 Christopher K. Salmon1*
Christopher K. Salmon1* Horia Pribiag1Claire Gizowski1
Horia Pribiag1Claire Gizowski1 W. Todd Farmer1
W. Todd Farmer1 Scott Cameron1
Scott Cameron1 Emma V. Jones1Vivek Mahadevan2Charles W. Bourque1
Emma V. Jones1Vivek Mahadevan2Charles W. Bourque1 David Stellwagen1
David Stellwagen1 Melanie A. Woodin2
Melanie A. Woodin2 Keith K. Murai1*
Keith K. Murai1*- 1Centre for Research in Neuroscience, Department of Neurology and Neurosurgery, Brain Repair and Integrative Neuroscience Program, The Research Institute of the McGill University Health Centre, Montreal General Hospital, Montreal, QC, Canada
- 2Department of Cell & Systems Biology, University of Toronto, Toronto, ON, Canada
γ-Aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the mature brain but has the paradoxical property of depolarizing neurons during early development. Depolarization provided by GABAA transmission during this early phase regulates neural stem cell proliferation, neural migration, neurite outgrowth, synapse formation, and circuit refinement, making GABA a key factor in neural circuit development. Importantly, depending on the context, depolarizing GABAA transmission can either drive neural activity or inhibit it through shunting inhibition. The varying roles of depolarizing GABAA transmission during development, and its ability to both drive and inhibit neural activity, makes it a difficult developmental cue to study. This is particularly true in the later stages of development when the majority of synapses form and GABAA transmission switches from depolarizing to hyperpolarizing. Here, we addressed the importance of depolarizing but inhibitory (or shunting) GABAA transmission in glutamatergic synapse formation in hippocampal CA1 pyramidal neurons. We first showed that the developmental depolarizing-to-hyperpolarizing switch in GABAA transmission is recapitulated in organotypic hippocampal slice cultures. Based on the expression profile of K+−Cl− co-transporter 2 (KCC2) and changes in the GABA reversal potential, we pinpointed the timing of the switch from depolarizing to hyperpolarizing GABAA transmission in CA1 neurons. We found that blocking depolarizing but shunting GABAA transmission increased excitatory synapse number and strength, indicating that depolarizing GABAA transmission can restrain glutamatergic synapse formation. The increase in glutamatergic synapses was activity-dependent but independent of BDNF signaling. Importantly, the elevated number of synapses was stable for more than a week after GABAA inhibitors were washed out. Together these findings point to the ability of immature GABAergic transmission to restrain glutamatergic synapse formation and suggest an unexpected role for depolarizing GABAA transmission in shaping excitatory connectivity during neural circuit development.
Introduction
γ-Aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the mature brain. However, GABA is paradoxically depolarizing during nervous system development. Many in vitro studies in rodents have shown that depolarizing GABAA transmission provides excitatory drive during gestation and early postnatal CNS development, driving early network oscillations (ENOs) thought to promote activity-dependent maturation of neural circuits (Ben-Ari et al., 2012). However, recent work suggests that despite providing local depolarization, immature GABAA transmission has inhibitory effects in vivo (Kirmse et al., 2015; Oh et al., 2016; Valeeva et al., 2016). This ability of GABA to be simultaneously depolarizing and inhibitory relies on shunting inhibition, which results from a decrease in input resistance and membrane time constant when GABAA receptors open, regardless of the direction of Cl− flux (Staley and Mody, 1992).
Depolarizing GABAA transmission is implicated in numerous neurodevelopmental processes in vertebrates, including neural stem cell proliferation (Liu et al., 2005), cell migration (Behar et al., 2000), neurite outgrowth (Cancedda et al., 2007), synapse formation, and circuit refinement (Akerman and Cline, 2006; Cancedda et al., 2007; Wang and Kriegstein, 2008). Critically, circuit activity supported by depolarizing GABAA transmission in vitro drives calcium influx thought to be important for glutamatergic synapse development (Leinekugel et al., 1995; Ben-Ari et al., 1997; Griguoli and Cherubini, 2017). Indeed, disrupting the depolarizing nature of GABAA transmission by interfering with chloride homeostasis alters glutamatergic synapse formation and maturation (Akerman and Cline, 2006; Wang and Kriegstein, 2008). However, the effects of GABAA transmission itself on glutamatergic synapse development and the timing of these effects remain poorly defined. This is partly due to the difficulty in manipulating depolarizing GABAA transmission in defined cell types and circuits with sufficient temporal resolution to specifically target the period when glutamatergic synapses are forming while sparing the preceding developmental roles of GABA. Several studies have prematurely hyperpolarized the reversal potential for chloride (ECl) by disrupting chloride homeostasis for more than a week during perinatal development, across a time span in which the targeted neurons terminally divide, migrate, extend neurites and are incorporated into the surrounding circuitry (Ge et al., 2006; Cancedda et al., 2007; Wang and Kriegstein, 2008). This work suggests that disrupting ECl alters neurite and synapse maturation, however, it has been noted that additional studies with the higher temporal resolution are needed (Akerman and Cline, 2007; Kirmse et al., 2018). Closing this gap in our understanding of how GABAA transmission and its transition from a depolarizing to a hyperpolarizing state impacts glutamatergic synapse development will help solve a now-classic problem in developmental neurobiology, and will likely be of clinical significance as disruptions of GABAA transmission during brain development are associated with neurodevelopmental disorders (El Marroun et al., 2014; He et al., 2014; Tyzio et al., 2014).
Here, we investigated the role of depolarizing GABAA transmission in glutamatergic synapse formation on hippocampal CA1 pyramidal cells. To perform temporally precise pharmacological manipulations of GABAA transmission during neural circuit development, we took advantage of the properties of the organotypic hippocampal slice culture. This preparation preserves the anatomy and the developmental progression of the hippocampus, including the time course of excitatory synapse formation (Buchs et al., 1993; Muller et al., 1993; De Simoni et al., 2003). This system enabled us to define a narrow time window during the first week of slice development in which GABAA transmission shifts from immature, depolarizing transmission, to hyperpolarizing transmission in CA1 pyramidal cells. Previous work suggests that blocking depolarizing GABAA transmission during development will remove the excitatory drive and decrease excitatory synapse formation and maturation (Ben-Ari et al., 2007; Wang and Kriegstein, 2008). Contrary to these predictions, we found that transient blockade of immature, depolarizing GABAA transmission increased glutamatergic synapse number and function on CA1 pyramidal cells. This unexpected effect was explained by the finding that, at this stage of development, depolarizing GABAA transmission provides shunting inhibition, which when blocked alleviated a restraint on activity-dependent synapse formation. Interestingly, the activity-dependent increase in glutamatergic synapses was stable for at least a week. Furthermore, the effect could not be reproduced by prematurely hyperpolarizing EGABA, and was independent of BDNF signaling. Our results, therefore, point to an important time window during hippocampal development when immature GABAA transmission can restrain excitatory synapse development, and demonstrate that interfering with GABAA transmission at this stage can have lasting effects on neural circuitry.
Materials and Methods
Animals
Experiments were approved by the Montreal General Hospital Facility Animal Care Committee and followed guidelines of the Canadian Council on Animal Care. Male and female C57BL6 mice kept on a 12:12 light-dark cycle were used to prepare organotypic cultures.
Slice Preparation
Organotypic hippocampal slices were prepared as described previously (Haber et al., 2006). Briefly, hippocampi were extracted from postnatal day five mice and cut into 300 μm slices with a McIllwain tissue chopper (Stoelting). Slices were cultured on semi-porous tissue culture inserts (Millipore, Cat. No. PICMORG50) that sat in culture medium composed of minimal essential medium (MEM) supplemented with Glutamax (Invitrogen, Cat. No. 42360032), 25% horse serum (Invitrogen, Cat. No. 26050088), 25% HBSS (Invitrogen, Cat. No. 14025092), 6.5 mg/ml D-glucose and 0.5% penicillin/streptomycin. Slices were cultured for 5–14 days with full medium changes every 2 days.
Labeling of CA1 Cells
Dendrites and spines of CA1 pyramidal cells were labeled using a Semliki Forest Virus (SFV)-mediated approach described in detail elsewhere (Haber et al., 2006). Briefly, SFV driving expression of enhanced green fluorescent protein, targeted to the cell membrane through a farnesylation sequence (EGFPf), was injected into the stratum oriens via a pulled glass pipette, broken to a diameter of approximately 50–100 μm. Glass pipettes were attached to a Picospritzer III (Parker Hannifin) and SFV was delivered with 10 ms pulses at 14–18 psi 18–20 h before fixation in 4% formaldehyde/0.1 M PO4^2- for 30 min.
Confocal Imaging and Spine Analysis
Imaging was performed using an Ultraview Spinning Disc confocal system (Perkin Elmer) attached to a Nikon TE-2000 microscope and an FV1000 laser scanning confocal microscope (Olympus). Z-stacks were acquired from approximately 100 μm of CA1 primary apical dendrites, just above the primary dendrite bifurcation. This dendritic subfield is consistently identifiable, fully formed by the period of interest, harbors the highest density of asymmetric synapses, and retains its native connectivity in organotypic slices (Megías et al., 2001; Amaral and Lavenex, 2007). Ten to forty z-stacks were acquired per animal. Experiments were comprised of cultures from animals originating from at least two litters. Two-dimensional spine counts and geometric measurements of spines were quantified using Reconstruct (Fiala, 2005) and a custom ImageJ macro. Three-dimensional spine classification was performed with NeuronStudio (Rodriguez et al., 2008). All spine analysis was performed by an investigator blinded to the experimental condition.
Western Blot Analysis
For Western blots, 4–6 organotypic slices were lifted from nylon culture inserts with a No. 10 scalpel blade, rinsed in cold PBS and incubated on ice in 100 μl of Triton lysis buffer (20 mM Tris pH 7.4, 137 mM NaCl, 2 mM EDTA, 1% Triton X-100 (TX-100), 0.1% SDS, 10% glycerol, with protease inhibitors and sodium orthovanadate) for 30 min. Lysates were centrifuged at high speed for 10 min and stored at −80°C in sample buffer. Supernatants were warmed to room temperature and run under standard SDS-PAGE conditions. Membranes were immunoblotted with anti-K+-Cl− co-transporter 2 (KCC2) 1:1,000 (N1/12, NeuroMab) and GAPDH 1:300,000 (MAB374, Millipore). KCC2 blots were run immediately after developmental time courses ended to reduce experimentally-induced aggregation of KCC2 oligomers, which we observe to increase with time at -80°C.
Electrophysiology
Gramicidin perforated patch whole-cell recordings were performed similarly to previously described (Acton et al., 2012). Briefly, current-voltage (IV) curves were generated by step depolarizing the membrane potential in 10 mV increments from ~−95 to −35 mV (Figure 1C) and during each increment GABAergic transmission was elicited via extracellular stimulation in the stratum radiatum. Pipettes had a resistance of 7–12 MΩ and were filled with an internal solution containing 150 mM KCl, 10 mM HEPES, and 50 mM μg/ml gramicidin (pH 7.4, 300 mOsm). We recorded EGABA in the current-clamp mode. The glutamatergic transmission was inhibited with CNQX.
