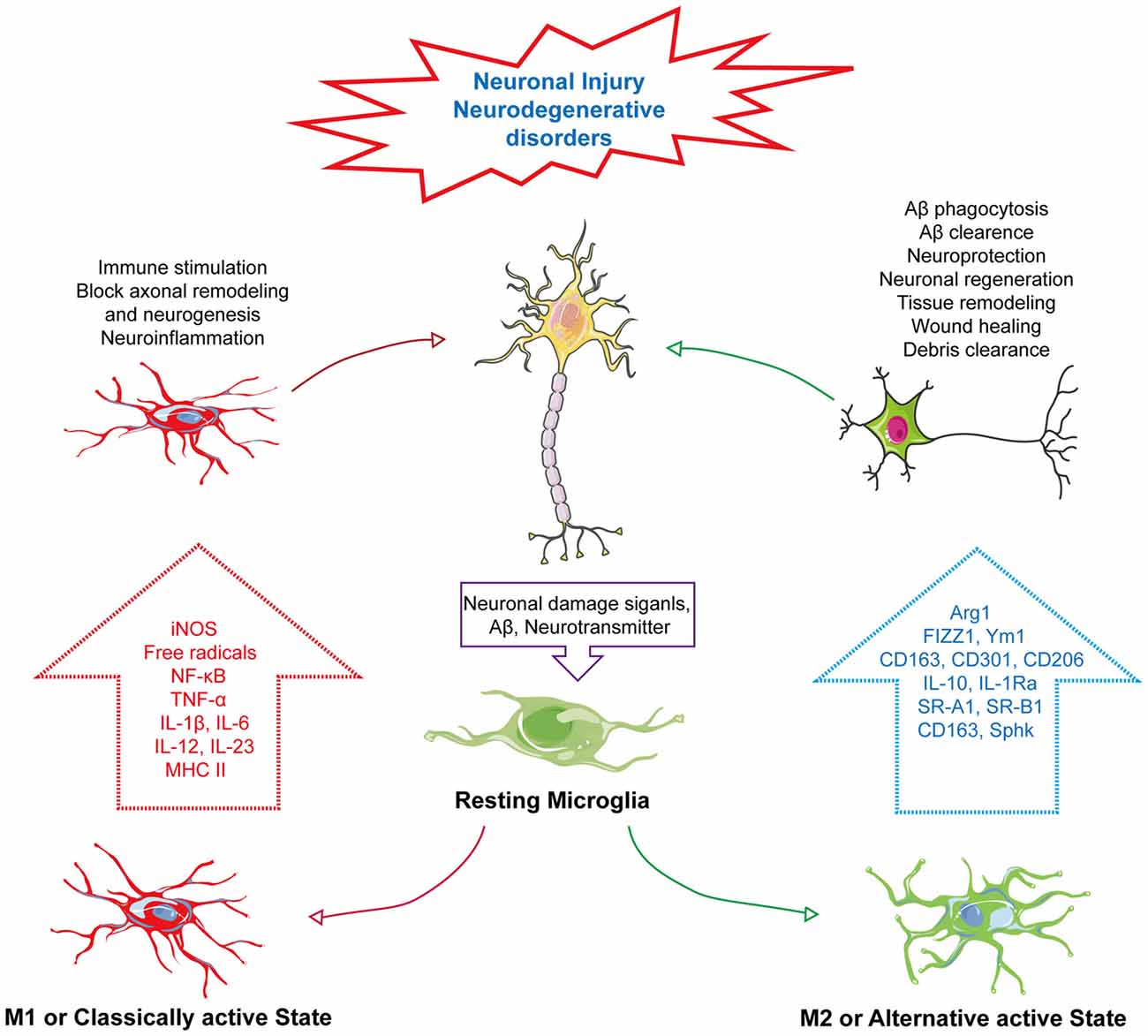
Figure 1. Phenotype change of microglia in neurodegenerative disorders. In response to neuronal damage, signals mediated from neurons, such as neurotransmitter or amyloid-β (Aβ), act as “on” or “off” signals for microglial activation. In response to “on” signals, there can be the alteration of its phenotype into two distinct states—M1: classically active state and M2: alternative active state. In the M1 state, microglia express iNOS and MHC II, activating the NF-κB pathway to produce several pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), IL-1β, IL-6, IL-12 and IL-23, and generate ROS and NO, which subsequently induce immune stimulation, neuroinflammation, block of axonal remodeling and prevent neurogenesis. Unlike the M1 active state, the M2 or alternative active state mediate neuroprotection through Aβ phagocytosis and clearance, modulate neuronal regeneration, and releases arginase 1 (Arg1) for tissue remodeling, wound healing and debris clearance through releasing M2 markers, Arg1, found in inflammatory zone 1 (FIZZ1), chitinase-like protein 1 (Ym1), triggering receptor also expressed on myeloid cells (TREM), CD163, CD301, CD206 and IL-1 receptor antagonist (IL-1Ra) and also expressing other markers, such as scavenger receptor class A1 (SR-A1), scavenger receptor class B1 (SR-B1) and sphingosine kinase (Sphk).
The GPCRs were first identified in 1986—there are almost 800 members of GPCRs within the human genome, which makes it not only the largest superfamily of cell surface receptors but also the largest protein family in vertebrates (Dixon et al., 1986; Gloriam et al., 2007; Ghanemi, 2015). More than 370 of these GPCRs are of the non-sensory origin, involved in mood, cognition, pain and appetite. Almost 90% of these GPCRs can bind to a wide range of neurotransmitters and neuromodulators, like dopamine, norepinephrine and serotonin in the brain (Gainetdinov et al., 2004; Doze and Perez, 2012). Moreover, GPCRs in the brain can react to a wide variety of extracellular stimuli and perturbation of these GPCRs’ function can result in the pathogenesis of many neurodegenerative disorders, such as Parkinson’s disease and AD (Huang et al., 2017). For example, β-arrestin 2 has been found to increase Aβ production upon binding with two GPCRs, the orphan GPCR, G Protein-Coupled Receptor 3 (GPR3) and β2 adrenergic receptor, in both in vitro and in vivo models of AD (Jiang et al., 2013; Thathiah et al., 2013). Additionally, recent findings suggest GPR3 activity is linked to amyloidogenic proteolysis of amyloid-β precursor protein (APP) and its loss of activity is connected with memory improvement in AD transgenic (ADtg) mouse models (Huang et al., 2015). Neprilysin, a peptidase capable of breaking down Aβ in the brain, has been described to decrease its Aβ proteolytic activity by somatostatin hormone through GPCR-mediated signaling (Iwata et al., 2005). There are several microglial GPCRs, such as formyl peptide receptor 2 (FPR2) that bind to Aβ and mediates various inflammatory markers while also regulating Aβ degradation and clearance by cellular phagocytosis (Yu and Ye, 2015). As GPCRs are the most abundantly expressed receptors in the CNS and are connected to different downstream signaling pathways, potentially modulating Aβ degradation and proteolysis of APP through modulating α, β and γ-secretases, these unique features of GPCRs have made them the one of the most promising therapeutic targets for neurodegenerative disorders (Thathiah and De Strooper, 2011; Komatsu, 2015; Huang et al., 2017). Surprisingly, GPCRs are already the target of 475 (~34%) Food and Drug Administration (FDA)-approved drugs available today (Hauser et al., 2018). Within two decades, despite the advances of therapeutics for neurodegenerative disorders, the treatments of AD are mostly based on symptoms rather than its root cause or underlying pathology. In fact, the most popular and current treatments for AD to date are acetylcholinesterase inhibitors (AChEI) and N-Methyl-D-aspartate (NMDA) receptor antagonists (Mota et al., 2014; Gao et al., 2016).
Here, we would like to evaluate the functional and mechanistic relationship of GPCRs with microglia activation and importance of this phenomenon in AD. First, we would discuss the role of GPCRs in the activation of the microglia. Second, based on current reports and findings, we tried to expand the implication of GPCR-mediated microglial activation in this context to the pathophysiology of AD. Finally, we will focus on the therapeutic perspective of GPCRs as emerging drug targets for the development of novel therapeutic agents to treat AD.
Microglial Activation and Neurodegeneration
Microglia, a motile phagocyte of our CNS. It is involved in neuronal cell defense from extremely harmful stimuli and capable of protecting cells from injury or death (Fu et al., 2014). On the other hand, microglia can change its activation to neurotoxic state. It’s because of the fact that microglia can switch their phenotype by a process called polarization (Hu et al., 2015). Polarization and changing of the phenotype are dependent on the types of CNS insults imposed on the brain and which type of mediator is produced in response (Hanisch and Kettenmann, 2007). It has been established for many decades that neuron cells are often the passive victims of microglia activation based on the accidental elimination of neurons when performing protective duties with respect to infection, damage or weakened selection pressures because of aging or neurodegenerative disorders (Brown and Vilalta, 2015). Microglia can shift to reactive states to deal with pathological contexts known as active states of microglia. However, many new studies have started to reveal the close intimacy of the microglia-neuron relationship regarding maintenance of the healthy state of the brain through bidirectional communication (Eyo and Wu, 2013). There is a probability that the cross-talk between these two cells can be achieved by neurotransmitters and their receiving receptors. We know that neurons can send different modulators to microglia requesting assistance to deal with pathological condition, though, on the other hand, microglia, upon receiving the signals, express diverse receptors to initiate feedback to maintain homeostasis (Peferoen et al., 2014; Wohleb, 2016). This wide array of signals triggers the activation of microglia through changing of microglia phenotype. These signals are basically of the “on” or “off” type; whereas the “on” signal is hypothesized to convert microglia to an activated state through a complex and multistage process, and through receptors, the “off” signal keeps microglia in the resting state (Liu et al., 2007). Activated microglia can migrate toward signals, proliferate and engulf injured cells (Kettenmann et al., 2011). There are two types of phenotypes of the active state of microglia—M1 and M2. The M1 phenotype is known as the classically activated state and M2 is referred to as an alternative active state that has other subpopulations (Mittelbronn, 2014). There many other subtypes of M2 states, like M2a, M2b, M2c and Mox, with each possessing unique characteristics with respect to different biological functions (Röszer, 2015). However, recent findings indicate that M1/M2 polarization is connected to several types of neurodegenerative diseases (Tang and Le, 2016). In response to neuronal injury, these two phenotypes have distinct characteristics. Upon the M1 activation state, microglia can release various proinflammatory cytokines, such as: IL-1β, TNF-α and IL-6, IL-12, IL-17 and IL-23; chemokines that include chemokine (C-C motif) ligand 2 (CCL2) and C-X-C motif chemokine 10 (CXCL10); express markers, cluster of differentiation 86 (CD86) and cluster of differentiation 16/32 (CD16/32); and free radicals through the NF-κB pathway that will adversely affect neuronal repair and regeneration (Mahad and Ransohoff, 2003; Kawanokuchi et al., 2006; Mosser and Edwards, 2008; Chhor et al., 2013; Davis et al., 2013; Franco and Fernández-Suárez, 2015; Subramaniam and Federoff, 2017; Che et al., 2018). Meanwhile, the M2 phenotype can act conversely by improving the brain’s repairing or regeneration processes by attenuating neuroinflammation. The M2 state of microglia is triggered by IL-4 or IL-13 that is activated by IL-10 or transforming growth factor-β (TGF-β; Ponomarev et al., 2007; Colton, 2009; Zhou et al., 2012; Subramaniam and Federoff, 2017). M2 increases phagocytosis activity and releases different markers, such as arginase 1 (Arg1) for tissue remodeling and wounding healing, chitinase-like protein 1 (Ym1), which prevents degradation of extracellular matrix components, Found in inflammatory zone 1 (FIZZ1), which blocks nerve growth factor-induced survival of the dorsal root ganglion neurons, CD163 hemoglobin-haptoglobin complex, triggering receptor also expressed on myeloid cells-2 (TREM2) for debris clearance, Dectin-1, leading to phagocytosis, CD301, for pathogen defense and is related to CD206, suppressor of cytokine signaling-3 (SOCS3), sphingosine kinase (Sphk), scavenger receptor class A1 (SR-A1), scavenger receptor class B1 (SR-B1), IL-1 receptor antagonist (IL-1Ra) and IL-10 (Figure 1; Holcomb et al., 2000; Willment et al., 2003; Raes et al., 2005; Morris, 2007; Neumann and Takahashi, 2007; Saba et al., 2009; Shechter et al., 2009; Chhor et al., 2013; Franco and Fernández-Suárez, 2015; Lisi et al., 2017; Subramaniam and Federoff, 2017).
Changes in microglial phenotype have a crucial impact on Aβ clearance during AD pathogenesis. In ADtg mice, it has been observed that acute intrahippocampal administration of lipopolysaccharide (LPS) in APP+PS1 transgenic mice reduces Aβ load (DiCarlo et al., 2001). In another study conducted on iNOS overexpression or iNOS mutant human β-amyloid precursor protein (hAPP) as well as human presenilin-1 (hPS1) expressing double transgenic mice to determine the role of iNOS, it was demonstrated that there are unpredictable reductions in Aβ levels as well as amyloid plaque formation with marked reduction in astrocytosis and microgliosis (Nathan et al., 2005). Changes in microglial phenotype were also reported in APP/PS1 mice. Shifting of the M2 state of microglia to the M1 state in the hippocampus of APP/PS1 mice was observed after 18 months of AD pathogenesis. Age-dependent shifting of microglial phenotype from alternative to classical was associated with increased downregulation of the M2 phenotypic marker Ym1 and TNF-α release (Jimenez et al., 2008). Similar results were found in an in vitro model that suggested that the shifting of the microglial state to the M1 phenotype ablated phagocytosis of Aβ. IL-4 or IL-10, two anti-inflammatory cytokines, are also involved in microglial polarization from M1 to M2 through affecting the Notch pathway, thereby increasing microglial Aβ phagocytosis (Koenigsknecht-Talboo and Landreth, 2005; Michelucci et al., 2009). In all, the aforementioned findings indicate a clear connection between microglial activation and its balance between different phenotypes playing a significant role in neurodegenerative disorders.
Microglial GPCR-Mediated Effect on Aβ in AD
Since the identification of microglia by Rio-Hortega (1932), microglia have become the centerpiece of a variety of infectious, inflammatory and neurodegenerative disorders of the CNS based on its dual characteristic in response to external stimuli to the brain (Rio-Hortega, 1932; Luo and Chen, 2012). In AD, Aβ can directly bind to microglia and activate them. Numerous studies have reported that GPCRs can directly influence the amyloid cascade by exploiting α, β, γ-secretase, APP proteolysis and Aβ degradation (Thathiah and De Strooper, 2011). On the other hand, microglia are also capable of clearance of Aβ by receptor-mediated phagocytosis (Table 1, Ries and Sastre, 2016; Clayton et al., 2017). Microglial expression of different membrane receptors is intended to respond to the disturbance in neuronal damage. GPCRs expressed by microglia have been shown to regulate distinct components of their activation process, including cell proliferation, migration and differentiation into M1 or M2 phenotypes (Fung et al., 2015). Among these diverse receptors types, GPCRs play a pivotal role in the modulation of various components of microglial activation (Table 1). With this, the involvement of GPCRs and its subtypes in neurodegenerative disorders have been implicated in many studies (Heng et al., 2013; Guerram et al., 2016). Moreover, there are many other uncharacterized GPCR subtypes that are featured in microglial activation and need to be investigated for their pharmacological and molecular activity in AD (Stella, 2010).
