Howard Hughes Medical Institute, Department of Psychiatry and Behavioral Sciences, Yerkes National Primate Research Center, Emory University, Atlanta, GA, USA
The use of conventional knockout technologies has proved valuable for understanding the role of key genes and proteins in development, disease states, and complex behaviors. However, these strategies are limited in that they produce broad changes in gene function throughout the neuroaxis and do little to identify the effects of such changes on neural circuits thought to be involved in distinct functions. Because the molecular functions of genes often depend on the specific neuronal circuit in which they are expressed, restricting gene manipulation to specific brain regions and times may be more useful for understanding gene functions. Conditional gene manipulation strategies offer a powerful alternative. In this report we briefly describe two conditional gene strategies that are increasingly being used to investigate the role of genes in behavior – the Cre/loxP recombination system and lentiviral vectors. Next, we summarize a number of recent experiments which have used these techniques to investigate behavior after spatial and/or temporal and gene manipulation. These conditional gene targeting strategies provide useful tools to study the endogenous mechanisms underlying complex behaviors and to model disease states resulting from aberrant gene expression.
The use of conventional knockout (KO) technologies has provided valuable information in identifying the role of key genes in neural development and plasticity. While these methods have been of value, they are often accompanied by a number of limitations that can restrict their usefulness. For example, if a gene of interest is essential for development, then gene KO can lead to gross developmental abnormalities or lethality that either preclude or complicate studies on adult animals (e.g., Asada et al., 1997
). Developmental defects and compensatory changes caused by mutations often make it difficult to assess whether phenotypic consequences result from the lack of normal gene expression in the adult animal or the lack of the normal expression during development. In addition, conventional methods eliminate the gene of interest in peripheral tissues as well as the central nervous system, and thus are not limited to alterations of brain molecular functions. Similarly, KO strategies produce broad changes in gene function throughout the neuroaxis and do little to identify the effects of such changes on specific brain regions or neural circuits thought to be involved in distinct functions. As a consequence, the complete deletion of a specific protein throughout the nervous system may prove ineffective toward understanding fine molecular processes in higher brain functions.
To overcome these drawbacks, researchers are increasingly making use of conditional gene manipulation strategies, which have the advantage of allowing temporal and spatial deletion of a gene of interest (Gaveriaux-Ruff and Kieffer, 2007
). One such approach is the Cre/loxP recombination system. The Cre/LoxP technique was first reported by Rajewsky and colleagues to KO the pol beta gene that encodes the DNA polymerase beta in T lymphocytes (Gu et al., 1994
), but has since been utilized as a powerful and popular approach to understanding the genetic and molecular basis of higher brain functions and their role in animal behavior models. Generally, the system requires crossbreeding of two lines of mice. One line carries a transgene that encodes the Cre recombinase gene under the control of a cell-specific promoter. Cre recombinase is an enzyme that catalyzes the site-specific, irreversible cleavage of DNA segments flanked by unique loxP sequences. The second line carries the target gene of interest bordered by the two requisite loxP recognition sites (Sauer and Henderson, 1988
; Tsien et al., 1996
). Each loxP site consists of two 13-bp palindromic sequences flanking an 8-bp core sequence. They are typically located in intronic sequences bordering exon(s) of the target gene to preserve gene transcription prior to deletion. Mice homozygous for this conditional ‘floxed’ (flanked by loxP) allele display a wild-type phenotype in the absence of Cre recombinase. However, crossbreeding with a Cre-expressing mouse line produces a null allele after Cre-mediated recombination.
In the Cre recombinase mouse, the spatio-temporal pattern of Cre activity can be assessed in vivo by using traditional immunohistochemistry or in situ hybridization for Cre recombinase or using sensitive Cre reporter mouse lines. Many reporter strains use different promoters to express lacZ, green fluorescent protein (GFP) or other detectible proteins in the presence of Cre recombinase (van der Neut, 1997
; Lobe et al., 1999
; Mao et al., 2001
; Srinivas et al., 2001
; Morozov et al., 2003
; Morozov, 2008
). It is important to note that there is great variability between the sensitivity of these reporter lines. If the reporter protein is not expressed in a particular cell-type, it does not prove that Cre is not expressed or that it would not be active on a different gene locus. Despite these limitations, this approach has proved quite successful in a large number of studies to date.
One commonly used reporter is the floxed-stop lacZ reporter mice (Soriano, 1999
).These mice possess a transgene integrated into the ROSA26 locus which consists of a floxed-stop sequence upstream of a lacZ gene (Gardner et al., 1996
; Takahashi et al., 2000
). When these RosaLacZ mice are crossed with Cre-expressing transgenic mice, the Cre-mediated excision of the floxed-stop sequence leads to lacZ expression. The expression of LacZ in these animals serves as an extremely sensitive measure, with very low background expression levels. In these animals, Cre expression can be detected by beta galactosidase (β-gal) immunohistochemistry, utilization of the β-gal colorimetric substrate X-gal, or by in situ hybridization.
In recent years numerous mouse lines that carry a Cre recombinase transgene have been engineered (see, Morozov et al., 2003
). Cre-expressing lines can be made both with traditional transgene technology as well as with knock-in lines that use the homologous recombination technique. In this case a Cre cassette is placed downstream of the endogenous gene promoter at the specific locus of that gene. In mice engineered using such an approach, the expression of Cre is consistent with what is known about the spatio-temporal expression pattern of the endogenous gene (e.g., Zhuang et al., 2005
). A number of studies investigating learning and memory in mice have utilized the Emx1-Cre and the CaMKII-Cre transgenic knock-in lines (Guo et al., 2000
; Casanova et al., 2001
). Both Emx1 and CaMKII-promoter driven Cre expression cause effective deletion in the cerebral cortex, hippocampus and other forebrain regions. When these transgenic Cre-expressing mice are crossed with Cre-sensitive reporter lines, such as the Rosa26 line containing lacZ flanked by LoxP, the high-efficiency and specificity of the expression pattern is observed. When these Cre-expressing mice are crossed with such ‘floxed’ mice, the targeted gene is effectively deleted throughout the cerebral cortex, hippocampus and other forebrain regions that are specific to the promoter driving Cre. Although there are many advantages to these knock-in approaches, there are also several drawbacks. One limitation is the risk of haplo-insufficiency when Cre recombinase replaces the gene of interest, thus leading to mice that express only a single allele of the targeted gene. One way of solving this issue has been to utilize the ‘internal ribosomal entry site’ (IRES) sequence, which was originally described from the picornavirus (Zhu et al., 2001
). This sequence, when placed between two coding sequences of DNA allows for co-expression of both genes off of a single transcribed mRNA. Thus, a knock-in may place an IRES-Cre or other expression marker downstream of the gene of interest, which in some cases solves the issue of haplo-insufficiency allowing equivalent expression of both genes from the single endogenous promoter.