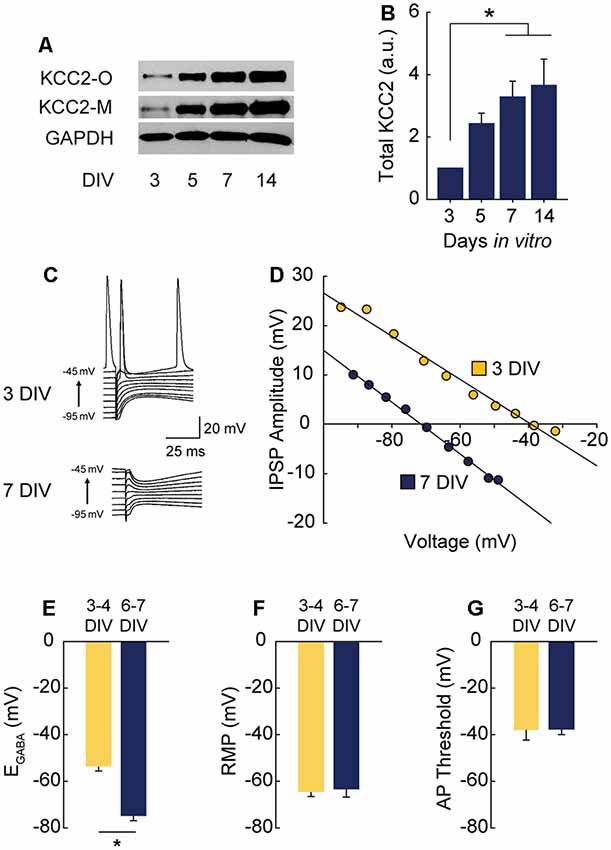
Figure 1. γ-Aminobutyric acid (GABA) reversal potential (EGABA) shifts from depolarizing to hyperpolarizing between 3 and 7 DIV. (A,B) Western blot of K+-CL− co-transporter 2 (KCC2) expression time course (A) and summary data, normalized to expression level at 3 DIV (B: 5 DIV 1.43 ± 0.34, 7 DIV 3.28 ± 0.51, 14 DIV 3.66 ± 0.84; n = 5; ANOVA p = 0.011; 3 vs. 5 DIV p = 0.25, 3 vs. 7 DIV p = 0.03, 3 vs. 14 DIV p = 0.01, Tukey post test). (C,D) Representative gramicidin perforated patch traces and representative IV curves from GABAergic responses at 3 DIV and 7 DIV. (E) EGABA summary plots (3/4 DIV: −53.3 ± 6.1 mV, n = 5; 6/7 DIV: −74.7 ± 6.4 mV, n = 5, p = 0.04). (F) Resting membrane potential (RMP) summary plots (3/4 DIV: −64.5 ± 2.3 mV, n = 5; 6/7 DIV: −63.4 ± 3.8 mV, n = 5). (G) Action potential (AP) threshold summary plot (3/4 DIV: −38.2 ± 4.2 mV, n = 5; 6/7 DIV-37.7 ± 2.3 mV, n = 5). *p < 0.05.
Miniature EPSCs (mEPSCs) were recorded using the whole-cell patch clamp configuration (Vh = −70 mV), at 30°C, in ACSF containing (in mM): 119 NaCl, 26.2 NaHCO3, 11 D-glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgCl2, 0.0002 TTX, 0.025 D-APV, 0.05 picrotoxin. Recording pipettes (2–5 MΩ) were filled with (in mM): 122 CsMeSO4, 8 NaCl, 10 D-glucose, 1 CaCl2, 10 EGTA, 10 HEPES, 0.3 Na3GTP, 2 MgATP, pH 7.2. Signals were low-pass filtered at 2 kHz, acquired at 10 kHz, and analyzed using Clampfit 10.3 (Molecular Devices).
For cell-attached recordings, ACSF and pipette solutions were as described above for mEPSC recordings, but ACSF lacked TTX, D-APV and picrotoxin. Low resistance recording pipettes (1–2 MΩ) were used to form loose patch seals (approximately 100–350 MΩ). Recordings were performed in I = 0 mode. GABA was diluted in ACSF to 100 μM and puffed in close proximity to the recorded cell using a glass pipette connected to a Picospritzer III (Parker Hannifin) delivering 10 ms duration air puffs at 14 psi. Electrically-evoked stimulations (1.3 V, 0.5 ms) were delivered by the recording amplifier via the recording pipette. Recorded signals were analyzed using threshold-based detection of spikes in Clampfit 10.3 (Molecular Devices).
Experiments comprised slices from at least three separate animals taken from at least two litters.
Pharmacology
Pharmacological agents (Tocris unless otherwise noted) were applied to the culture medium during a regular medium change. Gabazine (GBZ; 20 μM), bicuculline-methiodide (20 μM) and diazepam (5 μM) were used to manipulate GABAA transmission. GBZ was washed out by incubating slices in fresh medium for 30 min, then washing the top of the slices with equilibrated medium for 1–2 min before changing to fresh dishes and medium. Bumetanide (Bume, 10 μM), TrkB-Fc bodies (5 mg/mL, R&D Systems) and K252a (200 nM) were added to cultures 30 min before adding GBZ.
Quantitative Reverse Transcriptase PCR (qRT-PCR)
Six to eight organotypic slices per sample were lifted from nylon culture inserts with a No. 10 scalpel blade, washed briefly in ice-cold PBS and flash-frozen in microcentrifuge tubes in a 100% EtOH/dry ice slurry. Total RNA was extracted using the RNeasy Lipid Tissue Kit (Qiagen). cDNA libraries were created using the QuantiTect Reverse Transcription Kit (Qiagen). Quantitative PCR was performed using Sybr Green Master Mix (Applied Biosystems Systems) on a StepOne Plus thermocycler (Applied Biosystems). Relative levels of mRNA were calculated using the ΔΔCT method with GAPDH as the internal control. Primer sequences were as follows: GAPDH forward TTG AAG TCG CAG GAG ACA ACC; GAPDH reverse ATG TGT CCG TCG TGG ATC; BDNF forward GTG ACA GTA TTA GCG AGT GGG; BDNF reverse GGG ATT ACA CTT GGT CTC GTA G; Fos forward TCC CCA AAC TTC GAC CAT G; Fos reverse CAT GCT GGA GAA GGA GTC G.
Immunofluorescence
Slice cultures were fixed as described above, permeabilized for 30 min in 1% TritonX 100/PBS, blocked in 10% normal donkey serum (NDS, Jackson Immuno Research)/0.2% TX-100/PBS, and incubated with anti-c-Fos antibody (1:5,000, Cat. No. 226 003, Synaptic Systems) in 1% NDS/0.2% TX-100/PBS rocking at 4°C for 5–8 days. The primary antibody solution was washed with three rinses in 1% NDS/0.2% TX-100/PBS, followed by secondary antibodies at 1:1,000 for 2 h at room temp. TOPRO-3-iodide (Jackson Immuno Research) was applied at 1:10,000 for 10 min in the second of three washes following incubation with secondary antibodies. Quantification of Fos immunofluorescence intensity with background correction was performed with ImageJ. Full-field immunofluorescence within the CA1 stratum pyramidale was quantified and normalized within timepoint to the mean of the control.
Statistics
Data are presented as mean ± SEM, n sample size, N animals. Student t-tests were used except where noted that Mann–Whitney tests were used with datasets with non-normal distribution. Post hoc pairwise comparisons following ANOVA were performed with Tukey’s honestly significant difference (HSD) test. For mean comparisons: *p < 0.05, **p < 0.01, ***p < 0.001. For Kolmogorov–Smirnov tests: ***p < 0.0001.
Results
GABAA Transmission Switches From Depolarizing to Hyperpolarizing in CA1 Cells During the First Week in Hippocampal Slice Culture
Depolarizing GABAA transmission relies on relatively high intracellular chloride ([Cl−]i) during development. As neurons mature during the first weeks of postnatal CNS development, Na+-K+-Cl− cotransporter (NKCC1) expression is downregulated and KCC2 is upregulated, lowering [Cl−]i (Rivera et al., 1999; Yamada et al., 2004). GABAA receptors are largely permeable to Cl−, and to a lesser extent bicarbonate (HCO3−; Kaila, 1994; Staley and Proctor, 1999). When [Cl−]i lowers to the point at which the reversal potential for GABA (EGABA) hyperpolarizes below the resting membrane potential (RMP), GABAA transmission switches from depolarizing to hyperpolarizing. To pinpoint when this switch from depolarization to hyperpolarization occurs in CA1 pyramidal cells in hippocampal organotypic slices, we first assessed the timing of KCC2 upregulation across the first 2 weeks in vitro and found expression of KCC2 underwent a large and graded increase between 3 and 14 days in vitro (DIV), reaching near-maximal levels by 7 DIV (Figures 1A,B). Using this timeframe as a guide, we performed gramicidin perforated patch recordings to determine the GABAA reversal potential (EGABA) in CA1 pyramidal cells (exemplary traces and IV curves shown in Figures 1C,D). At 3–4 DIV, EGABA was depolarized with respect to RMP (Figures 1E–G). However, by 6–7 DIV EGABA was hyperpolarized with respect to RMP, indicating a switch to hyperpolarizing GABAA transmission by 6–7 DIV (Figures 1C–G), a timeframe similar to that reported previously for CA1 pyramidal cells (Swann et al., 1989). EGABA was more negative than action potential (AP) threshold at 3–4 DIV (Figures 1E,G), suggesting GABA is depolarizing but not capable of directly depolarizing neurons past AP threshold from rest at this stage.
Blocking Depolarizing GABAA Transmission Increases Glutamatergic Synapse Number and Function
Overexciting mature neurons by blocking hyperpolarizing GABAA transmission is known to cause a collapse of dendritic spines both in vivo (Zeng et al., 2007) and in vitro (Muller et al., 1993; Drakew et al., 1996; Jourdain et al., 2002; Zha et al., 2005). In particular, applying GABAA antagonists to organotypic hippocampal cultures at 5 or 23 DIV over a period of 2–3 days has been shown to cause a marked loss of spines (Drakew et al., 1996; Zha et al., 2005). Consistent with this, when we blocked GABAA transmission with the GABAAR antagonist, bicuculline (BIC) from 5 to 7 DIV [when GABAA transmission is hyperpolarizing (Figures 1C–G)], spine density decreased by 34% (Figures 2A–C). This suggests that by this stage, excitatory transmission causes overexcitation and spine loss in the absence of hyperpolarizing GABAA transmission.
