 Ilinca Untu1
Ilinca Untu1 Michael Davidson2
Michael Davidson2 Gabriela-Dumitrita Stanciu2*
Gabriela-Dumitrita Stanciu2* Jonathan Rabinowitz2,3
Jonathan Rabinowitz2,3 Romeo-Petru Dobrin1
Romeo-Petru Dobrin1 Diana-Sabina Vieru2
Diana-Sabina Vieru2 Bogdan-Ionel Tamba2,4
Bogdan-Ionel Tamba2,4- 1Department of Medicine III, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy of Iasi, Iași, Romania
- 2Advanced Research and Development Center for Experimental Medicine “Prof. Ostin C. Mungiu” CEMEX, “Grigore T. Popa” University of Medicine and Pharmacy of Iasi, Iași, Romania
- 3Bar Ilan University, Ramat Gan, Israel
- 4Department of Pharmacology, Clinical Pharmacology and Algesiology, “Grigore T. Popa” University of Medicine and Pharmacy of Iasi, Iași, Romania
Depression in Alzheimer’s disease (AD) dementia has become an increasingly recognized public health concern due to its high prevalence and substantial impact on patient outcomes. Despite extensive research having been conducted over the past decades, the precise causal mechanisms and the nature of the relationship between depression and AD dementia remain incompletely understood. This narrative review examines the bidirectional interaction between depression and Alzheimer’s disease, emphasizing shared neurobiological pathways, including neurotransmitter dysregulation, neuroinflammation, abnormalities in the hypothalamic–pituitary–adrenal (HPA) axis, and deficits in neuroplasticity. These mechanisms likely contribute to the acceleration of neurodegeneration in AD and the onset or worsening of depressive symptoms. Current therapeutic approaches remain largely nonspecific, with a lack of targeted therapies that address the unique pathophysiological context of depression in AD. While progress has been made, key research gaps remain, particularly in understanding the complex biological interactions between these two conditions. Future research should focus on identifying specific biomarkers and developing personalized treatment strategies tailored to the neurobiological features of both depression and AD. By addressing these neurobiological mechanisms, we can develop more effective and targeted interventions, ultimately improving patient outcomes and advancing clinical care for this dual pathology.
1 Introduction: depression in Alzheimer’s: a complex and unseen relationship
A growing body of epidemiological and longitudinal research has revealed a complex interplay between Alzheimer’s disease (AD) and depression. However, whether depression arises as a symptom of neurodegeneration or as a psychological reaction to cognitive impairment remains a subject of debate. Some researchers suggest that depression precedes AD, potentially serving as a risk factor, while others argue that it co-occurs with the disease, acting as an integral component of its progression (Galts et al., 2019; Diniz et al., 2013).
As global populations age, the prevalence of Alzheimer’s disease is projected to rise significantly. Aging remains the most well-established risk factor for AD, and demographic trends indicate a steady increase in the number of older adults worldwide (Hou et al., 2019; Liu et al., 2024). Currently, approximately 5% of individuals over the age of 65 are diagnosed with dementia, with prevalence doubling roughly every 5 years. Estimates suggest that over 55 million people globally are living with some form of dementia, with numbers expected to surge to 78 million by 2030 and 139 million by 2050. The sharpest rise is anticipated in developing nations, where the proportion of dementia cases is projected to grow from the current 60 to 71% by mid-century (Alzheimer’s Association, 2024; Liu et al., 2024).
Alzheimer’s disease is the leading cause of dementia, primarily affecting neurons in brain regions associated with memory, language, executive functioning, and other higher-order cognitive abilities. As a result, early symptoms typically manifest as episodic memory deficits, particularly difficulties in retaining newly acquired information. While these deficits may appear suddenly, the underlying neurodegenerative changes—such as β-amyloid deposition and tau pathology—begin nearly two decades before clinical symptoms become evident (Li et al., 2025). As the disease progresses, other cognitive domains become increasingly impaired, including attention, executive function (e.g., planning, decision-making), language (e.g., word-finding difficulties), and visuospatial abilities (e.g., disorientation in familiar places). This gradual deterioration transitions from mild cognitive impairment (MCI) to overt dementia, where patients often retain partial insight into their cognitive decline. This awareness can lead to frustration, anxiety or depression, further complicating the clinical presentation (Coughlan et al., 2018; Stanciu et al., 2020). Ultimately, these cognitive impairments result in a progressive loss of independence, severely affecting daily living and increasing reliance on caregivers.
The diagnosis of Alzheimer’s dementia is typically confirmed when symptoms progress to the point of significantly impairing an individual’s ability to function independently. As a progressive neurodegenerative disorder, AD worsens over time, with the rate of cognitive decline and the specific domains affected varying between individuals. As the disease advances, neuronal damage spreads across multiple brain regions, leading to severe cognitive and functional deterioration. In its later stages, AD leads to a complete loss of independence, requiring full-time care and support from family or caregivers (Langa and Levine, 2014; Coughlan et al., 2018).
Depression, a prevalent affective disorder, disproportionately affects older adults, particularly those receiving medical care, living in institutional settings, or requiring long-term assistance (Alzheimer’s Association, 2024). In late life, it exhibits higher recurrence rates than in middle-aged populations and represents an independent predictor of increased mortality (Mitchell and Subramaniam, 2005). Alarmingly, the risk of suicide is significantly elevated among older adults with depression, with reports indicating that up to 45% of individuals affected by both depression and AD experience suicidal ideation (Olin et al., 2002).
In the context of AD, depression often presents diagnostic challenges. Depression is regarded as a “syndrome” rather than a “disease” due to the absence of a definitive biomarker, making its diagnosis reliant on subjective questionnaire-based assessments. Many neuropsychiatric symptoms associated with depression—such as anxiety, irritability, aggression, and sleep disturbances—are also frequently observed in AD patients (Lyketsos, 2007), complicating differential diagnosis. Although it may share some features with major depressive disorder, depression in AD frequently presents atypically, manifesting as apathy, social withdrawal, or increased irritability (Table 1), rather than pervasive sadness or guilt (Alzheimer’s Association, 2024; Galts et al., 2019). These overlapping and atypical symptoms are often misattributed to the neurodegenerative process itself, which contributes to under recognition and undertreatment. Clinical data suggest that over half of individuals with AD experience comorbid depression, which not only diminishes quality of life but also exacerbates cognitive and functional decline. Depression has been particularly associated with impairments in instrumental activities of daily living (IADLs)—such as managing finances, maintaining social relationships, and performing complex self-care tasks (Giannouli and Tsolaki, 2021). Notably, emerging evidence suggests that depressive symptoms significantly compromise financial decision-making abilities even in patients with mild AD, underscoring the early impact of mood disturbances on executive functioning and autonomy. This cascade of cognitive and functional deterioration contributes to accelerated dependency and heightens caregiver burden (Giannouli and Tsolaki, 2022).

Table 1. Comparison of key neuropsychological aspects between depression and Alzheimer’s disease.
At the neurobiological level, converging evidence points to shared mechanisms underlying both conditions. Disruptions in neurotransmitter systems—including serotonin, norepinephrine, and dopamine pathways—have been implicated in the pathophysiology of both depression and AD. Neuroinflammation, characterized by elevated pro-inflammatory cytokines, is another key contributor to neuronal damage and mood dysregulation. Additionally, dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis has been observed in both disorders, potentially intensifying cognitive impairment and emotional instability (Zhan et al., 2024; Bajaj and Mahesh, 2024). Deficits in neuroplasticity, particularly within the hippocampus, further reinforce the pathological feedback loop between neurodegeneration and depressive symptoms (Stanciu et al., 2020).
Despite substantial clinical findings, depression associated with Alzheimer’s dementia remains frequently underdiagnosed and undertreated, mostly due to the absence of diagnostic criteria specifically tailored to this complex clinical context. The distinction between early affective disturbances often related to the patient’s awareness of emerging cognitive deficits and clinically significant depressive episodes is often ambiguous. This diagnostic ambiguity can delay or preclude appropriate therapeutic intervention. A key challenge lies in the considerable symptom overlap between depression and the cognitive and functional deterioration characteristic of dementia, compounded by the lack of standardized assessment tools validated for this population (Yun and Kim, 2021; Huang et al., 2024). Beyond neurobiology, increasing evidence points to the critical role of psychosocial and environmental determinants. Stressful life events, such as bereavement, transitions to institutional care, and perceived loss of autonomy, have been implicated as advancing or exacerbating factors in late-life depression, particularly in cognitively vulnerable populations (Giannouli, 2024). Sleep disturbances in particular—such as insomnia, fragmented sleep, or reduced slow-wave sleep—have been linked to cognitive decline not only in individuals with mild cognitive impairment (MCI), but also in those with established Alzheimer’s disease. Emerging evidence implicates altered sleep architecture in impaired memory consolidation, increased β-amyloid deposition and decreased lymphatic clearance, all of which may contribute to both neurodegeneration and affective dysregulation (Giannouli and Tsolaki, 2023). Within the broader category of lifestyle interventions, adherence to dietary models rich in neuroprotective and anti-inflammatory components has been consistently associated with a lower risk of both depressive disorders and neurodegenerative decline (Al Shamsi et al., 2024). Specifically, the Mediterranean (MeDi), Dietary Approaches to Stop Hypertension (DASH), and Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) diets have demonstrated favorable associations with cognitive preservation and emotional well-being. The MeDi and DASH diets emphasize high consumption of vegetables, fruits, whole grains, legumes, nuts, fish, and unsaturated fats (particularly from olive oil), while limiting intake of red meat, saturated fats, refined sugars, salt and alcohol (Tangney et al., 2014; Wengreen et al., 2013). The MIND diet, a hybrid of the MeDi and DASH frameworks, uniquely specifies olive oil as the principal source of dietary fat and prioritizes berry consumption as the sole fruit category, based on their high polyphenolic content. Nutritional components integral to these diets—such as omega-3 polyunsaturated fatty acids (n-3 PUFAs), flavonoids and other polyphenols—have been implicated in the maintenance of synaptic plasticity, reduction of oxidative stress, and modulation of neuroinflammation, all of which are processes relevant to both mood regulation and neurodegeneration in aging populations (Al Shamsi et al., 2024).
Collectively, these multifactorial influences, ranging from molecular alterations to psychosocial and behavioral factors, call for a comprehensive integrative framework. This narrative review aims to explore and present the current evidence on the complex bidirectional relationship between depression and Alzheimer’s disease. A more nuanced understanding of these overlapping and converging pathways may inform more precise diagnostic strategies and the development of targeted therapeutic strategies for patients suffering from both AD and comorbid depression.
2 Neurobiological mechanisms shared between depression and Alzheimer’s disease
Depression and Alzheimer’s disease share a complex, bidirectional relationship, underpinned by overlapping neurobiological mechanisms. Shared risk factors— including genetic, environmental, structural and neurochemical—may contribute to their frequent co-occurrence (Figure 1). The heterogeneity of depression characterized by diverse symptomatology and subtypes, may influence the variability in disease progression and association with Alzheimer’s pathology. These overlapping pathophysiological substrates not only help explain the frequent comorbidity of depression and Alzheimer’s disease but also point to potential shared biomarkers that could support earlier identification and differential diagnosis.