Alternatively, a line can be engineered by pronuclear injection of a fusion construct in which the Cre coding sequence is inserted downstream of a cell-specific promoter. In this approach, the transgene will be integrated into the genome randomly, thus the pattern of Cre expression, with some insertion events, may be somewhat different from that of the promoter’s endogenous gene pattern. Of note, these transgenic approaches can result in more than one insertion event as well as position effects on gene expression specificity and efficiency. Homologous recombination, ‘Knock-in’ strategies are significantly more effortful to produce, but are generally more assured of the expected expression pattern.
With Cre transgenic lines, as with Cre knock-in lines, genetic excision occurs as soon as the promoter driving Cre expression is active and may trigger developmental compensations if the promoter is active during embryogenesis. As an example, Chhatwal et al. (2007)
recently engineered a Cre-expressing line by subcloning the Cre coding sequence downstream of a promoter of the gene for cholecystokinin (CCK). The resulting plasmid (CCID) was linearized, purified and microinjected into the pronuclei of one-cell embryos, which were then implanted into pseudo-pregnant females to create a number of transgenic CCK-Cre driver mouse lines. When these animals were mated to Rosa-LacZ reporter mice, animals carrying both the CCID and Rosa-LacZ transgenes show varying expression patterns of β-gal in areas where endogenous CCK is expressed Figure 1
. Similarly, when the neocortex-specific CCID transgenic line was crossed with a floxed-stop GFP reporter line (Mao et al., 2001
; Muzumdar et al., 2007
), animals carrying both transgenes show GFP expression (limited to the neocortex with no expression in other areas like the hippocampus, thalamus, striatum, hypothalamus or amygdala; Figure 1
).
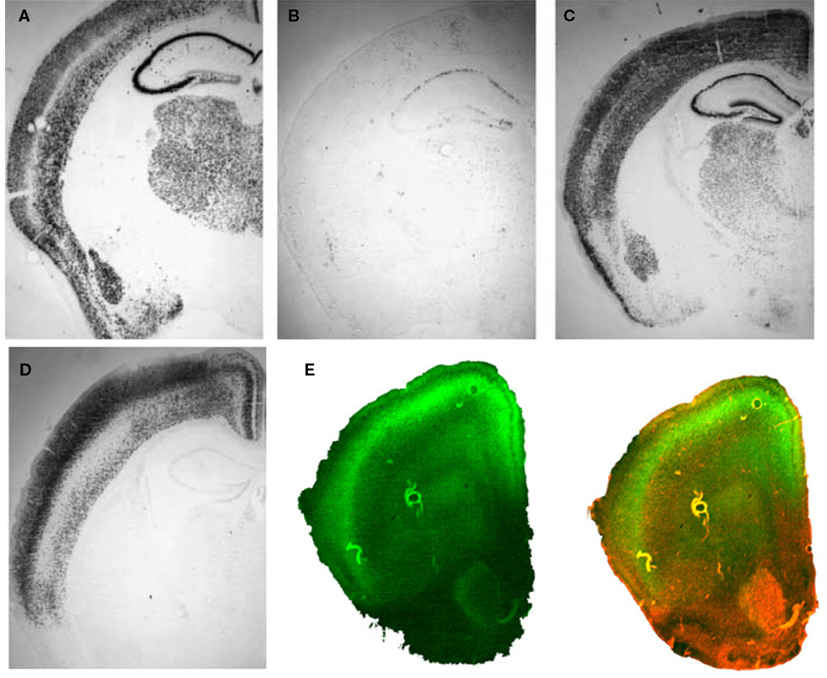
Figure 1. Cre recombinase expressing transgenic mice. A DNA template in which the coding sequence for Cre recombinase was placed under the control of the CCK promoter (CCID) was used to create transgenic mouse lines. Three founder animals, with distinct transgene insertion sites, were generated and bred to RosaLacZ animals to assess Cre expression as revealed by LacZ staining. (A) Normal patterns of CCK mRNA expression, revealed with in situ hybridization for CCK mRNA. (B) Animals from transgene line A show low levels of Cre expression, but in appropriate CCK-specific regions. (C) Animals from line B relatively widespread expression, which was, with the exception of the dentate, similar to endogenous CCK mRNA expression. (D) Animals from line C showed high levels of Cre expression in the cortex, in a manner that was qualitatively similar to observed patterns of CCK mRNA expression but with virtually no Cre expression in hippocampus or subcortical regions. (E) Animals from transgenic line C were crossed with Td tomato/floxed stop GFP, to assess Cre mediated GFP expression. These mice express strong red fluorescence in all tissues and cell types examined. When bred to Cre recombinase expressing mice, the resulting offspring have the stop cassette deleted in the Cre expressing tissue, deleting Td tomato expression and allowing expression of the membrane-targeted GFP (red = Td tomato, green = eGFP, blue = DAPI; Left – GFP expression, Right – merged GFP + TdTomato expression). Panels (A–D) from Chhatwal et al. (2007)
.
Because the molecular functions of proteins often depend on the specific neuronal circuit in which they are expressed, restricting gene manipulation to specific regions and time may be a more useful in understanding the function of genes in some complex behaviors. In such cases, the use of lentiviral vectors to deliver transgenes is a powerful tool. Lentivirus vectors integrate into both dividing and post-mitotic cells, generate little or no immune response, and express transgenes stably over several months (Naldini et al., 1996
; Lai and Brady, 2002
). Currently used HIV-derived lentiviruses contain only the necessary elements for gene transduction and include self-inactivating elements, which significantly improve the biosafety of vectors and greatly reduces the formation of replication competent viruses (Zufferey et al., 1998
; Abordo-Adesida et al., 2005
). Furthermore, in comparison to adeno-associated- and sindbis-based vectors, lentiviral vectors have a larger insert capacity. Thus, large transgenes can be subcloned into lentivirus vectors enabling the expression of genes encoding neurotransmitters (Azzouz et al., 2002
), neuropeptides (Keen-Rhinehart et al., 2009
), mutated receptors (Rattiner et al., 2004a
; Chhatwal et al., 2006
), small interfering RNAs (Tiscornia et al., 2003
), and promoters for the use of transgene expression in selected populations of cells (Chhatwal et al., 2007
). Lastly, lentiviral vectors, as compared to transgenic and knock-in mouse approaches, provide researchers with the choice of using rats or a variety of mouse strains at different ages. Note that age of the animal at the time of gene deletion may be particularly important in particular experimental designs or models.