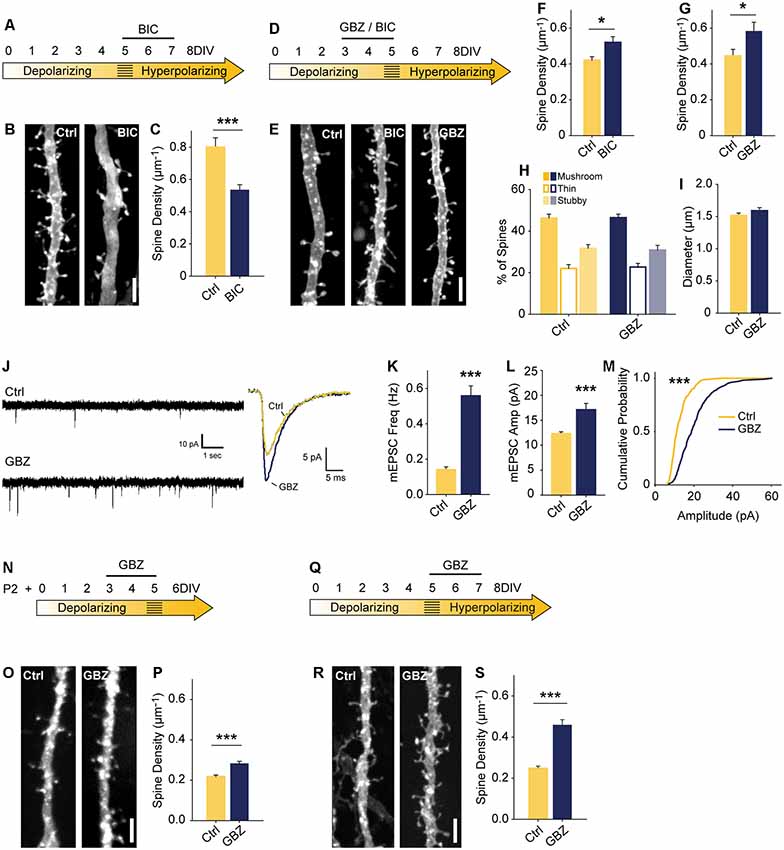
Figure 2. Blocking depolarizing GABAA transmission increases excitatory synapse number. (A) Time course of bicuculline (BIC) treatment for (B,C). (B,C) Spine density after 5–7 DIV BIC treatment (Control 0.80 ± 0.06 spines/μm, n = 36, BIC 0.53 ± 0.03, n = 50; N = 3; p < 0.001, Mann–Whitney). (D) Time course of pharmacological treatments for (E–M). (E–G) Spine density after 3–5 DIV GBZ (G: Control 0.44 ± 0.12 spines/μm, n = 145, GBZ 0.58 ± 0.17, n = 77; N = 11; p = 0.04) and BIC treatment (F: Control 0.42 ± 0.02 spines/μm, n = 55, BIC 0.52 ± 0.03 spines/μm, n = 41; N = 9; P = 0.027, Mann–Whitney). (H,I) Three dimensional spine morphology and dendrite diameter after GBZ. (J) Representative and mean traces of miniature EPSCs (mEPSCs). (K) mEPSC frequency summary plot (Control 0.14 ± 0.02 Hz, GBZ 0.56 ± 0.06 Hz, p < 0.001, Mann–Whitney). (L) mEPSC amplitude summary plot (Control 12.32 ± 0.37 pA, n = 8, GBZ 17.12 ± 1.27 pA, n = 10, p < 0.001, Mann–Whitney). (M) Cumulative distributions of amplitudes (p < 0.0001, Kolmogorov–Smirnov test). Scale bars 3 μm. (N) Time course of treatment of slices prepared from P2 pups. (O,P) Exemplary images and quantification of spine enhancing effect of GBZ when applied to slices from P2 pups (Ctrl 0.22 ± 0.008 μm−1, n = 217, GBZ 0.28 ± 0.01 spines/μm−1, n = 156; N = 3; p < 0.001, Mann Whitney). (Q) Time course of antibiotic-free GBZ treatment. (R,S) Exemplary images and quantification of the spine enhancing effect of GBZ on slices cultured in antibiotic-free culture medium (Ctrl 0.248 ± 0.0109 μm−1, n = 198, GBZ 0.458 ± 0.0264 μm−1, n = 70; N = 4; p < 0.001, Mann–Whitney). *p < 0.05, ***p < 0.001.
To assess the role of immature, depolarizing GABAA transmission on dendritic spine development, we inhibited GABAA transmission earlier, from 3 to 5 DIV (Figure 2D). Previous work suggests that inhibiting depolarizing GABAA transmission during development would decrease glutamatergic synapse formation and maturation (Ben-Ari et al., 1997; Hanse et al., 1997; Cancedda et al., 2007; Wang and Kriegstein, 2008). However, in contrast to these findings, BIC applied for 48 h from 3 to 5 DIV significantly increased dendritic spine density (25% increase; Figures 2E,F). This effect was fully reproducible with the GABAAR antagonist gabazine (GBZ; 31% increase; Figures 2E,G), which is a more specific antagonist of GABAARs (Heaulme et al., 1986) and blocks inhibition more consistently in hippocampal neurons (Sokal et al., 2000).
To assess whether the supernumerary spines induced by blocking depolarizing GABAA transmission showed structural differences, we analyzed spine morphology. GBZ treatment did not affect the proportions of mushroom, thin, and stubby spines (Figure 2H), 2-dimensional head area (Control: 0.32 ± 0.02 μm2; GBZ: 0.37 ± 0.04 μm2, p < 0.10), head diameter (Control: 0.58 ± 0.02 μm2; GBZ: 0.62 ± 0.03 μm2, p < 0.1), spine length (Control 1.66 ± 0.09 μm2; GBZ: 1.83 ± 0.08 μm2, p < 0.1) or dendrite diameter (Figure 2I).
We next asked whether the increased number of spines constituted an increase in bona fide glutamatergic synapses on CA1 cells by recording miniature EPSCs (mEPSCs). Consistent with the increase in dendritic spine density, GBZ treatment (3–5 DIV) increased mESPC frequency 3-fold (Figures 2J,K). Miniature EPSC amplitude also increased, indicating enhanced synaptic strength (Figures 2L,M). Together, these results suggest that immature GABAA transmission restrains glutamatergic synapse formation and maturation.
The narrow time window we examined raised the possibility that the spine-enhancing effect of GABAA blockade is limited to a short period directly prior to the depolarizing to hyperpolarizing shift in GABAA transmission. This would suggest that GABAA transmission restrains glutamatergic synapse formation only during a very short transition state. To test whether this was the case, we prepared slices 3 days earlier (P2) and applied GBZ at 3 DIV for 48 h (Figures 2N–P). We found that GABAAR blockade in these younger slices also caused a significant increase in spines (Figures 2O,P), suggesting that depolarizing GABAA transmission is capable of restraining synapse formation for an appreciable period during postnatal development.
We also verified that the presence of penicillin-streptomycin in the culture medium was not associated with the spine-enhancing effect of blocking depolarizing GABAA transmission by applying GBZ from 3 to 5 DIV in the absence of antibiotics, and found the same increase in dendritic spines (Figures 2Q–S).
Bumetanide Treatment Has No Effect on Spine Numbers
Previous work suggests that abrogating GABAergic depolarization by prematurely rendering GABA hyperpolarizing decreases glutamatergic synapse formation (Ge et al., 2006; Wang and Kriegstein, 2008). However, our data show that blocking depolarizing GABAA transmission increased glutamatergic synapse formation. These contrasting results raise the question of whether the depolarizing nature of GABAA transmission is important for the normal development of glutamatergic synapse number in our period of interest (3–5 DIV). To address this, we asked whether prematurely rendering EGABA hyperpolarizing could mimic the effect of GABAA blockade by treating slices with the NKCC1 blocker bumetanide (BUME) from 3 to 5 DIV. BUME is well established to lower EGABA in immature neurons (Dzhala et al., 2005) and prematurely render GABA hyperpolarizing (Wang and Kriegstein, 2011), and we verified that this was the case in the organotypic slice preparation (Figures 3A–C). We then applied BUME to organotypic slices from 3 to 5 DIV in the presence and absence of GBZ. BUME did not alter spine density on its own (Figures 3D,E), indicating that the depolarized nature of EGABA is not important for regulating spine numbers at this stage of development. Furthermore, BUME did not alter the effect of GBZ on spine density, indicating that if EGABA is prematurely decreased this does not change the role of GABAA transmission in regulating synapse formation at this stage.
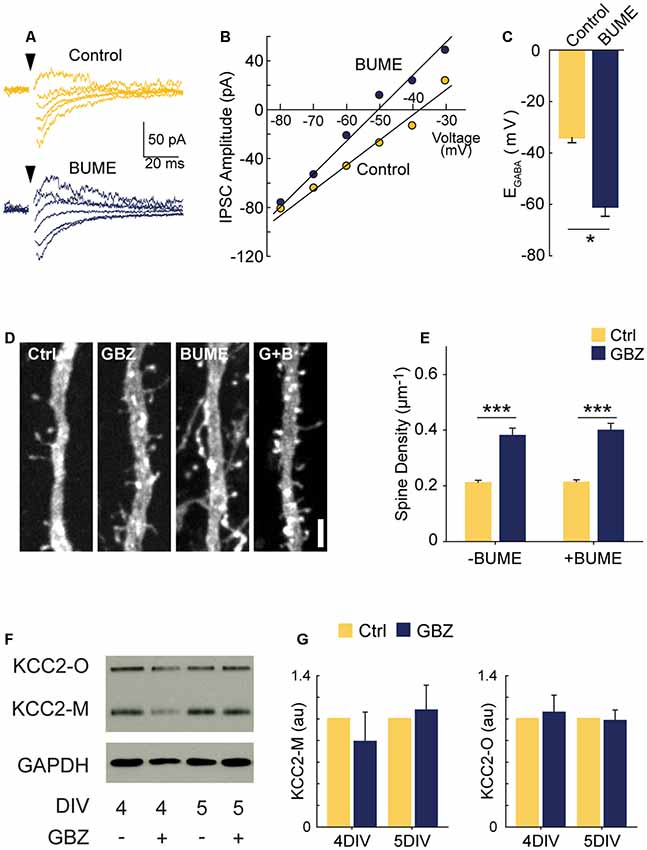
Figure 3. GBZ-induced increase in spines is not reproduced by bumetanide and is not associated with changes in KCC2 expression. (A–C) Sample traces from which IV curves were generated (A) and resulting sample IV curve (B), and summary plots showing BUME hyperpolarizes EGABA in organotypic slices (C, Ctrl −34.0 ± 2.0 mV, n = 3, GBZ −60.8 ± 3.8 mV, n = 3, p = 0.02). (D,E) Bumetanide does not increase spine density above control levels or change the effect of GBZ on spine density, (E, Control 0.21 ± 0.01 μm−1, n = 102; GBZ 0.38 ± 0.02 μm−1, n = 47; BUME 0.21 ± 0.02 μm−1, n = 88; BUME+ GBZ 0.40 ± 0.02 μm−1, n = 53; N = 3; two-way ANOVA indicated no significant interaction between GBZ and BUME treatment (p = 0.633). Tukey honestly significant difference (HSD) post-test indicates significant differences between Ctrl and GBZ in the absence of BUME (p < 0.001) and in the presence of BUME (p < 0.001). (F,G) Western blot (F) showing no changes in monomeric (KCC2-M) or oligomeric (KCC2-O) KCC2 expression following GBZ from 3 to 4 DIV (p = 0.52 and 0.77, respectively, one-sample t-test, n = 3) and 3–5 DIV (p = 0.76 and 0.87, respectively, one-sample t-test, n = 3) (G). Scale bar 3 μm. *p < 0.05, ***p < 0.001.
