 Ryszard Maleszka
Ryszard Maleszka- Research School of Biology, Australian National University, Canberra, ACT, Australia
Certain aspects of animal ageing can be quantified using molecular clocks or machine learning algorithms that are trained on specific omics data, with epigenetic clocks based on DNA methylation (DNAm) garnering the most attention. While the accuracy of epigenetic clocks has been established in mammals and several vertebrates, their applicability to invertebrates, which comprise 97% of all animal species, remains largely theoretical. In this context, we consider whether the relationship between chronological clocks, biological clocks, and DNA methylation is ancestral and evolutionarily conserved, potentially making it relevant beyond the vertebrate lineage. Evolutionary comparisons may help us determine whether epigenetic clocks are inherent mechanisms implemented during ageing or simply reflect the progressive erosion of epigenomic marks. These comparisons could also reveal the likely generality of the results from one type of epigenetic clock to another. We emphasise the substantial biological differences between invertebrates and mammals, all of which must be considered when evaluating the universality of epigenetic clocks. We conclude that mammalian-style DNAm epigenetic clocks are unlikely to be applicable to most invertebrates. We propose that quantitative approaches to ageing in non-vertebrate organisms should be specifically tailored to leverage the molecular mechanisms and distinct biology of different lineages.
Introduction
“Choosing the right organism for one’s research is as important as finding the right problems to work on (Sydney Brenner, Nobel Lecture, 2002).”
Over the past decade, research has documented age-related changes in the mammalian DNA methylome and identified specific cytosine methylation sites that, when combined, can be used to measure chronological and biological age. This concept is known as the DNA methylation (DNAm) clock or, more broadly, the epigenetic clock (Teschendorff and Horvath, 2025; Field et al., 2018). It is considered a valuable biomarker for distinguishing between healthy and unhealthy ageing and for assessing disease risks, such as late-onset cancers. Although some methylated CpG dinucleotides have been deemed potentially causal, it is not clear at present if the DNA methylation differences used for predicting age contribute to the ageing process or are just bystanders reflecting spatio-temporal erosion of flexible epigenomic marks (Ying et al., 2024; Yang et al., 2023; Bertucci-Richter and Parrott, 2023).
As with many intriguing discoveries in one group of organisms, numerous follow-up studies have been conducted to explore whether epigenetic clocks based on DNA methylation can measure chronological or biological age in non-mammalian species. While the premise of epigenetic clocks seems to hold in several vertebrates tested so far (Zoller et al., 2024), evidence supporting this process in invertebrates is contentious and remains hypothetical (Liu et al., 2025).
Since more than 97% of all animal species are invertebrates (May 1988), we ask whether the relationship between chronological clocks, biological clocks, and DNA methylation is ancestral and evolutionarily conserved and, thus, might be applicable outside the vertebrate lineage.
We highlight the important biological differences between mammals and invertebrates that must be considered in this context. These differences include their distinct DNA methylation machinery, with multiple examples of invertebrates missing this level of epigenomic modification (Kucharski et al., 2023), their varying and elaborate life cycles, and the lineage-specific epigenetic nature of relatively short lifespans (Maleszka and Kucharski, 2022; Yu et al., 2019; Heinze and Giehr, 2021). The long-lived reproductive females in eusocial insects are presented as an example of epigenetically controlled environmental influence on the genomic capacity to generate contrasting organismal outcomes, including extended longevity (Heinze and Giehr, 2021; Miklos and Maleszka, 2011; Blacher et al., 2017).
We conclude that the mechanisms driving mammalian epigenetic clocks, which are based on DNA methylation, cannot be directly applied to most invertebrates. Short-lived organisms represent a fundamentally different evolutionary strategy, and the idiosyncrasies influencing their ageing may not be comparable to those in longer-lived vertebrate models and to humans. We propose that innovative approaches, not confined to DNA methylation alone, are essential for developing algorithms that measure age in invertebrates based on molecular changes.
What makes the transferability of mammalian DNAm epigenetic clocks to invertebrates problematic?
Historically, invertebrate research has been at the forefront of biological science, paving the way for advancements in mammalian studies, including those related to human health. Model organisms like Drosophila melanogaster and C. elegans are regarded as gold standards in many spheres of discovery (Bertile et al., 2023; Miklos and Maleszka, 2000). However, in the context of methylomics, specifically DNAm epigenetic clocks, the process is reversed, with data from mammals being utilised to inform research on invertebrates. This approach carries inherent risks, as transferring data and concepts between mammals and evolutionarily older lineages can be problematic (Maleszka and Kucharski, 2022; Maleszka et al., 1998; Hardison, 2016; Setola and Roth, 2003).