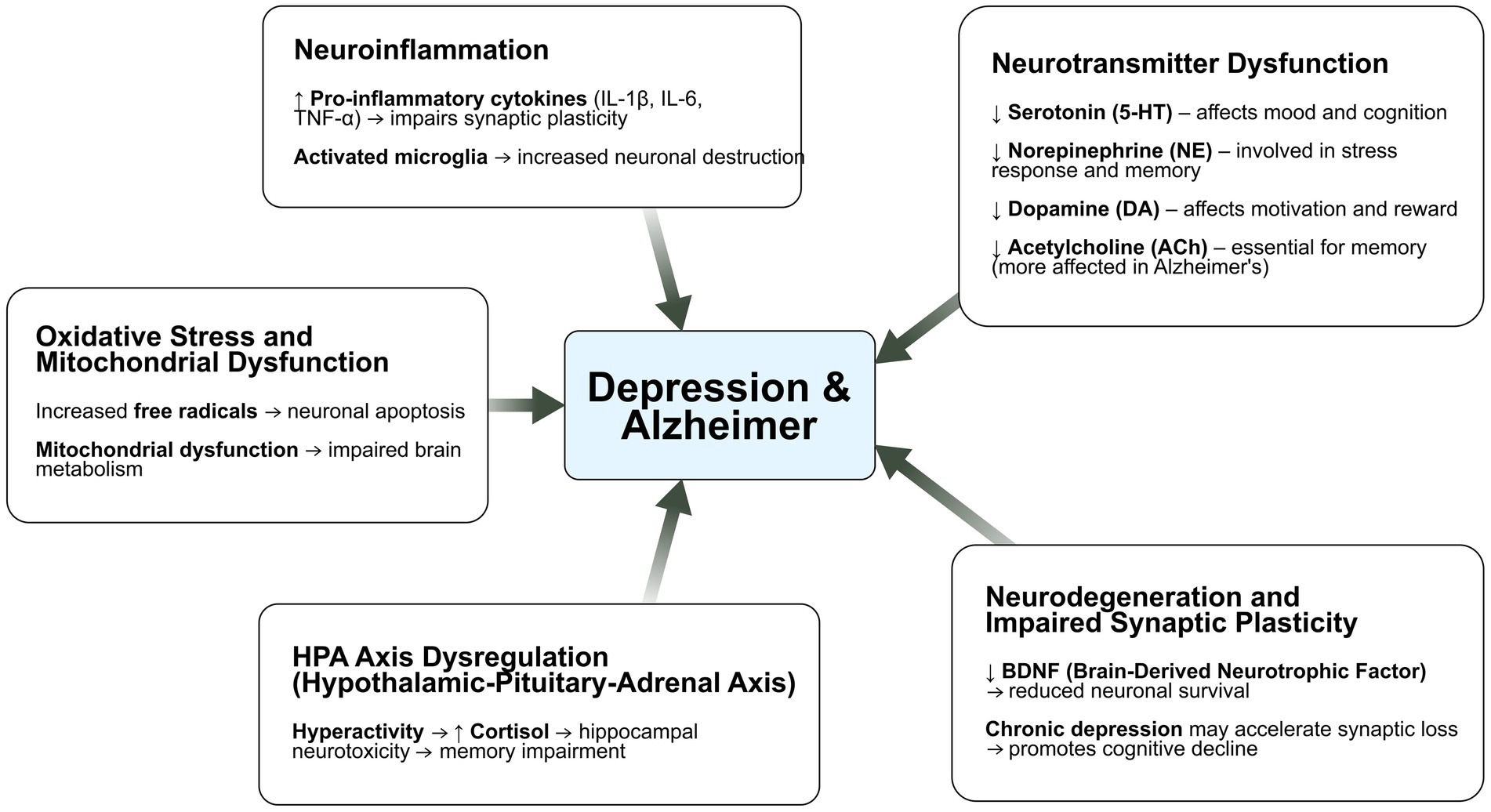
Figure 1. Shared mechanisms across depression and Alzheimer’s disease - key risk factors underlying their association. These include genetic predisposition, neurovascular dysfunction, impaired hippocampal neurogenesis, structural brain changes, and dysregulation of the cortisol-hippocampal pathway. Genetic factors may increase susceptibility to both disorders by influencing neuroinflammation, synaptic plasticity, and neuronal resilience. Neurovascular changes, such as reduced cerebral blood flow and blood–brain barrier disruption, may exacerbate neurodegeneration and mood disturbances. Impaired hippocampal neurogenesis and structural alterations, particularly in regions linked to memory and emotion regulation, further strengthen the connection between the two conditions. Finally, chronic activation of the hypothalamic–pituitary–adrenal (HPA) axis and elevated cortisol levels can accelerate hippocampal atrophy, a shared pathological feature of depression and Alzheimer’s disease. These interconnected mechanisms highlight potential targets for early detection and therapeutic interventions.
Clinical and neuropathological studies have demonstrated that Alzheimer’s patients with coexisting depression score lower on cognitive assessments, such as the CAMCOG test, suggesting that depressive symptoms may accelerate neurodegeneration (Milwain and Nagy, 2005; Huppert et al., 1995). Post-mortem studies further support this, revealing nearly double the number of amyloid plaques and neurofibrillary tangles in the hippocampus AD patients with comorbid depression compared to those without (Rapp et al., 2006). Longitudinal studies have also shown that depression may precede and even predict the onset of dementia, underscoring its potential role in accelerating neurodegeneration (Brunnström et al., 2013).
In addition to its association with cognitive decline, depression is a well-established risk factor for Alzheimer’s, even when the two conditions are separated by decades, a finding supported by a meta-analysis conducted by Ownby et al. (2006). Moreover, depression is highly prevalent among Alzheimer’s patients—affecting up to 75%, and up to 90% of female patients (Usman et al., 2010). This is particularly important, as depression not only worsens amyloid pathology but also accelerates the clinical progression of Alzheimer’s disease, contributing to faster cognitive decline and greater functional impairment (Zahodne et al., 2015).
2.1 Shared genetic mechanisms
Recent genetic research highlights significant commonalities between depression and AD, supporting the hypothesis of a shared genetic basis for both disorders. A recent genome-wide association study (GWAS) leveraging data from the UK Biobank identified 98 shared genetic variants between the two conditions, with TMEM106B emerging as a significant candidate. This gene has been linked to both depression and AD, as well as structural brain differences, including alterations in the corpus callosum and temporal cortex, suggesting a genetic bridge between affective symptoms and neurodegenerative changes (Monereo-Sánchez et al., 2021).
Among neurotrophic factors, brain-derived neurotrophic factor (BDNF) has received considerable attention due to its role in synaptic plasticity, neuronal survival, and mood regulation. Polymorphisms such as G196A (Val66Met) are more prevalent in AD patients with comorbid depression and have been associated with both the severity of depressive symptoms and response to antidepressant treatment (Zhang et al., 2011; Borroni et al., 2009; Arlt et al., 2013). In contrast, another SNP within the BDNF gene, C270T, does not appear to contribute significantly to depression in AD (Borroni et al., 2009; Grunblatt et al., 2009). Although BDNF is known to play a central role in depression, relatively few studies have explored its expression in AD patients with comorbid depressive symptoms. One such study found no significant differences in cerebrospinal fluid (CSF) BDNF levels between depressed and non-depressed AD patients (Pláteník et al., 2014). Beyond genetic polymorphisms, disruptions in BDNF signaling itself may contribute to Alzheimer’s pathology in a bidirectional manner. Impaired BDNF activity has been shown to facilitate the upregulation of amyloidogenic proteins such as amyloid precursor protein (APP) and presenilin-1 (PS1), leading to increased amyloid-β (Aβ) production in transgenic mice (Matrone et al., 2008). Conversely, Aβ has been demonstrated to inhibit neuroprotective signaling cascades—including the Raf-MAPK/ERK and PI3K-Akt pathways—by interfering with the interaction between BDNF and its receptor TrkB, resulting in synaptic dysfunction and neuronal stress (Tong et al., 2004). Notably, recent research using the 5xFAD mouse model of AD has shown that microglial repopulation restores BDNF expression and reactivates TrkB-mediated pathways, significantly improving synaptic plasticity and cognitive outcomes (Wang S. M. et al., 2023; Wang W. et al., 2023). To complement the role of BDNF, other neurotrophic factors have also been implicated in AD-related depression. For example, the CC genotype of transforming growth factor-beta 1 (TGF-β1) has been associated with an increased risk of depressive symptoms in AD patients (Caraci et al., 2012).
Other genetic contributors have also been implicated. The choline O-acetyltransferase (CHAT) gene, essential for cholinergic transmission, has been linked to depressive symptoms in AD, particularly in individuals carrying the ApoE ε4 allele—suggesting an interaction between cholinergic vulnerability and amyloid burden (Grunblatt et al., 2009). In contrast, genes related to dopamine transport, such as DAT1, have not shown consistent associations with depression in AD (Proitsi et al., 2012).
Emerging research has also highlighted the role of Sirtuin 2 (SIRT2) in neurodegeneration and affective disorders. The rs10410544 TT genotype appears to exert a protective effect against depression in AD (Porcelli et al., 2013), and experimental studies in aging mouse models have shown increased hippocampal SIRT2 protein expression with age, independent of mRNA levels—suggesting post-translational accumulation. Pharmacological inhibition of SIRT2 has led to cognitive improvement and reduced neuroinflammation in younger mice, although its effectiveness diminishes in more advanced aging stages (Diaz-Perdigon et al., 2020). Functional assays in SH-SY5Y cells further demonstrated that SIRT1 exerts a neuroprotective effect, counteracting Aβ42-induced toxicity, while SIRT2 exacerbates neuronal vulnerability, reducing cell survival (Li N. et al., 2023; Li Z. et al., 2023).
Recent findings indicates that late-life depression, particularly when it precedes the onset of AD by several years, may act as an independent risk factor for dementia. This association is especially strong in non-carriers of the ApoE ε4 allele, where depression may represent an independent contributor to neurodegeneration rather than a prodromal symptom (Sun et al., 2009). Animal models carrying mutations in the APP, in combination with human ApoE isoforms, confirm that the presence of the ApoE ε4 allele significantly enhances Aβ accumulation, largely through impaired clearance mechanisms, further substantiating the genetic interplay between mood disorders and AD pathophysiology (Bales et al., 2009; Castellano et al., 2011). Consistently, both neuropathological studies and neuroimaging in humans have revealed that ApoE ε4 carriers develop more extensive and earlier Aβ pathology than non-carriers, reinforcing the notion of genotype-specific vulnerability (Morris et al., 2010).