Since KCC2 overexpression can cause an increase in spines through its non-transport, scaffolding function (Li et al., 2007; Fiumelli et al., 2012), we also assessed KCC2 expression following GBZ treatment. GBZ did not significantly elevate the expression of KCC2 oligomers or monomers (Figures 3F,G).
Driving Depolarizing GABAA Transmission Does Not Alter Glutamatergic Synapse Number
Next, we investigated if increasing GABAA transmission over the 3–5 DIV period would have the opposite effect of GABA-blockade and reduce excitatory synapse number. Previous work has demonstrated that propofol, a positive allosteric modulator of GABAARs, decreases spine density in developing layer 2/3 principal cells of the somatosensory cortex when administered to rat pups over a 6 h period at postnatal day 10 when GABAA transmission is still depolarizing (Puskarjov et al., 2017). To test this in CA1 pyramidal cells, we pharmacologically enhanced depolarizing GABAA transmission from 3 to 5 DIV with diazepam (DZP). We first confirmed that bath applied-DZP caused the expected slower decay kinetics of mIPSCs (Figures 4A,B) and also that this led to enhanced charge transfer (Figure 4C). Miniature IPSC frequency and amplitude were unaffected by DZP, as expected (Not shown graphically; Frequency: Ctrl 0.27 ± 0.08 Hz, DZP 0.39 ± 0.2 Hz, p = 0.43; Amplitude: Ctrl 5.05 ± 1.11 pA, DZP 5.2 ± 1.21 pA, p = 0.55). However, contrary to the in vivo propofol administration (Puskarjov et al., 2017), DZP (5 μM) applied to organotypic slices from 3 to 5 DIV had no effect on spine density or mEPSCs (Figures 4D–I). Based on these results, increasing GABAA transmission was not sufficient to decrease glutamatergic synapse number or function, suggesting depolarizing GABAA transmission can only limit synapse formation up to a certain point at this stage of circuit development in our preparation. However, these results do not rule out the possibility that enhancing immature GABAA transmission on different timescales or in other systems decreases glutamatergic synapse formation (Puskarjov et al., 2017).
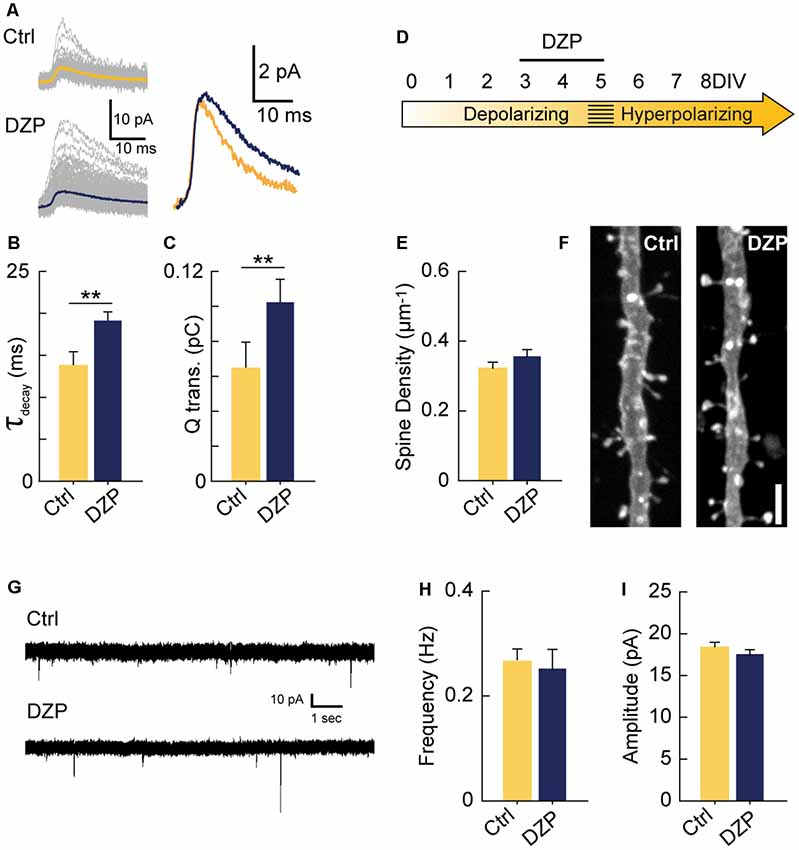
Figure 4. Driving depolarizing GABAA transmission does not decrease glutamatergic synapse numbers. (A) Sample mIPSC traces with superimposed mean traces in color for Ctrl (n = 104) and DZP (n = 197) conditions (left), and enlarged overlay of mean Ctrl and DZP mIPSCs (right). DZP was applied at 5 μM. (B,C) Summary data for mIPSC decay constant (Ctrl 13.7 ± 1.73 ms, DZP 19.1 ± 1.15 ms, p = 0.007, n = 3 cells) and charge transfer (Ctrl 0.063 ± 0.015 pC; DZP 0.101 ± 0.013 pC, p = 0.006, n = 3 cells). (D) Time course of DZP treatment in organotypic slices. (E,F) Spine density after 3–5 DIV DZP treatment (Ctrl 0.321 ± 0.02, n = 116; DZP 0.36 ± 0.02, n = 88; N = 6; p = 0.11, Mann–Whitney). (G) Representative traces of mEPSCs following 3–5 DIV treatment with DZP. (H) mEPSC frequency summary plot (Ctrl 0.27 ± 0.02 Hz, n = 9; DZP 0.25 ± 0.04 Hz, n = 8; p = 0.41 Mann–Whitney). (I) mEPSC amplitude summary plot (Ctrl 18.3 ± 0.7 pA, n = 9; DZP 17.5 ± 0.6 pA, n = 8; p = 0.39). **p < 0.01.
An Increase in Glutamatergic Synapses Following Blockade of Depolarizing GABAA Transmission is Activity-Dependent
Based on our recordings showing that at 3–4 DIV EGABA is depolarized relative to RMP, but lower than AP threshold (Figures 1E–G), we hypothesized that GABA is likely to mediate shunting inhibition despite being depolarizing at this stage (schematized in Figure 5A). To test this, we puffed GABA locally while recording spontaneous or electrically evoked firing. GABA inhibited both spontaneous (Figures 5B,C) and evoked spiking (Figures 5D,E), suggesting that although EGABA is depolarizing relative to RMP, GABAA transmission is inhibitory through shunting inhibition during the 3–5 DIV timeframe. Blocking this depolarizing but shunting GABAA transmission likely increased activity in our preparation, suggesting that the increase in glutamatergic synapses following GABAA-blockade at 3 DIV was driven by activity-dependent mechanisms (Balkowiec and Katz, 2002; Pérez-Gómez and Tasker, 2013). To begin to address this possibility, we measured levels of Bdnf and Fos mRNA, two activity-regulated genes associated with glutamatergic synapse formation (Vicario-Abejón et al., 1998, 2002; Tyler and Pozzo-Miller, 2003; Chapleau et al., 2009). Both transcripts were significantly upregulated following a 48-h blockade of depolarizing GABAA transmission from 3 to 5 DIV (Bdnf: 5-fold increase, Fos: 2.5-fold increase; Figure 5F). GABAA-blockade also significantly increased Fos protein expression by 2 h after commencing GBZ treatment at 3 DIV (Figure 5G). Furthermore, GBZ treatment elevated Fos expression relative to control across the 48 h treatment window, with a slow decay in the elevation later on (Figures 5H,I). Both the increased Fos expression and the partial decay of this expression over time are consistent with a sustained increase in neural activity as examined in previous studies (Tyssowski et al., 2018). Thus, the above data indicate that blocking immature depolarizing GABAA transmission at this point increased activity in CA1 pyramidal cells across the 3–5 DIV window. To test whether the increased synapse formation we observed following 3–5 DIV GABAA-blockade was activity-dependent, we treated slice cultures with GBZ and/or TTX, and found that while TTX alone had no effect on spine density, TTX blocked the GBZ-induced increase in spines (Figure 5J). From this, we conclude that depolarizing GABAA transmission limits activity-dependent glutamatergic synapse formation at this point in the development of hippocampal circuitry in slice culture.