For many of these reasons, the use of lentiviral vectors to deliver Cre recombinase is a useful alternative to the use of transgenic Cre-expressing mouse lines. Cre-expressing lentiviruses can be injected in a localized fashion directly into the brain of floxed mice, and the extent of the conditional genetic deletion will be restricted to the area of viral spread. For example, viral vectors used in a number of labs are derived from the lentivirus backbone pLV-CMV-GFP-U3Nhe (Naldini et al., 1996
; Tiscornia et al., 2003
; Jasnow et al., 2009
), which allows for virally mediated expression of GFP (LV-GFP) driven by a cytomegalovirus (CMV) promoter. For manipulation of gene expression using floxed mice, a Cre-recombinase expressing viral vector (LV-Cre) can be created by replacing the GFP coding sequence with the coding sequence for Cre-recombinase (Heldt et al., 2007
). In a typical experiment, mice are injected intra-cranially into specific brain areas with 1–2 μl of virus bilaterally, and allowed to recover for 7–14 days for stable integration and expression of the transgene within the host genome. Figure 2
shows the ability of LV-Cre to efficiently remove the floxed-stop sequences upstream of lacZ in the Rosa-LacZ reporter mice (129S-Gt(ROSA)26Sortm1Sor/J, Soriano, 1999
). LV-Cre injected into the striatum (Figures 2
A,B) or CA1/dentate gyrus (DG) region of the dorsal hippocampus (Figures 2
C–E) induces strong production of LacZ with no leakage in areas not infected with virus.

Figure 2. In vivo validation of LV-Cre lentivirus. Robust Cre recombinase expression as labeled by LacZ when LV-Cre is injected into RosaLacZ reporter mice. Sections were processed for lacZ histochemistry with β-gal 14 days after LV-Cre injections to visualize Cre-dependent recombination in vivo in the (A,B) striatum and (C,E) dorsal hippocampus seen at ×10 and ×20 magnification. LV-Cre-infected cells within (D) CA1 and (E) DG, illustrating the dense β-gal and intact morphology of hippocampal neurons.
Although this review is devoted to lentiviral vector approaches, we will say a few words about adeno-associated vectors and some of the reasons why one might choose one over the other system for genetic manipulation. Generally, the lentiviral vector system utilizes a significantly larger insert size, and is thus the optimal choice when larger gene inserts, more than one gene (poly-cistronic cassettes), and longer promoters are of primary importance. Additionally, lentiviruses are somewhat easier to concentrate and there is some evidence that they express for the lifetime of the cell/animal. In contrast AAV are easier to handle (do not require same level of BSL safety due to their not being a retrovirus-based backbone), and in some cases higher titers are possible with AAV. Several manuscripts have been published recently outlining important differences between AAV and lentiviral vectors for neuroscience applications (Nathanson et al., 2009
; Towne and Aebischer, 2009
; van den Pol et al., 2009
).
Many of the approaches discussed above have also been nicely demonstrated with AAV, along with several recent exciting innovations. For example novel combinations of Cre recombinase and channel rhodopsin (see below) have been used with AAV vectors to ‘tag’ neuronal populations for identification during in vivo electrophysiological recording (Lima et al., 2009
). Another novel use of Cre in an AAV system used a modified estrogen-inducible Cre recombinase that served as an in vivo ‘molecular switch’, allowing spatial and temporal control of transgene expression, thereby potentially increasing the safety of gene therapy (Li et al., 2006
). Additionally, the use of Cre-activated transgenes with AAV provide an additional assortment of possibilities for gene manipulation. For example, the ‘rAAV-FLEX-rev’ delivery approach has been demonstrated to lead to the expression of transgenes in a temporally and spatially restricted manner, using any Cre driver line as the template. (Atasoy et al., 2008
). Overall, as with lentiviruses, AAV vectors allow for rapid subcloning and engineering of novel approaches to manipulate gene expression within discrete brain regions and neuronal cell types.
Here we will illustrate several different in vivo uses of these vectors in approaching neuroscience questions. Lentiviruses have been particularly useful in demonstrating the role brain-derived neurotrophic factor (BDNF) and its receptor tyrosine kinase B (TrkB) in learning and memory, including the acquisition and extinction of conditioned fear. Although its functions in the adult brain are not entirely clear, BDNF undoubtedly plays a role in development, trophic support, neural plasticity, and neuroprotection after some brain insults (Lindvall et al., 1994
). There is increasing evidence that BDNF exerts acute effects on synaptic transmission and participates in long-term potentiation (LTP), an event related to memory processes (Patterson et al., 1992
; Korte et al., 1995
; Thoenen, 1995
; McAllister, 1999
).
However, understanding the role of the central BDNF-TrkB system in vivo within behaviorally relevant learning paradigms has been difficult (Minichiello et al., 2002
). In part, this difficulty is due to the lack of specific pharmacological antagonists for the TrkB receptor. In addition, homozygous BDNF KO mice display profound developmental abnormalities and often die prior to their third postnatal week (Ernfors et al., 1994
). Heterozygous BDNF KO mice are viable but develop with roughly half of wild-type BDNF levels throughout the brain which likely leads to alterations in development that alter normal functioning (Conover and Yancopoulos, 1997
). Given their time- and spatially limited gene expression characteristics, lentiviruses can be used to manipulate the BDNF-TrkB system after normal development, and thus, provide a more accurate assessment for the role of BDNF plasticity in learning and memory.