Vertebrates and invertebrates represent two extremely diverse classes of animals. When considering DNAm epigenetic clocks, several aspects of their respective biology must be considered.
First, while the accuracy for predicting chronological age is ±1–3 years (Zhang et al., 2019), impressive for mammals, this error range encompasses the entire lifespan of most invertebrates, such as nematodes and insects. Although some species, including sponges, jellyfish, and annelids, exhibit remarkably long lifespans, sometimes referred to as “immortal”, their unusual longevity is not necessarily linked to the presence of DNA methylation machinery (see discussion below).
Second, the age-related changes in DNA methylation are subtle, typically involving only 2%–5% of methylated CpG dinucleotides over many decades (Teschendorff and Horvath, 2025; Seale et al., 2024). Thus, in addition to the timeframe that does not apply to most invertebrates, discovering rare predictive CpGs in their sparsely methylated genomes poses an entirely different challenge. Another feature of mammalian epigenetic clocks that is not compatible with most invertebrates’ lifespans is the 24-h periodicity with fluctuations up to 5 years within a single day (Koncevičius et al., 2024).
Third, various advanced tools, such as Illumina bead array technology and sophisticated algorithms trained on specific omics, can achieve the precision of DNA methylation changes observed in mammals (Bell et al., 2019). These technologies have not yet been developed for use in other lineages.
Finally, the solution to the problem of the universality of epigenetic clocks is not limited to methyl-cytosines; it must also include a better understanding of demethylation and methyl-binding systems, both of which are under-researched in invertebrates (Cramer et al., 2017; Wojciechowski et al., 2014).
Longevity, ageing and DNA methylation in invertebrates
DNA methylation is an evolutionarily ancient feature found in basal metazoans (Dabe et al., 2015; Ying et al., 2022), including sponges, which are regarded as the most ancient extant metazoan lineage that diverged from other metazoans over 600 million years ago (Yi et al., 2015). However, there is no clear correlation between lifespans, senescence, and the presence of DNA methylation biochemistry across the tree of life (Table 1) (Maleszka and Kucharski, 2022; Lewis et al., 2020). Among the most ancient metazoans, the distribution of the DNA methylation toolkit is not uniform (Sarkies, 2022). In the four basal phyla, Porifera (sponges), Cnidaria (sea anemones, corals, and jellyfish), Ctenophora (comb jellies), and Placozoa, some species possess the core enzymes of the DNA methylation toolkit, namely, both types of DNA methyltransferases (DNMTs), DNMT1 and DNMT3, and ten-eleven translocation (TET) methylcytosine dioxygenases, while others do not. Notably, this characteristic is not linked to longevity or senescence. Long-lived sponges and all comb-jellies possess this enzymology, whereas all four extant species of Placozoa do not have this level of epigenomic modification (Wedd et al., 2022). In the phylum Cnidaria, the “immortal” jellyfish Turritopsis dohrnii lacks both DNMTs and TET enzymes, whereas another Cnidarian, Hydra vulgaris, which exhibits no apparent senescence, possesses DNMT1 and DNMT3, as well as TET.