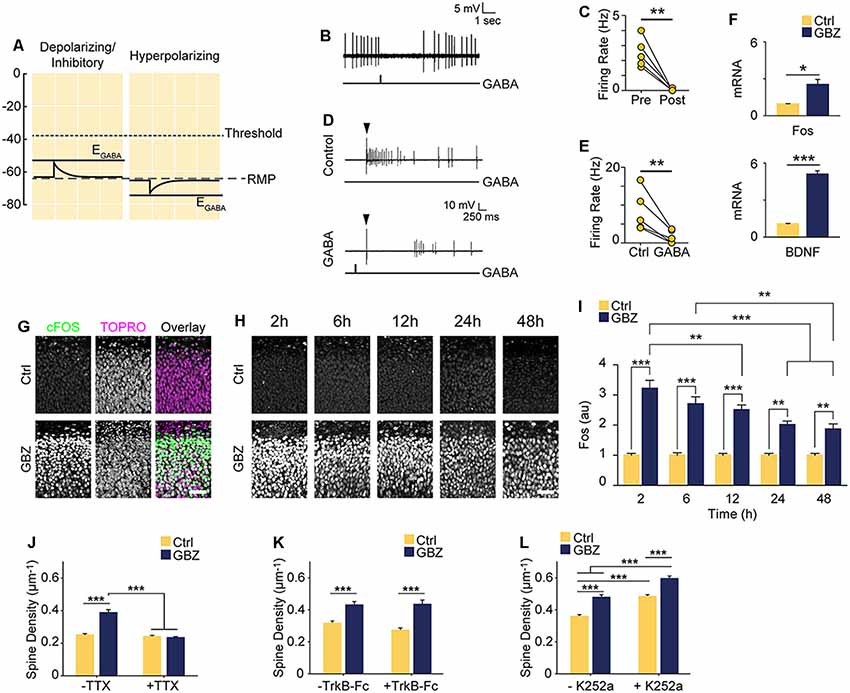
Figure 5. Increased spine density following blockade of depolarizing GABAA transmission is activity-dependent but does not rely on BDNF signaling. (A) Schematic demonstrating the likely shunting and hence inhibitory nature of depolarizing GABAA transmission at 3–4 DIV due to the relative values of AP Threshold < EGABA < RMP. The scale in (A) aligns with that of Figures 1E–G such that the threshold, RMP and EGABA values are represented accurately relative to each other. (B) Sample trace of spontaneous activity inhibited by puffing on GABA. The line trace below indicates the time of GABA puff. (C) Summary plots of spontaneous activity pre- and post-GABA puff. (D) Sample traces from the same cell demonstrating that activity could be evoked electrically (Control) and that puffed GABA inhibited electrically evoked activity (GABA). The arrow above the traces denotes the timing of electrical stimulation, while the line trace below denotes the timing of the GABA puff. (E) Summary plots of electrically evoked activity in the absence and presence of puffed GABA. (F) Fos and BDNF transcript levels following GBZ from 3 to 5 DIV (BDNF: Ctrl 1.07 ± 0.04, GBZ 5.08 ± 0.3, N = 3, p < 0.001; Fos: Ctrl 0.94 ± 0.04, GBZ 2.52 ± 0.4, N = 3, p = 0.02). (G) Fos immunofluorescence 2 h after GBZ treatment beginning at 3 DIV. Images depict the top of the stratum pyramidale, including the lower extremity of the stratum oriens, in area CA1. TOPRO-3-Iodide was used to visualize nuclei. (H,I) GBZ significantly increased Fos immunofluorescence after 2 h (Ctrl 1 ± 0.06 au, n = 13, GBZ 3.21 ± 0.26 au, n = 13), 6 h (Ctrl 1 ± 0.08 au, n = 14, GBZ 2.70 ± 0.23 au, n = 14), 12 h (Ctrl 1 ± 0.07 au, n = 15, GBZ 2.51 ± 0.16 au, n = 14), 24 h (Ctrl 1 ± 0.06 au, n = 10, GBZ 2.00 ± 0.12 au, n = 11) and 48 h (Ctrl 1 ± 0.06 au, n = 10, GBZ 1.86 ± 0.18 au, n = 10; two-way ANOVA, Tukey post test, p < 0.001 for population comparisons and interaction). (J) Quantification of spine density following GBZ and/or TTX treatment beginning at 3 DIV (Ctrl 0.25 ± 0.01 μm−1, n = 196, GBZ 0.39 ± 0.01 μm−1, n = 110, TTX 0.24 ± 0.01 μm−1, n = 166, GBZ + TTX 0.23 ± 0.01 μm−1, n = 154; N = 5). Two-way ANOVA indicates a significant interaction between GBZ and TTX conditions, p < 0.001. Significant differences between GBZ and all other conditions, p < 0.001, Tukey post-test. (K) Quantification of spine density following GBZ and/or TrkB-Fc treatment (Ctrl 0.31 ± 0.02, n = 86, GBZ 0.42 ± 0.02, n = 68, TrkB-Fc 0.27 ± 0.02, n = 96, TrkB-Fc + GBZ 0.43 ± 0.02, n = 61; N = 3; two-way ANOVA, no interaction, Tukey post-test). (L) Quantification of spine density following GBZ and/or K252a treatment (Ctrl 0.35 ± 0.01, n = 198, GBZ 0.49 ± 0.03, n = 144, K252a 0.47 ± 0.02, n = 216, K252a+GBZ 0.58 ± 0.04, n = 185; all significant differences <0.001, two-way ANOVA, no interaction, Tukey post-test). Scale bar 60 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
BDNF is known to regulate activity-dependent synapse formation (Park and Poo, 2013). We therefore asked whether BDNF signaling was responsible for the increase in spines following blockade of depolarizing GABAA transmission. We inhibited BDNF signaling during the 3–5 DIV GBZ treatment using TrkB-Fc bodies or K252a (Ji et al., 2010; Puskarjov et al., 2015), however, neither manipulation blocked the increase in spine density (Figures 5K,L), suggesting that BDNF signaling is not necessary for the observed increase in spines.
Blocking Depolarizing GABAA Transmission Leads to a Sustained Increase in Glutamatergic Synapse Number
The observed increase in spine density induced by blocking depolarizing GABAA transmission may only lead to a transient alteration without a longer-lasting effect on glutamatergic synapses. To determine whether blockade of GABAA transmission caused a temporary or sustained increase in glutamatergic synapses, we treated slices with GBZ from 3 to 5 DIV and allowed them to recover for an additional 5–9 days in the absence of GBZ (Figure 6A). This temporary GABAA blockade resulted in a 37% increase in spine density after a 5-day recovery period (Figures 6B,C). Furthermore, after this recovery period, CA1 cells had more thin spines than mushroom spines, a difference not present in the control condition (Figure 6D). No changes in dendrite diameter were observed (Figure 6E). To determine if transient GBZ treatment led to long-term functional changes in glutamatergic synapses, we recorded mEPSC frequency and amplitude after 8–9 days of recovery. We found that mEPSC frequency was enhanced by 79%, while mEPSC amplitude was unchanged at this stage (Figures 6F–I). Together these data suggest that inhibiting depolarizing GABAA transmission during a narrow time window can lead to persistent changes in glutamatergic synapse number in the hippocampus.
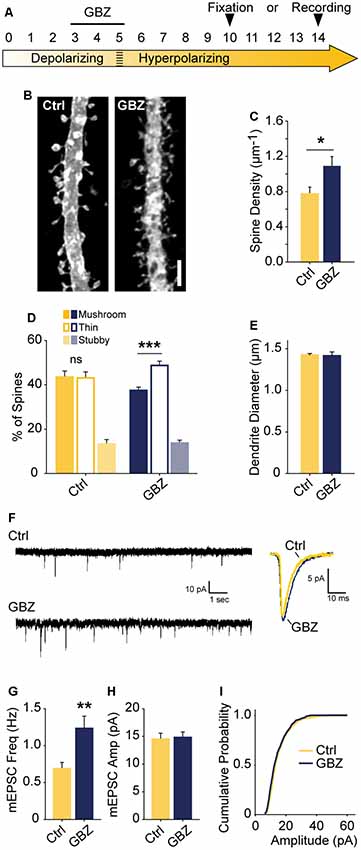
Figure 6. Transient blockade of depolarizing GABAA transmission causes a lasting increase in excitatory synapse number and alters spine morphology. (A) Schematic time course of GBZ treatment and experimental endpoints. (B,C) Spine density after 3–5 DIV GBZ treatment and 5 days of recovery (Control 0.78 ± 0.08 spines/μm, n = 127; GBZ washout 1.07 ± 0.07 spines/μm, n = 112; N = 6; p = 0.024). (D) 3D spine morphology after 5 days of recovery (***p < 0.001, critical level 0.05, two-way ANOVA with Holm Sidak Post Test). (E) Dendrite diameter after recovery (p = 0.86). (F) Representative mEPSC traces from slices after 8–9 days of recovery. (G) mEPSC frequency summary plot (Control: 0.70 ± 0.08 Hz, n = 10 GBZ: 1.23 ± 0.17 Hz, n = 10, p = 0.009). (H) mEPSC amplitude summary plot (Control: 14.50 ± 1.07 pA, n = 10, GBZ: 14.80 ± 1.00 pA, n = 10, p = 0.84). (I) Cumulative mEPSC distributions (p = 0.58, Kolmogorov–Smirnov test). Scale bar 3 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Immature, depolarizing GABAA transmission is believed to promote glutamatergic synapse formation and maturation (Ben-Ari et al., 1997; Hanse et al., 1997; Wang and Kriegstein, 2009; Chancey et al., 2013). However, when and how GABA affects glutamatergic synapse formation remains to be fully understood. Indeed, several groups have noted that tools and approaches for manipulating depolarizing GABAA transmission with higher temporal and spatial precision are needed to resolve this question (Akerman and Cline, 2007; Chancey et al., 2013; Kirmse et al., 2018). We, therefore, sought to address the role of GABAA transmission in glutamatergic synapse formation by performing precisely timed pharmacological manipulations in hippocampal slice cultures. We first mapped the depolarizing-to-hyperpolarizing shift of GABAA transmission in CA1 cells. This was followed by a structural and electrophysiological analysis which showed that blocking immature, depolarizing GABAA transmission enhanced glutamatergic synapse function and number. Interestingly, the enhanced synapse number was stable following a recovery period. These results suggest that immature GABAA transmission restrains glutamatergic synapse formation during an early phase of hippocampal circuit development. Using slice cultures allowed for more temporally precise manipulations that revealed this effect, though limitations of this model system must be considered when interpreting our results. In particular, exuberant glutamatergic synapse formation has been observed in slice cultures and has been attributed to increases in distal dendritic branching (De Simoni et al., 2003). However, we minimized this confound by focusing on primary apical dendrites, which are fully formed by the time of pharmacological treatment. Thus, while further work will be required to extend our findings to other systems, the results of this study show that immature, depolarizing GABAA transmission is capable of restraining glutamatergic synapse formation in certain contexts, and that the removal of this restraint by interfering with GABAA transmission during development may cause a long-term increase in glutamatergic synapses.
An Unpredicted Role for Immature GABAA Transmission in Restraining Glutamatergic Synapse Formation
In the time window we examined, GABAA transmission provides subthreshold depolarization and shunting inhibition, which when blocked alleviates a brake on glutamatergic synapse development. Taken in the context of previous work, our results suggest a couple of models for how immature GABAA transmission affects hippocampal excitatory connectivity (Figure 7). Firstly, the GABA-mediated restraint on glutamatergic synapse formation may be a short-lived feature of a “depolarizing but inhibitory” transition state that GABA passes through as ECl matures from depolarizing and excitatory to hyperpolarizing (Model 1, Figures 7A–C). However, recent work suggests GABA may be inhibitory throughout most or all of postnatal development. Therefore, in a second model, depolarizing but inhibitory GABAA transmission may inhibit circuit activity from birth onward (Model 2, Figures 7B,C), thus restraining glutamatergic synapse formation across development. In both of these cases we specify “depolarizing but inhibitory” rather than simply shunting, since shunting inhibition occurs in conjunction with both depolarizing and hyperpolarizing GABA transmission.