The mammalian hippocampus is the brain region containing the highest BDNF mRNA and protein levels (Conner et al., 1997
). BDNF is known to have a functional role in the neuronal plasticity associated with hippocampal LTP (Korte et al., 1995
). Although hippocampal circuits are quite plastic and clearly involved in learning and memory, the contribution of these circuits to the behaviors under study is often unclear. Correlational studies show that BDNF is increased in its expression following hippocampal dependent tasks (Hall et al., 2000
; Mizuno et al., 2000
). Genetic manipulation studies suggest that animals with decreased levels of BDNF or its receptor, TrkB, are deficient in behavioral tasks thought to be hippocampally dependent (Linnarsson et al., 1997
; Minichiello et al., 1999
; Gorski et al., 2003
; Monteggia et al., 2004
; Duman and Monteggia, 2006
). However, all of these studies are limited, in that none of them examined the effects of BDNF deletion only within the hippocampus of adults while sparing BDNF in other forebrain regions. Having deletions of BDNF limited to only the hippocampus and only during a time-limited period in adulthood may also lead to a number of differences compared with the developmental and spatially broader BDNF deletions studied earlier.
To address this issue, we examined the consequence of hippocampus-specific BDNF deletion in adult animals on the acquisition and extinction of fear, object recognition memory, and a task strongly dependent on hippocampal functioning – the Morris water maze (MWM, Morris et al., 1982
). Hippocampal BDNF was deleted by performing bilateral injections of LV-Cre into the dorsal hippocampus of BDNF-floxed mice (Heldt et al., 2007
). Initial verification of this approach was performed with unilateral injections followed 14 days later with in situ hybridization for the Cre-recombinase or BDNF mRNA. Figure 3
A reveals BDNF mRNA expression, which was specifically deleted within the hippocampus where Cre recombinase was now expressed.
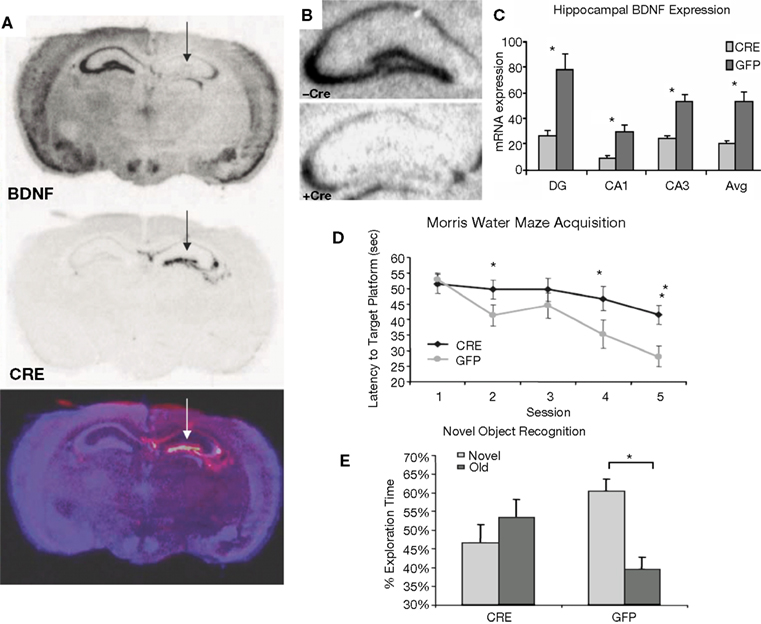
Figure 3. Inducible BDNF deletion in hippocampus with Cre lentivirus. (A) BDNF (top) and Cre-recombinase (middle) mRNA expression visualized with in situ hybridization 2 weeks after LV-Cre infection into BDNF-floxed mice. The bottom figure represents a pseudocolor overlay of the two in situ sections, demonstrating that Cre, but not BDNF, is now expressed where BDNF was previously expressed. (B) Qualitative figure showing BDNF in situ hybridization of dorsal hippocampus following a sham injection (top, −Cre) or following LV-Cre injection (bottom, +Cre). (C) Relative mRNA expression in dentate gyrus (DG), and CA1 and CA3 regions and the average of all regions (Avg) of dorsal hippocampus in LV-Cre (Cre)- or LV-GFP (GFP)-infected mice. (D) Morris water maze acquisition, measured as the average latency to find the platform over daily sessions of training. LV-Cre-infected mice demonstrated significantly slower acquisition (impairment) compared with LV-GFP-infected controls. (E) Percent of time spent exploring the new vs. old object during the test day for novel object recognition. LV-GFP-infected mice spent significantly more time exploring the novel compared to the previously habituated object. The LV-Cre-infected animals did not differentiate between the two, demonstrating their impairment on this task. Panels adapted from Heldt et al. (2007)
.
In animals used for behavioral tests, both qualitative and quantitative analyses of mRNA expression levels reveal that the bilateral deletion of BDNF in the mice was largely limited to the hippocampus. Figure 3
B shows the qualitative decrease in BDNF expression within the dorsal hippocampus following LV-Cre infection. Quantitative analyses of levels of BDNF revealed that, with reference to GFP control animals, the relative expression level of remaining BDNF within the dorsal hippocampus of LV-Cre-infected animals was 35, 30 and 45% for DG, CA1 and CA3, respectively (Figure 3
C).
In the MWM, BDNF deletion within dorsal hippocampus caused deficits in escape latencies during acquisition as well as during probe trials when compared to the performance of control animals. BDNF hippocampal deletions also reduced the amount of time spent with the novel compared to the habituated object in the novel object recognition task (Figures 3
D,E). Past studies indicate heterozygous and forebrain-specific BDNF KO mice likewise have deficits in MWM learning and other hippocampal dependent tasks (Mizuno et al., 2000
; Gorski et al., 2003
). However, these KO lines lack BDNF during development throughout cortical and subcortical areas as well as hippocampus. Together these data support the concept that BDNF expression in adults is required for normal acquisition and expression of spatial learning tasks, and that these previously reported effects were not due to developmental alterations or long-term BDNF effects on hippocampal neuronal survival or morphology.
Interestingly, hippocampal BDNF deletion also disrupted the extinction of conditioned fear without effecting the initial acquisition of fear. LV-Cre and LV-GFP mice given tone CS + footshocks during Pavlovian fear training showed equivalent levels of conditioned fear during testing. However, when groups were subsequently tested for fear over the course of next 6 days, LV-Cre mice displayed less fear extinction than controls, suggesting that hippocampal BDNF is required for the neural plasticity underlying the acquisition or consolidation of extinction memories (Figure 4
). Impaired extinction resulting from decreased hippocampal BDNF offers new avenues for understanding emotion regulation in patients with anxiety disorders.