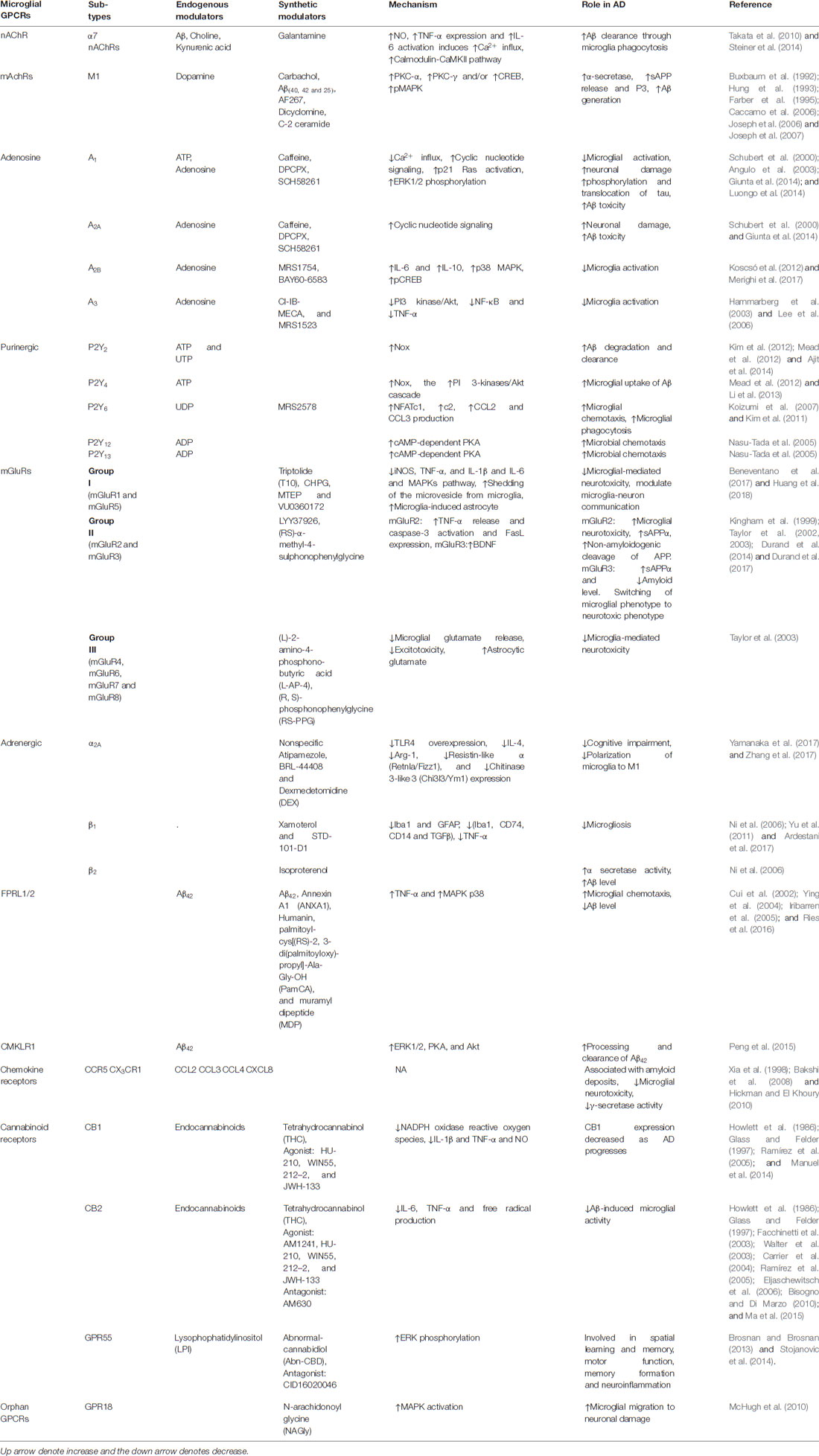
Table 1. Reported microglial-G protein-coupled receptors (GPCRs) and their role in Alzheimer’s disease (AD).
Purinergic Receptors
ATP acts as an extracellular signal taking part in neuromodulation and neurotransmission through activating purinergic receptors (P2) and mediating purinergic signaling in the CNS. P2 receptors are categorized into two receptor families—ATP-gated ion channels (P2X) and GPCRs (P2Y; Burnstock, 2017). P2X receptors are further subdivided into seven subtypes: P2X1, P2X2, P2X3, P2X4, P2X5, P2X6 and P2X7 receptors as well as P27. GPCRs, P2Y receptors, are further subdivided into eight subtypes, comprising five Gq-coupled receptors—P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11—and three Gi-coupled receptors—P2Y12, P2Y13 and P2Y14 (Peterson et al., 2010). A handful of evidence indicates that ATP release is linked to Aβ generation. Numerous studies have also reported the involvement of P2Y receptors in Aβ generation and depletion and neuroinflammation mediated by Aβ toxicity in AD (Erb et al., 2015). Extracellular ATP generated from damaged tissue and astrocytes tends to attract microglial response to brain injury-activating P2Y receptors and connexin channels (Davalos et al., 2005). The microglial P2Y2 receptor has been implicated to enhance Aβ degradation via microglial activation. To find a relationship between Aβ clearance and P2Y2 receptor a study was carried out in an AD mice model using TgCRND8 transgenic mice with homozygous or heterozygous P2Y2 receptor deletion. The study demonstrated that APP mice with homozygous deletion of P2Y2 receptors showed comparatively more Aβ clearance and a greater survival rate with mild expression of the microglial marker, CD11b than heterozygous P2Y2 receptor deletion (Ajit et al., 2014). Another study in P2Y2 knockout mice confirmed the involvement of microglial P2Y2 receptors in Aβ clearance. In that investigation, primary microglia cells treated with ATP and uridine diphosphate (UDP) exhibited a marked increase in Aβ1–42 uptake whereas it shows no increase in primary cells isolated from P2Y2R knock-out mice. The ablation of Aβ1–42 uptake was speculated to be based on the inhibition of the αV integrin-, Src- and Rac-mediated P2Y2R signaling pathways (Kim et al., 2012).
In addition, it has been claimed that microglial P2Y4R is linked to previously unreported microglial pinocytosis mediated uptake of Aβ. The pinocytic effect of microglia was modulated by the phosphatidylinositol 3-kinase/Akt cascade in the absence of ATP (Li et al., 2013). Exceptionally, the P2Y4 receptor in humans is seen to be antagonized by ATP in contrast to rat P2Y4 receptors. Stability of P2Y4 receptors in both human and rat astrocytoma cells were measured by intracellular Ca2+ levels. Although these same receptors in two different species share 83% identical amino acid sequences, they have contrasting pharmacological activity (Kennedy et al., 2000). Depending on the expression of receptors microglia superoxide production through Nox activation can change microglial migration to either neurotoxic or neuroprotective phenotype. Nox can affect the microglial response to external stimuli depending on its agonist. However, P2Y(2/4) receptors are integral to the activation of Nox and the neurotoxic profile of microglia in response to changes in neurodegenerative stimuli (Mead et al., 2012). When hippocampal neurons are damaged, they release UDP. In response to UDP activating glial cells, which rapidly produce CCL2 and CCL3, as a result, chemokine levels rise by activation of the P2Y6 receptor. In addition, UDP also activates NFATc1 and c2, two calcium-activated transcription factors. In two different studies, it was demonstrated that attenuating the P2Y6 receptor-mediated pathway by inhibiting phospholipase C and calcium or administering P2Y6-specific antagonists (MRS2578) led to a significant decrease in chemokine expression (Kim et al., 2011). Moreover, kainic acid administration elevated the release of UDP with markedly enhanced P2Y6 receptor activation, which subsequently led to neuronal cell death both in vivo and in vitro, which suggests that UDP may act as a sensor for microglia-mediated phagocytosis in response to neuronal damage in neurodegenerative disorders (Koizumi et al., 2007). Furthermore, the role of Gi-coupled receptors—P2Y12 and P2Y13 mediating microglial chemotaxis by ADP and ATP has been reported in many studies (Honda et al., 2001; Haynes et al., 2006). According to Nasu-Tada et al. (2005), ADP induces microbial chemotaxis in the presence of fibronectin and in a β1 integrin-dependent manner, whereas cAMP-dependent PKA positively regulates β1 integrin-induced microglial proliferation, which was reduced by purinergic signals from P2Y12/13 (Nasu-Tada et al., 2005).
Adenosine Receptors
Adenosine is a neurotransmitter expressed in different cells of the CNS, including glia, and plays a pivotal role in the neuronal excitation, synaptic transmission and neuronal excitability (Ribeiro and Sebastiao, 2010). There are four subtypes of adenosine receptors: A1, A2A, A2B and A3 receptors, which belong to purinergic GPCR family. The imbalance of adenosine receptor subtypes is associated with cognitive functional issues in AD (Yan et al., 2014). In the human brain, high expression of A1 and A2A receptors is seen in the frontal cortex (Albasanz et al., 2008). The A2A receptor is generally expressed in a striatal neuron in healthy brains, but in AD patients, it is expressed in the glial cells in the hippocampus and cerebral cortex region (Angulo et al., 2003). Microglia treated with ATP showed upregulation of A1 receptors and upon selective modulation of A1, suppressed microglial activation through decreasing Ca2+ influx (Luongo et al., 2014). In addition, the rise of extracellular adenosine levels have been reported to mediate both A1 and A2A receptor activation and cyclic nucleotide signaling to counteract glial cell-mediated neuronal damage in AD (Schubert et al., 2000). A1 receptors are abundantly present in the CA1 region of the hippocampus of the normal human brain. Activation of A1 receptors subsequently causes p21 Ras activation and ERK1/2 phosphorylation, which results in phosphorylation and translocation of tau in an ERK-dependent manner (Angulo et al., 2003). Suppressing the activity of these (A1 and A2A) has been found to prevent Aβ toxicity in vitro (Giunta et al., 2014). A3 receptor activation in the murine microglial cell has been reported. Activation of the A3 receptor by adenosine and Cl-IB-MECA, a selective adenosine A3 receptor agonist, tend to inhibit PI3 kinase/Akt and NF-κB and eventually production of TNF-α in BV-2 microglia cells. On the other hand, MRS1523, a selective A3 receptor antagonist, reverses such an effect (Hammarberg et al., 2003; Lee et al., 2006). In a microglia-mediated neuroinflammation model, A2B receptors have been reported to stimulate IL-6 and IL-10 production through the p38 mitogen-activated protein kinase (MAPK) pathway and phosphorylation of cAMP response element-binding protein (CREB; Koscsó et al., 2012; Merighi et al., 2017).
The capability of adenosine to regulate microglial activation proliferation and chemotaxis through A2A receptors has made the A2A receptor an anticipated therapeutic target for treating the diseases that are linked to microglial activation (Santiago et al., 2014). In addition, 2’,3’-cAMP and its metabolites (3’-AMP, 2’-AMP and adenosine), precursors of adenosine involved in the extracellular adenosine pathway, have been reported to suppress different cytokines and chemokines, like TNF-α and CXCL10, being released from the activated microglia (Newell et al., 2015). As cytokines or chemokines are found predominantly in the transition of the microglial phenotype from M1 to M2, the role of A2A receptors should receive more attention (Franco and Fernández-Suárez, 2015). Spatial recognition memory improvement has been observed in mice lacking A2AR while the opposite was seen in transgenic mice overexpressing A2A receptors (Wang et al., 2006; Giménez-Llort et al., 2007). Preladenant, a selective antagonist of adenosine A2A receptors, has been reported to restore the microglial response towards cellular damage and plaque-associated microglial function in AD (Gyoneva et al., 2016). Caffeine, an antagonist of both A1 and A2A receptors, has been widely studied against Aβ-induced neurotoxicity and cognitive impairments in both in vivo and in vitro models of AD and exhibited much potential in the management of cognitive dysfunction (Querfurth et al., 1997; Arendash et al., 2006; Dall’Igna et al., 2007). It seems clear that A2A receptor plays a vital role in the activation of microglia in AD, therefore designing a selective antagonist of this receptor will prove a breakthrough in AD treatment (Abbracchio and Cattabeni, 1999; Nobre et al., 2010).
Metabotropic Glutamate Receptors
Glutamate is abundantly found throughout the CNS—it is an excitatory neurotransmitter that features prominently in maintaining communication between neurons, microglia, astroglia and oligodendrocytes (Brosnan and Brosnan, 2013; Stojanovic et al., 2014). Imbalance of glutamate signaling plays an important role in AD pathogenesis (Lan et al., 2014). There are two types of glutamate receptors (GluRs)—ionotropic and metabotropic, and microglia express both types of receptors (Noda, 2016). Based on sequence homology, coupling with G proteins and ligands, metabotropic glutamate receptors (mGluRs) are further divided into three subgroups: Group I consists of mGlu1 and mGlu5 receptors; Group II consists of mGlu2 and mGlu3 receptors; and Group III consists of mGlu4, mGlu6, mGlu7 and mGlu8 receptors (Niswender and Conn, 2010). Recently, the function of mGlu5, a Group I mGluR in microglia activation, has been investigated (Xue et al., 2014; Zhang et al., 2015). In an LPS-induced neuroinflammation model, Triptolide (T10), a potent inhibitor of microglia activation has been reported to attenuate inflammation and show immunosuppression through mGlu5 receptor upregulation. In both BV-2 microglia and primary microglia cells, blocking or knocking down of mGlu5 receptors abolishes the anti-inflammatory effect of T10. T10 also blocks the LPS-induced expression of iNOS, TNF-α, IL-1β and IL-6 through modulating the MAPK pathway (Huang et al., 2018). These findings suggest the role of mGlu5 in neuroprotection against microglial activation-mediated neurotoxicity. Another striking finding illustrates mGlu5 receptors’ role in microglial communication with neurons through microglial macrovesicles (MV) shed. MV shedding from BV-2 microglia cells was produced upon activation of purinergic receptor P2X7 by benzoyl-ATP, which is elevated by treatment upon CHPG, a mGlu5 receptor agonist and ablated by LPS treatment. The results of this study demonstrated that MV produced from CHPG-treated BV-2 microglia significantly rose rotenone-induced neurotoxicity in SHSY5Y cells. An increased level of miR146a, a miRNA only found in MV produced from CHPG-treated BV-2 cells, has been postulated to be responsible for mGlu5-mediated neurotoxicity, which proves the involvement of microglial mGlu5 receptors in neuronal death (Beneventano et al., 2017). However, further research is required to clarify the underlying mechanism in mGlu5 receptor-mediated MV shedding and its possible implication in neurotoxicity. On the other hand, group II mGluRs are involved in the switching of microglial phenotypes to a neurotoxic phenotype upon activation by Chromogranin A (CGA). In contrast, a specific antagonist, (RS)-α-methyl-4-sulphonophenylglycine, of these receptors diminishes microglial activation and neurotoxicity (Kingham et al., 1999; Taylor et al., 2002). The neurotoxic effect of mGluR2 upon activation by L-2-amino-4-phosphono-butyric acid, a specific group III receptor agonist, is associated with TNF-α release and caspase-3 activation, which was potentiated by a death receptor ligand, Fas (Taylor et al., 2003). Durand et al. (2014) reported the link between mGluR3-mediated microglial activation in the pathogenesis of AD. Their work provided evidence that activation of mGluR3 by its agonist, LYY379268, increased soluble amyloid Precursor Protein-α (sAPPα) by facilitating the non-amyloidogenic cleavage of APP (Durand et al., 2014). This finding was further validated by their very recent investigation proving mGluR3 activation in glial cells has a dual function, such as increasing BDNF and sAPPα levels and reducing amyloid from cells by phagocytosis (Durand et al., 2017). Altogether, the evidence suggests the multifunctional role of mGluR2- and mGluR3-mediated microglial phagocytosis, switching of microglial phenotype, and non-amyloidogenic cleavage of APP. These findings indicate their potential value as therapeutic targets for AD. Besides the well-documented role of microglial group II mGluRs in AD, very little is knowabout the involvement of microglial group III mGluRs in AD. In neurodegenerative diseases the activation of microglial group III mGluRs can modulate the release of stable neurotoxins from microglia and protects neurons from microglial neurotoxicity (Taylor et al., 2003). In addition, activation of group III mGluRs can encourage microglia to adopt neurotrophic phenotype. Microglia adopt neurotrophic phenotypes through decreasing the glutamate release and suppressing excitotoxicity as a result of astrocytic glutamate release being elevated. (Williams and Dexter, 2014). Two agonists of group III mGlu receptors—(L)-2-amino-4-phosphono-butyric acid (L-AP-4) and (R,S)-phosphonophenylglycine (RS-PPG)—showed a reduction of microglial neurotoxicity induced by LPS, CGA or Aβ(25–35) (Taylor et al., 2003). Therefore, selectively targeting microglial group III mGluRs may prove beneficial to understand microglial neurotoxicity in AD and to design effective therapeutic agents.