2.2 Neurotransmitter dysregulation
The serotonergic system itself bridges depression and AD due to serotonin’s critical role in mood regulation and cognitive function. Serotonin has seven families of receptors (5-HT1–7), which are all G-protein-coupled except for the ionotropic 5-HT3 receptor (Grunblatt et al., 2009; Micheli et al., 2006; Pritchard et al., 2007; Proitsi et al., 2012; Liu et al., 2001; Mohammad-Zadeh et al., 2008). The most prescribed antidepressants, selective serotonin reuptake inhibitors (SSRIs), target serotonin restoration in the central nervous system. SSRIs are foundational in the serotonin hypothesis of depression, which attributes depression and its complications to reduced serotonergic neurotransmission (Fakhoury, 2016). Besides the serotonin transporter (SERT), three serotonin receptor subtypes (5-HT1A, 5-HT1B, and 5-HT2A), predominantly localized in the limbic system, are particularly important in depression. In AD, serotonin and its metabolites are found in reduced quantities in frontal and temporal cortices, highlighting a potential link to the disease’s pathology. Amyloidogenic activity also increases in the post-menopausal period, possibly due to decreased serotonergic signaling stemming from lower estrogen levels, which is consistent with the higher prevalence of AD in women (Bethea et al., 2017). The expression of 5-HT1A receptors, particularly in the hippocampus, is reduced in Alzheimer’s patients, correlating with worsened clinical symptoms (Kepe et al., 2006; Lanctôt et al., 2007). Similarly, 5-HT2 receptors decrease by up to 69%, documented by reduced binding of 5-HT2 ligands, such as setoperone and altanserin, and corresponding with cognitive deficits (Blin et al., 1993). Moreover, recently discovered serotonin receptors, 5-HT4 and 5-HT6, play critical roles in depression and AD pathophysiology. 5-HT4 receptor activation has been shown to have an antidepressant-like effect and restore cognitive abilities in depression (Mendez-David et al., 2014; Darcet et al., 2016). Conversely, 5-HT6 receptor inhibition has been associated with antidepressant-like effects and could help improve memory and learning deficits in Alzheimer’s patients (Geldenhuys and Van der Schyf, 2011). Further supporting this, experimental studies have demonstrated that pharmacological targeting of 5-HT6 receptors can attenuate AD-related pathology. In an AD mouse model, the 5-HT6 antagonist SB271036 reversed memory impairments, likely through γ-secretase inhibition—leading to reduced amyloid-beta accumulation—and through the suppression of glial cell activation (Yun et al., 2015). The compound also restored serotonin levels, potentially via indirect GABAergic modulation of serotonergic neurons. Moreover, SSRIs such as fluoxetine have shown neuroprotective and cognition-enhancing effects in both in vitro and in vivo models of AD. In a familial AD model, cells expressing the Swedish APP mutation demonstrated altered responsiveness of the serotonergic system, particularly involving the 5-HT1B receptor (Tajeddinn et al., 2016). In double transgenic AD mouse models, fluoxetine prevented neuronal loss, improved spatial memory, and delayed synaptic deterioration (Ma et al., 2017; Zhou et al., 2018).
Other neurotransmitter systems, including the dopaminergic and cholinergic pathways, have also been explored in the context of AD-related depression. While one study found no significant association between the dopamine D4 receptor (D4DR) polymorphism and depressive symptoms in AD patients (Grunblatt et al., 2009), a larger cohort study involving over 1,000 individuals with AD identified a significant correlation between the dopamine receptor D3BalI polymorphism and depression. These findings underscore a potential role for the dopaminergic system in the neuropsychiatric manifestations of Alzheimer’s disease. Expanding on these findings, preclinical research using the TgF344-AD rat model has revealed early alterations in serotonergic–dopaminergic connectivity. At 6 months of age, prior to amyloid plaque accumulation, these animals exhibited reduced striatal dopamine release following 5-HT2A receptor antagonism, despite unaltered receptor binding, indicating functional disconnection rather than receptor loss. Enhanced sensitivity of both postsynaptic D2 receptors and presynaptic D2 autoreceptors was also observed in the absence of changes in receptor density or neuroinflammation markers. Interestingly, administration of the SSRI citalopram modified 5-HT2A–D2 receptor functional dynamics without affecting D2 autoreceptors hypersensitivity. In aged animals, amyloid pathology and increased translocator protein expression were found in dopaminergic regions, coinciding with decreased 5-HT2A receptor density in the nucleus accumbens and substantia nigra, likely mediated by astrocytic receptor expression (Ceyzériat et al., 2021).
Cholinergic dysfunction is a well-established feature of Alzheimer’s disease (AD), where impaired cholinergic signaling is believed to contribute to learning and memory deficits—an effect that can be experimentally reproduced using anticholinergic drugs. The cholinergic hypothesis of AD posits that cognitive impairments largely stem from reduced cholinergic transmission, a theory supported by the partial efficacy of acetylcholinesterase inhibitors in alleviating symptoms in some patients (Craig et al., 2011). However, the limited response observed in a subset of individuals complicates this framework and suggests additional mechanisms may be involved. In depression, the cholinergic system also plays a crucial, albeit more complex, role. Whereas diminished cholinergic activity is linked to cognitive decline in AD, evidence points toward cholinergic hyperactivity in depression (Dilsaver and Coffman, 1989). Over recent decades, research has suggested that excessive cholinergic tone may underlie depressive symptoms (Bassi et al., 2015). For instance, antagonizing nicotinic receptors can produce antidepressant-like effects, while cholinergic overactivation is associated with depressive behaviors. Interestingly, activation of specific nicotinic receptor subtypes—particularly the α7 receptor—has been shown to alleviate depressive symptoms by enhancing hippocampal function (Zhao et al., 2017). These findings suggest that the cholinergic system exerts divergent effects in AD and depression, with the hippocampus playing a central modulatory role. Expanding on this, recent findings indicate that Aβ1–42 forms a high-affinity, stable complex with the α7 nicotinic acetylcholine receptor (α7 nAChR) in vitro and co-localizes with neuritic plaques in the AD brain, suggesting a direct modulatory effect on cholinergic neurotransmission (Wang et al., 2000). In the PDAPP transgenic mouse model of AD with microdialysis revealed significant Aβ-dependent disruptions in both basal and evoked acetylcholine (ACh) release compared to wild-type controls. These deficits were associated with Aβ interactions not only with α7 nAChRs but also with the high-affinity choline transporter, potentially impairing both steady-state and activity-dependent ACh release. Notably, administration of the anti-Aβ antibody m266 fully restored hippocampal ACh release and choline uptake, while also improving habituation learning—a behavioral deficit typical of this model. These results suggest that soluble Aβ species can directly induce cholinergic dysfunction and cognitive impairment in early AD, even in the absence of overt neurodegeneration. Anti-Aβ immunotherapy may therefore represent a promising approach for the rapid reversal of early cholinergic deficits (Bales et al., 2006).
2.3 Hypothalamic–pituitary–adrenal axis dysregulation and stress response
A pathophysiological link between depression and dementia could be mediated by dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis (Caraci et al., 2010; Krishnan and Nestler, 2008; Crochemore et al., 2005; Galts et al., 2019). Studies comparing cortisol levels in individuals with depression or dementia to a control group show that those with both dementia and comorbid depression exhibit the highest cortisol levels. Additionally, in patients with both conditions, cortisol levels are correlated with cognitive function, suggesting an impact on cognitive decline (Barca et al., 2019). However, HPA dysfunction in AD may develop as the disease progresses, and at present, plasma cortisol concentrations do not serve as a reliable biomarker to distinguish between depression and dementia (Galts et al., 2019; Chi et al., 2014).
The dysfunction of the HPA axis, leading to excessive glucocorticoid release, has been observed in both chronic depression and AD patients. Elevated glucocorticoid levels in plasma and CSF are commonly found in both conditions (Popp et al., 2009). However, the pattern of hypercortisolemia differs between the two: in AD, cortisol elevation is phasic, whereas in depression, it tends to be chronic. Further evidence suggests distinct pathophysiological mechanisms underlying depression in AD compared to idiopathic depression (Notarianni, 2013). Studies examining CSF cortisol levels in depressed and non-depressed AD patients found no significant differences, although cortisol levels in AD patients were notably higher than in controls (Hoogendijk et al., 2006). Similarly, plasma cortisol levels in AD patients, with or without depressive symptoms, showed no significant variation, reinforcing the idea of a different underlying mechanism of depression in AD compared to primary depression (Zverova et al., 2013).
In young 3xTg-AD mice, an activated HPA axis is observed during the early stages of neuropathology, despite normal glucocorticoid levels. This activation is detectable even when the mice exhibit only mild behavioral alterations, indicating that neuroendocrine regulation occurs prior to the onset of severe AD-like pathology and more pronounced behavioral deficits. Furthermore, when these animals undergo periodic cognitive stimulation, both corticosterone levels and Aβ pathology increase. These findings suggest that dysfunction in the HPA axis may transform typically stimulating environments into stress-inducing ones, thus contributing to a feedback loop that promotes Aβ release and plaque formation (Sharan and Vellapandian, 2024).
2.4 Neuroinflammation and vascular contributions to neurodegeneration
Vascular risk factors, such as hypertension, diabetes, cardiovascular diseases, and stroke, have been linked to both late-life depression and AD (Regan et al., 2006). Many AD patients exhibit mixed types of vascular dementia, and cerebrovascular disease has been suggested to contribute to a form of depression known “Vascular Depression.” This form of depression is linked to several mechanisms, including brain circuit disconnections, inflammation and hypoperfusion, which are interrelated and complementary (Taylor et al., 2013). Studies indicate that a history of stroke is associated with an increased risk of depression and apathy in AD patients, possibly due to disrupted neural circuitry in brain regions involved in mood regulation (Treiber et al., 2008). Moreover, hypertension and asymptomatic stroke have been found to exacerbate depressive symptoms in AD, suggesting the role of cerebrovascular damage in mood disorders (Moon et al., 2013). Additionally, spontaneous cerebral emboli, which damage the fronto-striatal pathways, have been linked to clinically relevant depressive symptoms in patients with both AD and vascular dementia (Purandare et al., 2006). These findings suggest that ischemia and white matter lesions may disrupt neural connections, contributing to depression in AD patients, thus supporting the vascular depression theory. However, some studies have not found vascular risk factors to modify depression in AD, which raises controversy on the exact nature of their relationship (Steinberg et al., 2014; Luchsinger et al., 2008).
On a different note, neurovascular dysfunction in mood-regulating brain regions, associated with increased blood–brain barrier (BBB) permeability, may serve as a link between chronic stress and depression. This dysfunction may involve the VEGF/VEGFR2 pathway (Matsuno et al., 2022) abnormal blood vessel morphology, and peripheral cytokine infiltration into the brain (Menard et al., 2017). Research has shown that the deletion of Lrp1 in brain capillaries exacerbates AD pathology and cognitive impairment (Storck et al., 2016; Nation et al., 2019). Interestingly, BBB breakdown in the hippocampus does not appear to be directly related to Aβ or tau biomarkers in individuals with early cognitive decline, highlighting the complexity of the relationship between vascular dysfunction and AD pathology (Nation et al., 2019). Further contributing to neurovascular compromise, reduced activity and expression of the glucose transporter GLUT-1 at the BBB suggests impaired glucose uptake and utilization by the brain. This metabolic deficiency may aggravate cerebrovascular degeneration and further promote BBB disruption. In animal models overexpressing APP, GLUT-1 dysfunction has been linked to intensified Aβ deposition, neurodegeneration, and the emergence of cognitive deficits (Winkler et al., 2015). Microglia detect Aβ deposits through surface receptors, including Toll-like receptors (TLRs) and receptors for advanced glycation end-products (RAGE). Activation of these receptors triggers an inflammatory cascade designed to eliminate toxic components. This process underscores the dual nature of microglial activation, which can either mitigate or exacerbate amyloid pathology depending on the stage of the disease (Wu et al., 2021). During the phagocytosis of Aβ, microglia release several pro-inflammatory cytokines, including interleukin-1 beta (IL-1 beta), tumor necrosis factor-alpha (TNF-a), interleukin-6 (IL-6), which promote local inflammation, cause neurodegenerative effects and contribute to synaptic disfunction. This prolonged process leads to production of reactive oxygen species (ROS) and nitric oxide (NO), which increase oxidative stress and neuronal damage, contributing to the indirect pathway of neuronal loss and impaired synaptic function (Wu et al., 2021).