Figure 4. Impaired fear extinction in mice with hippocampus-specific BDNF deletions. (A) Acquisition of cue conditioned fear, as measured with freezing after the onset of the auditory CS, during the conditioned fear acquisition session. There was no difference in acquisition of fear in animals receiving bilateral hippocampal injections of LV-Cre or LV-GFP. (B) Animals were tested within the same context in which training occurred for the presence of contextual fear as measured with freezing. There was no difference between the groups on level of contextual fear. (C,D) Extinction of conditioned fear is impaired in mice with dorsal hippocampus BDNF deletions. (C) Percent FPS is graphed for the first post-fear training test (pre-extinction) vs. the last test (post-extinction). Mice infected with LV-GFP demonstrate significant decreases in their level of conditioned fear as measured with %FPS compared with LV-Cre-infected mice. (D) Impaired extinction of fear, measured with %FPS, is stable across multiple testing sessions. Panels adapted from Heldt et al. (2007)
.
In addition to the hippocampus and mPFC, it is well established that fear conditioning is dependent on the basolateral amygdala (BLA) (Davis et al., 1993
; Fanselow and LeDoux, 1999
). Previous findings using in situ hybridization have revealed temporally specific increases in BDNF mRNA in the BLA after associative fear conditioning but not after exposure to an equal number of CS-alone or US alone presentations (Ressler et al., 2002
; Rattiner et al., 2004a
,b
). Fear conditioning also results in activation of the TrkB receptor in the amygdala, as indicated by increased receptor phosphorylation during consolidation period (Rattiner et al., 2004a
). In addition to the increased production of amygdala BDNF mRNA during the consolidation of fear conditioning, levels of BDNF mRNA transcripts are also increased following the extinction of conditioned fear (Chhatwal et al., 2006
).
TrkB is a classical receptor tyrosine kinase, which is activated by BDNF binding, leading to receptor homodimerization, transphosphorylation and subsequent activation for downstream signaling required for neuroplasticity processes. The homodimerization of TrkB makes them particularly amenable to study with dominant-negative truncated recombinant proteins (Klein et al., 1991
). Truncated TrkB receptors have extracellular domains which are identical to the full-length receptors, allowing for ligand binding, but also contain a shortened intracellular domain that inhibits BDNF signaling by blocking Trk autophosphorylation (Saarelainen et al., 2000
; Haapasalo et al., 2001
). Truncated TrkB receptor genes can be delivered in a spatial, temporal, and discrete manner using lentiviral vectors, and in turn, these vectors can be used to evaluate the role of central BDNF-TrkB mechanisms in behaviorally relevant learning paradigms.
The use of a viral gene-delivery system to express a dominant-negative truncated TrkB receptor (TrkB.t1) has been successfully employed to examine the role of BLA TrkB receptors in the acquisition and consolidation of conditioned fear and fear extinction, as assessed by fear-potentiated startle (FPS, Chhatwal et al., 2006
). For the acquisition of fear, this involved giving rats bilateral infusions of a lentivirus expressing TrkB.t1 (LV-TrkB.T1) or LV-GFP into the BLA 2 weeks before training and testing. To examine whether acquisition deficits were due to impaired expression, a second group of animals received BLA infusions of LV-TrkB.t1 or LV-GFP 4 days after training (Rattiner et al., 2004a
). This ability to manipulate gene expression before vs. after the learning paradigm illustrates one of the strengths of viral vector approaches.
To confirm the applicability of these vectors in vivo, animals injected with LV-TrkB.t1 or LV-GFP were sacrificed after behavioral procedures. In LV-GFP infected rats, a large number of GFP-positive cells were identified in the amygdala, whereas rats infected with LV-TrkB.t1 showed a large number of TrkB.t1-positive cells in the amygdala, indicating that cells in the amygdala were successfully infected with these lentiviruses (Figures 5
A–C). As seen in Figure 5
D, pretraining infusion of LV-TrkB.t1 caused impaired fear learning when animals were tested 48 h after the last training session. In contrast, post-training infusion of LV-TrkB.t1 produced no significant deficits (Figure 5
E), indicating that genetic blockade of the TrkB receptors did not prevent the expression of fear.
To examine the role of amygdala TrkB mechanisms in the formation of extinction memories, rats were fear-conditioned and matched into two groups with equivalent levels of conditioned fear before virus infusion of LV-GFP or LV-TrkB.t1 into the BLA bilaterally. When tested 2 days after extinction training, rats infected with the TrkB.t1 dominant-negative virus expressed more fear than those with the GFP virus, suggesting that blockade of the TrkB receptors prevented the retention of extinction memories (Figure 5
F).

Figure 5. Effect of TrkB.T1 in the amygdala on the acquisition of conditioned fear and extinction. (A–C) Histological examination of viral infection in the BLA following behavioral studies. (A) No amygdala damage was seen following infection as visualized with Cresyl violet staining. LA, lateral amygdala; CeA, central amygdala. (B) Expression of TrkB.t1 was assessed using immunocytochemistry (ICC) for a hemagglutinin (HA) epitope tag incorporated into the TrkB.t1 coding sequence. (C) GFP expression directly visualizing under an epifluorescence microscope (scale bar, 1 mm). Level of fear-potentiated startle (FPS) following (D,E) acquisition and (F,G) extinction of fear. (D) Mean startle amplitude on startle-alone trials, light-startle trials, and the difference between the two are shown for animals receiving lentivirus infusion into the amygdala. Mean difference scores of LV-TrkB.T1-infused animals were significantly lower than difference scores of LV-GFP-infused animals. (E) Effect of amygdala infection with LV-TrkB.T1 on the expression of fear-potentiated startle. When LV-TrkB.T1 is present during expression, but not acquisition, of fear learning there is no difference between FPS with LV-TrkB.T1 animals compared with LV-GFP animals. (F) Averaging across all trials, TrkB.t1-infected rats showed a deficit in extinction as compared to GFP-infected rats. (G) Examining extinction within the testing session suggested that the TrkB.t1-infected rats had normal within-session extinction, but lacked extinction retention across the 2-day interval between tests TrkB.t1 and GFP groups. Panels (A–C, F,G) adapted from Chhatwal et al. (2006) and panels (D,E) from Rattiner et al. (2004a)
.