Adrenergic Receptors
Noradrenaline neurotransmitters has been reported to influence microglial activation in response to pathogenic conditions (Gyoneva and Traynelis, 2013). They operate by an adrenergic receptor which is divided into two groups, namely α and β adrenergic receptors, and further subdivided into several subtypes, including α1, α2 (subtypes α2A-, α2B- and α2C), β1, β2 and β3 (Ciccarelli et al., 2017). This distinct family of GPCRs couples with Gs, Gi and Gq G proteins and modulates their signaling by adenylate cyclase activity and generating diacylglycerol and inositol 1,4,5-triphosphate through phospholipase C stimulation (Shryock and Belardinelli, 1997). Expression of all five adrenergic receptors has been investigated in the different parts of AD patients and the expression of α2 receptors in particular was markedly increased in the cerebral cortex (Russo-Neustadt and Cotman, 1997). However, to investigate the link between microglia, adrenergic receptors and neuroinflammation in AD several studies have been conducted. Injection of a selective neurotoxin, N-(2-chloroethyl)-N-ethyl-2 bromobenzylamine (DSP4), into a mice model of AD has been demonstrated to increase iNOS and IL-β levels in microglia in Aβ-treated rats. Reversal of iNOS and IL-6 concentration elevations has been observed with co-administration of noradrenalin or isoproterenol, an adrenergic receptor agonist, proving that loss of noradrenaline is connected to Aβ-induced neuroinflammation and consequent death in an animal model of AD (Heneka et al., 2002). In another study, α2 levels were found to be significantly increased in the different locations of the brain in AD patients, such as prefrontal cortex and hippocampus cerebral microvessels (Kalaria et al., 1989). In recent times, the α2A-adrenergic receptor has been reported to modulate a novel role in norepinephrine release and response control by APP. APP has been found to disrupt the recruitment of arrestin 3, which modulates α2A-adrenergic endocytosis (Zhang et al., 2017). Moreover, the α2A-adrenergic receptor has been reported to play a crucial role in dexmedetomidine (DMED)-induced improvement in systemic inflammation (SI)-induced cognitive dysfunction. The protective effect of DMED against SI-induced microglial hyperactivation, cognitive impairment and hippocampal neuroinflammation was mediated by the α2A-AR signaling pathway, and its involvement was confirmed by blocking its activity by nonspecific α2A receptor antagonist, atipamezole, or the specific antagonist of α2A-AR, BRL-44408. However, the results showed that DMED treatment is only effective if treated during the SI (Yamanaka et al., 2017). Furthermore, DMED has been reported to play a crucial role in α2-adrenoceptor-mediated microglial polarization in 6-hydroxy dopamine (6-OHDA)-treated BV-2 cells. The 6-OHDA treatment can induce polarization of microglia to the M1 state in BV-2 cells, which was prevented by DMED. Pretreatment with DMED seemed to attenuate 6-OHDA-induced release of different proinflammatory markers, such as IL-6, IL-1β and TNF-α as well as expression of IL-10 and IL-13 along with TGF-β2. Furthermore, DMED also suppressed IL-4-mediated microglial polarization and subsequent expression of microglial M2 markers, Arg-1, resistin-like α (Retnla/Fizz1) and chitinase 3-like 3 (Chi3l3/Ym1; Zhang et al., 2017).
The role of β2 receptors in the pathogenesis of AD has gained the most interest among all other adrenergic receptors and is also linked with Aβ generation (Ni et al., 2006; Yu et al., 2011). In addition, chronic treatment with β2 adrenergic receptor agonist, isoproterenol, has been implicated to increase Aβ concentrations in murine models of AD accumulation through increasing the activity of γ secretase (Ni et al., 2006). Very recently, in a 5XFAD transgenic mouse model of AD, chronic administration of biased and selective β1 adrenergic receptor agonist, xamoterol, reduced Aβ toxicity-associated neuroinflammation and tau by reducing microgliosis and astrogliosis markers, allograft inflammatory factor 1 (Iba1) and glial fibrillary acidic protein (GFAP), as well as inflammatory markers, Iba1, CD74, CD14 and TGFβ. In primary microglia cell cultures, xamoterol diminished TNF-α produced by LPS treatment (Ardestani et al., 2017). Lately, a biased and selective β1 adrenergic receptor partial agonist, STD-101-D1, has been shown to suppress TNF-α production and LPS-induced neuroinflammation in both primary microglia and C57Bl/6J mice with high brain permeability (Yi et al., 2017).
Muscarinic Acetylcholine Receptors
Acetylcholine was one of the first discovered neurotransmitters. In the CNS, there are two types of acetylcholine receptors—nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs). Both nAChRs and mAChRs neurotransmitters mediate neurotransmission by acetylcholine and are involved in learning and cognition (Zhao et al., 2016). These two receptors are widely expressed in neurons and glial cells, whereas expression of mAChrRs on microglia deserves more investigation (Wessler et al., 1998; Ragheb et al., 2001). Further, most of the therapeutic drugs approved by FDA for the symptomatic treatment of AD belongs to the class of AChEI (Clark and Karlawish, 2003). mAChRs are one of the subfamilies of GPCRs involved in many fundamental neurological functions (Kruse et al., 2014). There are five subtypes of mAChRs (M1–M5) reported thus far. They have been implicated in many CNS disorders, including AD (Bonner et al., 1987; Jiang et al., 2014). Based on many expert opinions, M1 mAChRs has been suggested as a potential drug target for AD (Langmead et al., 2008; Melancon et al., 2013). It has been claimed that the G-protein coupling of mAChR M1 in the neocortex of AD patients is connected with cognition, short-term memory, and memory consolidation (Anagnostaras et al., 2003; Tsang et al., 2006). Jiang et al. (2014) verified the uncoupling the mAChR M1/G-proteins as one of the causes of cognitive impairment in AD. Therefore, M1 can be a potential target to develop therapeutic interventions to remedy cognitive deficits. Moreover, it has been described that human microglial mAChR responses to cholinergic agonists induce rapid changes based on activation of Ca2+, which acts as a secondary messenger initiating a downstream cascade of signaling pathways in CNS disorders (Zhang et al., 1998; Hasselmo, 2006). Unfortunately, responsiveness to ACh by different microglial mAChRs in various locations of brain has not been broadly studied nor received much attention in the literature (Zhang et al., 1998; Hirayama and Kuriyama, 2001; Nyakas et al., 2011; Lee et al., 2014). Despite many reports surrounding microglial mAChRs in different CNS disorders, there are very few studies related to mAChR-mediated microglial activation in AD (Zhang et al., 1998; Kondo et al., 1999; Gnatek et al., 2012; Shin et al., 2015). Recently, Pannell et al. (2016) studied the functional expression of mAChR in microglia isolated from a mouse model of AD. They noted that nearly 25% of isolated microglia and 60% from a stroke model responded to carbachol, which is an agonist of mAChRs. The result was obtained through using an anti-M3 antibody in fluorescence-activated cell sorting (FACS) analysis (Pannell et al., 2016). Additionally, Joseph et al. (2006) carried out several experiments regarding the effect of the blueberry extract (BBE) on oxidative stress-mediated signaling on Aβ, which induced a change in cognitive function while aging and in dementia in cultured primary hippocampal neuronal cells (HNC). This suggests that BBE suppresses oxidative stress by lowering pCREB and pPKC γ activated by dopamine in M1-transfected cells (Joseph et al., 2006, 2007). A similar result was described for C-2 ceramide-induced stress signaling in COS-7 cells (Joseph et al., 2010).
Recent evidence shows that there is a relationship between APP and M1 and M2 mAChRs from slices of rat cortex, hippocampus, striatum and cerebellum. M1 mAChR increases APP release and M2 mAChR decreases the formation of APPs in vitro (Farber et al., 1995). In another study, an increase in amyloidogenic APP processing was observed in M1 mAChR knock-out mice (Davis et al., 2010). In addition, M1 and M2 mAChR activation was also associated with a rise in α-secretase, which can be antagonized by PKC inhibitors or phorbol esters that will activate PKC and increase sAPP release and P3, and as such, Aβ generation will be diminished (Buxbaum et al., 1992; Hung et al., 1993; Farber et al., 1995). Caccamo et al. (2006) reported that M1 mAChR antagonist, dicyclomine, can upregulate BACE1 levels in ADtg mice, whereas AF267B, an M1 mAChR agonist, remarkably decreases Aβ and tau levels in the hippocampus and cortex region of the brain. The deletion of the M1 mAChR gene has also seemed to markedly elevate Aβ plaque formation. Its expression in neurons is sufficient for non-amyloidogenic APP processing. and nonselective inhibition of mAChRs blocks the beneficial effects of M2 and M4 receptors (Farber et al., 1995; Davis et al., 2010). Although there is much less work on microglial mAChRs related to AD progression, this data offers hints for further evaluation to locate other mAChRs, such as M2 on microglia. Since that discovery, the M1 mAChR agonist exerts the most selective and specific attenuation of the pathological hallmarks of AD, and developing a drug based on selective agonists of M1 mAChR for both neurons and microglia, like Xanomeline, hold possibilities for the treatment of AD (Bodick et al., 1997).
Cannabinoid Receptors
Cannabinoid receptors are G-protein coupled receptors found ubiquitously throughout the brain and the other parts of the body. Their endogenous ligand is endocannabinoid and external ligand is tetrahydrocannabinol (THC), which both inhibit cAMP accumulation. CB receptor signaling pathways couple with inhibitory G-protein (Gi), and their binding with Gs has also been reported (Howlett et al., 1986; Glass and Felder, 1997). They comprise of three receptors, namely CB1, CB2 and GPR55 receptors. Recent evidences suggest that endocannabinoid system is more complicated and it contains additional receptor (Ryberg et al., 2007). In this regards GPR55, previously an orphan receptor which can also be activated many classical cannabinoids has been added as a third cannabinoid receptor (Moriconi et al., 2010). These receptors mediate many psychoactive effects of cannabinoids (Mackie, 2008). The expression of cannabinoid receptors depends on the microglial activation state (Becher and Antel, 1996). CB1 and CB2 are both expressed on microglia and are especially overexpressed in neuroinflammatory disorders (Ma et al., 2015). Expression of CB1 receptors in human microglia is not well documented (Stella, 2010). In rat microglia, CB1 receptor has been found to inhibit NO release from endotoxin or cytokine-activated cortical microglia cells. Cells treated with both high and low-affinity binding enantiomers of cannabinoid CP ((+)-CP56667 and (−)-CP55940) showed dose-dependent inhibition of NO release by LPS stimulation. This attenuation of NO was reversed by pretreatment with pertussis toxin (Gαi/Gαo protein inactivator) or cholera toxin (Gαs activator). The presence of CB1 receptors in rat microglia was demonstrated by immunoblot assaying with CB1 receptor amine terminal domain-specific antibody and mutagenic reverse transcription-polymerase chain reaction. Moreover, colocalization in microglia was also confirmed by a microglial marker (Waksman et al., 1999). Besides this, CB1 has been reported to play a neuroprotective role in MPTP-induced neurotoxicity in Parkinson’s disease model. Activation of CB1 receptors showed marked improvement in motor function within animal models and survival rate of DA neurons in both the substantia nigra (SN) and striatum. Supposedly, this result is based on suppression of the production of NADPH oxidase reactive oxygen species by microglia and reduction in proinflammatory cytokines, IL-1β and TNF-α, from activated microglia (Chung et al., 2011).
CB2 receptors are highly expressed in inflammatory and primed macrophages and the functional sates of macrophages are also sensitive to the action of CB2 receptors in response to both endogenous and exogenous ligands (Carlisle et al., 2002). Their activation in microglia modulates cell proliferation and migration while also increasing the accumulation of less harmful microglia at sites of injury while also reducing IL-6, TNF-α and free radical production (Facchinetti et al., 2003; Walter et al., 2003; Carrier et al., 2004; Eljaschewitsch et al., 2006; Bisogno and Di Marzo, 2010). To assess the upregulation of CB2 receptors in activated microglia, microglia cells were cultured with microglial activators, IFN-γ and granulocyte macrophage-colony stimulating factor (GM-CSF). Activated microglia were tested for increases in the expression of CB2 receptors, which was an 8- to 10-fold increase over non-treated microglia cells. In another study, it was seen that activation of microglia by both LPS and IFNγ was attenuated by treatment with a cannabinoid CB2 receptor agonist, AM1241, which shifted the microglial state from M1 to M2. To confirm the role of CB2 receptors on the attenuation of microglial activation, microglial cells were treated with a CB2 receptor antagonist, AM630, or chelerythrine, a PKC inhibitor that completely reversed the effect of the agonist (Ma et al., 2015).
In the brain of AD patients, the expression of cannabinoid receptors has been implicated to provide neuroprotection and prevention of neurodegeneration. The expression of cannabinoid receptors was investigated in the hippocampus and entorhinal cortex sections of postmortem brains of AD patients. The expression of the CB2 receptors in the neurite plaques of microglia and astrocytes was very high and selective, but the expression of CB1 receptors was indifferent (Benito et al., 2003). The expression of CB1 in the postmortem brain samples of various AD patients during the clinical deteriorating stage of AD has been described. CB1 expression was initially higher in the selective hippocampal areas and gradually decreased as AD progressed, clearly indicating the involvement of CB1 receptors during the earlier stages of AD (Manuel et al., 2014). In Aβ1–40-induced microglial activation within an animal model featuring AD administration of synthetic cannabinoids (HU-210, WIN55, 212-2 and JWH-133), a selective CB2 agonist inhibited Aβ-induced microglial activity with marked changes in microglial activation markers and cell morphology with a release of pro-inflammatory cytokines (TNF-α; Ramírez et al., 2005). In summary, microglial CB1 and CB2 receptors are able to modulate microglial activation in both in vitro and in vivo models of AD, which provides a potential avenue to develop and design therapeutic agents for the treatment of AD.
Furthermore, the third cannabinoid receptor GPR55 is a novel cannabinoid receptor highly expressed in various regions of brain, especially in the striatum, hippocampus, hypothalamus, frontal cortex, cerebellum, and brain stem (Ryberg et al., 2007). GPR55 can bind to both cannabinoid compounds, such as abnormal-cannabidiol (Abn-CBD), although its endogenous ligand is lysophophatidylinositol (LPI; Shore and Reggio, 2015). The potential of GPR55 in neurodegenerative disorders has been reported in many studies (Shore and Reggio, 2015; Celorrio et al., 2017; Alavi et al., 2018). It is significantly expressed in both primary and BV-2 microglia cells. Stimulation of LPS in BV-2 exhibits downregulation of GPR55 expression upon treatment with LPI. LPI increases ERK phosphorylation in BV-2 cells after IFN-γ treatment, which suggests the involvement of GPR55 in microglia activation and modulating inflammation (Pietr et al., 2009). In addition, how microglial GPR55 mediates neuroprotection by LPI was demonstrated in rat organotypic hippocampal slice cultures (OHSC). Treatment with LPI protects dentate gyrus granule cells from NMDA-induced lesion. The involvement of GPR55 in neuroprotection was confirmed by the deletion of GPR55 through siRNA (Kallendrusch et al., 2013). Furthermore, GPR55 knockout mice exhibited impaired movement coordination in comparison with GPR55 null mutant mice (Wu et al., 2013). Administration of LPI onto the hippocampus of rats showed a reduction in the capability of spatial navigation strategy and worsened the capability of finding the escape tunnel in the barnes-maze (BM), a test that performed to assess spatial learning and memory (memory to place objects). In contrast, rats receiving CID16020046, a GPR55 antagonist, spent less time in the target zone during the test, suggesting the involvement of hippocampal CA1 GPR55 in spatial learning and memory (Marichal-Cancino et al., 2018). Therefore, all these findings indicate a direct connection of GPR55 to motor function, memory formation, and neuroinflammation, but additional research is merited for its role in microglial activation in neurodegeneration. Uncovering the GPR55 pathway in microglia may provide a better understanding of AD.