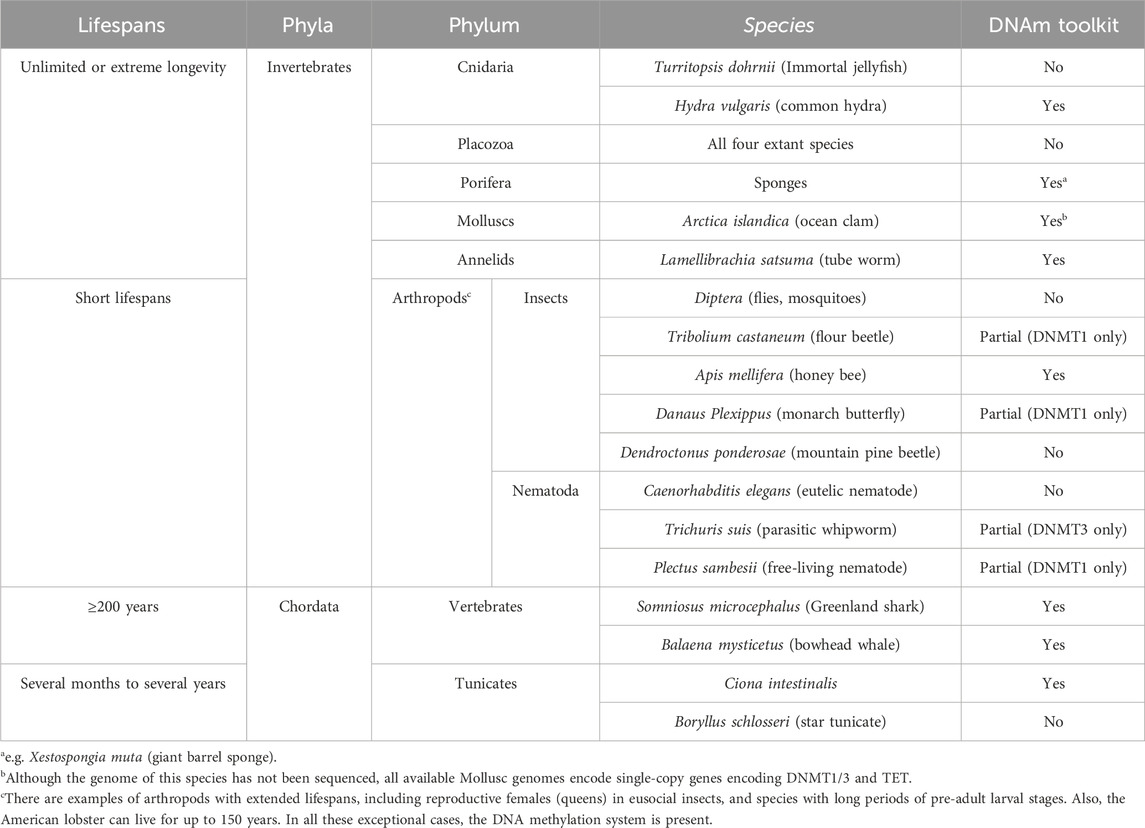
Table 1. Longevity and DNA methylation systems in selected organisms. These examples illustrate the lack of correlation between maximum lifespans and the presence of highly diverse DNA methylation machinery across various taxa.
Insects have the most diverse complements of DNMTs with no apparent relationship to their lifespans, developmental strategies, or life cycles (Kucharski et al., 2023; Bewick et al., 2017). All dipterans (e.g., flies, mosquitoes) have lost their DNA methylation toolkits, and DNMT-less species are found across other large orders, such as Coleoptera (beetles), Strepsiptera (twisted-wing parasites), and Neuroptera (net-winged insects), among others. While there is nothing different or unusual about the lifespans of insects without DNA methylation, DNAm epigenetic clocks cannot be used to measure their ageing. In insects that methylate their genomes, most have only DNMT1, although the copy number varies from 1 to 3. In these species, DNMT1 likely possesses both de novo and maintenance capacities, although its catalytic activity has not been experimentally investigated. In approximately 25% of insects, both DNMT1 and DNMT3 have been identified, with variable copy numbers (Kucharski et al., 2023). While these proteins show sequence similarity to their mammalian counterparts, their enzymatic activities have been tested in vitro only in the honey bee Apis mellifera (Wang et al., 2006). Interestingly, in the order Hymenoptera (bees, wasps, and ants), multiple copies of DNMT1 and DNMT3 have been identified, along with an unusual duplication of the functionally essential PWWP domain in DNMT3, which binds to H3K36me2 and H3K27me chromatin modifications, in contrast to the mammalian PWWP domain that only binds to H3K36me (Kucharski et al., 2023; Maleszka, 2024).
Insect DNA methylation toolkits exemplify how evolution can create diverse epigenomic layers that confer lineage-specific advantages. This raises several important questions: What benefits do additional DNMTs or duplicated PWWP domains in DNMT3 offer to many species? How does a partial toolkit with only one DNMT1 and no DNMT3 function in most insects? It is essential to consider the unique ways in which insects and other invertebrates employ either full or partial DNA methylation toolkits, especially when developing non-mammalian epigenetic clocks.