Inflammation is a common feature of multiple psychiatric disorders. In depression, the Maastricht study has found that elevated serum biomarkers of inflammation such as the acute phase reactant C-reactive protein (CRP) and TNF-a were associated in both major and minor depressive episodes (van Dooren et al., 2016). Inflammation in depression may be linked to altered connectivity between brain regions. A resting state MRI study on 48 medication-free patients with major depression revealed serum levels of CRP were associated with reduced connectivity between the ventral striatum and prefrontal cortex (correlating with increased anhedonia), the dorsal striatum and prefrontal cortex (correlating with psychomotor slowing). Inflammatory cytokines like IL-6, IL-1 beta and IL-1 receptor antagonist were also linked to diminished connectivity between the striatum and prefrontal cortex (van Dooren et al., 2016). A longitudinal MRI analysis conducted on transgenic J20 mice, compared to wild-type littermates, identified disruptions in neural networks of J20 mice, particularly between posterior and anterior brain regions, with age-related exacerbation of these changes. Reduced fractional anisotropy in the corpus callosum of these AD mouse models revealed white matter microstructural changes, correlated with axonal degeneration and myelin disruption associated with AD, which may be detectable in the pre-symptomatic stages. The paper highlights key features of the disconnection syndrome hypothesis in AD, in which the progression of symptoms is related to functional and/ or structural disconnection between regions, rather than localized changes in specific isolated brain areas (Magalhães et al., 2024).
Alterations in white matter are frequently observed in patients with depression, potentially underlying the dysfunction of fronto-cortico-limbic neural circuits that are crucial to the pathogenesis of depression, especially in the context of emotion dysregulation symptoms (Price and Drevets, 2010; Buckner et al., 2008). In comparison to healthy controls, individuals with major depression exhibited increased functional connectivity (FC) in the anterior default mode network (DMN) and between the anterior DMN and the salience network (Wang S. M. et al., 2023; Wang W. et al., 2023), while showing decreased FC in the posterior DMN and between the posterior DMN and the central executive network. This pattern of dissociation could be attributed to a reduction in the integrity of white matter within the cingulate bundle, which connects the two regions (Charlton et al., 2014). In patients with late-onset depression, abnormal FC was found to correlate not only with the severity of depression but also with the accumulation of cerebral Aβ (Wang et al., 2021) and generalized cognitive dysfunction. Changes in connectivity within the DMN may offer a potential neural mechanism linking depression, Aβ pathology and the progression of Alzheimer’s disease (Yin et al., 2015; Guan et al., 2022).
Despite the complexity and ongoing controversies, these findings, as summarized in Table 2, emphasize the need for an integrated approach to understanding depression in AD. Future research should focus on elucidating the precise molecular and neurobiological mechanisms that link these conditions, as well as identifying biomarkers that could facilitate early diagnosis and targeted interventions. Clinically, the recognition of depression as an early marker or comorbid condition in AD may offer opportunities for more effective treatment strategies that address both mood symptoms and cognitive decline. By targeting modifiable risk factors, such as vascular health and neurotrophic support, interventions may help slow AD progression and improve the quality of life for affected individuals.
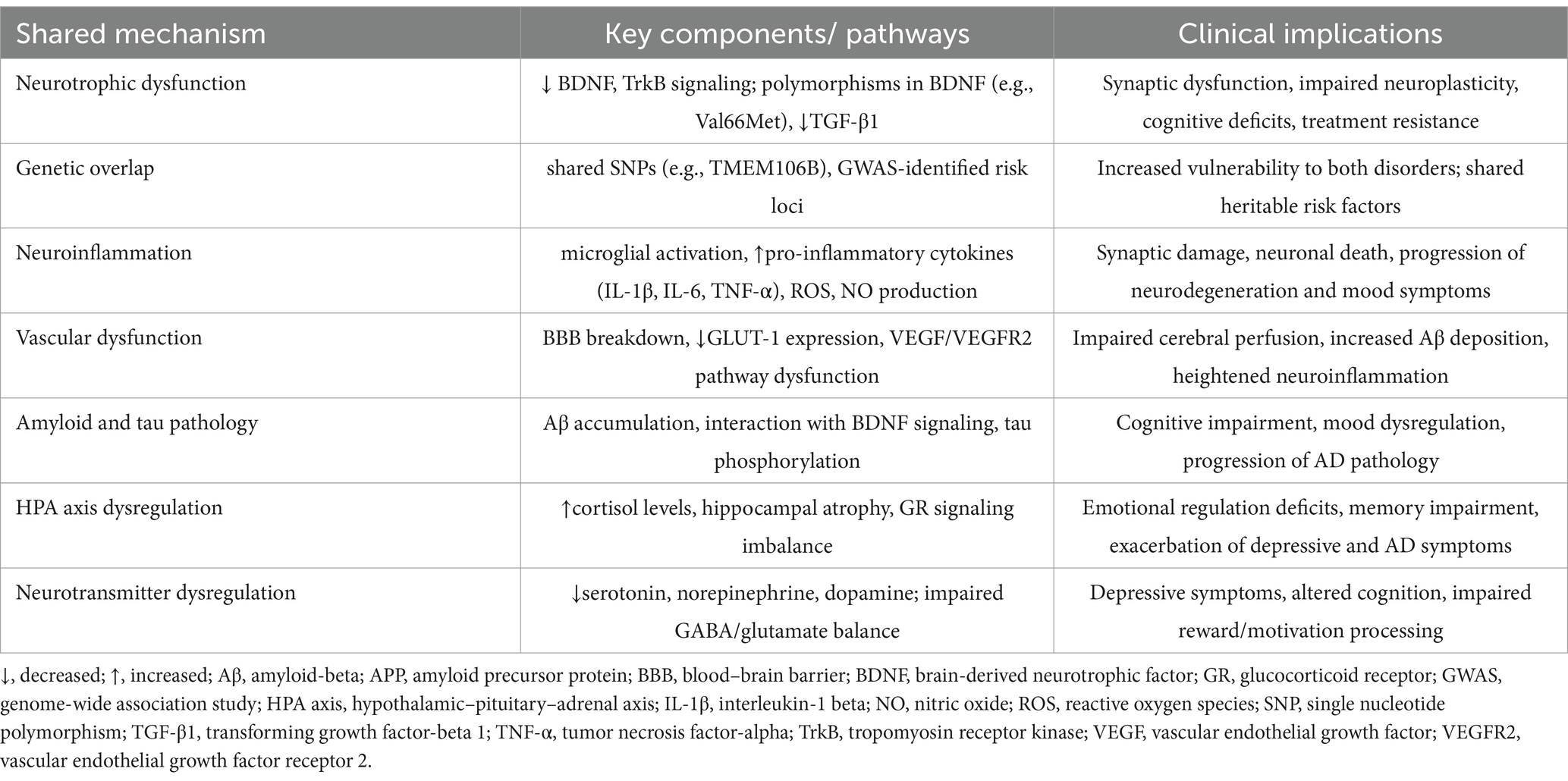
Table 2. Summary of shared neurobiological mechanisms between depression and Alzheimer’s disease and their clinical implications.
3 Therapeutic strategies for depression associated with AD
Depression is commonly observed in individuals with AD, and strategies for managing this condition remain a subject of ongoing debate. The challenge of diagnosing depression in the context of dementia, given the overlap between depressive and cognitive symptoms, underscores the need for personalized and effective therapeutic approaches.
Current treatment guidelines recommend antidepressants as first-line therapy for depression associated with AD, prioritizing medications with minimal side effects. Among the most commonly prescribed options are Selective Serotonin Reuptake Inhibitors (SSRIs) and Serotonin-Norepinephrine Reuptake Inhibitors (SNRIs), frequently cited in the literature (Bartels et al., 2018). In recent years, several systematic reviews and meta-analyses have investigated the efficacy of these treatments in AD-related depression. While findings suggest potential benefits, results remain limited and sometimes contradictory (Hsu et al., 2021; Sanchez et al., 2015; Huang et al., 2022; Li N. et al., 2023; Li Z. et al., 2023) (Table 3). One systematic review and meta-analysis focusing on serotonergic antidepressants (Hsu et al., 2021) found that these agents significantly improved neuropsychiatric symptoms (NPS) and agitation (with small effect sizes), alleviated depressive symptoms, reduced care burden, and slightly enhanced cognitive function. Subgroup analyses revealed that both SSRIs (particularly citalopram/ escitalopram) and non-SSRIs effectively reduced agitation and depressive symptoms, though only SSRIs showed broader effects on NPS and care burden. Furthermore, benefits were more pronounced among patients with AD-related depression compared to those with pure AD. These medications were generally well-tolerated, with no significant differences in attrition rates or adverse events compared to placebo (Hsu et al., 2021). In addition to serotonergic agents, vortioxetine, a newer antidepressant with multimodal pharmacological actions, has been evaluated in two recent meta-analyses. Vortioxetine was shown to significantly improve cognitive function and depressive symptoms, with greater effects at higher doses, as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS). These findings suggest that vortioxetine may support cognitive recovery even when depressive symptoms persist. However, its potential role in preventing dementia requires further longitudinal investigation (Huang et al., 2022; Li N. et al., 2023; Li Z. et al., 2023). In clinical practice, vortioxetine appears to be effective, well-tolerated, and safe, contributing to improved cognitive and functional outcomes.

Table 3. The interplay between depression treatment and Alzheimer’s disease progression: insights from pharmacological and psychological therapies.
While these medications are proven effective for major depressive disorder (MDD), their benefits in depression associated with AD may be more subtle, and the therapeutic response can vary from patient to patient. A key factor in this discrepancy lies in the distinct molecular mechanisms underlying depression in AD versus MDD (Thompson et al., 2007; Guan et al., 2022). In AD, depression is often closely linked to neurodegenerative processes such as neuronal death in the hippocampus and prefrontal cortex, which disrupt brain function and cognitive abilities. These processes lead to significant cholinergic deficits and neuropsychiatric disturbances associated with NMDA receptor dysfunction, which contribute to the symptoms of depression. Conversely, in MDD, depression is primarily associated with neurotransmitter imbalances, particularly involving serotonin, norepinephrine, and dopamine, which affect mood regulation, reward processing, and other psychological functions. Nevertheless, despite these differences, SSRIs and SNRIs can alleviate depressive symptoms in AD patients, although these medications do not directly address the underlying neurodegenerative processes specific to AD, such as cholinergic deficits and NMDA receptor dysfunction (Orgeta et al., 2017; Liu et al., 2023). Serotonergic neurons, which originate from the median and dorsal raphe nuclei, play a crucial role in regulating mood, motor activity, sleep, and aggression by innervating regions such as the limbic system and cortex, particularly the forebrain (Bartels et al., 2018). The wide-ranging projections of these neurons contribute to the antidepressant effects of serotonergic medications, which, while effective in treating depressive symptoms in AD, do not modify the fundamental neuropathological changes characteristic of the disease (Thompson et al., 2007).
In addition to pharmacological treatments for depression, antidementia medications play a significant role in treating patients with concurrent AD and depression (Table 3). Medications targeting neurodegenerative processes, such as cholinesterase inhibitors (e.g., donepezil) and NMDA antagonists (e.g., memantine), can contribute to improving both cognitive and depressive symptoms (Weiner et al., 2000; Gauthier et al., 2002; Cummings et al., 2006). These drugs may help reduce neuropsychiatric disturbances associated with AD and modulate the chemical balance in the brain, which can support the management of depressive symptoms (Hermann, 2005; Rosler et al., 1998; Maidment et al., 2008).