Chemokine Receptors
Chemokine receptors are mostly located on microglial membrane and linked with diverse physiological functions, such as neuronal migration, synaptic activity, cell proliferation and even neuronal death. However, different chemokine receptors are reported to be involved in neurodegenerative diseases (Cartier et al., 2005; Mines et al., 2007). There are almost 50 types of chemokines, 18 of which are signal transducing and five acting as decoy/scavenger receptors. They are further grouped into two major categories (Proudfoot, 2002). Four chemokine receptors, CCR3, CCR5, CXCR2 and CXCR3, and their ligands have been described to be present in the brain of AD patients. Yet, CCR3 and CCR5 are predominantly found in microglia of both control and AD brains, expressing reactive microglia, and MIP-1β expression (ligands for CCR5 and/or CCR3) of reactive astrocytes were seen to be associated with amyloid deposits (Xia et al., 1998). Moreover, with the administration of Aβ peptide in microglia isolated from the brain of AD and control patients, CXCL8, CCL2, and CCL3 levels increased in a dose-dependent manner (Cartier et al., 2005). Another report on microglia isolated from postmortem human brains showed upregulation of CXCL8, CCL2, CCL3 and CCL4 to a lesser extent after incubation with Aβ (Walker et al., 2001). Furthermore, it has been noted by Cardona et al. (2006) that the CCR3, CCR5, CX3CR1, CXCR2 and CXCR3 expressed in microglia were associated with senile plaques, whereas expression of CX3CR1 was determined as high in microglia and its ligand CX3CL1 in neurons. They also reported the involvement of CX3CR1 signaling to protect against microglial neurotoxicity in three different in vivo models (Hickman and El Khoury, 2010). Besides that, CXCR2 has been investigated in order to establish its involvement in Aβ production and γ-secretase activity. The findings suggest that CXCR2-mediated Aβ production is regulated by diminishing γ-secretase activity (Bakshi et al., 2008). Overall, this evidence supports the idea there is involvement of chemokine receptors in the recruitment and accumulation of microglia in AD. Determining the precise chemokine receptors and understanding the underlying mechanism of chemokine receptor-mediated signaling will aid in develop therapeutic strategies to treat AD.
G-Protein-Coupled Formyl Peptide Receptor-Like 1 (FPRL1)
G-protein-coupled formyl peptide receptor-like 1 (FPRL1) and its mouse homolog, FPR2, binds Aβ42 and activates microglia, maintaining its chemotactic activity in AD (Iribarren et al., 2005). FPR2 binds to Aβ and mediates Aβ uptake in glia-infiltrating senile plaques. To determine the relationship between Aβ and resolution of inflammation, microglia cells were treated with Aβ42, and the result showed that FPR2, α7nAChR, and (PPAR)-δ was downregulated by Aβ42 treatment, indicating the expression of FPR2 is correlated with resolution of inflammation and dysfunction in AD (Zhu et al., 2015). Annexin A1 (ANXA1), an anti-inflammatory mediator, has been reported to suppress activation of microglial activation through formyl peptide receptor-like 1 (FPRL1/FPR2) signaling (Gavins and Hickey, 2012). In a more contemporary work, ANXA1 was investigated as an anti-inflammatory agent and whether it had any effect on Aβ clearance and degradation in both human patients and a 5XFAD mice model. The result showed that FPR2 levels rose in both human patient and mice models. The Aβ concentration was significantly reduced by enzymatic degradation in N2a cells followed by treatment with ANXA1. It was claimed that this effect was based on the FRP2 receptor (Ries et al., 2016). Humanin, a neuroprotective peptide, has been reported to ameliorate pathological changes and cognitive deficits in AD models induced by Aβ-peptide (Niikura et al., 2011; Chai et al., 2014). In addition, humanin also suppresses Aβ-induced aggregation and fibrillary formation in mononuclear phagocytes. Moreover, humanin is reported to exert neuroprotective effects by inducing chemotaxis of mononuclear phagocytes by both human FPRL1 and its murine counterpart, FPR2 (Ying et al., 2004). In primary mouse microglia cells, N9 treatment and incubation with TNF-α was shown to increase FPR2 gene expression and decrease the chemotactic response against Aβ42. Meanwhile, the effect of TNF-α on increasing FRP2 expression was based on MAPK p38 and dependent on the p55 TNF-α receptor (Cui et al., 2002). On the contrary, IL-4 and TGFβ were observed to inhibit mFPR2 in mice (Iribarren et al., 2005). Additionally, TNF-α’s synergistic effect was observed through IL-10 (Iribarren et al., 2007). Palmitoyl-cys[(RS)-2, 3-di(palmitoyloxy)-propyl]-Ala-Gly-OH (PamCAG), a TLR2 muramyl dipeptide (MDP) ligand featuring an intracellular receptor nucleotide-binding oligomerization domain 2 (NOD2) ligand has been reported to upregulate mouse mFPR2 (Chen et al., 2008). With this, receptors for advanced glycation endproducts (RAGE) and both FPR2 and FPRL1 are thought to play important roles in Aβ1–42-mediated signal transduction in glial cells. The interaction between RAGE and FPRs are further investigated by co-immunoprecipitation and fluorescence microscopy (Slowik et al., 2012). This finding revealed the missing link and clarifies the explanation of the broad ligand spectrum of FPRs and a clear mechanism underlying their anti-inflammatory and neuroprotective activity. However, to completely understand FPR1/2-mediated chemotactic activity in AD, further investigation is necessary.
Chemokine-Like Receptor 1 (CMKLR1)
The chemerin receptor chemokine-like receptor 1 (CMKLR1), a homolog of FPR2, is also a functional receptor of Aβ42. Although these two receptors bind to the same Aβ, they have a different response. Unlike FPR2, CMKLR1 acts through different pathways, like activation of ERK1/2, PKA, and Akt, and facilitates processing and clearance of Aβ42. CMKLR1 also lacks Aβ-induced Ca2+ mobilization in CMKLR1-RBL cells (Peng et al., 2015). This lack of secondary messenger is connected with many signaling pathways responsible for downstream activation of GPCRs. Very little is known about microglial CMKLR1 receptors and the pathway associated with its binding with Aβ42, further detailed investigation is needed to establish its potential as a therapeutic target for AD.
Orphan GPCRs
There are almost 140 GPCRs lacking any known endogenous ligands. These GPCRs are thus termed orphan GPCRs. Despite the sophisticated advancement of GPCR-based drug design and discovery, orphan receptors remain less popular therapeutic drug targets (Stockert and Devi, 2015). The orphan GPCR, GPR3, has been reported to be a potential therapeutic target for AD treatment (Ruiz-Medina et al., 2011). Internalized GPR3 with β arrestin 2 promotes Aβ production. Moreover, GPR3-mediated Aβ production is regulated by the cleavage of APP (Nelson and Sheng, 2013). The overexpression of GPR3 in a transgenic mouse model confirmed the claim of the generation of Aβ42 and Aβ40. The generation of Aβ was based on the cleavage of APP by γ-secretase without any alteration to the expression of γ-secretase subunits. On the other hand, genetic depletion of GPR3 in mice showed a marked decrease in both Aβ40 and Aβ42 generation (Thathiah et al., 2009; Huang et al., 2015). Although there is a very little or no work of orphan GPCRs in microglial activation, it has been speculated that previously unidentified abnormal cannabidiol (Abn-CBD) receptors, GPR18, is linked with abnormal CBD-mediated microglial migration. In addition, it was noted that microglia abundantly express GPR18 on the cell surface. N-arachidonoyl glycine (NAGly), a GPR18 agonist, also mimics abnormal CBD activity and potentiates microglial migration through MAPK activation in response to CNS damage (McHugh et al., 2010). This exquisite finding suggests the involvement of orphan GPCRs in microglial migration to a pre- or pro-inflammatory state through lipid-based signaling mechanisms in terms of CNS stimuli.
G-Protein-Coupled Receptor Kinase
G-protein-coupled receptor kinase is responsible for homologous desensitization of retarded GPCRs, prolongation of GPCR activity and is widely assumed to selectively phosphorylate GPCRs (Li et al., 2015). Microglial GRK2 is well-known for its cell-specific regulation severity and transition of pain from acute to chronic (Eijkelkamp et al., 2010; Kavelaars et al., 2011). Suo et al. (2004) documented that at prodromal and earlier stages of the development of AD, Aβ and GPCR kinase-2/5 (GRK2/5) dysfunction at sub-threshold levels is relevant. An in vitro study of Aβ treatment noted reduced GRK2/5 levels in the membrane of murine microglia cells, and an in vivo study in ADtg mice (CRND8) showed similar results. These findings indicated a novel link between Aβ accumulation and GRK at an early stage of AD development (Suo et al., 2004). Since, Aβ-induced neurotoxicity and inflammation plays a pivotal role in the progression of AD, an in-depth understanding of the role of GRKs in Aβ-induced neurotoxicity may provide critical knowledge of AD pathogenesis.
Perspective and Conclusions
After decades of research and clinical trials, still, there is no definitive treatment option for AD (Godyn et al., 2016; Hung and Fu, 2017). Seeing the conventional strategy for selecting a therapeutic agent is based on the characteristics of prevention, clearance and degradation of Aβ peptides, these can also be achieved by targeting microglial GPCRs involved in different stages from Aβ generation, degradation, and clearance in the brain. Targeting specific microglial GPCRs at a specific stage of AD progression will not only offer better options for symptomatic treatment, but also a therapeutically potential disease modification. In this article, we have thoroughly discussed GPCR-linked microglial activation in AD, which also sheds light onto the potential of microglial GPCRs for strategic therapeutic intervention for AD. Changes in receptors as well as pre- and pro- inflammatory markers expressed during the various stages of microglia activation are a potential research avenue to completely understand microglial characteristics in many neurodegenerative disorders (Hanisch, 2013). The role of different microglia GPCRs, such as adenosine receptors (A1, A2A, A2B and A3) has been found to be directly linked with microglia activation. Modulation of these receptors has been found to suppress microglia activation and induce Aβ toxicity and neuronal damage. In addition, microglial adrenergic α2A receptors have impact on microglial polarization towards the M1 state. Moreover, both microglial adenosine and adrenergic receptor’s role in Aβ toxicity and cognitive dysfunction show promise. Different microglial GPCRs are linked with Aβ generation. Microglial β2 adrenergic receptor and mAChRs, especially M1 have distinct roles in Aβ generation. Activation of microglial M1 mAChR increases α-secretase and sAPP production. Although the presence of M2 and M4 subtypes on microglia requires further evidence, the M1 receptor acts in concert with these two mAChR subtypes (Farber et al., 1995). The microglial mGluRs are also involved in Aβ generation through modulating non-amyloidogenic cleavage of APP and sAPPα activity. Group II microglial mGluR activation can elevate sAPPα and decrease Aβ concentrations. They also promote non-amyloidogenic cleavage of APP, thus decreasing amyloid levels in AD brain. Besides the role of GPCRs in microglial activation, polarization and migration towards neuronal damage, degradation of Aβ and microglia-mediated neurotoxicity is prominently featured in Aβ-mediated neurotoxicity. Microglial P2 (described earlier) and α7 nAChRs have been found to play a crucial role in Aβ degradation and clearance through microglial phagocytosis and chemotaxis. Similarly, two microglial Aβ receptors, FPRL1 and CMKLR1, are responsible for microglial chemotaxis and microglia-mediated Aβ degradation and clearance. Very recently, the orphan receptor, GPR18, has been reported to promote microglial migration towards neuronal damage, though more experimental support is necessary to clarify its role in AD progression. Furthermore, a few GPCRs are related to microglia-mediated neurotoxicity and microglia-neuronal bidirectional communication, which plays a vital part in AD pathology, such as mGluRs. At present, the involvement of microglial cannabinoid receptors and chemokine receptors are gaining much attention regarding Aβ toxicity and Aβ-induced microglial activity. Therefore, targeting cannabinoid receptors and chemokine receptors may serve as additional promising AD therapeutic targets in the future.
Generally, symptoms associated with AD are ambiguous in pathological origin and also linked with different individual GPCRs. Aβ can bind to a single microglial GPCR or with other co-receptors. Depending on the binding of Aβ to different microglial GPCRs GPCR-Aβ-mediated signaling cascade may vary. Therefore, knowledge of these co-receptors or receptor complexes is a prerequisite to understanding Aβ-mediated neurotoxicity. Therefore, targeting more than one GPCR or GPCR with other receptors involved in AD pathogenicity can be a strategic approach to design and develop new therapeutic agents that can counter the multifaceted pathogenesis of AD. For the better understanding of AD pathophysiology, more advanced and sophisticated animal models are required to mimic specific pathologic conditions for AD, such as specific GPCR knockout mice, different microglial cells representing various activated phenotypes for specific targets and incorporating different gene editing tools may also prove effective. Microglia are generally restless—they frequently change their phenotype. Recently, it has been speculated that microglia may show heterogenicity in their activity depending on their location in the brain and in accordance with the type of stimuli (Town et al., 2005; Arcuri et al., 2017).
The success rate of GPCR-targeted therapeutic agents in 2013–2017 reported by FDA in phases I, II and III in terms of clinical trials are 78%, 39% and 29%, respectively. This is comparatively higher than the FDA’s average for other drug candidates currently on trial (Hauser et al., 2017). The major limitation of developing therapeutic agents for AD is the lack of mechanistic selection criteria for drug targets based on biological markers that can meet clinical efficacy standards. To do so, classification and selection of GPCRs based on functional activity rather than conventional structural similarity will reduce the burden of screening and designing GPCR selective drugs. In addition, to identify novel ligands for GPCRs, DNA-encoded libraries may be useful. Potential candidates for GPCRs and their chemical and biological data can be found from reported research and evaluation. However, the chemical and biological data of potential GPCR candidates are not well-organized in online libraries and databases. Yet, this data can be organized and made available through online libraries, such as GPCRdb and CheMBL to facilitate GPCR-based ligand design. A very efficient concept of biased ligand can also be adopted. Biased ligands can stabilize the subsets of receptor conformation to produce novel therapeutic outcomes. The increasing popularity of this ligand concept lies in its capability for designing more safe, tolerable drugs with high efficacy (Violin et al., 2014). Another concept is allosteric modulators—they can remotely modulate receptor activity by endogenous or physiological ligands that induce activity at the binding site. This novel and attractive mechanism can offers superior target selectivity over conventional modulators of GPCRs (Hauser et al., 2017). Another concern regarding targeting GPCRs on microglia is heteromerization of receptors. Heteromerization of receptors is associated with crosstalk between two extracellular signals. Recently, it has been established that appropriate heteromers of GPCRs are important and modification of the pharmacology and signaling properties of drugs depends of selection of unique GPCR heteromers (Albizu et al., 2010). The presence of heterodimers or oligomeric complexes of GPCRs on different cells is now widely known and accepted (Albizu et al., 2010). Although heterodimerization may have implications in the pharmacology of therapeutic agents, the knowledge of regarding the molecular basis of such heteromerization still remains in its initial stage (Milligan, 2004). However, several heteromers of various GPCRs on different tissues have been reported to possess therapeutic relevance to drug design. Co-internalization or coexpression of different receptors, such as α1a/α1b adrenoceptors, A1/A2A, A1/mGlu1α and A2/mGlu5, CB1/A2A in different types of cells has been reported in both in vitro and in vivo studies related to a variety of neurological disorders (Ciruela et al., 2001, 2006; Stanasila et al., 2003; Kachroo et al., 2005; Carriba et al., 2007; Tebano et al., 2009). Many drugs possess unique pharmacology and affinity towards GPCR heterodimers, such as anti-parkinsonian drugs featuring more affinity to D3/D2 than its homodimers (Yoshioka et al., 2001; Maggio et al., 2003). These findings suggest that heterodimers are worthy of more attention as drug targets for neurological disorders. Meanwhile, there is still lack of studies on microglial GPCR heteromerization related to Aβ generation, clearance, and degradation, which should be a prime concern for developing bivalent ligands, such as agonist-agonist, agonist-antagonist or one linked with amino acid spacers. These bivalent ligands can target multiple heteromers at a time and have superior efficacy than single ligand-based therapy (Zhang et al., 2007). Therefore, biased ligands and allosteric modulators, both novel concepts, offer a potential strategy to design GPCR-based drugs for AD.