A separate cohort of animals was used to determine whether the extinction deficit in LV-TrkB.t1 rats was a result of the deficiencies in initial encoding or the consolidation of extinction memories. After fear-conditioned, matching and viral surgery, rats were tested for fear potentiated startle over multiple days to measure both within- and between session extinction of fear (Figure 5
G). Testing revealed no difference between the groups in the rate of within-session extinction as observed by the decreased levels of FPS across test trials. However, rats expressing TrkB.t1 demonstrated significantly less extinction across several days of extinction testing, as compared to LV-GFP rats which showed marked extinction across days. This suggests that these rats may initially encode the extinction memory within each session but are not able to consolidate it. Together, these data suggest that the TrkB receptor is not involved in the process of within-session encoding of fear extinction, but instead is required for the normal consolidation of extinction as measured during the retention test.
In addition to using lentiviral vectors to KO the gene or inhibit the protein of interest, lentiviruses can also over-express a specific protein as a means of elucidating protein function. Constitutive gene expression may be particular useful to model disease states resulting from an overactive gene or as a means of gene replacement for recovery of function experiments. An example of this application is contained in a recent study which sought to over-express corticotropin-releasing factor (CRF) in an effort to model physiological and behavioral changes observed in stress-related pathologies (Keen-Rhinehart et al., 2009
). Past research suggests that CRF is upregulated in central nucleus of the amygdala (CeA) during chronic stress (Makino et al., 1999
) and is related with the upregulation of adrenocorticotropic hormone release from the pituitary which is crucial for the adaptation to prolonged exposure to stressors (Herman et al., 2003
). A dysregulation of this control may be involved in the development of a maladaptive response to chronic stress and may produce many of the alterations observed in several stress-induced pathologies including PTSD, anxiety, and depression (Arborelius et al., 1999
; Swaab et al., 2005
). Transgenic mice that over-express CRF indeed exhibit HPA dysregulation (Groenink et al., 2002
). However, this approach likely produces developmental compensation within the system that does not likely mimic the consequences of region-specific increased CRF expression resulting from chronic stress. Using lentiviral vectors, Keen-Rhinehart et al. (2009)
recently examined whether constitutive expression of CRF in the CeA of rats produced changes in the regulation of the HPA axis and behaviors shown previously to be indicative of chronic stress.
In this study, a lentivirus construct was engineered to over-express CRF by subcloning the CRF cDNA sequence downstream of the constitutively active CMV promoter. After injections of either the LV-CRF or LV-GFP control virus, female rats were administered a battery of tests, including the Porsolt forced swim test and the acoustic startle response which is elevated in fear and anxiety states (Davis et al., 1993
). The dexamethasone (DEX) suppression test was conducted to assess the consequences of increased CRF release from CeA on glucocorticoid negative feedback.
Histological analysis after testing indicated that the CRF protein concentration was significantly elevated in the CeA of rats that received LV-CRF when compared with LV-GFP animals, indicating that the LV-CRF site-specifically increased synthesis of CRF in CeA (Figure 6
). Prior testing revealed that LV-CRF animals showed a number of physiologic and behavioral signs typically exhibited by animals subjected to chronic stress (Figure 7
). In the DEX suppression test, plasma corticosterone levels were significantly elevated in LV-CRF animals compared to controls by 6 h post injection, indicating that the LV-CRF animals had escaped from glucocorticoid negative feedback. As revealed in behavioral tests, the baseline acoustic startle response was significantly greater in LV-CRF compared with LV-GFP-injected rats, suggesting that basal levels of anxiety are increased in the LV-CMV-CRF group. In the Porsolt forced swim test, LV-CRF animals displayed less time attempting to escape and more immobile floating time than controls, behaviors indicative of an increased depression-like state in rats.
Together, the data from this study demonstrate that lentiviral over-expression of CRF in CeA of rats induces phenotypic behaviors reminiscent of traits associated with a state of chronic stress. Moreover, it revealed the value of using lentiviral vectors to constitutively express genes to model disease states resulting from an overactive gene.
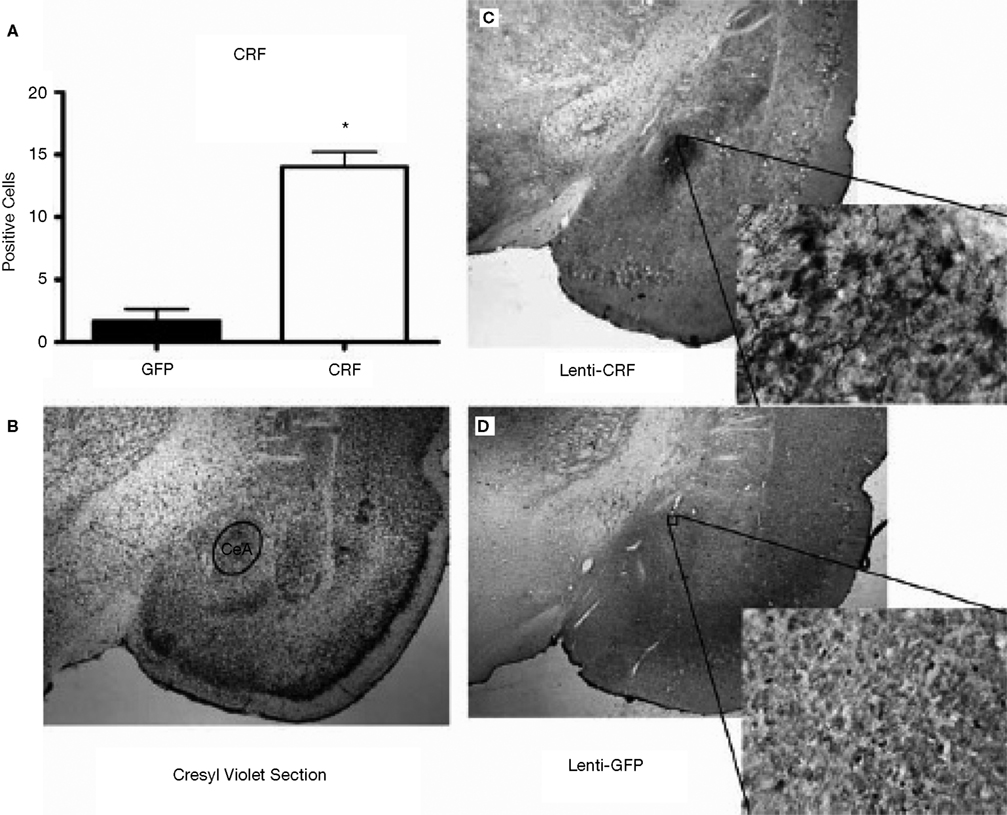
Figure 6. LV-CRF injection into the central nucleus of the amygdala (CeA) significantly increased corticotrophin releasing hormone (CRF) protein production site-specifically. (A) Number of positively labeled CRF cells in CeA of LV-GFP (closed bars) and LV-CRF treated rats (open bars) determined by immunohistochemistry. (B) Cresyl violet stained section representing a section parallel to the sections used to quantify the number of CRF positive neurons in CeA. (C) Representative section with an additional ×20 magnification inset showing the effects of LV-CRF injection into CeA on the number of positively labeled CRF neurons. (D) Representative section with an additional ×20 magnification inset showing the amount of CRF staining observed in the control, LV-GFP-treated rats. *P < 0.05. Panels adapted from Keen-Rhinehart et al. (2009)
.