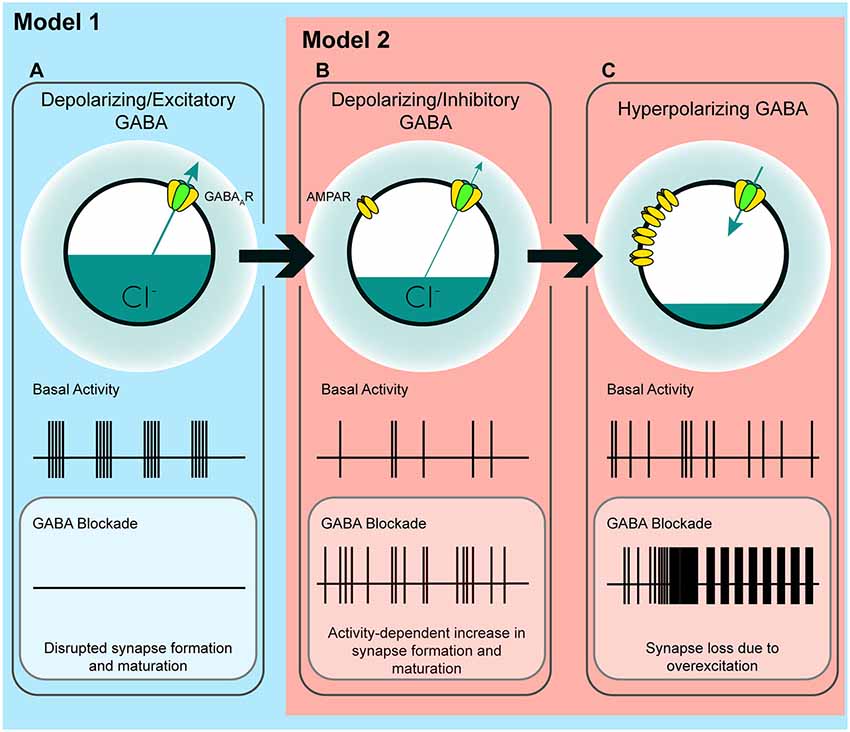
Figure 7. A model of the possible roles of GABAA transmission in glutamatergic synapse formation as chloride homeostasis matures. (A) Work performed in acute slices suggests that depolarizing GABAA transmission provides the initial excitatory drive required for activity- and calcium-dependent formation and maturation of glutamatergic synapses. The in vitro work supporting a phase in which GABA drives network activity suggests that blocking GABAA transmission at this stage eliminates early network oscillations (ENOs). (B) Our work suggests a possible transition state wherein blocking depolarizing but inhibitory transmission alleviates an inhibitory restraint on circuit activity, allowing for activity-dependent formation of glutamatergic synapses. Such a transition state would likely rely on a still underdeveloped glutamatergic system that is not yet capable of pathological levels of overexcitation. Conversely, recent in vivo work suggests that GABA may inhibit circuit activity throughout postnatal development, indicating that blocking GABAA transmission might enhance circuit activity and glutamatergic synapse formation from birth until GABA becomes fully hyperpolarizing (Model 2; although the basal activity here is depicted as uncoordinated to clearly differentiate (B) from (A), the activity pattern in this transition state, as well as in (C), may very well be oscillatory depending on the state of the system being studied). (C) When ECl and the glutamatergic system are mature, blocking hyperpolarizing GABAA transmission causes overexcitation and loss of glutamatergic synapses.
The first model is based on evidence from acute slices suggesting that immature GABAA transmission is capable of driving excitation (Gulledge and Stuart, 2003) and that depolarizing GABAA transmission drives ENOs, which in turn promote glutamatergic synapse formation and unsilencing, and circuit refinement (Hanse et al., 1997; Ben-Ari, 2002; Wang and Kriegstein, 2009; Griguoli and Cherubini, 2017). Disrupting ECl or GABAA transmission in this phase of development is hypothesized to interfere with synapse formation (Figure 7A), and this has been borne out by experimentally lowering ECl across the postmitotic period in immature neurons (Ge et al., 2006; Cancedda et al., 2007; Wang and Kriegstein, 2008). Assuming this model is correct, incorporating our results refines the model and accounts for the role of GABAA transmission in circuit development as it transitions from a depolarizing and excitatory to a hyperpolarizing state. Our work suggests that following an initial depolarizing phase in which GABA promotes excitation, as ECl progressively matures, GABAA transmission passes through a transient but developmentally relevant depolarizing but inhibitory phase (Figure 7B). Such a transition phase is hinted at in the literature, as certain studies have shown that blocking depolarizing GABAA transmission can silence ENOs (Ben-Ari et al., 1989; Garaschuk et al., 1998; Mohajerani and Cherubini, 2005), while others show that blocking depolarizing GABAA transmission increases circuit activity, eliciting interictal discharges or paroxysmal activity (Khazipov et al., 1997; Khalilov et al., 1999; Lamsa et al., 2000; Wells et al., 2000; Le Magueresse et al., 2006; Ben-Ari et al., 2007). This latter group of studies indicates a role for depolarizing GABA in inhibiting circuit activity, as GABA transitions from depolarizing and excitatory to hyperpolarizing. Our results suggest that during the transition phase, depolarizing but inhibitory GABAA transmission restrains glutamatergic synapse formation. Blocking GABAA transmission at this time alleviates the restraint, allowing for activity-dependent synapse formation (Figure 7B). Following this transition phase, GABAA transmission becomes fully hyperpolarizing, and the glutamatergic system becomes capable of overexcitation. The result of GABAA blockade at this stage is the loss of spines (Figures 2, 7C; Swann et al., 1989; Drakew et al., 1996; Zeng et al., 2007). Crucially, in the present study, a similar spine loss following blockade of depolarizing but inhibitory GABAA transmission at 3 DIV does not occur, despite the fact that GABA is inhibitory at this stage. This may be explained by a glutamatergic system that is not yet mature enough to drive overexcitation capable of causing pathological collapse of synapse numbers similar to that seen in models of epilepsy (Zha et al., 2005; Zeng et al., 2007).
Alternatively, in the second model, it is possible that depolarizing GABAA transmission provides shunting inhibition throughout the postnatal period, thereby restraining synapse formation and circuit activity during development (Model 2, Figures 7B,C). Indeed, emerging evidence suggests that depolarizing GABAA transmission exerts inhibitory effects on ENOs in vivo, from at least P3 onward (Kirmse et al., 2015; Valeeva et al., 2016; Che et al., 2018). Consistent with this, our results in slices cultured from younger mice (Figures 2N–P) show that GABAA transmission restrains synapse formation over a period of at least 5 days of hippocampal circuit development. While previous work has admittedly demonstrated that prematurely rendering GABAA transmission hyperpolarizing in vivo decreases glutamatergic synapse formation (Ge et al., 2006; Cancedda et al., 2007; Wang and Kriegstein, 2008, 2011), it is noteworthy that these earlier studies manipulated ECl over extended periods that spanned multiple phases of postmitotic neuronal development, including cell migration, axonal/dendritic growth, synapse formation, and circuit refinement. Depolarizing GABAA transmission is thought to play important roles in all of these processes (Owens and Kriegstein, 2002), and hence the observed effects of prematurely reducing ECl on synapses may be secondary to other alterations in neuronal and circuit development. Indeed, soma size and dendritic branching are altered when GABA is prematurely rendered hyperpolarizing over an extended time period (Cancedda et al., 2007; Wang and Kriegstein, 2008). More temporally precise manipulations of GABAA transmission and ECl are therefore essential for clarifying the roles of GABA during critical phases of synapse formation in vivo. Interestingly, the finding that propofol administered to postnatal day 10 rats decreased spine number supports the notion that there is a developmental period in vivo during which immature GABAA transmission restrains glutamatergic synapse formation (Puskarjov et al., 2017).
When considering these two models, it is important to note that an inhibitory effect of depolarizing GABAA transmission does not preclude a role for GABA in driving ENOs, as it has been demonstrated that depolarizing chloride currents are only involved in the initial generation of ENOs in acute slices, after which they inhibit the continuation of the same ENOs (Khalilov et al., 2015). Thus, depolarizing GABAA transmission may simultaneously aid in generating ENOs, while also maintaining control of wider circuit activity, thereby limiting runaway glutamatergic synapse formation. These dichotomous effects of GABA may rely on where GABAergic inputs impinge on the postsynaptic neuron. Gulledge and Stuart (2003) showed that in young rats, puffing GABA on distal dendrites of Layer 5 pyramidal cells facilitated firing while puffing GABA on the cell body inhibited firing. Thus, different GABAergic interneuron subtypes may be responsible for driving ENOs vs. restraining glutamatergic synapse formation. Furthermore, despite the evidence suggesting GABA is inhibitory throughout most of the postnatal development in vivo, it has been shown that high frequency uncaging or stimulated release of GABA onto dendrites of layer 2/3 pyramidal cells in the neocortex can elicit formation of glutamatergic and GABAergic synapses during development in vivo (Oh et al., 2016). Although it remains to be seen whether endogenous patterns of GABA release can have similar effects, it appears there may be a local trophic role for depolarizing GABAA transmission, which may promote synapse formation even as its circuit-wide inhibitory effects restrain the same process as we have demonstrated. More work is needed to dissect the possible roles of GABA in local synapse formation and more global circuit development, and to understand how the role of GABAA transmission changes across development.
Depolarizing GABAA Transmission and Sustained Changes in Glutamatergic Synapses
Remarkably, we found that a transient blockade of depolarizing GABAA transmission led to a sustained increase in both the number of glutamatergic synapses and the proportion of thin spines, indicating that transient manipulations of immature GABAA transmission can profoundly alter hippocampal connectivity (Figure 6). Importantly, the observed changes in synapse number may elicit compensatory homeostatic responses. For instance, increased synapse number can be compensated for by decreasing overall dendritic length (Tripodi et al., 2008), however, the increased mEPSC frequency we observed after GBZ washout suggests that overall synapse number was indeed elevated at the time point examined. Using slice cultures allowed for more temporally precise manipulations that revealed this effect, though it remains to be seen if the phenomenon persists in vivo. These questions are clinically relevant, as a role for GABA in restraining synapse formation may change how we understand and mitigate the effects of anticonvulsants, anesthetics and drugs of abuse on neonatal, as well as fetal development, as GABA is believed to be depolarizing mainly in late gestation in humans (Vanhatalo et al., 2005; Sedmak et al., 2016). Furthermore, both the persistent increase in synapses and spines and the shift in spine morphologies we observed after recovery from transient GBZ treatment are reminiscent of “spinopathies” seen in intellectual disabilities including Fragile X syndrome and autism spectrum disorders (Lacey and Terplan, 1987; Irwin et al., 2000, 2001; Kaufmann and Moser, 2000; Fiala et al., 2002; Hutsler and Zhang, 2010). Importantly, such neurodevelopmental disorders are often associated with altered excitatory/inhibitory (E/I) balance, thus when testing the findings of the current study in vivo, it will be important to examine excitatory and inhibitory synapse development in parallel. Interestingly, there are a number of potential molecular targets that are thought to both limit glutamatergic synapse formation and regulate E/I balance, such as the SRGAP2s (Fossati et al., 2016; Schmidt et al., 2019) and the activity-regulated MEF2C (Harrington et al., 2016). These potential mechanisms should be investigated as the role of depolarizing GABAA transmission in synapse formation continues to be more finely dissected.