Author Contributions
MEH, D-KC and I-SK conceptualized and designed the study. D-KC also supervised and corresponded. MEH reviewed the literature and wrote the manuscript, drew the figures and made the tables. MJ and MA contributed to the arrangement of data. I-SK contributed to drawing the figures and helped in the revision of the article. All authors read and approved the final manuscript.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2018R1C1B6005129 and Grant no. 2017R1A2A2A07001035).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
6-OHDA, 6-hydroxy dopamine; Aβ, Amyloid-β peptide; Abn-CBD, abnormal cannabidiol; AChE, Acetylcholinesterase; AChEI, Acetylcholinesterase inhibitors; AD, Alzheimer disease; APL, Allosterically potentiating ligand; APP, Amyloid-β precursor protein; Arg1, Arginase 1; β-APP, Amyloid-β precursor protein; BACE1, β-site amyloid precursor protein cleaving enzyme 1; CCL2, Chemokine (C-C motif); CD16/32, Cluster of differentiation 16/32; CGA, Chromogranin A; CNS, Central nervous system; CREB, cAMP response element-binding protein; CXCL10, C-X-C motif chemokine 10; DMED, Dexmedetomidine; FACS, Fluorescence-activated cell sorting; FDA, Food and Drug Administration; FPRL1, G-protein-coupled formyl peptide receptor-like 1; GFAP, Glial fibrillary acidic protein; GPR3, G-Protein-Coupled Receptor 3; GRK, G-protein-coupled receptor kinase; hAPP, Human beta-amyloid precursor protein; hPS1, Human presenilin-1; Iba1, Allograft inflammatory factor 1; IL-1Ra, IL-1 receptor antagonist; LPS, Lipopolysaccharide; mAChRs, muscarinic acetylcholine receptor; MAPK, Mitogen-activated protein kinase; mGluRs, metabotropic glutamate receptors; nAChRs, nicotinic acetylcholine receptor; NAGly, N-arachidonoyl glycine; NFT, Immunoreactive neurofibrillary tangles; PI3 kinase/Akt, phosphatidylinositol 3-kinase/Akt; RAGE, Receptor for advanced glycation endproducts; sAPP, Soluble amyloid Precursor Protein; SR-A1, Scavenger receptor class A1; SR-B1, Scavenger receptor class B1; TGF-β, Transforming growth factor-β; TREM-2, Triggering receptor also expressed on myeloid cells-2; UDP, Uridine 5’-diphosphate; Ym1, Chitinase-like protein 1.
References
Abbracchio, M. P., and Cattabeni, F. (1999). Brain adenosine receptors as targets for therapeutic intervention in neurodegenerative diseases. Ann. N Y Acad. Sci. 890, 79–92. doi: 10.1111/j.1749-6632.1999.tb07983.x
Ajit, D., Woods, L. T., Camden, J. M., Thebeau, C. N., El-Sayed, F. G., Greeson, G. W., et al. (2014). Loss of P2Y2 nucleotide receptors enhances early pathology in the TgCRND8 mouse model of Alzheimer’s disease. Mol. Neurobiol. 49, 1031–1042. doi: 10.1007/s12035-013-8577-5
Alavi, M. S., Shamsizadeh, A., Azhdari-Zarmehri, H., and Roohbakhsh, A. (2018). Orphan G protein-coupled receptors: the role in CNS disorders. Biomed. Pharmacother. 98, 222–232. doi: 10.1016/j.biopha.2017.12.056
Albasanz, J. L., Perez, S., Barrachina, M., Ferrer, I., and Martín, M. (2008). Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol. 18, 211–219. doi: 10.1111/j.1750-3639.2007.00112.x
Albizu, L., Moreno, J. L., González-Maeso, J., and C Sealfon, S. (2010). Heteromerization of G protein-coupled receptors: relevance to neurological disorders and neurotherapeutics. CNS Neurol. Disord. Drug Targets 9, 636–650. doi: 10.2174/187152710793361586
Amihăesei, I., Cojocarut, E., and Mungiu, O. (2013). Alzheimer—certitudes and hypotheses. Rev. Med. Chir. Soc. Med. Nat. Iasi 117, 119–126.
Anagnostaras, S. G., Murphy, G. G., Hamilton, S. E., Mitchell, S. L., Rahnama, N. P., Nathanson, N. M., et al. (2003). Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat. Neurosci. 6, 51–58. doi: 10.1038/nn992
Angulo, E., Casadó, V., Mallol, J., Canela, E. I., Viñals, F., Ferrer, I., et al. (2003). A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer’s disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 13, 440–451. doi: 10.1111/j.1750-3639.2003.tb00475.x
Arcuri, C., Mecca, C., Bianchi, R., Giambanco, I., and Donato, R. (2017). The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 10:191. doi: 10.3389/fnmol.2017.00191
Ardestani, P. M., Evans, A. K., Yi, B., Nguyen, T., Coutellier, L., and Shamloo, M. (2017). Modulation of neuroinflammation and pathology in the 5XFAD mouse model of Alzheimer’s disease using a biased and selective β-1 adrenergic receptor partial agonist. Neuropharmacology 116, 371–386. doi: 10.1016/j.neuropharm.2017.01.010
Arendash, G., Schleif, W., Rezai-Zadeh, K., Jackson, E., Zacharia, L., Cracchiolo, J., et al. (2006). Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain β-amyloid production. Neuroscience 142, 941–952. doi: 10.1016/j.neuroscience.2006.07.021
Bakshi, P., Margenthaler, E., Laporte, V., Crawford, F., and Mullan, M. (2008). Novel role of CXCR2 in regulation of γ-secretase activity. ACS Chem. Biol. 3, 777–789. doi: 10.1021/cb800167a
Bard, F., Cannon, C., Barbour, R., Burke, R.-L., Games, D., Grajeda, H., et al. (2000). Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919. doi: 10.1038/78682
Becher, B., and Antel, J. P. (1996). Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia 18, 1–10. doi: 10.1002/(sici)1098-1136(199609)18:1<1::aid-glia1>3.3.co;2-i
Beneventano, M., Spampinato, S. F., Merlo, S., Chisari, M., Platania, P., Ragusa, M., et al. (2017). Shedding of microvesicles from microglia contributes to the effects induced by metabotropic glutamate receptor 5 activation on neuronal death. Front. Pharmacol. 8:812. doi: 10.3389/fphar.2017.00812
Benito, C., Nunez, E., Tolon, R. M., Carrier, E. J., Rabano, A., Hillard, C. J., et al. (2003). Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 23, 11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003
Bisogno, T., and Di Marzo, V. (2010). Cannabinoid receptors and endocannabinoids: role in neuroinflammatory and neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 9, 564–573. doi: 10.2174/187152710793361568
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Bodick, N. C., Offen, W. W., Levey, A. I., Cutler, N. R., Gauthier, S. G., Satlin, A., et al. (1997). Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol. 54, 465–473. doi: 10.1001/archneur.1997.00550160091022
Bonner, T. I., Buckley, N., Young, A., and Brann, M. (1987). Identification of a family of muscarinic acetylcholine receptor genes. Science 237, 527–532. doi: 10.1126/science.3037705
Brosnan, J. T., and Brosnan, M. E. (2013). Glutamate: a truly functional amino acid. Amino Acids 45, 413–418. doi: 10.1007/s00726-012-1280-4
Brown, G. C., and Vilalta, A. (2015). How microglia kill neurons. Brain Res. 1628, 288–297. doi: 10.1016/j.brainres.2015.08.031
Burnstock, G. (2017). Purinergic signalling and neurological diseases: an update. CNS Neurol. Disord. Drug Targets 16, 257–265. doi: 10.2174/1871527315666160922104848
Buxbaum, J. D., Oishi, M., Chen, H. I., Pinkas-Kramarski, R., Jaffe, E. A., Gandy, S. E., et al. (1992). Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer β/A4 amyloid protein precursor. Proc. Natl. Acad. Sci. U S A 89, 10075–10078. doi: 10.1073/pnas.89.21.10075
Caccamo, A., Oddo, S., Billings, L. M., Green, K. N., Martinez-Coria, H., Fisher, A., et al. (2006). M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 49, 671–682. doi: 10.1016/j.neuron.2006.01.020
Cardona, A. E., Huang, D., Sasse, M. E., and Ransohoff, R. M. (2006). Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 1, 1947–1951. doi: 10.1038/nprot.2006.327
Carlisle, S., Marciano-Cabral, F., Staab, A., Ludwick, C., and Cabral, G. (2002). Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int. Immunopharmacol. 2, 69–82. doi: 10.1016/s1567-5769(01)00147-3
Carriba, P., Ortiz, O., Patkar, K., Justinova, Z., Stroik, J., Themann, A., et al. (2007). Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology 32, 2249–2259. doi: 10.1038/sj.npp.1301375
Carrier, E. J., Kearn, C. S., Barkmeier, A. J., Breese, N. M., Yang, W., Nithipatikom, K., et al. (2004). Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol. Pharmacol. 65, 999–1007. doi: 10.1124/mol.65.4.999
Cartier, L., Hartley, O., Dubois-Dauphin, M., and Krause, K.-H. (2005). Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res. Rev. 48, 16–42. doi: 10.1016/j.brainresrev.2004.07.021
Celorrio, M., Rojo-Bustamante, E., Fernández-Suárez, D., Sáez, E., Estella-Hermoso de Mendoza, A., Müller, C. E., et al. (2017). GPR55: a therapeutic target for Parkinson’s disease? Neuropharmacology 125, 319–332. doi: 10.1016/j.neuropharm.2017.08.017
Chai, G.-S., Duan, D.-X., Ma, R.-H., Shen, J.-Y., Li, H.-L., Ma, Z.-W., et al. (2014). Humanin attenuates Alzheimer-like cognitive deficits and pathological changes induced by amyloid β-peptide in rats. Neurosci. Bull. 30, 923–935. doi: 10.1007/s12264-014-1479-3
Che, Y., Hou, L., Sun, F., Zhang, C., Liu, X., Piao, F., et al. (2018). Taurine protects dopaminergic neurons in a mouse Parkinson’s disease model through inhibition of microglial M1 polarization. Cell Death Dis. 9:435. doi: 10.1038/s41419-018-0468-2
Chen, K., Zhang, L., Huang, J., Gong, W., Dunlop, N. M., and Wang, J. M. (2008). Cooperation between NOD2 and Toll-like receptor 2 ligands in the up-regulation of mouse mFPR2, a G-protein-coupled Aβ42 peptide receptor, in microglial cells. J. Leukoc. Biol. 83, 1467–1475. doi: 10.1189/jlb.0907607
Chhor, V., Le Charpentier, T., Lebon, S., Oré, M.-V., Celador, I. L., Josserand, J., et al. (2013). Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 32, 70–85. doi: 10.1016/j.bbi.2013.02.005
Chung, Y. C., Bok, E., Huh, S. H., Park, J. Y., Yoon, S. H., Kim, S. R., et al. (2011). Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J. Immunol. 187, 6508–6517. doi: 10.4049/jimmunol.1102435
Ciccarelli, M., Sorriento, D., Coscioni, E., Iaccarino, G., and Santulli, G. (2017). “Adrenergic receptors,” in Endocrinology of the Heart in Health and Disease, (New York, NY: Elsevier), 285–315. doi: 10.1016/B978-0-12-803111-7.00011-7
Ciruela, F., Casadó, V., Rodrigues, R. J., Luján, R., Burgueño, J., Canals, M., et al. (2006). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers. J. Neurosci. 26, 2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006
Ciruela, F., Escriche, M., Burgueno, J., Angulo, E., Casado, V., Soloviev, M. M., et al. (2001). Metabotropic glutamate 1α and adenosine A1 receptors assemble into functionally interacting complexes. J. Biol. Chem. 276, 18345–18351. doi: 10.1074/jbc.M006960200
Clark, C. M., and Karlawish, J. H. (2003). Alzheimer disease: current concepts and emerging diagnostic and therapeutic strategies. Ann. Intern. Med. 138, 400–410. doi: 10.7326/0003-4819-138-5-200303040-00010
Clayton, K. A., Van Enoo, A. A., and Ikezu, T. (2017). Alzheimer’s disease: the role of microglia in brain homeostasis and proteopathy. Front. Neurosci. 11:680. doi: 10.3389/fnins.2017.00680
Colton, C. A. (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418. doi: 10.1007/s11481-009-9164-4
Craig-Schapiro, R., Fagan, A. M., and Holtzman, D. M. (2009). Biomarkers of Alzheimer’s disease. Neurobiol. Dis. 35, 128–140. doi: 10.1016/j.nbd.2008.10.003
Crouch, P. J., Harding, S.-M. E., White, A. R., Camakaris, J., Bush, A. I., and Masters, C. L. (2008). Mechanisms of Aβ mediated neurodegeneration in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 40, 181–198. doi: 10.1016/j.biocel.2007.07.013
Cui, Y. H., Le, Y., Zhang, X., Gong, W., Abe, K., Sun, R., et al. (2002). Up-regulation of FPR2, a chemotactic receptor for amyloid β 1–42 (Aβ42), in murine microglial cells by TNFα. Neurobiol. Dis. 10, 366–377. doi: 10.1006/nbdi.2002.0517
Dall’Igna, O. P., Fett, P., Gomes, M. W., Souza, D. O., Cunha, R. A., and Lara, D. R. (2007). Caffeine and adenosine A2a receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Exp. Neurol. 203, 241–245. doi: 10.1016/j.expneurol.2006.08.008
Datta, M., Staszewski, O., Raschi, E., Frosch, M., Hagemeyer, N., Tay, T. L., et al. (2018). Histone deacetylases 1 and 2 regulate microglia function during development, homeostasis, and neurodegeneration in a context-dependent manner. Immunity 48, 514.e6–529.e6. doi: 10.1016/j.immuni.2018.02.016
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Davis, A. A., Fritz, J. J., Wess, J., Lah, J. J., and Levey, A. I. (2010). Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J. Neurosci. 30, 4190–4196. doi: 10.1523/JNEUROSCI.6393-09.2010
Davis, M. J., Tsang, T. M., Qiu, Y., Dayrit, J. K., Freij, J. B., Huffnagle, G. B., et al. (2013). Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio 4:e00264-13. doi: 10.1128/mbio.00264-13
DiCarlo, G., Wilcock, D., Henderson, D., Gordon, M., and Morgan, D. (2001). Intrahippocampal LPS injections reduce Aβ load in APP+PS1 transgenic mice. Neurobiol. Aging 22, 1007–1012. doi: 10.1016/s0197-4580(01)00292-5
Dixon, R. A., Kobilka, B. K., Strader, D. J., Benovic, J. L., Dohlman, H. G., Frielle, T., et al. (1986). Cloning of the gene and cDNA for mammalian β-adrenergic receptor and homology with rhodopsin. Nature 321, 75–79. doi: 10.1038/321075a0
Doze, V. A., and Perez, D. M. (2012). G-protein-coupled receptors in adult neurogenesis. Pharmacol. Rev. 64, 645–675. doi: 10.1124/pr.111.004762
Dukhinova, M., Kopeikina, E., and Ponomarev, E. D. (2018). “Usage of multiparameter flow cytometry to study microglia and macrophage heterogeneity in the central nervous system during neuroinflammation and neurodegeneration,” in Cellular Heterogeneity: Methods in Molecular Biology, eds N. Barteneva and I. Vorobjev (New York, NY: Humana Press), 167–177.