Figure 7. Effect of LV-CRF injection on HPA axis feedback, anxiety-and depression-related behaviors. (A) HPA axis feedback as assessed by the dexamethasone suppression test. Corticosterone levels before and following a dexamethasone injection (shown by arrow) for LV-GFP control (closed symbol) and LV-CRF rats (open symbol). (B–D) LV-GFP-injected control (closed bars) and LV-CRF-injected rats (open bars). (B) Baseline acoustic startle response, (C) amount of time animals spent actively trying to escape and (D) and time spent floating in the forced swim test. Mean ± SEM. *P < 0.05. Panels adapted from Keen-Rhinehart et al. (2009)
.
An essential characteristic of most viral vectors is that they do not require a region-specific promoter. At present many lentiviruses use the constitutive CMV promoter which drives the ubiquitous expression of a downstream transgene not confined to just one subtype of cells. Thus, it is difficult to attribute whether phenotypic alterations caused by transgene transcription are the consequence of intended targeted manipulation in desired cells or unintended effects on non-target cells. This complexity highlights the value of restricting transgene expression within specific types of cells. For this reason, restricting expression to specific cell populations through the use of a cell type-specific promoter is particularly attractive in future research.
Promoters are generally defined as regions a few hundred to thousand base pairs located upstream of a gene’s transcriptional initiation site; however, more distal regions and 5′ and 3′ untranslated regions (UTR), may also contain various regulatory elements that govern transcription and cell-type specificity. As such, transgenes containing full-length promoter/enhancer regions often exceed the limited insert capacity of lentiviral vectors. Thus, for lentivirus-based applications, it is essential to identify a minimal promoter region that is important for cell-type specificity which efficiently drives gene expression and is also small enough to be packaged efficiently as a viral vector.
The process of determining minimal promoter lengths usually involves the screening of several short versions of putative promoter/enhancer regions by cloning the core or minimal proximal regions of a promoter and assessing their activity and appropriate expression specificity in vitro and in vivo. An example of the in vivo screening process has been reported by Chhatwal et al. (2007)
who recently engineered a minimal promoter of the gene for CCK, an abundant neuropeptide of significant interest in psychiatry and neuroscience (Fink et al., 1998
). In this study, lentivirus constructs were engineered to express either a Cre recombinase or GFP under control of an upstream a 3-kb promoter region of the CCK gene that included 5′ UTR sequences. The resulting CCK-Cre and CCK-GFP lentiviral constructs were assessed in vivo by comparing the expression pattern of constructs to endogenous CCK mRNA expression after microinjection of packaged lentivirus.
Both CCK-Cre and -GFP viruses (LV-CCK-Cre, LV-CCK-GFP) were injected into the CA1 and DG regions of the hippocampus which possess a large number of CCK mRNA-positive cells located close to CCK-negative cells. In the case of LV-CCK-Cre, injections were performed on RosaLacZ mice and the resulting expression of LacZ, as detected by β-gal staining with X-gal, was co-localized with CCK mRNA by means of in situ hybridization. Both β-gal staining and GFP expression patterns closely paralleled CCK mRNA expression in the dentate, CA1, and fasciola cinereum regions of the hippocampus (Figure 8
). Taken together, these observations suggest that minimal CCK promoters are capable of producing highly selective expression in the appropriate sub-population of cells. Furthermore, when used as promoters in lentivirus constructs, similar approaches will allow for in vivo cell-type specific expression of transgenes that are biologically relevant in the study of complex behaviors.
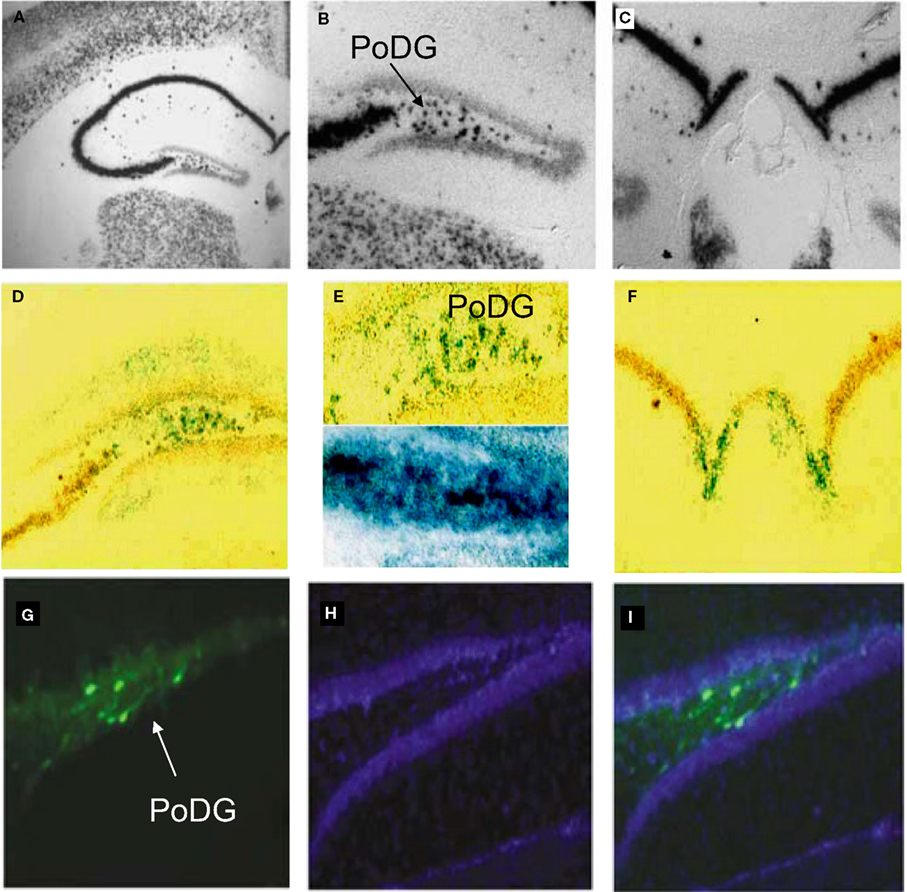
Figure 8. In vivo validation of cell-type specific LV-CCK lentivirus. CCK mRNA expression. CCK mRNA was examined using in situ hybridization, demonstrating high levels of expression within the hippocampal formation [low power (A), high power (B,C)]. (B) Intense CCK mRNA expression is normally present within the CA3 subfield and within the interneuron-rich region (PoDG) separating the granule cell layers of the dentate gyrus, and (C) in the fasciola cinereum medial to CA1. (D) Low power image showing LacZ expression parallels CCK mRNA expression in RosaLacZ Cre-reporter mice injected with LV-CCK-Cre. LacZ expression (blue precipitate) and CCK mRNA expression (silver grains) were assessed in the same sections. (E) High-power images showing a high degree of overlap in mRNA expression was observed in the polymorph layer of the dentate (high power). High and low panels depict images of low and high CCK mRNA expression in the polymorph layer of the dentate, with correspondingly low and high numbers of LacZ-positive cells. (F) Similar co-expression was seen in the fasciola cinereum. (G–I) Virus encoding CCK-GFP injected into the dentate gyrus of adult mice. (G) Fluorescence was assessed on sectioned, fixed tissue. (H) Hoechst-stained photomicrographs of the same section shown in (G). (I) Overlays of (G) and (H). Panels adapted from Chhatwal et al. (2007)