Numerous models of ASDs are associated with a delay in the depolarizing to hyperpolarizing shift in EGABA (He et al., 2014; Tyzio et al., 2014; Leonzino et al., 2016). Such a delayed transition to hyperpolarized EGABA is likely associated with a delay in the onset of adequate shunting inhibition when GABA is still depolarizing, which may increase glutamatergic synapse formation in a manner similar to that which we observed when blocking depolarizing GABAA transmission. Furthermore, mutation of the β3 GABAA receptor subunit, the expression of which peaks during development when GABA is depolarizing, has been observed in ASD (Menold et al., 2001; Buxbaum et al., 2002; Chen et al., 2014). The findings presented in the current study may provide a causal link between these mutations and the hyperconnectivity observed in ASDs. Thus, further investigation is required to understand if impairments of depolarizing GABAA transmission contribute to the lasting alterations of spines and synapses in these conditions. Finally, the possibility that GABA bidirectionally controls synapse formation may yield novel clinical approaches for correcting synaptic deficits in neurodevelopmental disorders.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
All procedures were performed in accordance to guidelines established by the Canadian Council on Animal Care and by the Montreal General Hospital Facility Animal Care Committee.
Author Contributions
CS and KM conceived of the project. CS, HP, SC, DS, CB, MW, and KM designed experiments. CS, HP, CG, SC, EJ, VM, MW, and WF performed and analyzed experiments. CS and KM wrote the manuscript.
Funding
This work was supported by the Canadian Institutes of Health Research (OGB 111152, PJT148569, 156247 to KM) and Natural Sciences and Engineering Research Council of Canada (482622 to MW and 408044-2011 and 69404 to KM). CS was supported through a CGS-M from the CIHR (201002GSM-222639-198785) and a Doctoral Award from the Fonds de Recherche du Québec-Santé (FF6-15D).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Edward Ruthazer for valuable input throughout the course of this study, as well as Dr. Andrew Greenhalgh and Andy YL Gao for a critical review of the manuscript. A version of this article has been released as a bioRxiv preprint at: https://doi.org/10.1101/742148 (Salmon et al., 2019).
References
Acton, B. A., Mahadevan, V., Mercado, A., Uvarov, P., Ding, Y., Pressey, J., et al. (2012). Hyperpolarizing GABAergic transmission requires the KCC2 C-terminal ISO domain. J. Neurosci. 32, 8746–8751. doi: 10.1523/JNEUROSCI.6089-11.2012
Akerman, C. J., and Cline, H. T. (2006). Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J. Neurosci. 26, 5117–5130. doi: 10.1523/JNEUROSCI.0319-06.2006
Akerman, C. J., and Cline, H. T. (2007). Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 30, 382–389. doi: 10.1016/j.tins.2007.06.002
Amaral, D., and Lavenex, P. (2007). “Hippocampal neuroanatomy,” in The Hippocampus Book, eds P. Andersen, R. Morris, D. Amaral, T. Bliss and J. O’Keefe (Oxford: Oxford University Press), 37–115.
Balkowiec, A., and Katz, D. M. (2002). Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J. Neurosci. 22, 10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002
Behar, T. N., Schaffner, A. E., Scott, C. A., Greene, C. L., and Barker, J. L. (2000). GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb. Cortex 10, 899–909. doi: 10.1093/cercor/10.9.899
Ben-Ari, Y. (2002). Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325. doi: 10.1113/jphysiol.1989.sp017762
Ben-Ari, Y., Gaiarsa, J.-L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Ben-Ari, Y., Khazipov, R., Leinekugel, X., Caillard, O., and Gaiarsa, J. (1997). GABAA, NMDA and AMPA receptors: a developmentally regulated ‘ménage à trois.’. Trends Neurosci. 20, 523–529. doi: 10.1016/s0166-2236(97)01147-8
Ben-Ari, Y., Woodin, M. A., Sernagor, E., Cancedda, L., Vinay, L., Rivera, C., et al. (2012). Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever!. Front. Cell. Neurosci. 6:35. doi: 10.3389/fncel.2012.00035
Buchs, P.-A., Stoppini, L., and Muller, D. (1993). Structural modifications associated with synaptic development in area CA1 of rat hippocampal organotypic cultures. Dev. Brain Res. 71, 81–91. doi: 10.1016/0165-3806(93)90108-m
Buxbaum, J. D., Silverman, J. M., Smith, C. J., Greenberg, D. A., Kilifarski, M., Reichert, J., et al. (2002). Association between a GABRB3 polymorphism and autism. Mol. Psychiatry 7, 311–316. doi: 10.1038/sj.mp.4001011
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007
Chancey, J. H., Adlaf, E. W., Sapp, M. C., Pugh, P. C., Wadiche, J. I., and Overstreet-Wadiche, L. S. (2013). GABA depolarization is required for experience-dependent synapse unsilencing in adult-born neurons. J. Neurosci. 33, 6614–6622. doi: 10.1523/JNEUROSCI.0781-13.2013
Chapleau, C. A., Larimore, J. L., Theibert, A., and Pozzo-Miller, L. (2009). Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J. Neurodev. Disord. 1, 185–196. doi: 10.1007/s11689-009-9027-6
Che, A., Babij, R., Iannone, A. F., Fetcho, R. N., Ferrer, M., Liston, C., et al. (2018). Layer I interneurons sharpen sensory maps during neonatal development. Neuron 99, 98.e7–116.e7. doi: 10.1016/j.neuron.2018.06.002
Chen, C.-H., Huang, C.-C., Cheng, M.-C., Chiu, Y.-N., Tsai, W.-C., Wu, Y.-Y., et al. (2014). Genetic analysis of GABRB3 as a candidate gene of autism spectrum disorders. Mol. Autism 5:36. doi: 10.1186/2040-2392-5-36
De Simoni, A., Griesinger, C. B., and Edwards, F. A. (2003). Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J. Physiol. 550, 135–147. doi: 10.1113/jphysiol.2003.039099
Drakew, A., Müller, M., Gahwiler, B., Thompson, S., and Frotscher, M. (1996). Spine loss in experimental epilepsy: quantitative light and electron miscroscopic analysis of intracellularly stained CA3 pyramidal cells in hippocampal slice cultures. Neuroscience 70, 31–45. doi: 10.1016/0306-4522(95)00379-w
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., et al. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213. doi: 10.1038/nm1301
El Marroun, H., White, T., Verhulst, F. C., and Tiemeier, H. (2014). Maternal use of antidepressant or anxiolytic medication during pregnancy and childhood neurodevelopmental outcomes: a systematic review. Eur. Child Adolesc. Psychiatry 23, 973–992. doi: 10.1007/s00787-014-0558-3
Fiala, J. C. (2005). Reconstruct: a free editor for serial section microscopy. J. Microsc. 218, 52–61. doi: 10.1111/j.1365-2818.2005.01466.x
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Rev. 39, 29–54. doi: 10.1016/s0165-0173(02)00158-3
Fiumelli, H., Briner, A., Puskarjov, M., Blaesse, P., Belem, B. J., Dayer, A. G., et al. (2012). An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb. Cortex 23, 378–388. doi: 10.1093/cercor/bhs027
Fossati, M., Pizzarelli, R., Schmidt, E. R., Kupferman, J. V., Stroebel, D., Polleux, F., et al. (2016). SRGAP2 and its human-specific paralog co-regulate the development of excitatory and inhibitory synapses. Neuron 91, 356–369. doi: 10.1016/j.neuron.2016.06.013
Garaschuk, O., Hanse, E., and Konnerth, A. (1998). Developmental profile and synaptic origin of early network oscillations in the CA1 region of rat neonatal hippocampus. J. Physiol. 507, 219–236. doi: 10.1111/j.1469-7793.1998.219bu.x
Ge, S., Goh, E. L. K., Sailor, K. A., Kitabatake, Y., Ming, G., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593. doi: 10.1038/nature04404
Griguoli, M., and Cherubini, E. (2017). Early correlated network activity in the hippocampus: its putative role in shaping neuronal circuits. Front. Cell. Neurosci. 11:255. doi: 10.3389/fncel.2017.00255
Gulledge, A. T., and Stuart, G. J. (2003). Excitatory actions of GABA in the cortex. Neuron 37, 299–309. doi: 10.1016/s0896-6273(02)01146-7
Haber, M., Zhou, L., and Murai, K. K. (2006). Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J. Neurosci. 26, 8881–8891. doi: 10.1523/JNEUROSCI.1302-06.2006
Hanse, E., Durand, G. M., Garaschuk, O., and Konnerth, A. (1997). Activity-dependent wiring of the developing hippocampal neuronal circuit. Semin. Cell Dev. Biol. 8, 35–42. doi: 10.1006/scdb.1996.0119
Harrington, A. J., Raissi, A., Rajkovich, K., Berto, S., Kumar, J., Molinaro, G., et al. (2016). MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife 5:e20059. doi: 10.7554/eLife.20059
He, Q., Nomura, T., Xu, J., and Contractor, A. (2014). The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 34, 446–450. doi: 10.1523/JNEUROSCI.4447-13.2014
Heaulme, M., Chambon, J. P., Leyris, R., Molimard, J. C., Wermuth, C. G., and Biziere, K. (1986). Biochemical characterization of the interaction of three pyridazinyl-GABA derivatives with the GABAA receptor site. Brain Res. 384, 224–231. doi: 10.1016/0006-8993(86)91158-3
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Irwin, S. A., Galvez, R., and William, T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044. doi: 10.1093/cercor/10.10.1038
Irwin, S. A., Patel, B., Idupulapati, M., Harris, J. B., Crisostomo, R. A., Larsen, B. P., et al. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 98, 161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b
Ji, Y., Lu, Y., Yang, F., Shen, W., Tang, T. T. T., Feng, L., et al. (2010). Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat. Neurosci. 13, 302–309. doi: 10.1038/nn.2505
Jourdain, P., Nikonenko, I., Alberi, S., and Muller, D. (2002). Remodeling of hippocampal synaptic networks by a brief anoxia—hypoglycemia. J. Neurosci. 22, 3108–3116. doi: 10.1523/JNEUROSCI.22-08-03108.2002
Kaila, K. (1994). Ionic basis of GABAA receptor channel function in the nervous system. Prog. Neurobiol. 42, 489–537. doi: 10.1016/0301-0082(94)90049-3
Kaufmann, W. E., and Moser, H. W. (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 10, 981–991. doi: 10.1093/cercor/10.10.981
Khalilov, I., Dzhala, V., Ben-Ari, Y., and Khazipov, R. (1999). Dual role of GABA in the neonatal rat hippocampus. Dev. Neurosci. 21, 310–319. doi: 10.1159/000017380