Durand, D., Carniglia, L., Beauquis, J., Caruso, C., Saravia, F., and Lasaga, M. (2014). Astroglial mGlu3 receptors promote α-secretase-mediated amyloid precursor protein cleavage. Neuropharmacology 79, 180–189. doi: 10.1016/j.neuropharm.2013.11.015
Durand, D., Carniglia, L., Turati, J., Ramírez, D., Saba, J., Caruso, C., et al. (2017). Amyloid-β neurotoxicity and clearance are both regulated by glial group II metabotropic glutamate receptors. Neuropharmacology 123, 274–286. doi: 10.1016/j.neuropharm.2017.05.008
Eijkelkamp, N., Heijnen, C. J., Willemen, H. L., Deumens, R., Joosten, E. A., Kleibeuker, W., et al. (2010). GRK2: a novel cell-specific regulator of severity and duration of inflammatory pain. J. Neurosci. 30, 2138–2149. doi: 10.1523/JNEUROSCI.5752-09.2010
Eljaschewitsch, E., Witting, A., Mawrin, C., Lee, T., Schmidt, P. M., Wolf, S., et al. (2006). The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 49, 67–79. doi: 10.1016/j.neuron.2005.11.027
Erb, L., Cao, C., Ajit, D., and Weisman, G. A. (2015). P2Y receptors in Alzheimer’s disease. Biol. Cell 107, 1–21. doi: 10.1111/boc.201400043
Eyo, U. B., and Wu, L.-J. (2013). Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. 2013:456857. doi: 10.1155/2013/456857
Facchinetti, F., Del Giudice, E., Furegato, S., Passarotto, M., and Leon, A. (2003). Cannabinoids ablate release of TNFα in rat microglial cells stimulated with lypopolysaccharide. Glia 41, 161–168. doi: 10.1002/glia.10177
Farber, S. A., Nitsch, R. M., Schulz, J. G., and Wurtman, R. (1995). Regulated secretion of β-amyloid precursor protein in rat brain. J. Neurosci. 15, 7442–7451. doi: 10.1523/JNEUROSCI.15-11-07442.1995
Franco, R., and Fernández-Suárez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Fu, R., Shen, Q., Xu, P., Luo, J. J., and Tang, Y. (2014). Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49, 1422–1434. doi: 10.1007/s12035-013-8620-6
Fung, S., Cherry, A. E., Xu, C., and Stella, N. (2015). Alkylindole-sensitive receptors modulate microglial cell migration and proliferation. Glia 63, 1797–1808. doi: 10.1002/glia.22845
Gainetdinov, R. R., Premont, R. T., Bohn, L. M., Lefkowitz, R. J., and Caron, M. G. (2004). Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27, 107–144. doi: 10.1146/annurev.neuro.27.070203.144206
Gao, L.-B., Yu, X.-F., Chen, Q., and Zhou, D. (2016). Alzheimer’s disease therapeutics: current and future therapies. Minerva Med. 107, 108–113.
Gavins, F. N. E., and Hickey, M. J. (2012). Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 3:354. doi: 10.3389/fimmu.2012.00354
Gentleman, S. (2013). Microglia in protein aggregation disorders: friend or foe? Neuropathol. Appl. Neurobiol. 39, 45–50. doi: 10.1111/nan.12017
Ghanemi, A. (2015). Targeting G protein coupled receptor-related pathways as emerging molecular therapies. Saudi Pharm. J. 23, 115–129. doi: 10.1016/j.jsps.2013.07.007
Giménez-Llort, L., Schiffmann, S. N., Shmidt, T., Canela, L., Camón, L., Wassholm, M., et al. (2007). Working memory deficits in transgenic rats overexpressing human adenosine A2A receptors in the brain. Neurobiol. Learn. Mem. 87, 42–56. doi: 10.1016/j.nlm.2006.05.004
Giunta, S., Andriolo, V., and Castorina, A. (2014). Dual blockade of the A1 and A2A adenosine receptor prevents amyloid β toxicity in neuroblastoma cells exposed to aluminum chloride. Int. J. Biochem. Cell Biol. 54, 122–136. doi: 10.1016/j.biocel.2014.07.009
Giunti, D., Parodi, B., Cordano, C., Uccelli, A., and Kerlero de Rosbo, N. (2014). Can we switch microglia’s phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology 141, 328–339. doi: 10.1111/imm.12177
Glass, M., and Felder, C. C. (1997). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 17, 5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997
Gloriam, D. E., Fredriksson, R., and Schiöth, H. B. (2007). The G protein-coupled receptor subset of the rat genome. BMC Genomics 8:338. doi: 10.1186/1471-2164-8-338
Gnatek, Y., Zimmerman, G., Goll, Y., Najami, N., Soreq, H., and Friedman, A. (2012). Acetylcholinesterase loosens the brain’s cholinergic anti-inflammatory response and promotes epileptogenesis. Front. Mol. Neurosci. 5:66. doi: 10.3389/fnmol.2012.00066
Godyn, J., Jonczyk, J., Panek, D., and Malawska, B. (2016). Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 68, 127–138. doi: 10.1016/j.pharep.2015.07.006
Guerram, M., Zhang, L.-Y., and Jiang, Z.-Z. (2016). G-protein coupled receptors as therapeutic targets for neurodegenerative and cerebrovascular diseases. Neurochem. Int. 101, 1–14. doi: 10.1016/j.neuint.2016.09.005
Gyoneva, S., Swanger, S. A., Zhang, J., Weinshenker, D., and Traynelis, S. F. (2016). Altered motility of plaque-associated microglia in a model of Alzheimer’s disease. Neuroscience 330, 410–420. doi: 10.1016/j.neuroscience.2016.05.061
Gyoneva, S., and Traynelis, S. F. (2013). Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J. Biol. Chem. 288, 15291–15302. doi: 10.1074/jbc.M113.458901
Hammarberg, C., Schulte, G., and Fredholm, B. B. (2003). Evidence for functional adenosine A3 receptors in microglia cells. J. Neurochem. 86, 1051–1054. doi: 10.1046/j.1471-4159.2003.01919.x
Hanisch, U.-K. (2013). Functional diversity of microglia-how heterogeneous are they to begin with? Front. Cell. Neurosci. 7:65. doi: 10.3389/fncel.2013.00065
Hanisch, U.-K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hasselmo, M. E. (2006). The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 16, 710–715. doi: 10.1016/j.conb.2006.09.002
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B., and Gloriam, D. E. (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842. doi: 10.1038/nrd.2017.178
Hauser, A. S., Chavali, S., Masuho, I., Jahn, L. J., Martemyanov, K. A., Gloriam, D. E., et al. (2018). Pharmacogenomics of GPCR drug targets. Cell 172, 41.e19–54.e19. doi: 10.1016/j.cell.2017.11.033
Haynes, S. E., Hollopeter, G., Yang, G., Kurpius, D., Dailey, M. E., Gan, W.-B., et al. (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519. doi: 10.1038/nn1805
Heneka, M. T., Galea, E., Gavriluyk, V., Dumitrescu-Ozimek, L., Daeschner, J., O’Banion, M. K., et al. (2002). Noradrenergic depletion potentiates β-amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J. Neurosci. 22, 2434–2442. doi: 10.1523/JNEUROSCI.22-07-02434.2002
Heng, B. C., Aubel, D., and Fussenegger, M. (2013). An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 31, 1676–1694. doi: 10.1016/j.biotechadv.2013.08.017
Hickman, S. E., and El Khoury, J. (2010). Mechanisms of mononuclear phagocyte recruitment in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 9, 168–173. doi: 10.2174/187152710791011982
Hirayama, M., and Kuriyama, M. (2001). MK-801 is cytotoxic to microglia in vitro and its cytotoxicity is attenuated by glutamate, other excitotoxic agents and atropine: Possible presence of glutamate receptor and muscarinic receptor on microglia. Brain Res. 897, 204–206. doi: 10.1016/s0006-8993(01)02114-x
Holcomb, I. N., Kabakoff, R. C., Chan, B., Baker, T. W., Gurney, A., Henzel, W., et al. (2000). FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 19, 4046–4055. doi: 10.1093/emboj/19.15.4046
Honda, S., Sasaki, Y., Ohsawa, K., Imai, Y., Nakamura, Y., Inoue, K., et al. (2001). Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci. 21, 1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001
Howlett, A., Qualy, J. M., and Khachatrian, L. L. (1986). Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol. Pharmacol. 29, 307–313.
Hu, X., Leak, R. K., Shi, Y., Suenaga, J., Gao, Y., Zheng, P., et al. (2015). Microglial and macrophage polarization—new prospects for brain repair. Nat. Rev. Neurol. 11, 56–64. doi: 10.1038/nrneurol.2014.207
Huang, Y., Skwarek-Maruszewska, A., Horré, K., Vandewyer, E., Wolfs, L., Snellinx, A., et al. (2015). Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer’s disease mouse models. Sci. Transl. Med. 7:309r.a164. doi: 10.1126/scitranslmed.aab3492
Huang, Y., Todd, N., and Thathiah, A. (2017). The role of GPCRs in neurodegenerative diseases: avenues for therapeutic intervention. Curr. Opin. Pharmacol. 32, 96–110. doi: 10.1016/j.coph.2017.02.001
Huang, Y.-Y., Zhang, Q., Zhang, J.-N., Zhang, Y.-N., Gu, L., Yang, H.-M., et al. (2018). Triptolide up-regulates metabotropic glutamate receptor 5 to inhibit microglia activation in the lipopolysaccharide-induced model of Parkinson’s disease. Brain Behav. Immun. 71, 93–107. doi: 10.1016/j.bbi.2018.04.006
Hung, S.-Y., and Fu, W.-M. (2017). Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 24:47. doi: 10.1186/s12929-017-0355-7
Hung, A., Haass, C., Nitsch, R., Qiu, W. Q., Citron, M., Wurtman, R., et al. (1993). Activation of protein kinase C inhibits cellular production of the amyloid β-protein. J. Biol. Chem. 268, 22959–22962.
Iribarren, P., Chen, K., Gong, W., Cho, E. H., Lockett, S., Uranchimeg, B., et al. (2007). Interleukin 10 and TNFα synergistically enhance the expression of the G protein-coupled formylpeptide receptor 2 in microglia. Neurobiol. Dis. 27, 90–98. doi: 10.1016/j.nbd.2007.04.010
Iribarren, P., Chen, K., Hu, J., Zhang, X., Gong, W., and Wang, J. M. (2005). IL-4 inhibits the expression of mouse formyl peptide receptor 2, a receptor for amyloid β1–42, in TNF-α-activated microglia. J. Immunol. 175, 6100–6106. doi: 10.4049/jimmunol.175.9.6100
Iribarren, P., Zhou, Y., Hu, J., Le, Y., and Wang, J. M. (2005). Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol. Res. 31, 165–176. doi: 10.1385/ir:31:3:165
Iwata, N., Higuchi, M., and Saido, T. C. (2005). Metabolism of amyloid-β peptide and Alzheimer’s disease. Pharmacol. Ther. 108, 129–148. doi: 10.1016/j.pharmthera.2005.03.010
Jiang, S., Li, Y., Zhang, C., Zhao, Y., Bu, G., Xu, H., et al. (2014). M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neurosci. Bull. 30, 295–307. doi: 10.1007/s12264-013-1406-z
Jiang, T., Yu, J.-T., Tan, M.-S., Zhu, X.-C., and Tan, L. (2013). β-arrestins as potential therapeutic targets for Alzheimer’s disease. Mol. Neurobiol. 48, 812–818. doi: 10.1007/s12035-013-8469-8
Jimenez, S., Baglietto-Vargas, D., Caballero, C., Moreno-Gonzalez, I., Torres, M., Sanchez-Varo, R., et al. (2008). Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 28, 11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008
Joseph, J. A., Bielinski, D. F., and Fisher, D. R. (2010). Blueberry treatment antagonizes C-2 ceramide-induced stress signaling in muscarinic receptor-transfected COS-7 cells. J. Agric. Food Chem. 58, 3380–3392. doi: 10.1021/jf9039155
Joseph, J., Carey, A., Brewer, G., Lau, F., and Fisher, D. (2007). Dopamine and Aβ-induced stress signaling and decrements in Ca2+ buffering in primary neonatal hippocampal cells are antagonized by blueberry extract. J. Alzheimers Dis. 11, 433–446. doi: 10.3233/jad-2007-11404
Joseph, J., Fisher, D., and Bielinski, D. (2006). Blueberry extract alters oxidative stress-mediated signaling in COS-7 cells transfected with selectively vulnerable muscarinic receptor subtypes. J. Alzheimers Dis. 9, 35–42. doi: 10.3233/jad-2006-9103
Kachroo, A., Orlando, L. R., Grandy, D. K., Chen, J.-F., Young, A. B., and Schwarzschild, M. A. (2005). Interactions between metabotropic glutamate 5 and adenosine A2A receptors in normal and parkinsonian mice. J. Neurosci. 25, 10414–10419. doi: 10.1523/JNEUROSCI.3660-05.2005
Kalaria, R., Andorn, A., and Harik, S. (1989). Alterations in adrenergic receptors of frontal cortex and cerebral microvessels in Alzheimer’s disease and aging. Prog. Clin. Biol. Res. 317, 367–374.
Kallendrusch, S., Kremzow, S., Nowicki, M., Grabiec, U., Winkelmann, R., Benz, A., et al. (2013). The G protein-coupled receptor 55 ligand l-α-lysophosphatidylinositol exerts microglia-dependent neuroprotection after excitotoxic lesion. Glia 61, 1822–1831. doi: 10.1002/glia.22560
Karran, E., Mercken, M., and De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Kavelaars, A., Eijkelkamp, N., Willemen, H. L., Wang, H., Carbajal, A. G., and Heijnen, C. J. (2011). Microglial GRK2: a novel regulator of transition from acute to chronic pain. Brain Behav. Immun. 25, 1055–1060. doi: 10.1016/j.bbi.2011.03.019
Kawanokuchi, J., Mizuno, T., Takeuchi, H., Kato, H., Wang, J., Mitsuma, N., et al. (2006). Production of interferon-γ by microglia. Mult. Scler. J. 12, 558–564. doi: 10.1177/1352458506070763
Kennedy, C., Qi, A.-D., Herold, C. L., Harden, T. K., and Nicholas, R. A. (2000). ATP, an agonist at the rat P2Y4 receptor, is an antagonist at the human P2Y4 receptor. Mol. Pharmacol. 57, 926–931.