In addition to pharmacological treatments, non-pharmacological strategies (Table 3) have garnered increasing attention in the management of depression associated with AD. These approaches aim to address the psychological, social and neurobiological aspects of the condition, offering a complementary avenue to conventional therapies. Although antidepressants remain the first-line treatment for depression in AD, their limited efficacy and the potential side effects highlight the need for alternative, personalized interventions. One such non-pharmacological intervention is cognitive-behavioral therapy (CBT), which has been widely studied for its effectiveness in managing depressive symptoms in various populations. A recent meta-analysis has indicated that CBT may help AD patients improve coping strategies, regulate emotions and manage stress (Orgeta et al., 2022). However, its application in AD is complicated by cognitive impairments, and further research is required to adapt CBT techniques for this population, ensuring their feasibility and effectiveness. Another promising intervention is music therapy, which has been shown to improve mood, reduce anxiety, and enhance cognitive function in individuals with AD (Tonga et al., 2021). Music therapy may facilitate emotional expression and foster social interaction, thus providing both psychological and social benefits. Studies suggest that structured music interventions can promote relaxation and decrease agitation, symptoms commonly observed in AD patients. However, as with other non-pharmacological treatments, the variability in patient responses necessitates further randomized controlled trials to substantiate these findings and establish standardized protocols.
Physical activity is also gaining recognition as a valuable intervention for managing depression in AD. Regular exercise has been associated with improved mood, cognitive function, and overall well-being in older adults, including those with dementia (Baruch et al., 2019). Physical activity can enhance neuroplasticity, reduce neuroinflammation, and promote the release of neurotrophic factors, which are critical for brain health. While these benefits are promising, the optimal type, intensity, and frequency of exercise interventions remain unclear, and additional studies are needed to determine the most effective exercise regimens for AD patients. Moreover, emerging evidence supports the use of neurostimulation therapies, including transcranial magnetic stimulation (TMS) and electroconvulsive therapy (ECT), in treating depression in AD. TMS has been shown to have positive effects on mood and cognitive function, potentially by modulating brain activity in regions implicated in mood regulation. Although the use of TMS in AD is still in its early stages, a pilot randomized, double-blind, parallel, sham-controlled study suggests its potential as a non-invasive and well-tolerated option for patients with slow cognitive decline (LoBue et al., 2025). ECT, although more invasive, remains a viable treatment option for severe depression that is refractory to other therapies. Recent research into the use of ECT in AD patients is have shown symptomatic benefits in managing behavioral symptoms in individuals with dementia, particularly those with major depressive episodes (Bachu et al., 2024).
In conclusion, while non-pharmacological treatments such as CBT, music therapy, physical activity, and neurostimulation hold promise in the management of depression in AD, the evidence supporting their efficacy remains preliminary. Given the heterogeneous nature of depression in AD and the complexity of the disorder, a personalized, multimodal treatment approach that incorporates both pharmacological and non-pharmacological strategies is essential. Future research should focus on large-scale randomized controlled trials to clarify the long-term benefits, risks, and optimal protocols for these interventions. Additionally, investigating how these therapies can be integrated into routine clinical practice is vital to improving outcomes for patients with AD and comorbid depression.
4 Future perspectives and research directions
The treatment of depression in Alzheimer’s disease remains a multifaceted challenge, necessitating a combined effort to address the significant gaps in current understanding. While substantial progress has been made, several critical areas remain poorly understood, and future research should focus more explicitly on these unexplored or underexplored aspects.
One major gap is the neurobiological mechanisms linking depression with Alzheimer’s pathology. Specifically, the role of neurotransmitter systems such as dopamine, GABA, and neuroimmune signaling pathways remains unclear and warrants further investigation. Although some studies have explored the role of serotonin and norepinephrine, the contribution of other neurotransmitters in the pathophysiology of depression in AD is still unknown. Moreover, the interaction between neuroinflammation and cognitive decline needs further exploration, particularly regarding how inflammatory processes in the brain may exacerbate mood disturbances and accelerate Alzheimer’s progression. This knowledge could be pivotal in developing targeted therapeutic strategies aimed at modulating these pathways.
Another unresolved question is the variable efficacy of current antidepressant treatments in AD. The underlying causes of these inconsistent results, especially in relation to different stages of the disease and symptom heterogeneity, require in-depth study. Understanding the mechanisms behind the differential response to antidepressant therapies could lead to more personalized and effective treatment options. Additionally, research must focus on the development of more refined diagnostic criteria for depression in AD, as the overlap between mood disturbances and cognitive decline complicates accurate identification.
There is also an emerging need to investigate alternative pharmacological and non-pharmacological treatments that may better address the specific neurobiological features of depression in AD. While current treatments, such as selective serotonin reuptake inhibitors (SSRIs), show some promise, their inconsistent effects suggest that more targeted interventions are needed. Exploring combinations of antidepressant therapies with antidementia treatments, such as cholinesterase inhibitors and NMDA receptor antagonists, could help develop more effective, multimodal treatment regimens. Understanding the potential synergistic effects of such combined therapies could improve both cognitive and mood symptoms. Moreover, the efficacy of non-pharmacological interventions like cognitive-behavioral therapy (CBT), physical exercise, and other psychosocial approaches requires further rigorous evaluation in large-scale, randomized controlled trials. Preliminary evidence is promising, but more robust data are needed to confirm the long-term benefits and effectiveness of these therapies in diverse patient populations. Clearly defined guidelines for integrating these treatments into standard clinical practice are essential.
Lastly, the move toward personalized medicine in AD offers significant promise. Given the heterogeneity of depression in AD, integrating genetic, biomarker, and clinical data could allow for better patient stratification, leading to more effective treatments. Identifying subgroups of patients based on specific genetic predispositions, disease stages, or biomarker profiles could enhance therapeutic outcomes and reduce adverse effects.
In conclusion, while substantial progress has been made in understanding and treating depression in Alzheimer’s disease, many critical questions remain unanswered. Future research must prioritize understanding the neurobiological mechanisms of depression in AD, refining diagnostic tools, and developing more effective, personalized interventions. Addressing these gaps will not only improve the quality of care for individuals with AD but also open new avenues for therapeutic strategies that target both cognitive decline and mood disturbances.
Author contributions
IU: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. MD: Conceptualization, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. G-DS: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. JR: Conceptualization, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. R-PD: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. D-SV: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. B-IT: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by Romania’s National Recovery and Resilience Plan (PNRR), Pylon III, Section I8. Development of a Program to Attract Highly Specialized Human Resources from Abroad in Research, Development and Innovation Activities, PNRR-III-C9-2023-I8, Project “Modelling negative symptom domains neurobiology: a transdiagnostic, translational study,” code CF 46/ 28.07.2023, and by a grant of the Ministry of Research, Innovation and Digitization, CNCS - UEFISCDI, project number PN-IV-P2-2.1-TE-2023-0879, within PNCDI IV.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al Shamsi, H. S. S., Rainey-Smith, S. R., Gardener, S. L., Sohrabi, H. R., Canovas, R., Martins, R. N., et al. (2024). The relationship between diet, depression, and Alzheimer’s disease: a narrative review. Mol. Nutr. Food Res. 68:2300419. doi: 10.1002/mnfr.202300419
Alzheimer’s Association (2024). Alzheimer’s disease facts and figures. Alzheimers Dement. 20, 3708–3821. doi: 10.1002/alz.13809
Arlt, S., Demiralay, C., Tharun, B., Geisel, O., Storm, N., Eichenlaub, M., et al. (2013). Genetic risk factors for depression in Alzheimer’s disease patients. Curr. Alzheimer Res. 10, 72–81. doi: 10.2174/1567205011310010010
Bachu, A. K., Kotapati, V. P., Kainth, T., Patel, R., Youssef, N. A., and Tampi, R. R. (2024). Electroconvulsive therapy in individuals with dementia/major NCD presenting with behavioral symptoms: a systematic review. Int. Psychogeriatr. 36, 864–879. doi: 10.1017/S104161022300039X
Bajaj, S., and Mahesh, R. (2024). Converged avenues: depression and Alzheimer’s disease– shared pathophysiology and novel therapeutics. Mol. Biol. Rep. 51:225. doi: 10.1007/s11033-023-09170-1