.
Many other potential uses of viral vector approaches to gene manipulation have not been discussed due to space constraints. It is important for the reader to be aware of the multitude of new approaches including gene silencing. In neuroscience, antisense oligonucleotide approaches were used with increasing frequency for about a decade (Van Oekelen et al., 2003
), but there remained significant concern that the mechanism of action and the specificity of gene knockdown were unknown. In recent years, it has become increasingly appreciated that these antisense approaches likely were successful because of the previously unknown mechanisms of cellular heteronuclear RNA silencing mechanisms, which utilize the DICER complex to prevent translation or lead to targeted mRNA degradation. This field has grown exponentially in recent years, and is far beyond the scope of this brief review. Suffice it to say that small heteronuclear RNAs have been successfully encoded in lentiviral and AAV vectors in neuroscience applications to silence genes (Janas et al., 2006
; Harper and Gonzalez-Alegre, 2008
), and this approach will undoubtedly continue to grow rapidly.
Another area of unprecedented growth and excitement concerns light-activated ion channels which have been utilized with tremendous success by Deisseroth and colleagues (Boyden et al., 2005
; Arenkiel et al., 2007
). Several excellent reviews have been written about this new approach and the burgeoning field of ‘optogenetics’ that has resulted (Airan et al., 2007
; Gradinaru et al., 2007
). In brief, these approaches have shown that specific ion channels from green algae which respond to light can be expressed in the brain via both transgenic and viral vector approaches. Different channels have been identified and engineered which respond to different frequencies of light as well as produce different channel characteristics (e.g., excitation vs. inhibition). By combining localized infection with viral vectors with implanted fiber-optics, Deisseroth and colleagues have elegantly shown that they can produce cell-type specific activation within defined neural pathways. These approaches are expanding quickly, and they have the promise of revolutionizing systems neurobiology.
In summary, viral vectors (particularly lentivirus) and Cre/loxP mediated deletion provide powerful methods to inducibly manipulate gene expression in the brain. When combined, they provide for the ability to over-express and delete genes of interest in a temporal, spatial, and cell-type specific fashion. Such inducible genetic approaches will allow for eventual understanding of the differential roles of genes in specific neural pathways which underlie the enormous complexity of brain function and behavior.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Azzouz, M., Martin-Rendon, E., Barber, R. D., Mitrophanous, K. A., Carter, E. E., Rohll, J. B., Kingsman, S. M., Kingsman, A. J., and Mazarakis, N. D. (2002). Multicistronic lentiviral vector-mediated striatal gene transfer of aromatic L-amino acid decarboxylase, tyrosine hydroxylase, and GTP cyclohydrolase I induces sustained transgene expression, dopamine production, and functional improvement in a rat model of Parkinson’s disease. J. Neurosci. 22, 10302–10312.
Keen-Rhinehart, E., Michopoulos, V., Toufexis, D. J., Martin, E. I., Nair, H., Ressler, K. J., Davis, M., Owens, M. J., Nemeroff, C. B., and Wilson, M. E. (2009). Continuous expression of corticotropin-releasing factor in the central nucleus of the amygdala emulates the dysregulation of the stress and reproductive axes. Mol. Psychiatry 14, 37–50.
Li, X. G., Okadam T., Kodera, M., Nara, Y., Takino, N., Muramatsu, C., Ikeguchi, K., Urano, F., Ichinose, H., Metzger, D., Chambon, P., Nakano, I., Ozawa, K., and Muramatsu, S. (2006). Viral-mediated temporally controlled dopamine production in a rat model of Parkinson disease. Mol. Ther. 13, 160–166.
Makino, S., Shibasaki, T., Yamauchi, N., Nishioka, T., Mimoto, T., Wakabayashi, I., Gold, P. W., and Hashimoto, K. (1999). Psychological stress increased corticotropin-releasing hormone mRNA and content in the central nucleus of the amygdala but not in the hypothalamic paraventricular nucleus in the rat. Brain Res. 850, 136–143.
van den Pol, A. N., Ozduman, K., Wollmann, G., Ho, W. S., Simon, I., Yao, Y., Rose, J. K., and Ghosh, P. (2009). Viral strategies for studying the brain, including a replication-restricted self-amplifying delta-G vesicular stomatis virus that rapidly expresses transgenes in brain and can generate a multicolor golgi-like expression. J. Comp. Neurol. 516, 456–481.