Kettenmann, H., Hanisch, U.-K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kim, H. J., Ajit, D., Peterson, T. S., Wang, Y., Camden, J. M., Gibson Wood, W., et al. (2012). Nucleotides released from Aβ1–42-treated microglial cells increase cell migration and Aβ1–42 uptake through P2Y2 receptor activation. J. Neurochem. 121, 228–238. doi: 10.1111/j.1471-4159.2012.07700.x
Kim, B., Jeong, H.-K., Kim, J.-H., Lee, S. Y., Jou, I., and Joe, E.-H. (2011). Uridine 5′-diphosphate induces chemokine expression in microglia and astrocytes through activation of the P2Y6 receptor. J. Immunol. 186, 3701–3709. doi: 10.4049/jimmunol.1000212
Kingham, P., Cuzner, M., and Pocock, J. (1999). Apoptotic pathways mobilized in microglia and neurones as a consequence of chromogranin A-induced microglial activation. J. Neurochem. 73, 538–547. doi: 10.1046/j.1471-4159.1999.0730538.x
Koenigsknecht-Talboo, J., and Landreth, G. E. (2005). Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 25, 8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005
Koffie, R. M., Meyer-Luehmann, M., Hashimoto, T., Adams, K. W., Mielke, M. L., Garcia-Alloza, M., et al. (2009). Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U S A 106, 4012–4017. doi: 10.1073/pnas.0811698106
Koizumi, S., Shigemoto-Mogami, Y., Nasu-Tada, K., Shinozaki, Y., Ohsawa, K., Tsuda, M., et al. (2007). UDP acting at P2Y 6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095. doi: 10.1038/nature05704
Komatsu, H. (2015). Novel therapeutic GPCRs for psychiatric disorders. Int. J. Mol. Sci. 16, 14109–14121. doi: 10.3390/ijms160614109
Kondo, Y., Asanuma, M., Iwata, E., Kondo, F., Miyazaki, I., and Ogawa, N. (1999). Early treatment with cyclosporin A ameliorates the reduction of muscarinic acetylcholine receptors in gerbil hippocampus after transient forebrain ischemia. Neurochem. Res. 24, 9–13. doi: 10.1023/A:1020915727119
Koscsó, B., Csóka, B., Selmeczy, Z., Himer, L., Pacher, P., Virág, L., et al. (2012). Adenosine augments IL-10 production by microglial cells through an A2B adenosine receptor-mediated process. J. Immunol. 188, 445–453. doi: 10.4049/jimmunol.1101224
Kruse, A. C., Kobilka, B. K., Gautam, D., Sexton, P. M., Christopoulos, A., and Wess, J. (2014). Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat. Rev. Drug Discov. 13, 549–560. doi: 10.1038/nrd4295
Lan, Y.-L., Zhao, J., and Li, S. (2014). Estrogen receptors’ neuroprotective effect against glutamate-induced neurotoxicity. Neurol. Sci. 35, 1657–1662. doi: 10.1007/s10072-014-1937-8
Langmead, C. J., Watson, J., and Reavill, C. (2008). Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 117, 232–243. doi: 10.1016/j.pharmthera.2007.09.009
Lannes, N., Eppler, E., Etemad, S., Yotovski, P., and Filgueira, L. (2017). Microglia at center stage: a comprehensive review about the versatile and unique residential macrophages of the central nervous system. Oncotarget 8, 114393–114413. doi: 10.18632/oncotarget.23106
Lee, J. Y., Jhun, B. S., Oh, Y. T., Lee, J. H., Choe, W., Baik, H. H., et al. (2006). Activation of adenosine A 3 receptor suppresses lipopolysaccharide-induced TNF-α production through inhibition of PI 3-kinase/Akt and NF-κB activation in murine BV2 microglial cells. Neurosci. Lett. 396, 1–6. doi: 10.1016/j.neulet.2005.11.004
Lee, J.-S., Kim, H.-G., Han, J.-M., Kim, D.-W., Yi, M.-H., Son, S.-W., et al. (2014). Ethanol extract of astragali radix and salviae miltiorrhizae radix, myelophil, exerts anti-amnesic effect in a mouse model of scopolamine-induced memory deficits. J. Ethnopharmacol. 153, 782–792. doi: 10.1016/j.jep.2014.03.048
Li, H.-Q., Chen, C., Dou, Y., Wu, H.-J., Liu, Y.-J., Lou, H.-F., et al. (2013). P2Y4 receptor-mediated pinocytosis contributes to amyloid β-induced self-uptake by microglia. Mol. Cell. Biol. 33, 4282–4293. doi: 10.1128/MCB.00544-13
Li, L., Homan, K. T., Vishnivetskiy, S. A., Manglik, A., Tesmer, J. J., Gurevich, V. V., et al. (2015). G protein-coupled receptor kinases of the GRK4 protein subfamily phosphorylate inactive G protein-coupled receptors (GPCRs). J. Biol. Chem. 290, 10775–10790. doi: 10.1074/jbc.M115.644773
Lindberg, C., Selenica, M.-L. B., Westlind-Danielsson, A., and Schultzberg, M. (2005). β-amyloid protein structure determines the nature of cytokine release from rat microglia. J. Mol. Neurosci. 27, 1–12. doi: 10.1385/jmn:27:1:001
Lisi, L., Ciotti, G., Braun, D., Kalinin, S., Currò, D., Dello Russo, C., et al. (2017). Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 645, 106–112. doi: 10.1016/j.neulet.2017.02.076
Liu, Z., Fan, Y., Won, S. J., Neumann, M., Hu, D., Zhou, L., et al. (2007). Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke 38, 146–152. doi: 10.1161/01.str.0000251791.64910.cd
Luna, S., Cameron, D. J., and Ethell, D. W. (2013). Amyloid-β and APP deficiencies cause severe cerebrovascular defects: important work for an old villain. PLoS One 8:e75052. doi: 10.1371/journal.pone.0075052
Luo, X.-G., and Chen, S.-D. (2012). The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 1:9. doi: 10.1186/2047-9158-1-9
Luongo, L., Guida, F., Imperatore, R., Napolitano, F., Gatta, L., Cristino, L., et al. (2014). The A1 adenosine receptor as a new player in microglia physiology. Glia 62, 122–132. doi: 10.1002/glia.22592
Ma, L., Jia, J., Liu, X., Bai, F., Wang, Q., and Xiong, L. (2015). Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 458, 92–97. doi: 10.1016/j.bbrc.2015.01.073
Mackie, K. (2008). Cannabinoid receptors: where they are and what they do. J. Neuroendocrinol. 20, 10–14. doi: 10.1111/j.1365-2826.2008.01671.x
Maggio, R., Scarselli, M., Novi, F., Millan, M. J., and Corsini, G. U. (2003). Potent activation of dopamine D3/D2 heterodimers by the antiparkinsonian agents, S32504, pramipexole and ropinirole. J. Neurochem. 87, 631–641. doi: 10.1046/j.1471-4159.2003.02038.x
Mahad, D. J., and Ransohoff, R. M. (2003). The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin. Immunol. 15, 23–32. doi: 10.1016/s1044-5323(02)00125-2
Manuel, I., González de San Román, E., Giralt, M. T., Ferrer, I., and Rodríguez-Puertas, R. (2014). Type-1 cannabinoid receptor activity during Alzheimer’s disease progression. J. Alzheimers Dis. 42, 761–766. doi: 10.3233/JAD-140492
Marichal-Cancino, B. A., Fajardo-Valdez, A., Ruiz-Contreras, A. E., Méndez-Díaz, M., and Prospéro-García, O. (2018). Possible role of hippocampal GPR55 in spatial learning and memory in rats. Acta Neurobiol. Exp. 78, 41–50. doi: 10.21307/ane-2018-001
McGeer, P., Itagaki, S., Boyes, B., and McGeer, E. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1285. doi: 10.1212/wnl.38.8.1285
McGeer, P. L., and McGeer, E. G. (2013). The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 126, 479–497. doi: 10.1007/s00401-013-1177-7
McHugh, D., Hu, S. S., Rimmerman, N., Juknat, A., Vogel, Z., Walker, J. M., et al. (2010). N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 11:44. doi: 10.1186/1471-2202-11-44
Mead, E. L., Mosley, A., Eaton, S., Dobson, L., Heales, S. J., and Pocock, J. M. (2012). Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J. Neurochem. 121, 287–301. doi: 10.1111/j.1471-4159.2012.07659.x
Melancon, B. J., Tarr, J. C., Panarese, J. D., Wood, M. R., and Lindsley, C. W. (2013). Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discov. Today 18, 1185–1199. doi: 10.1016/j.drudis.2013.09.005
Merighi, S., Bencivenni, S., Vincenzi, F., Varani, K., Borea, P. A., and Gessi, S. (2017). A2B adenosine receptors stimulate IL-6 production in primary murine microglia through p38 MAPK kinase pathway. Pharmacol. Res. 117, 9–19. doi: 10.1016/j.phrs.2016.11.024
Michelucci, A., Heurtaux, T., Grandbarbe, L., Morga, E., and Heuschling, P. (2009). Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-β. J. Neuroimmunol. 210, 3–12. doi: 10.1016/j.jneuroim.2009.02.003
Milligan, G. (2004). G protein-coupled receptor dimerization: function and ligand pharmacology. Mol. Pharmacol. 66, 1–7. doi: 10.1124/mol.104.000497.
Mines, M., Ding, Y., and Fan, G.-H. (2007). The many roles of chemokine receptors in neurodegenerative disorders: emerging new therapeutical strategies. Curr. Med. Chem. 14, 2456–2470. doi: 10.2174/092986707782023686
Mittelbronn, M. (2014). The M1/M2 immune polarization concept in microglia: a fair transfer. Neuroimmunol. Neuroinflamm. 1, 6–7. doi: 10.4103/2347-8659.135567
Mondragón-Rodríguez, S., Perry, G., Luna-Muñoz, J., Acevedo-Aquino, M., and Williams, S. (2014). Phosphorylation of tau protein at sites Ser396–404 is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 40, 121–135. doi: 10.1111/nan.12084
Moriconi, A., Cerbara, I., Maccarrone, M., and Topai, A. (2010). GPR55: current knowledge and future perspectives of a purported “Type-3” cannabinoid receptor. Curr. Med. Chem. 17, 1411–1429. doi: 10.2174/092986710790980069
Morris, S. M. Jr. (2007). Arginine metabolism: boundaries of our knowledge. J. Nutr. 137, 1602S–1609S. doi: 10.1093/jn/137.6.1602S
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Mota, S. I., Ferreira, I. L., and Rego, A. C. (2014). Dysfunctional synapse in Alzheimer’s disease—a focus on NMDA receptors. Neuropharmacology 76, 16–26. doi: 10.1016/j.neuropharm.2013.08.013
Nagele, R. G., Wegiel, J., Venkataraman, V., Imaki, H., Wang, K.-C., and Wegiel, J. (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674. doi: 10.1016/j.neurobiolaging.2004.01.007
Nasu-Tada, K., Koizumi, S., and Inoue, K. (2005). Involvement of β1 integrin in microglial chemotaxis and proliferation on fibronectin: different regulations by ADP through PKA. Glia 52, 98–107. doi: 10.1002/glia.20224
Nathan, C., Calingasan, N., Nezezon, J., Ding, A., Lucia, M. S., La Perle, K., et al. (2005). Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J. Exp. Med. 202, 1163–1169. doi: 10.1084/jem.20051529
Nelson, C. D., and Sheng, M. (2013). Gpr3 stimulates Aβ production via interactions with APP and β-arrestin2. PLoS One 8:e74680. doi: 10.1371/journal.pone.0074680
Neumann, H., and Takahashi, K. (2007). Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 184, 92–99. doi: 10.1016/j.jneuroim.2006.11.032
Newell, E. A., Exo, J. L., Verrier, J. D., Jackson, T. C., Gillespie, D. G., Janesko-Feldman, K., et al. (2015). 2′, 3′-cAMP, 3′-AMP, 2′-AMP and adenosine inhibit TNF-α and CXCL10 production from activated primary murine microglia via A2A receptors. Brain Res. 1594, 27–35. doi: 10.1016/j.brainres.2014.10.059
Ni, Y., Zhao, X., Bao, G., Zou, L., Teng, L., Wang, Z., et al. (2006). Activation of β2-adrenergic receptor stimulates γ-secretase activity and accelerates amyloid plaque formation. Nat. Med. 12, 1390–1396. doi: 10.1038/nm1485
Niikura, T., Sidahmed, E., Hirata-Fukae, C., Aisen, P. S., and Matsuoka, Y. (2011). A humanin derivative reduces amyloid β accumulation and ameliorates memory deficit in triple transgenic mice. PLoS One 6:e16259. doi: 10.1371/journal.pone.0016259
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. doi: 10.1146/annurev.pharmtox.011008.145533
Nobre, H. V. Jr., Cunha, G. M., de Vasconcelos, L. M., Magalhães, H. I. F., Oliveira Neto, R. N., Maia, F. D., et al. (2010). Caffeine and CSC, adenosine A2A antagonists, offer neuroprotection against 6-OHDA-induced neurotoxicity in rat mesencephalic cells. Neurochem. Int. 56, 51–58. doi: 10.1016/j.neuint.2009.09.001
Noda, M. (2016). Dysfunction of glutamate receptors in microglia may cause neurodegeneration. Curr. Alzheimer Res. 13, 381–386. doi: 10.2174/1567205013666151116125810
Nyakas, C., Granic, I., Halmy, L. G., Banerjee, P., and Luiten, P. G. (2011). The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-β42 with memantine. Behav. Brain Res. 221, 594–603. doi: 10.1016/j.bbr.2010.05.033
Ohsawa, K., Irino, Y., Nakamura, Y., Akazawa, C., Inoue, K., and Kohsaka, S. (2007). Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia 55, 604–616. doi: 10.1002/glia.20489
Pannell, M., Meier, M. A., Szulzewsky, F., Matyash, V., Endres, M., Kronenberg, G., et al. (2016). The subpopulation of microglia expressing functional muscarinic acetylcholine receptors expands in stroke and Alzheimer’s disease. Brain Struct. Funct. 221, 1157–1172. doi: 10.1007/s00429-014-0962-y
Peferoen, L., Kipp, M., van der Valk, P., van Noort, J. M., and Amor, S. (2014). Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 141, 302–313. doi: 10.1111/imm.12163
Peng, L., Yu, Y., Liu, J., Li, S., He, H., Cheng, N., et al. (2015). The chemerin receptor CMKLR1 is a functional receptor for amyloid-β peptide. J. Alzheimers Dis. 43, 227–242. doi: 10.3233/JAD-141227
Peterson, T. S., Camden, J. M., Wang, Y., Seye, C. I., Wood, W., Sun, G. Y., et al. (2010). P2Y2 nucleotide receptor-mediated responses in brain cells. Mol. Neurobiol. 41, 356–366. doi: 10.1007/s12035-010-8115-7
Pietr, M., Kozela, E., Levy, R., Rimmerman, N., Lin, Y. H., Stella, N., et al. (2009). Differential changes in GPR55 during microglial cell activation. FEBS Lett. 583, 2071–2076. doi: 10.1016/j.febslet.2009.05.028
Ponomarev, E. D., Maresz, K., Tan, Y., and Dittel, B. N. (2007). CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 27, 10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007
Preissler, J., Grosche, A., Lede, V., Le Duc, D., Krügel, K., Matyash, V., et al. (2015). Altered microglial phagocytosis in GPR34-deficient mice. Glia 63, 206–215. doi: 10.1002/glia.22744
Proudfoot, A. E. (2002). Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2, 106–115. doi: 10.1038/nri722
Puzzo, D., Gulisano, W., Palmeri, A., and Arancio, O. (2015). Rodent models for Alzheimer’s disease drug discovery. Expert Opin. Drug Discov. 10, 703–711. doi: 10.1517/17460441.2015.1041913
Querfurth, H. W., Jiang, J., Geiger, J. D., and Selkoe, D. J. (1997). Caffeine stimulates amyloid β-peptide release from β-amyloid precursor Protein-transfected HEK293 cells. J. Neurochem. 69, 1580–1591. doi: 10.1046/j.1471-4159.1997.69041580.x
Raes, G., Van den Bergh, R., De Baetselier, P., and Ghassabeh, G. H. (2005). Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J. Immunol. 174, 6561–6562. doi: 10.4049/jimmunol.174.11.6561
Ragheb, F., Molina-Holgado, E., Cui, Q. L., Khorchid, A., Liu, H. N., Larocca, J. N., et al. (2001). Pharmacological and functional characterization of muscarinic receptor subtypes in developing oligodendrocytes. J. Neurochem. 77, 1396–1406. doi: 10.1046/j.1471-4159.2001.00356.x
Ramírez, B. G., Blázquez, C., Gómez del Pulgar, T., Guzmán, M., and de Ceballos, M. L. (2005). Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J. Neurosci. 25, 1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005
Ribeiro, J. A., and Sebastiao, A. M. (2010). Caffeine and adenosine. J. Alzheimers Dis. 20, S3–S15. doi: 10.3233/JAD-2010-1379
Ries, M., Loiola, R., Shah, U. N., Gentleman, S. M., Solito, E., and Sastre, M. (2016). The anti-inflammatory Annexin A1 induces the clearance and degradation of the amyloid-β peptide. J. Neuroinflammation 13:234. doi: 10.1186/s12974-016-0692-6
Ries, M., and Sastre, M. (2016). Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 8:160. doi: 10.3389/fnagi.2016.00160
Rio-Hortega, D. P. (1932). “Microglia,” in Cytology and Cellular Pathology of the Nervous System, ed. W. Penfield (New York, NY: Paul B Hoeber), 482–534.