Understanding the diverse roles of DNA methylation across all taxonomic groups is essential for advancing invertebrate methylomics. Genome defence and regulatory function are considered the ancient roles of cytosine methylation, with the regulatory aspect lost in species with low rates of cellular turnover, which may include eutelic organisms with a fixed cell number (Maleszka and Kucharski, 2022; Regev et al., 1998). In Hydra and several other Cnidarians, DNA methylation predominantly targets transposons, especially the evolutionarily youngest ones (Ying et al., 2022). There is a notable preference for methylation to occur in longer and more highly active genes. Interestingly, as transposons age, their levels of methylation tend to decline. This decline helps mitigate the potentially harmful mutagenic effects associated with CpG methylation. The relationship between the extent of DNA methylation and the content of transposons, observed in Cnidaria and other invertebrates, may provide valuable insights into the context of ageing.
The mosaic distribution of DNMT1 and DNMT3 in invertebrates (Kucharski et al., 2023; Bewick et al., 2017; Engelhardt et al., 2022) suggests that their roles vary significantly across different species, and these functions are only beginning to be understood in selected cases. For instance, in a clonal ant species, the knockout of DNMT1 leads to a decrease in DNA methylation and results in sterility (Ivasyk et al., 2023). In honey bees, silencing DNMT3 through RNA interference (RNAi) results in a higher proportion of females exhibiting the queen phenotype (Kucharski et al., 2008). Recent research has identified that a transposon-derived microRNA, known as miR-3721, post-transcriptionally regulates DNMT3 (Jiang et al., 2025). Treating larvae with agomir-3721 produces phenotypic effects similar to those observed in the RNAi experiment. Additionally, the role of DNA methylation in regulating gene expression and alternative splicing has been demonstrated in several studies (Li-Byarlay et al., 2013; Foret et al., 2012; Flores et al., 2012; Xu et al., 2021; Elango et al., 2009). However, knowledge about demethylation processes in invertebrates remains limited, with only one study showing such a role for a single TET enzyme in honey bees (Wojciechowski et al., 2014). In the insect with no DNA methylation, D. melanogaster, a TET homolog seems to mediate N6-methyladenine demethylation and 5 mC demethylation in DNA and mRNA, respectively (Zhang et al., 2015).
Invertebrates generally have only a single MBD protein, MBD2/3, that does not always contain appropriate residues for selectively binding methylated DNA. However, the sponge Ephydatia muelleri has genes for each of the NuRD core components, including an EmMBD2/3 that selectively binds methylated DNA. NMR analyses reveal a remarkably conserved binding mode. These data support a model in which the MBD2/3 methylation-dependent functional role emerged with the earliest multicellular organisms and has been maintained to varying degrees across animal evolution (Cramer et al., 2017).
Can long-lived insects be used to develop models for invertebrate epigenetic clocks?
Maximum lifespans can vary greatly among different species, even those that are closely related (Cohen, 2018; Jones et al., 2014). Several factors influence the lifespans of animals, including body size, metabolism, and environmental conditions, which are considered the most significant. Generally, smaller animals with higher metabolic rates tend to have shorter lifespans. However, the environment is crucial in affecting lifespans, even among closely related species.
While the vast majority of insects live for a few weeks to several months, cicadas, which are known for their periodic emergence, can live underground for 17 years as nymphs, and splendour beetles can reach 25–30 years living as larvae in a host tree. In such cases, the pre-adult stages exist in a comfortable niche that is fully protected from environmental insults, highlighting the importance of the environment in longevity.
Although transferring data from model organisms to humans is not straightforward, especially when considering the influence of social environments on healthy ageing (Mason et al., 2017), eusocial insects, such as ants, termites, and honey bees, may offer valuable insights into the epigenetics of longevity (Promislow et al., 2022; Omholt and Amdam, 2004; Keller and Jemielity, 2006). These insects live in highly organised societies that affect their lifespan, with social cues enabling older individuals to exhibit more youthful behaviour (Amdam, 2011). While queens of eusocial insects do not display the same behavioural flexibility as non-reproductive workers, they can reach an age of 20–30 years with continuing high fecundity (Kramer et al., 2016). This is a compelling example of how different epigenetic interpretations of a single genome can result in contrasting phenotypic outcomes, including variations in ageing (Miklos and Maleszka, 2011). The social structures within these insect communities support an evolutionary theory of ageing, as purely mechanistic explanations for senescence do not account for the relationship between social structure and ageing outcomes (Mason et al., 2017).