Bales, K. R., Liu, F., Wu, S., Lin, S., Koger, D., DeLong, C., et al. (2009). Human APOE isoformdependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009
Bales, K. R., Tzavara, E. T., Wu, S., Wade, M. R., Bymaster, F. P., Paul, S. M., et al. (2006). Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-a beta antibody. J. Clin. Invest. 116, 825–832. doi: 10.1172/JCI27120
Barca, M. L., Eldholm, R. S., Persson, K., Bjørkløf, G. H., Borza, T., Telenius, E., et al. (2019). Cortisol levels among older people with and without depression and dementia. Int. Psychogeriatr. 31, 597–601. doi: 10.1017/S1041610218001199
Bartels, C., Wagner, M., Wolfsgruber, S., Ehrenreich, H., and Schneider, A. (2018). Alzheimer’s Disease Neuroimaging Initiative impact of SSRI therapy on risk of conversion from mild cognitive impairment to Alzheimer’s dementia in individuals with previous depression. Am. J. Psychiatry 175, 232–241. doi: 10.1176/appi.ajp.2017.17040404
Baruch, N., Burgess, J., Pillai, M., and Allan, C. L. (2019). Treatment for depression comorbid with dementi. Evid. Based Ment. Health 22, 167–171. doi: 10.1136/ebmental-2019-300113
Bassi, S., Seney, M. L., Argibay, P., and Sibille, E. (2015). Elevated hippocampal cholinergic neurostimulating peptide precursor protein (HCNP-pp) mRNA in the amygdala in major depression. J. Psychiatr. Res. 63, 105–116. doi: 10.1016/j.jpsychires.2015.02.006
Bethea, C. L., Reddy, A. P., and Christian, F. L. (2017). How studies of the derotonin system in macaque models of menopause relate to Alzheimer’s disease. J. Alzheimers Dis. 57, 1001–1015. doi: 10.3233/JAD-160601
Blin, J., Baron, J. C., Dubois, B., Crouzel, C., Fiorelli, M., Attar-Lévy, D., et al. (1993). Loss of brain 5-HT 2 receptors in Alzheimer’s disease. Brain 116, 497–510. doi: 10.1093/brain/116.3.497
Borroni, B., Grassi, M., Archetti, S., Costanzi, C., Bianchi, M., Caimi, L., et al. (2009). BDNF genetic variations increase the risk of Alzheimer’s disease-related depression. J. Alzheimers Dis. 18, 867–875. doi: 10.3233/JAD-2009-1191
Brunnström, H., Passant, U., Englund, E., and Gustafson, L. (2013). History of depression prior to Alzheimer’s disease and vascular dementia verified post-mortem. Arch. Gerontol. Geriatr. 56, 80–84. doi: 10.1016/j.archger.2012.10.008
Buckner, R. L., Andrews-Hanna, J. R., and Schacter, D. L. (2008). The brain's default network: anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci. 1124, 1–38. doi: 10.1196/annals.1440.011
Cammisuli, D. M., Cipriani, G., Giusti, E. M., and Castelnuovo, G. (2022). Effects of reminiscence therapy on cognition, depression and quality of life in elderly people with Alzheimer's disease: a systematic review of randomized controlled trials. J. Clin. Med. 11:5752. doi: 10.3390/jcm11195752
Caraci, F., Copani, A, Nicoletti, F., and Drago, F. (2010). Depression and c Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur J Pharmacol. 626:64–71. doi: 10.1016/j.ejphar.2009.10.022
Caraci, F., Bosco, P., Signorelli, M., Spada, R. S., Cosentino, F. I., Toscano, G., et al. (2012). The CC genotype of transforming growth factor-B1 increases the risk of late-onset Alzheimer’s disease and is associated with AD-related depression. Eur. Neuropsychopharmacol. 22, 281–289. doi: 10.1016/j.euroneuro.2011.08.006
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human ApoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Ceyzériat, K., Gloria, Y., Tsartsalis, S., Fossey, C., Cailly, T., Fabis, F., et al. (2021). Alterations in dopamine system and in its connectivity with serotonin in a rat model of Alzheimer's disease. Brain Commun. 3:fcab029. doi: 10.1093/braincomms/fcab029
Charlton, R. A., Lamar, M., Zhang, A., Yang, S., Ajilore, O., and Kumar, A. (2014). White-matter tract integrity in late-life depression: associations with severity and cognition. Psychol. Med. 44, 1427–1437. doi: 10.1017/S0033291713001980
Chi, S., Yu, J.-T., Tan, M.-S., and Tan, L. (2014). Depression in Alzheimer’s disease: epidemiology, mechanisms, and management. J. Alzheimers Dis. 42, 739–755. doi: 10.3233/JAD-140324
Cirrito, J. R., Wallace, C. E., Yan, P., Davis, T. A., Gardiner, W. D., Doherty, B. M., et al. (2020). Effect of escitalopram on aβ levels and plaque load in an Alzheimer mouse model. Neurology 95, e2666–e2674. doi: 10.1212/WNL.0000000000010733
Clerici, F., Vanacore, N., Elia, A., Spila-Alegiani, S., Pomati, S., Da Cas, R., et al. (2012). Memantine effects on behaviour in moderately severe to severe Alzheimer’s disease: a post-marketing surveillance study. Neurol. Sci. 33, 23–31. doi: 10.1007/s10072-011-0618-0
Coughlan, G., Laczó, J., Hort, J., Minihane, A. M., and Hornberger, M. (2018). Spatial navigation deficits—overlooked cognitive marker for preclinical Alzheimer disease? Nat. Rev. Neurol. 14, 496–506. doi: 10.1038/s41582-018-0031-x
Craig, L. A., Hong, N. S., and McDonald, R. J. (2011). Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 35, 1397–1409. doi: 10.1016/j.neubiorev.2011.03.001
Crochemore, C., Lu, J., Wu, Y., Liposits, Z., Sousa, N., Holsboer, F., et al. (2005). Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol. Psychiatry 10, 790–798. doi: 10.1038/sj.mp.4001679
Cummings, J. L., McRae, T., and Zhang, R.Donepezil-Sertraline Study, Group (2006). Effects of donepezil on neuropsychiatric symptoms in patients with dementia and severe behavioral disorders. Am. J. Geriatr. Psychiatry 14, 605–612. doi: 10.1097/01.JGP.0000221293.91312.d3
Darcet, F., Gardier, A. M., David, D. J., and Guilloux, J.-P. (2016). Chronic 5-HT4 receptor agonist treatment restores learning and memory deficits in a neuroendocrine mouse model of anxiety/depression. Neurosci. Lett. 616, 197–203. doi: 10.1016/j.neulet.2016.01.055
Diaz-Perdigon, T., Belloch, F. B., Ricobaraza, A., Elboray, E. E., Suzuki, T., Tordera, R. M., et al. (2020). Early Sirtuin 2 inhibition prevents age-related cognitive decline in a senescence-accelerated mouse model. Neuropsychopharmacology 45, 347–357. doi: 10.1038/s41386-019-0503-8
Dilsaver, S. C., and Coffman, J. A. (1989). Cholinergic hypothesis of depression: a reappraisal. J. Clin. Psychopharmacol. 9, 173–179. doi: 10.1097/00004714-198906000-00003
Diniz, B. S., Butters, M. A., Albert, S. M., Dew, M. A., and Reynolds, C. F. (2013). Late-life depression and risk of vascular dementia and Alzheimer’s disease: systematic review and meta-analysis of community-based cohort studies. Br. J. Psychiatry 202, 329–335. doi: 10.1192/bjp.bp.112.118307
Dudas, R., Malouf, R., McCleery, J., and Dening, T. (2018). Antidepressants for treating depression in dementia. Cochrane Database Syst. Rev. 2018:CD003944. doi: 10.1002/14651858.CD003944.pub2
Fakhoury, M. (2016). Revisiting the serotonin hypothesis: implications for major depressive disorders. Mol. Neurobiol. 53, 2778–2786. doi: 10.1007/s12035-015-9152-z
Galts, C. P. C., Bettio, L. E. B., Jewett, D. C., Yang, C. C., Brocardo, P. S., Rodrigues, A. L. S., et al. (2019). Depression in neurodegenerative diseases: common mechanisms and current treatment options. Neurosci. Biobehav. Rev. 102, 56–84. doi: 10.1016/j.neubiorev.2019.04.002
Gauthier, S., Feldman, H., Hecker, J., Vellas, B., Ames, D., Subbiah, P., et al. (2002). Efficacy of donepezil on behavioral symptoms in patients with moderate to severe Alzheimer’s disease. Int. Psychogeriatr. 14, 389–404. doi: 10.1017/s104161020200858x
Gauthier, S., Loft, H., and Cummings, J. (2008). Improvement in behavioural symptoms in patients with moderate to severe Alzheimer’s disease by memantine: a pooled data analysis. Int. J. Geriatr. Psychiatry 23, 537–545. doi: 10.1002/gps.1949
Geldenhuys, W. J., and Van der Schyf, C. J. (2011). Role of serotonin in Alzheimer’s disease. CNS Drugs 25, 765–781. doi: 10.2165/11590190-000000000-00000
Giannouli, V. (2024). “The impact of stressful life events in Alzheimer’s disease” in Handbook of the behavior and psychology of disease. eds. C. R. Martin, V. R. Preedy, V. B. Patel, and R. Rajendram (Cham: Springer).
Giannouli, V., and Tsolaki, M. (2021). Mild Alzheimer disease, financial capacity, and the role of depression: eyes wide shut? Alzheimer Dis. Assoc. Disord. 35, 360–362. doi: 10.1097/WAD.0000000000000427
Giannouli, V., and Tsolaki, M. (2022). Is negative affect associated with deficits in financial capacity in nondepressed older adults? A preliminary study. J. Affect. Disord. Rep. 10:100391. doi: 10.1016/j.jadr.2022.100391
Giannouli, V., and Tsolaki, M. (2023). In the hands of hypnos: associations between sleep, cognitive performance and financial capacity in aMCI and mild AD. Sleep Sci 16, 231–236. doi: 10.1055/s-0043-1770796
González-Martín, A. M., Aibar Almazán, A., Rivas Campo, Y., Rodríguez Sobrino, N., and Castellote Caballero, Y. (2023). Addressing depression in older adults with Alzheimer’s through cognitive behavioral therapy: systematic review and meta-analysis. Front. Aging Neurosci. 15:1222197. doi: 10.3389/fnagi.2023.1222197
Grunblatt, E., Zehetmayer, S., Bartl, J., Loffler, C., Wichart, I., Rainer, M. K., et al. (2009). Genetic risk factors and markers for Alzheimer’s disease and/or depression in the VITA study. J. Psychiatr. Res. 43, 298–308. doi: 10.1016/j.jpsychires.2008.05.008
Guan, C., Amdanee, N., Liao, W., Zhou, C., Wu, X., Zhang, X., et al. (2022). Altered intrinsic default mode network functional connectivity in patients with remitted geriatric depression and amnestic mild cognitive impairment. Int. Psychogeriatr. 34, 703–714. doi: 10.1017/S1041610221001174
Guétin, S., Portet, F., Picot, M. C., Pommié, C., Messaoudi, M., Djabelkir, L., et al. (2009). Effect of music therapy on anxiety and depression in patients with Alzheimer’s type dementia: randomised, controlled study. Dement Geriatr Cogn Disord. 28:36–46. doi: 10.1159/000229024
He, Y., Li, H., Huang, J., Huang, S., Bai, Y., Li, Y., et al. (2021). Efficacy of antidepressant drugs in the treatment of depression in Alzheimer disease patients: a systematic review and network meta-analysis. J. Psychopharmacol. 35, 901–909. doi: 10.1177/02698811211030181
Hermann, K. (2005) The influence of social self-efficacy, self-esteem, and personality differences on loneliness and depression. Ohio State University, Doctoral dissertation.