Khalilov, I., Minlebaev, M., Mukhtarov, M., and Khazipov, R. (2015). Dynamic changes from depolarizing to hyperpolarizing GABAergic actions during giant depolarizing potentials in the neonatal rat hippocampus. J. Neurosci. 35, 12635–12642. doi: 10.1523/JNEUROSCI.1922-15.2015
Khazipov, R., Leinekugel, X., Khalilov, I., Gaiarsa, J., and Ben-Ari, Y. (1997). Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J. Physiol. 483, 763–772. doi: 10.1113/jphysiol.1997.sp021900
Kirmse, K., Hübner, C. A., Isbrandt, D., and Witte, O. W. (2018). GABAergic transmission during brain development: multiple effects at multiple stages. Neuroscientist 24, 36–53. doi: 10.1177/1073858417701382
Kirmse, K., Kummer, M., Kovalchuk, Y., Witte, O. W., Garaschuk, O., and Holthoff, K. (2015). GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 6:7750. doi: 10.1038/ncomms8750
Lacey, D. J., and Terplan, K. (1987). Abnormal cerebral cortical neurons in a child with maternal PKU syndrome. J. Child Neurol. 2, 201–204. doi: 10.1177/088307388700200306
Lamsa, K. P., Palva, J. M., Ruusuvuori, E., Kaila, K., and Taira, T. (2000). Synaptic GABAA activation inhibits AMPA-kainate receptor—mediated bursting in the newborn (P0—P2) rat hippocampus. J. Neurophysiol. 83, 359–366. doi: 10.1152/jn.2000.83.1.359
Le Magueresse, C., Safiulina, V., Changeux, J.-P., and Cherubini, E. (2006). Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. J. Physiol. 576, 533–546. doi: 10.1113/jphysiol.2006.117572
Leinekugel, X., Tseeb, V., Ben-Ari, Y., and Bregestovski, P. (1995). Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. J. Physiol. 487, 319–329. doi: 10.1113/jphysiol.1995.sp020882
Leonzino, M., Busnelli, M., Antonucci, F., Verderio, C., Mazzanti, M., Chini, B., et al. (2016). The timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor report the timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 15, 96–103. doi: 10.1016/j.celrep.2016.03.013
Li, H., Khirug, S., Cai, C., Ludwig, A., Blaesse, P., Kolikova, J., et al. (2007). KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033. doi: 10.1016/j.neuron.2007.10.039
Liu, X., Wang, Q., Haydar, T. F., and Bordey, A. (2005). Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci. 8, 1179–1187. doi: 10.1038/nn1522
Megías, M., Emri, Z., Freund, T. F., and Gulyás, A. I. (2001). Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102, 527–540. doi: 10.1016/s0306-4522(00)00496-6
Menold, M. M., Shao, Y., Wolpert, C. M., Donnelly, S. L., Raiford, K. L., Martin, E. R., et al. (2001). Association analysis of chromosome 15 gabaa receptor subunit genes in autistic disorder. J. Neurogenet. 15, 245–259. doi: 10.3109/01677060109167380
Mohajerani, M. H., and Cherubini, E. (2005). Spontaneous recurrent network activity in organotypic rat hippocampal slices. Eur. J. Neurosci. 22, 107–118. doi: 10.1111/j.1460-9568.2005.04198.x
Muller, D., Buchs, P.-A., and Stoppini, L. (1993). Time course of synaptic development in hippocampal organotypic cultures. Dev. Brain Res. 71, 93–100. doi: 10.1016/0165-3806(93)90109-n
Oh, W. C., Lutzu, S., Castillo, P. E., and Kwon, H. B. (2016). De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 353, 1037–1040. doi: 10.1126/science.aaf5206
Owens, D. F., and Kriegstein, A. R. (2002). Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 3, 715–727. doi: 10.1038/nrn919
Park, H., and Poo, M. M. (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23. doi: 10.1038/nrn3379
Pérez-Gómez, A., and Tasker, R. A. (2013). Transient domoic acid excitotoxicity increases BDNF expression and activates both MEK- and PKA-dependent neurogenesis in organotypic hippocampal slices. BMC Neurosci. 14:72. doi: 10.1186/1471-2202-14-72
Puskarjov, M., Ahmad, F., Khirug, S., Sivakumaran, S., Kaila, K., and Blaesse, P. (2015). BDNF is required for seizure-induced but not developmental up-regulation of KCC2 in the neonatal hippocampus. Neuropharmacology 88, 103–109. doi: 10.1016/j.neuropharm.2014.09.005
Puskarjov, M., Fiumelli, H., Briner, A., Bodogan, T., Demeter, K., Lacoh, C., et al. (2017). K-Cl Cotransporter 2-mediated Cl− extrusion determine developmental stage-dependent impact of propofol mediated anesthesia on dendritic spines. Anesthesiology 126, 855–867. doi: 10.1097/aln.0000000000001587
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rodriguez, A., Ehlenberger, D. B., Dickstein, D. L., Hof, P. R., and Wearne, S. L. (2008). Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3:e1997. doi: 10.1371/journal.pone.0001997
Salmon, C. K., Pribiag, H., Farmer, W. T., Cameron, S., Jones, E. V., Mahadevan, V., et al. (2019). Depolarizing GABA transmission restrains activity-dependent glutamatergic synapse formation in the developing hippocampal circuit. bioRxiv [Preprint]. doi: 10.1101/596940
Schmidt, E. R., Kupferman, J. V., Stackmann, M., and Polleux, F. (2019). The human-specific paralogs SRGAP2B and SRGAP2C differentially modulate SRGAP2A-dependent synaptic development. bioRxiv [Preprint]. doi: 10.1101/74214810.1101/596940
Sedmak, G., Jovanov-Milošević, N., Puskarjov, M., Ulamec, M., Krušlin, B., Kaila, K., et al. (2016). Developmental expression patterns of KCC2 and functionally associated molecules in the human brain. Cereb. Cortex 26, 4574–4589. doi: 10.1093/cercor/bhv218
Sokal, D. M., Mason, R., and Parker, T. L. (2000). Multi-neuronal recordings reveal a differential effect of thapsigargin on bicuculline- or gabazine-induced epileptiform excitability in rat hippocampal neuronal networks. Neuropharmacology 39, 2408–2417. doi: 10.1016/s0028-3908(00)00095-2
Staley, K. J., and Mody, I. (1992). Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J. Neurophysiol. 68, 197–212. doi: 10.1152/jn.1992.68.1.197
Staley, K. J., and Proctor, W. R. (1999). Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3− transport. J. Physiol. 519, 693–712. doi: 10.1111/j.1469-7793.1999.0693n.x
Swann, J. W., Brady, R. J., and Martin, D. L. (1989). Postnatal development of GABA-mediated synaptic inhibition in rat hippocampus. Neuroscience 28, 551–561. doi: 10.1016/0306-4522(89)90004-3
Tripodi, M., Evers, J. F., Mauss, A., Bate, M., and Landgraf, M. (2008). Structural homeostasis: compensatory adjustments of dendritic arbor geometry in response to variations of synaptic input. PLoS Biol. 6:e260. doi: 10.1371/journal.pbio.0060260
Tyler, W. J., and Pozzo-Miller, L. (2003). Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J. Physiol. 553, 497–509. doi: 10.1113/jphysiol.2003.052639
Tyssowski, K. M., DeStefino, N. R., Cho, J.-H., Dunn, C. J., Poston, R. G., Carty, C. E., et al. (2018). Different neuronal activity patterns induce different article different neuronal activity patterns induce different gene expression programs. Neuron 98, 530.e11–546.e11. doi: 10.1016/j.neuron.2018.04.001
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Valeeva, G., Tressard, T., Mukhtarov, M., Baude, A., and Khazipov, R. (2016). An optogenetic approach for investigation of excitatory and inhibitory network GABA actions in mice expressing channelrhodopsin-2 in GABAergic neurons. J. Neurosci. 36, 5961–5973. doi: 10.1523/jneurosci.3482-15.2016
Vanhatalo, S., Palva, J. M., Andersson, S., Rivera, C., Voipio, J., and Kaila, K. (2005). Slow endogenous activity transients and developmental expression of K+-Cl− cotransporter 2 in the immature human cortex. Eur. J. Neurosci. 22, 2799–2804. doi: 10.1111/j.1460-9568.2005.04459.x
Vicario-Abejón, C., Collin, C., McKay, R. D., and Segal, M. (1998). Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J. Neurosci. 18, 7256–7271. doi: 10.1523/jneurosci.18-18-07256.1998
Vicario-Abejón, C., Owens, D., McKay, R., and Segal, M. (2002). Role of neurotrophins in central synapse formation and stabilization. Nat. Rev. Neurosci. 3, 965–974. doi: 10.1038/nrn988
Wang, D. D., and Kriegstein, A. R. (2008). GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 28, 5547–5558. doi: 10.1523/jneurosci.5599-07.2008
Wang, D. D., and Kriegstein, A. R. (2009). Defining the role of GABA in cortical development. J. Physiol. 587, 1873–1879. doi: 10.1113/jphysiol.2008.167635
Wang, D. D., and Kriegstein, A. R. (2011). Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb. Cortex 21, 574–587. doi: 10.1093/cercor/bhq124
Wells, J. E., Porter, J. T., Agmon, A., and Virginia, W. (2000). GABAergic inhibition suppresses paroxysmal network activity in the neonatal rodent hippocampus and neocortex. J. Neurosci. 20, 8822–8830. doi: 10.1523/jneurosci.20-23-08822.2000
Yamada, J., Okabe, A., Toyoda, H., Kilb, W., Luhmann, H. J., and Fukuda, A. (2004). Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 557, 829–841. doi: 10.1113/jphysiol.2004.062471
Zeng, L., Xu, L., Rensing, N. R., Sinatra, P. M., Rothman, S. M., and Wong, M. (2007). Kainate seizures cause acute dendritic injury and actin depolymerization in vivo. J. Neurosci. 27, 11604–11613. doi: 10.1523/jneurosci.0983-07.2007
Keywords: synapse formation, hippocampus, GABA transmission, dendritic spines, chloride homeostasis, KCC2, circuit development, autism
Citation: Salmon CK, Pribiag H, Gizowski C, Farmer WT, Cameron S, Jones EV, Mahadevan V, Bourque CW, Stellwagen D, Woodin MA and Murai KK (2020) Depolarizing GABA Transmission Restrains Activity-Dependent Glutamatergic Synapse Formation in the Developing Hippocampal Circuit. Front. Cell. Neurosci. 14:36. doi: 10.3389/fncel.2020.00036
Received: 29 September 2019; Accepted: 05 February 2020;
Published: 25 February 2020.
Edited by:
Kimberly M. Huber, UT Southwestern Medical Center, United StatesReviewed by:
Shiva K. Tyagarajan, University of Zurich, SwitzerlandKnut Holthoff, Friedrich Schiller University Jena, Germany
Copyright © 2020 Salmon, Pribiag, Gizowski, Farmer, Cameron, Jones, Mahadevan, Bourque, Stellwagen, Woodin and Murai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher K. Salmon, Y2hyaXMuc2FsbW9uQG1haWwubWNnaWxsLmNh; Keith K. Murai a2VpdGgubXVyYWlAbWNnaWxsLmNh