Röszer, T. (2015). Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015:816460. doi: 10.1155/2015/816460
Ruiz-Medina, J., Ledent, C., and Valverde, O. (2011). GPR3 orphan receptor is involved in neuropathic pain after peripheral nerve injury and regulates morphine-induced antinociception. Neuropharmacology 61, 43–50. doi: 10.1016/j.neuropharm.2011.02.014
Russo-Neustadt, A., and Cotman, C. W. (1997). Adrenergic receptors in Alzheimer’s disease brain: selective increases in the cerebella of aggressive patients. J. Neurosci. 17, 5573–5580. doi: 10.1523/JNEUROSCI.17-14-05573.1997
Ryberg, E., Larsson, N., Sjögren, S., Hjorth, S., Hermansson, N. O., Leonova, J., et al. (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 152, 1092–1101. doi: 10.1038/sj.bjp.0707460
Saba, K., Denda-Nagai, K., and Irimura, T. (2009). A C-type lectin MGL1/CD301a plays an anti-inflammatory role in murine experimental colitis. Am. J. Pathol. 174, 144–152. doi: 10.2353/ajpath.2009.080235
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. doi: 10.1038/nm.4397
Santiago, A. R., Baptista, F. I., Santos, P. F., Cristóvão, G., Ambrósio, A. F., Cunha, R. A., et al. (2014). Role of microglia adenosine A2A receptors in retinal and brain neurodegenerative diseases. Mediators Inflamm. 2014:465694. doi: 10.1155/2014/465694
Schubert, P., Morino, T., Miyazaki, H., Ogata, T., Nakamura, Y., Marchini, C., et al. (2000). Cascading glia reactions: a common pathomechanism and its differentiated control by cyclic nucleotide signaling. Ann. N Y Acad. Sci. 903, 24–33. doi: 10.1111/j.1749-6632.2000.tb06346.x
Serrano-Pozo, A., Muzikansky, A., Gómez-Isla, T., Growdon, J. H., Betensky, R. A., Frosch, M. P., et al. (2013). Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol. Exp. Neurol. 72, 462–471. doi: 10.1097/NEN.0b013e3182933788
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shechter, R., London, A., Varol, C., Raposo, C., Cusimano, M., Yovel, G., et al. (2009). Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 6:e1000113. doi: 10.1371/journal.pmed.1000113
Shin, H., Kim, J., and Song, J.-H. (2015). Clozapine and olanzapine inhibit proton currents in BV2 microglial cells. Eur. J. Pharmacol. 755, 74–79. doi: 10.1016/j.ejphar.2015.03.003
Shore, D. M., and Reggio, P. H. (2015). The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 6:69. doi: 10.3389/fphar.2015.00069
Shryock, J. C., and Belardinelli, L. (1997). Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am. J. Cardiol. 79, 2–10. doi: 10.1016/s0002-9149(97)00256-7
Slowik, A., Merres, J., Elfgen, A., Jansen, S., Mohr, F., Wruck, C. J., et al. (2012). Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)—and amyloid β 1–42-induced signal transduction in glial cells. Mol. Neurodegener. 7:55. doi: 10.1186/1750-1326-7-55
Stanasila, L., Perez, J.-B., Vogel, H., and Cotecchia, S. (2003). Oligomerization of the α1a-and α1b-adrenergic receptor subtypes. Potential implications in receptor internalization. J. Biol. Chem. 278, 40239–40251. doi: 10.1074/jbc.M306085200
Steiner, L., Gold, M., Mengel, D., Dodel, R., and Bach, J.-P. (2014). The endogenous α7 nicotinic acetylcholine receptor antagonist kynurenic acid modulates amyloid-β-induced inflammation in BV-2 microglial cells. J. Neurol. Sci. 344, 94–99. doi: 10.1016/j.jns.2014.06.032
Stella, N. (2010). Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 58, 1017–1030. doi: 10.1002/glia.20983
Stockert, J. A., and Devi, L. A. (2015). Advancements in therapeutically targeting orphan GPCRs. Front. Pharmacol. 6:100. doi: 10.3389/fphar.2015.00100
Stojanovic, I. R., Kostic, M., and Ljubisavljevic, S. (2014). The role of glutamate and its receptors in multiple sclerosis. J. Neural Transm. 121, 945–955. doi: 10.1007/s00702-014-1188-0
Subramaniam, S. R., and Federoff, H. J. (2017). Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front. Aging Neurosci. 9:176. doi: 10.3389/fnagi.2017.00176
Suo, Z., Wu, M., Citron, B. A., Wong, G. T., and Festoff, B. W. (2004). Abnormality of G-protein-coupled receptor kinases at prodromal and early stages of Alzheimer’s disease: an association with early β-amyloid accumulation. J. Neurosci. 24, 3444–3452. doi: 10.1523/JNEUROSCI.4856-03.2004
Takata, K., Kitamura, Y., Saeki, M., Terada, M., Kagitani, S., Kitamura, R., et al. (2010). Galantamine-induced amyloid-β clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J. Biol. Chem. 285, 40180–40191. doi: 10.1074/jbc.M110.142356
Takata, K., Kitamura, Y., Umeki, M., Tsuchiya, D., Kakimura, J.-I., Taniguchi, T., et al. (2003). Possible involvement of small oligomers of amyloid-β peptides in 15-deoxy-Δ12, 14 prostaglandin J2-sensitive microGlial activation. J. Pharmacol. Sci. 91, 330–333. doi: 10.1254/jphs.91.330
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Taylor, D., Diemel, L., Cuzner, M., and Pocock, J. (2002). Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin A, a peptide up-regulated in Alzheimer’s disease. J. Neurochem. 82, 1179–1191. doi: 10.1046/j.1471-4159.2002.01062.x
Taylor, D. L., Diemel, L. T., and Pocock, J. M. (2003). Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J. Neurosci. 23, 2150–2160. doi: 10.1523/JNEUROSCI.23-06-02150.2003
Tebano, M. T., Martire, A., Chiodi, V., Pepponi, R., Ferrante, A., Domenici, M. R., et al. (2009). Adenosine A2A receptors enable the synaptic effects of cannabinoid CB1 receptors in the rodent striatum. J. Neurochem. 110, 1921–1930. doi: 10.1111/j.1471-4159.2009.06282.x
Thathiah, A., and De Strooper, B. (2011). The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat. Rev. Neurosci. 12, 73–87. doi: 10.1038/nrn2977
Thathiah, A., Horré, K., Snellinx, A., Vandewyer, E., Huang, Y., Ciesielska, M., et al. (2013). β-arrestin 2 regulates Aβ generation and γ-secretase activity in Alzheimer’s disease. Nat. Med. 19, 43–49. doi: 10.1038/nm.3023
Thathiah, A., Spittaels, K., Hoffmann, M., Staes, M., Cohen, A., Horré, K., et al. (2009). The orphan G protein-coupled receptor 3 modulates amyloid-β peptide generation in neurons. Science 323, 946–951. doi: 10.1126/science.1160649
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619. doi: 10.1074/jbc.R800019200
Town, T., Nikolic, V., and Tan, J. (2005). The microglial “activation” continuum: from innate to adaptive responses. J. Neuroinflammation 2:24. doi: 10.1186/1742-2094-2-24
Tsang, S. W., Lai, M. K., Kirvell, S., Francis, P. T., Esiri, M. M., Hope, T., et al. (2006). Impaired coupling of muscarinic M1 receptors to G-proteins in the neocortex is associated with severity of dementia in Alzheimer’s disease. Neurobiol. Aging 27, 1216–1223. doi: 10.1016/j.neurobiolaging.2005.07.010
Violin, J. D., Crombie, A. L., Soergel, D. G., and Lark, M. W. (2014). Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci. 35, 308–316. doi: 10.1016/j.tips.2014.04.007
Waksman, Y., Olson, J. M., Carlisle, S. J., and Cabral, G. A. (1999). The central cannabinoid receptor (CB1) mediates inhibition of nitric oxide production by rat microglial cells. J. Pharmacol. Exp. Ther. 288, 1357–1366.
Walker, D. G., Lue, L.-F., and Beach, T. G. (2001). Gene expression profiling of amyloid β peptide-stimulated human post-mortem brain microglia. Neurobiol. Aging 22, 957–966. doi: 10.1016/s0197-4580(01)00306-2
Walter, L., Franklin, A., Witting, A., Wade, C., Xie, Y., Kunos, G., et al. (2003). Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 23, 1398–1405. doi: 10.1523/jneurosci.23-04-01398.2003
Wang, J. H., Ma, Y. Y., and van den Buuse, M. (2006). Improved spatial recognition memory in mice lacking adenosine A2A receptors. Exp. Neurol. 199, 438–445. doi: 10.1016/j.expneurol.2006.01.005
Wessler, I., Kirkpatrick, C. J., and Racké, K. (1998). Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol. Ther. 77, 59–79. doi: 10.1016/s0163-7258(97)00085-5
Wilcock, D. M., DiCarlo, G., Henderson, D., Jackson, J., Clarke, K., Ugen, K. E., et al. (2003). Intracranially administered anti-αβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J. Neurosci. 23, 3745–3751. doi: 10.1523/jneurosci.23-09-03745.2003
Williams, C. J., and Dexter, D. T. (2014). Neuroprotective and symptomatic effects of targeting group III mGlu receptors in neurodegenerative disease. J. Neurochem. 129, 4–20. doi: 10.1111/jnc.12608
Willment, J. A., Lin, H.-H., Reid, D. M., Taylor, P. R., Williams, D. L., Wong, S. Y., et al. (2003). Dectin-1 expression and function are enhanced on alternatively activated and GM-CSF-treated macrophages and are negatively regulated by IL-10, dexamethasone, and lipopolysaccharide. J. Immunol. 171, 4569–4573. doi: 10.4049/jimmunol.171.11.6297-b
Wohleb, E. S. (2016). Neuron-microglia interactions in mental health disorders: “for better, and for worse”. Front. Immunol. 7:544. doi: 10.3389/fimmu.2016.00544
Wu, C.-S., Chen, H., Sun, H., Zhu, J., Jew, C. P., Wager-Miller, J., et al. (2013). GPR55, a G-protein coupled receptor for lysophosphatidylinositol, plays a role in motor coordination. PLoS One 8:e60314. doi: 10.1371/journal.pone.0060314
Xia, M., Qin, S., Wu, L., Mackay, C. R., and Hyman, B. T. (1998). Immunohistochemical study of the β-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am. J. Pathol. 153, 31–37. doi: 10.1016/S0002-9440(10)65542-3
Xue, F., Stoica, B. A., Hanscom, M., V Kabadi, S., and Faden, A. I. (2014). Positive allosteric modulators (PAMs) of metabotropic glutamate receptor 5 (mGluR5) attenuate microglial activation. CNS Neurol. Disord. Drug Targets 13, 558–566. doi: 10.2174/18715273113126660199
Yamanaka, D., Kawano, T., Nishigaki, A., Aoyama, B., Tateiwa, H., Shigematsu-Locatelli, M., et al. (2017). Preventive effects of dexmedetomidine on the development of cognitive dysfunction following systemic inflammation in aged rats. J. Anesth. 31, 25–35. doi: 10.1007/s00540-016-2264-4
Yan, R., Hu, Z.-Y., Zhou, W.-X., Wang, Q., and Zhang, Y.-X. (2014). Roles of adenosine receptors in Alzheimer’s disease. Yao Xue Xue Bao 49, 751–756.
Yang, S., Sun, R., Zhou, Z., Zhou, J., Liang, J., and Mu, H. (2014). Expression of amyloid-β protein and amyloid-β precursor protein after primary brain-stem injury in rats. Am. J. Forensic Med. Pathol. 35, 201–205. doi: 10.1097/PAF.0000000000000103
Yi, B., Jahangir, A., Evans, A. K., Briggs, D., Ravina, K., Ernest, J., et al. (2017). Discovery of novel brain permeable and G protein-biased β-1 adrenergic receptor partial agonists for the treatment of neurocognitive disorders. PLoS One 12:e0180319. doi: 10.1371/journal.pone.0180319
Ying, G., Iribarren, P., Zhou, Y., Gong, W., Zhang, N., Yu, Z.-X., et al. (2004). Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 172, 7078–7085. doi: 10.4049/jimmunol.172.11.7078
Yoshioka, K., Saitoh, O., and Nakata, H. (2001). Heteromeric association creates a P2Y-like adenosine receptor. Proc. Natl. Acad. Sci. U S A 98, 7617–7622. doi: 10.1073/pnas.121587098
Yu, J.-T., Wang, N.-D., Ma, T., Jiang, H., Guan, J., and Tan, L. (2011). Roles of β-adrenergic receptors in Alzheimer’s disease: Implications for novel therapeutics. Brain Res. Bull. 84, 111–117. doi: 10.1016/j.brainresbull.2010.11.004
Yu, Y., and Ye, R. D. (2015). Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 35, 71–83. doi: 10.1007/s10571-014-0101-6
Zempel, H., and Mandelkow, E. (2014). Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 37, 721–732. doi: 10.1016/j.tins.2014.08.004
Zhang, F., Gannon, M., Chen, Y., Zhou, L., Jiao, K., and Wang, Q. (2017). The amyloid precursor protein modulates α2A-adrenergic receptor endocytosis and signaling through disrupting arrestin 3 recruitment. FASEB J. 31, 4434–4446. doi: 10.1096/fj.201700346R
Zhang, A., Liu, Z., and Kan, Y. (2007). Receptor dimerization–rationale for the design of bivalent ligands. Curr. Top. Med. Chem. 7, 343–345. doi: 10.2174/156802607779941279
Zhang, P., Li, Y., Han, X., Xing, Q., and Zhao, L. (2017). Dexmedetomidine regulates 6-hydroxydopamine-induced microglial polarization. Neurochem. Res. 42, 1524–1532. doi: 10.1007/s11064-017-2209-9
Zhang, L., McLarnon, J. G., Goghari, V., Lee, Y. B., Kim, S. U., and Krieger, C. (1998). Cholinergic agonists increase intracellular Ca2+ in cultured human microglia. Neurosci. Lett. 255, 33–36. doi: 10.1016/s0304-3940(98)00706-x
Zhang, Z.-Y., Sun, B.-L., Liu, J.-K., Yang, M.-F., Li, D.-W., Fang, J., et al. (2015). Activation of mGluR5 attenuates microglial activation and neuronal apoptosis in early brain injury after experimental subarachnoid hemorrhage in rats. Neurochem. Res. 40, 1121–1132. doi: 10.1007/s11064-015-1572-7
Zhao, J., Deng, Y., Jiang, Z., and Qing, H. (2016). G protein-coupled receptors (GPCRs) in Alzheimer’s disease: a focus on BACE1 related GPCRs. Front. Aging Neurosci. 8:58. doi: 10.3389/fnagi.2016.00058
Zhou, X., Spittau, B., and Krieglstein, K. (2012). TGFβ signalling plays an important role in IL4-induced alternative activation of microglia. J. Neuroinflammation 9:210. doi: 10.1186/1742-2094-9-210
Keywords: GPCR, microglia, Alzheimer’s disease, amyloid beta, acetylcholine receptors, adrenergic receptors, dopamine receptors, purinergic receptors
Citation: Haque ME, Kim I-S, Jakaria M, Akther M and Choi D-K (2018) Importance of GPCR-Mediated Microglial Activation in Alzheimer’s Disease. Front. Cell. Neurosci. 12:258. doi: 10.3389/fncel.2018.00258
Received: 30 April 2018; Accepted: 30 July 2018;
Published: 21 August 2018.
Edited by:
Rocío Martínez De Pablos, Universidad de Sevilla, SpainReviewed by:
Dimitrios Davalos, Cleveland Clinic Lerner College of Medicine, United StatesJavier Fernàndez-Ruiz, Complutense University of Madrid, Spain
Nicholas D. Mazarakis, Imperial College London, United Kingdom
Copyright © 2018 Haque, Kim, Jakaria, Akther and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong-Kug Choi, Y2hvaWRrQGtrdS5hYy5rcg==