Herrmann, N., Rabheru, K., Wang, J., and Binder, C. (2005). Galantamine treatment of problematic behavior in Alzheimer disease: post-hoc analysis of pooled data from three large trials. Am. J. Geriatr. Psychiatry 13, 527–534. doi: 10.1176/appi.ajgp.13.6.527
Hoogendijk, W. J., Meynen, G., Endert, E., Hofman, M. A., and Swaab, D. F. (2006). Increased cerebrospinal fluid cortisol level in Alzheimer’s disease is not related to depression. Neurobiol. Aging 27, 780.e1–780.e2. doi: 10.1016/j.neurobiolaging.2005.07.017
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Hsu, T. W., Stubbs, B., Liang, C. S., Chen, T. Y., Yeh, T. C., Pan, C. C., et al. (2021). Efficacy of serotonergic antidepressant treatment for the neuropsychiatric symptoms and agitation in dementia: a systematic review and meta-analysis. Ageing Res. Rev. 69:101362. doi: 10.1016/j.arr.2021.101362
Huang, I. C., Chang, T. S., Chen, C., and Sung, J. Y. (2022). Effect of vortioxetine on cognitive impairment in patients with major depressive disorder: a systematic review and meta-analysis of randomized controlled trials. Int. J. Neuropsychopharmacol. 25, 969–978. doi: 10.1093/ijnp/pyac054
Huang, Y. Y., Gan, Y. H., Yang, H., Cheng, H., and Yu, J. T. (2024). Depression in Alzheimer’s disease: epidemiology, mechanisms, and treatment. Biol. Psychiatry 95, 992–1005. doi: 10.1016/j.biopsych.2023.10.008
Huppert, F. A., Brayne, C., Gill, C., Paykel, E. S., and Beardsall, L. (1995). CAMCOG – a concise neuropsychological test to assist dementia diagnosis: socio-demographic determinants in an elderly population sample. Br. J. Clin. Psychol. 34, 529–541. doi: 10.1111/j.2044-8260.1995.tb01487.x
Kepe, V., Barrio, J. R., Huang, S.-C., Ercoli, L., Siddarth, P., Shoghi-Jadid, K., et al. (2006). Serotonin 1A receptors in the living brain of Alzheimer’s disease patients. Proc. Natl. Acad. Sci. 103, 702–707. doi: 10.1073/pnas.0510237103
Krishnan, V., and Nestler, E. J. (2008). The molecular neurobiology of depression. Nature 455, 894–902. doi: 10.1038/nature07455
Lanctôt, K. L., Hussey, D. F., Herrmann, N., Black, S. E., Rusjan, P. M., Wilson, A. A., et al. (2007). A positron emission tomography study of 5-hydroxytryptamine-1A receptors in Alzheimer disease. Am. J. Geriatr. Psychiatry 15, 888–898. doi: 10.1097/JGP.0b013e3180488325
Langa, K. M., and Levine, D. A. (2014). The diagnosis and management of mild cognitive impairment: a clinical review. JAMA 312, 2551–2561. doi: 10.1001/jama.2014.13806
Li, N., Bai, N., Zhao, X., Cheng, R., Wu, X., Jiang, B., et al. (2023). Cooperative effects of SIRT1 and SIRT2 on APP acetylation. Aging Cell 22:e13967. doi: 10.1111/acel.13967
Li, Z., Liu, S., Wu, Q., Li, J., Yang, Q., Wang, X., et al. (2023). Effectiveness and safety of vortioxetine for the treatment of major depressive disorder in the real world: a systematic review and meta-analysis. Int. J. Neuropsychopharmacol. 26, 373–384. doi: 10.1093/ijnp/pyad018
Li, S., Zhang, Q., Liu, J., Zhang, N., Li, X., Liu, Y., et al. (2025). Bibliometric analysis of Alzheimer's disease and depression. Curr. Neuropharmacol. 23, 98–115. doi: 10.2174/1570159X22666240730154834
Liu, H. C., Hong, C. J., Liu, C. Y., Lin, K. N., Tsai, S. J., Liu, T. Y., et al. (2001). Association analysis of the 5-HT6 receptor polymorphism C267T with depression in patients with Alzheimer’s disease. Psychiatry Clin. Neurosci. 55, 427–429. doi: 10.1046/j.1440-1819.2001.00886.x
Liu, M., Xie, X., Xie, J., Tian, S., Du, X., Feng, H., et al. (2023). Early-onset Alzheimer’s disease with depression as the first symptom: a case report with literature review. Front Psychiatry. 14:1192562. doi: 10.3389/fpsyt.2023.1192562
Liu, Y., Tan, Y., Zhang, Z., Yi, M., Zhu, L., and Peng, W. (2024). The interaction between ageing and Alzheimer's disease: insights from the hallmarks of ageing. Transl. Neurodegener. 13:7. doi: 10.1186/s40035-024-00397-x
LoBue, C., Chiang, H. S., Salter, A., McClintock, S., Nguyen, T. P., Logan, R., et al. (2025). High-definition transcranial direct current stimulation as an intervention for cognitive deficits in Alzheimer’s dementia: a randomized controlled trial. J. Prev Alzheimers Dis. 12:100023. doi: 10.1016/j.tjpad.2024.100023
Luchsinger, J. A., Honig, L. S., Tang, M. X., and Devanand, D. P. (2008). Depressive symptoms, vascular risk factors, and Alzheimer’s disease. Int. J. Geriatr. Psychiatry 23, 922–928. doi: 10.1002/gps.2006
Lyketsos, C. G. (2007). Neuropsychiatric symptoms (behavioral and psychological symptoms of dementia) and the development of dementia treatments. Int. Psychogeriatr. 19, 409–420. doi: 10.1017/S104161020700484X
Ma, J., Gao, Y., Jiang, L., Chao, F. L., Huang, W., Zhou, C. N., et al. (2017). Fluoxetine attenuates the impairment of spatial learning ability and prevents neuron loss in middle-aged APPswe/PSEN1dE9 double transgenic Alzheimer’s disease mice. Oncotarget 8, 27676–27692. doi: 10.18632/oncotarget.15398
Magalhães, R., Marques, F., Selingue, E., Boumezbeur, F., Mériaux, S., and Sousa, N. (2024). A longitudinal MRI analysis reveals altered brain connectivity and microstructural changes in a transgenic mouse model of Alzheimer's disease. Neurobiol. Dis. 201:106679. doi: 10.1016/j.nbd.2024.106679
Maidment, I. D., Fox, C. G., Boustani, M., Rodriguez, J., Brown, R. C., and Katona, C. L. (2008). Efficacy of memantine on behavioral and psychological symptoms related to dementia: a systematic meta-analysis. Ann. Pharmacother. 42, 32–38. doi: 10.1345/aph.1K372
Matrone, C., Ciotti, M. T., Mercanti, D., Marolda, R., and Calissano, P. (2008). NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 105, 13139–13144. doi: 10.1073/pnas.0806133105
Matsuno, H., Tsuchimine, S., O’Hashi, K., Sakai, K., Hattori, K., Hidese, S., et al. (2022). Association between vascular endothelial growth factor-mediated blood-brain barrier dysfunction and stress-induced depression. Mol. Psychiatry 27, 3822–3832. doi: 10.1038/s41380-022-01618-3
Menard, C., Pfau, M. L., Hodes, G. E., Kana, V., Wang, V. X., Bouchard, S., et al. (2017). Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 20, 1752–1760. doi: 10.1038/s41593-017-0010-3
Mendez-David, I., David, D. J., Darcet, F., Wu, M. V., Kerdine-Römer, S., Gardier, A. M., et al. (2014). Rapid anxiolytic effects of a 5-HT4 receptor agonist are mediated by a neurogenesis-independent mechanism. Neuropsychopharmacology 39, 1366–1378. doi: 10.1038/npp.2013.332
Micheli, D., Bonvicini, C., Rocchi, A., Ceravolo, R., Mancuso, M., Tognoni, G., et al. (2006). No evidence for allelic association of serotonin 2A receptor and transporter gene polymorphisms with depression in Alzheimer disease. J. Alzheimers Dis. 10, 371–378. doi: 10.3233/jad-2006-10405
Miller, M. D., and Reynolds, C. F. (2007). Expanding the usefulness of interpersonal psychotherapy (IPT) for depressed elders with co-morbid cognitive impairment. Int. J. Geriatr. Psychiatry 22, 101–105. doi: 10.1002/gps.1699
Milwain, E. J., and Nagy, Z. (2005). Depressive symptoms increase the likelihood of cognitive impairment in elderly people with subclinical Alzheimer pathology. Dement. Geriatr. Cogn. Disord. 19, 46–50. doi: 10.1159/000080971
Mitchell, A. J., and Subramaniam, H. (2005). Prognosis of depression in old age compared to middle age: a systematic review of comparative studies. Am. J. Psychiatry 162, 1588–1601. doi: 10.1176/appi.ajp.162.9.1588
Modrego, P. J. (2010). Depression in Alzheimer’s disease. Pathophysiology, diagnosis, and treatment. J. Alzheimers Dis. 21, 1077–1087. doi: 10.3233/jad-2010-100153
Mohammad-Zadeh, L. F., Moses, L., and Gwaltney-Brant, S. M. (2008). Serotonin: a review. J. Vet. Pharmacol. Ther. 31, 187–199. doi: 10.1111/j.1365-2885.2008.00944.x
Monereo-Sánchez, J., Schram, M. T., Frei, O., O’Connell, K., Shadrin, A. A., Smeland, O. B., et al. (2021). Genetic overlap between Alzheimer’s disease and depression mapped onto the brain. Front. Neurosci. 15:653130. doi: 10.3389/fnins.2021.653130
Moon, Y., Kim, H., Ok Kim, J., and Han, S. H. (2013). Vascular factors are associated with the severity of the neuropsychiatric symptoms in Alzheimer’s disease. Int. J. Neurosci. 124, 512–517. doi: 10.3109/00207454.2013.856902
Morris, J. C., Roe, C. M., Xiong, C., Fagan, A. M., Goate, A. M., Holtzman, D. M., et al. (2010). APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 67, 122–131. doi: 10.1002/ana.21843
Nation, D. A., Sweeney, M. D., Montagne, A., Sagare, A. P., D’Orazio, L. M., Pachicano, M., et al. (2019). Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 25, 270–276. doi: 10.1038/s41591-018-0297-y
Notarianni, E. (2013). Hypercortisolemia and glucocorticoid receptor-signaling insufficiency in Alzheimer’ s disease initiation and development. Curr. Alzheimer Res. 10, 714–731. doi: 10.2174/15672050113109990137
Olin, J. T., Katz, I. R., Meyers, B. S., Schneider, L. S., and Lebowitz, B. D. (2002). Provisional diagnostic criteria for depression of Alzheimer disease: rationale and background. Am. J. Geriatr. Psychiatry 10, 129–141. doi: 10.1097/00019442-200203000-00004
Orgeta, V., Tabet, N., Nilforooshan, R., and Howard, R. (2017). Efficacy of antidepressants for depression in Alzheimer’s disease: systematic review and meta-analysis. J. Alzheimers Dis. 58, 725–733. doi: 10.3233/JAD-161247
Orgeta, V., Leung, P., Del-Pino-Casado, R., Qazi, A., Orrell, M., Spector, A. E., et al. (2022). Psychological treatments for depression and anxiety in dementia and mild cognitive impairment. Cochrane Database Syst Rev. ; 4:CD009125. doi: 10.1002/14651858.CD009125.pub3
Ownby, R. L., Crocco, E., Acevedo, A., John, V., and Loewenstein, D. (2006). Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch. Gen. Psychiatry 63, 530–538. doi: 10.1001/archpsyc.63.5.530
Pláteník, J., Fišar, Z., Buchal, R., Jirák, R., Kitzlerová, E., Zvěřová, M., et al. (2014). GSK3β, CREB, and BDNF in peripheral blood of patients with Alzheimer\u0027s disease and depression. Prog Neuropsychopharmacol Biol Psychiatry. ; 50:83–93. doi: 10.1016/j.pnpbp.2013.12.001
Popp, J., Schaper, K., Kolsch, H., Cvetanovska, G., Rommel, F., Klingmuller, D., et al. (2009). CSF cortisol in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 30, 498–500. doi: 10.1016/j.neurobiolaging.2007.07.007
Porcelli, S., Salfi, R., Politis, A., Atti, A. R., Albani, D., Chierchia, A., et al. (2013). Association between Sirtuin 2 gene rs10410544 polymorphism and depression in Alzheimer’s disease in two independent European samples. J. Neural Transm. 120, 1709–1715. doi: 10.1007/s00702-013-1045-6
Price, J. L., and Drevets, W. C. (2010). Neurocircuitry of mood disorders. Neuropsychopharmacology 35, 192–216. doi: 10.1038/npp.2009.104
Pritchard, A. L., Pritchard, C. W., Bentham, P., and Lendon, C. L. (2007). Role of serotonin transporter polymorphisms in the behavioural and psychological symptoms in probable Alzheimer disease patients. Dement. Geriatr. Cogn. Disord. 24, 201–206. doi: 10.1159/000107081
Proitsi, P., Lupton, M. K., Reeves, S. J., Hamilton, G., Archer, N., Martin, B. M., et al. (2012). Association of serotonin and dopamine gene pathways with behavioral subphenotypes in dementia. Neurobiol. Aging 33, 791–803. doi: 10.1016/j.neurobiolaging.2010.06.011
Purandare, N., Voshaar, R. C., Hardicre, J., Byrne, J., McCollum, C., and Burns, A. (2006). Cerebral emboli and depressive symptoms in dementia. Br. J. Psychiatry 189, 260–263. doi: 10.1192/bjp.bp.105.016188
Quintana-Hernández, D. J., Rojas-Hernández, J., Santana-del Pino, A., Céspedes Suarez, C., Pellejero Silva, M., Miró-Barrachina, M. T., et al. (2023). Mindfulness prevents depression and psychopathology in elderly people with mild to moderate Alzheimer’s disease: a randomized clinical trial. J. Alzheimers Dis. 91, 471–481. doi: 10.3233/JAD-220889
Rapp, M. A., Schnaider-Beeri, M., Grossman, H. T., Sano, M., Perl, D. P., Purohit, D. P., et al. (2006). Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch. Gen. Psychiatry 63:161. doi: 10.1001/archpsyc.63.2.161
Regan, C., Katona, C., Walker, Z., Hooper, J., Donovan, J., and Livingston, G. (2006). Relationship of vascular risk to the progression of Alzheimer disease. Neurology 67, 1357–1362. doi: 10.1212/01.wnl.0000240129.46080.53
Rosler, M., Retz, W., Retz-Junginger, P., and Dennler, H. J. (1998). Effects of two-year treatment with the cholinesterase inhibitor rivastigmine on behavioural symptoms in Alzheimer’s disease. Behav. Neurol. 11, 211–216. doi: 10.1155/1999/168023
Sanchez, C., Asin, K. E., and Artigas, F. (2015). Vortioxetine, a novel antidepressant with multimodal activity: review of preclinical and clinical data. Pharmacol. Ther. 145, 43–57. doi: 10.1016/j.pharmthera.2014.07.001
Sharan, P., and Vellapandian, C. (2024). Hypothalamic-pituitary-adrenal (HPA) Axis: unveiling the potential mechanisms involved in stress-induced Alzheimer's disease and depression. Cureus 16:e67595. doi: 10.7759/cureus.67595
Sheline, Y. I., Snider, B. J., Beer, J. C., Seok, D., Fagan, A. M., Suckow, R. F., et al. (2020). Effect of escitalopram dose and treatment duration on CSF aβ levels in healthy older adults: a controlled clinical trial. Neurology 95, e2658–e2665. doi: 10.1212/WNL.0000000000010725
Stanciu, G. D., Luca, A., Rusu, R. N., Bild, V., Beschea Chiriac, S. I., Solcan, C., et al. (2020). Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomol. Ther. 10:40. doi: 10.3390/biom10010040
Steinberg, M., Hess, K., Corcoran, C., Mielke, M. M., Norton, M., Breitner, J., et al. (2014). Vascular risk factors and neuropsychiatric symptoms in Alzheimer’s disease: the Cache County study. Int. J. Geriatr. Psychiatry 29, 153–159. doi: 10.1002/gps.3980
Storck, S. E., Meister, S., Nahrath, J., Meißner, J. N., Schubert, N., Di Spiezio, A., et al. (2016). Endothelial LRP1 transports amyloid-β(1–42) across the blood-brain barrier. J. Clin. Invest. 126, 123–136. doi: 10.1172/JCI81108
Sun, X., Chiu, C. C., Liebson, E., Crivello, N. A., Wang, L., Claunch, J., et al. (2009). Depression and plasma amyloid β peptides in the elderly with and without the apolipoprotein E4 allele. Alzheimer Dis. Assoc. Disord. 23, 238–244. doi: 10.1097/WAD.0b013e31819cb3ac
Tajeddinn, W., Persson, T., Calvo-Garrido, J., Seed Ahmed, M., Maioli, S., Vijayaraghavan, S., et al. (2016). Pharmacological modulations of the serotonergic system in a cell-model of familial Alzheimer’s disease. J. Alzheimers Dis. 53, 349–361. doi: 10.3233/JAD-160046
Tangney, C. C., Li, H., Wang, Y., Barnes, L., Schneider, J. A., Bennett, D. A., et al. (2014). Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology 83, 1410–1416. doi: 10.1212/WNL.0000000000000884
Taylor, W. D., Aizenstein, H. J., and Alexopoulos, G. S. (2013). The vascular depression hypothesis: mechanisms linking vascular disease with depression. Mol. Psychiatry 18, 963–974. doi: 10.1038/mp.2013.20
Thompson, S., Herrmann, N., Rapoport, M. J., and Lanctot, K. L. (2007). Efficacy and safety of antidepressants for treatment of depression in Alzheimer’s disease: a meta-analysis. Can. J. Psychiatr. 52, 248–255. doi: 10.1177/070674370705200407
Tong, L., Balazs, R., Thornton, P. L., and Cotman, C. W. (2004). Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J. Neurosci. 24, 6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004
Tonga, J. B., Šaltytė Benth, J., Arnevik, E. A., Werheid, K., Korsnes, M. S., and Ulstein, I. D. (2021). Managing depressive symptoms in people with mild cognitive impairment and mild dementia with a multicomponent psychotherapy intervention: a randomized controlled trial. Int. Psychogeriatr. 33, 217–231. doi: 10.1017/S1041610220000216
Treiber, K. A., Lyketsos, C. G., Corcoran, C., Steinberg, M., Norton, M., Green, R. C., et al. (2008). Vascular factors and risk for neuropsychiatric symptoms in Alzheimer’s disease: the Cache County study. Int. Psychogeriatr. 20, 538–553. doi: 10.1017/S1041610208006704
Usman, S., Chaudhary, H. R., Asif, A., and Yahya, M. I. (2010). Severity and risk factors of depression in Alzheimer’s disease. J. Coll. Physicians Surg. Pak. 20, 327–330.
van Dooren, F. E., Schram, M. T., Schalkwijk, C. G., Stehouwer, C. D., Henry, R. M., Dagnelie, P. C., et al. (2016). Associations of low-grade inflammation and endothelial dysfunction with depression—the Maastricht study. Brain Behav. Immun. 56, 390–396. doi: 10.1016/j.bbi.2016.03.004
Wang, S. M., Kang, D. W., Um, Y. H., Kim, S., Lee, C. U., and Lim, H. K. (2023). Depression is associated with the aberration of resting state default mode network functional connectivity in patients with amyloid-positive mild cognitive impairment. Brain Sci. 13:1111. doi: 10.3390/brainsci13071111
Wang, S. M., Kim, N. Y., Um, Y. H., Kang, D. W., Na, H. R., Lee, C. U., et al. (2021). Default mode network dissociation linking cerebral beta amyloid retention and depression in cognitively normal older adults. Neuropsychopharmacology 46, 2180–2187. doi: 10.1038/s41386-021-01072-9
Wang, H.-Y., Lee, D. H., D’Andrea, M. R., Paterson, P. A., Shank, R. P., and Reitz, A. B. (2000). Β-Amyloid 1-42 binds to α7 nicotinic acetylcholine receptor with high affinity. J. Biol. Chem. 275, 5626–5632. doi: 10.1074/jbc.275.8.5626
Wang, W., Li, Y., Ma, F., Sheng, X., Chen, K., Zhuo, R., et al. (2023). Microglial repopulation reverses cognitive and synaptic deficits in an Alzheimer's disease model by restoring BDNF signaling. Brain Behav. Immun. 113, 275–288. doi: 10.1016/j.bbi.2023.07.011
Weiner, M. F., Martin-Cook, K., Foster, B. M., Saine, K., Fontaine, C. S., and Svetlik, D. A. (2000). Effects of donepezil on emotional/ behavioral symptoms in Alzheimer’s disease patients. J. Clin. Psychiatry 61, 487–492. doi: 10.4088/jcp.v61n0705
Wengreen, H., Munger, R. G., Cutler, A., Quach, A., Bowles, A., Corcoran, C., et al. (2013). Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am. J. Clin. Nutr. 98, 1263–1271. doi: 10.3945/ajcn.112.051276
Winkler, E. A., Nishida, Y., Sagare, A. P., Rege, S. V., Bell, R. D., Perlmutter, D., et al. (2015). GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 18, 521–530. doi: 10.1038/nn.3966
Wu, K. M., Zhang, Y. R., Huang, Y. Y., Dong, Q., Tan, L., and Yu, J. T. (2021). The role of the immune system in Alzheimer’s disease. Ageing Res. Rev. 70:101409. doi: 10.1016/j.arr.2021.101409
Yin, Y., Hou, Z., Wang, X., Sui, Y., and Yuan, Y. (2015). Association between altered resting-state cortico-cerebellar functional connectivity networks and mood/cognition dysfunction in late-onset depression. J. Neural Transm. (Vienna) 122, 887–896. doi: 10.1007/s00702-014-1347-3
Yun, J. Y., and Kim, Y. K. (2021). Graph theory approach for the structural-functional brain connectome of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 111:110401. doi: 10.1016/j.pnpbp.2021.110401
Yun, H. M., Park, K. R., Kim, E. C., Kim, S., and Hong, J. T. (2015). Serotonin 6 receptor controls Alzheimer's disease and depression. Oncotarget 6, 26716–26728. doi: 10.18632/oncotarget.5777
Zahodne, L. B., Ornstein, K., Cosentino, S., Devanand, D. P., and Stern, Y. (2015). Longitudinal relationships between Alzheimer disease progression and psychosis, depressed mood, and agitation/aggression. Am. J. Geriatr. Psychiatry 23, 130–140. doi: 10.1016/j.jagp.2013.03.014
Zhan, Q., Kong, F., Shao, S., Zhang, B., and Huang, S. (2024). Pathogenesis of depression in Alzheimer’s disease. Neurochem. Res. 49, 548–556. doi: 10.1007/s11064-023-04061-0
Zhang, L., Fang, Y., Zeng, Z., Lian, Y., Wei, J., Zhu, H., et al. (2011). BDNF gene polymorphisms are associated with Alzheimer’s disease-related depression and antidepressant response. J. Alzheimers Dis. 26, 523–530. doi: 10.3233/JAD-2011-110113
Zhao, D., Xu, X., Pan, L., Zhu, W., Fu, X., Guo, L., et al. (2017). Pharmacologic activation of cholinergic alpha7 nicotinic receptors mitigates depressive-like behavior in a mouse model of chronic stress. J. Neuroinflammation 14:234. doi: 10.1186/s12974-017-1007-2
Zhou, C. N., Chao, F. L., Zhang, Y., Jiang, L., Zhang, L., Fan, J. H., et al. (2018). Fluoxetine delays the cognitive function decline and synaptic changes in a transgenic mouse model of early Alzheimer’s disease. J. Comp. Neurol. 527, 1378–1387. doi: 10.1002/cne.24616
Keywords: Alzheimer’s disease, depression, shared neurobiological mechanisms, depression–Alzheimer’s disease comorbidity, bidirectional relationship
Citation: Untu I, Davidson M, Stanciu G-D, Rabinowitz J, Dobrin R-P, Vieru D-S and Tamba B-I (2025) Neurobiological and therapeutic landmarks of depression associated with Alzheimer’s disease dementia. Front. Aging Neurosci. 17:1584607. doi: 10.3389/fnagi.2025.1584607
Edited by:
Maria Luisa Soto-Montenegro, Instituto de Investigación Sanitaria Gregorio Marañón, SpainReviewed by:
Vaitsa Giannouli, Aristotle University of Thessaloniki, GreeceMaria Luisa Garo, Mathsly Research, Italy
Laura Vegas Gómez, University of Malaga, Spain
Copyright © 2025 Untu, Davidson, Stanciu, Rabinowitz, Dobrin, Vieru and Tamba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela-Dumitrita Stanciu, Z2FicmllbGEtZHVtaXRyaXRhLnNAdW1maWFzaS5ybw==