When considering female castes of eusocial insects as experimental models for designing approaches to study DNAm epigenetic clocks, it is essential to account for their elaborate life cycles. In holometabolous insects, which represent about 80% of all insect species, the preadult stages are often as long, or even longer, than the adult stages. The larval feeding stage is particularly susceptible to external influences. Most larval tissues, except for certain parts of the nervous system and small clusters of progenitor cells known as imaginal discs (ImDs), are destroyed during metamorphosis (Maleszka and Kucharski, 2022; Beira and Paro, 2016). During the pupation stage, these undifferentiated but committed ImDs give rise to the adult structures. These progenitor cells represent the only continuity at the cellular level in these insects and may provide valuable insights into the epigenomic changes that occur during post-embryonic development. Significant life span extension observed in eusocial queens can be achieved by different feeding, which in honey bees involves a potent diet called royal jelly (Miklos and Maleszka, 2011). Additionally, these highly fecund females spend their lives in a protected environment, and it is known that their longevity is negatively affected when they are outside of their optimal conditions. For example, leaf-cutter queens can live up to 8 years in the wild, but in a laboratory setting, they can reach up to 18 years (Figure 1). This scenario underscores the adverse effects of environmental stressors, such as seasonal climatic changes and fluctuations in food availability or quality, on longevity. The developmental differentiation between functionally sterile female workers and reproductive queens is epigenetically controlled, particularly in honey bees, where it is dependent on DNA methylation (Kucharski et al., 2008), and may involve other epigenomic regulatory layers, in particular histone modifications and microRNAs (Jiang et al., 2025; Wojciechowski et al., 2018; Dickman et al., 2013; Ashby et al., 2016). However, the complexities of the interactions between environmental factors and the epigenome remain largely unexplored. Understanding these epigenetic processes could be highly valuable in the context of epigenetic clocks.

Figure 1. A young leaf-cutter queen (Atta sexdens) and her founding fungus garden with workers. The queen is approximately 8 months old. In the field, adult nests reach 8 years, but this species can live up to 18 years in the lab, underscoring the impact of environmental stressors like climatic seasonal changes or food availability/quality on longevity. The workers’ lifespan is 10–20 times shorter. Photo courtesy of Daniela Römer and Flavio Roces.
The research conducted thus far does not support epigenetic DNAm clocks in invertebrates
So far, only one report, deposited in bioRxiv, claims the discovery of an epigenetic clock in an insect, namely, the parasitic wasp Nasonia vitripennis (Brink et al., 2023). However, in this study’s published, peer-reviewed version, the authors have toned down their claims, including the title, opting for an exploration of “… the ageing methylome in the model insect, N. vitripennis” (Brink et al., 2024). Indeed, their approach using 19 pre-selected CpGs was questioned as likely to lead to overfitting to the experimental samples (Liu et al., 2025). In another Hymenopteran, the honey bee (Apis mellifera), no significant loss of age-related DNA methylation has been found in several genes using ultra-deep amplicon sequencing (Kucharski and Maleszka, 2020). These results are particularly significant because, unlike most insects, Hymenoptera possess a complete DNA methylation toolkit that is more comparable to the mammalian toolkit (Kucharski et al., 2023).
Regarding other arthropods, age-dependent DNA methylation changes have been reported in two species: the aquatic crustacean Daphnia magna (Hearn et al., 2021) and the European lobster Homarus gammarus (Fairfield et al., 2021). The D. magna epigenetic clock was built using 12 CpGs and two age groups. However, in a recent study, attempts to reproduce age-related changes in Daphnia across multiple life stages and to construct an epigenetic clock using machine learning models were unsuccessful (Liu et al., 2025). The authors conclude that, due to the overall low DNA methylation levels and lack of robust age-associated methylation changes, age-associated methylomics in D. magna should focus on environmental factors to reveal methylation dynamics (Liu et al., 2025).
The study of European lobsters is particularly important because it can enhance the accurate assessment of their population dynamics, which is essential for sustainable fisheries management. Notably, these lobsters have a relatively long lifespan, 31 years for males and 54 years for females, allowing epigenetic clocks to address the challenges posed by their indeterminate growth and the shedding of their exoskeleton throughout their lives. The initial research, which examined ribosomal DNA methylation in lobsters aged between 0 and 51 months, established a linear correlation between age and rDNA methylation. This correlation was successfully applied to individuals whose ages were unknown (Fairfield et al., 2021).
Can other epigenetic modifications and cellular mechanisms be utilised to construct ageing clocks with comparable accuracy to DNA methylation clocks?
Given the apparent difficulties of using DNA methylation as an ageing indicator in invertebrates, it may be prudent to consider other cellular mechanisms as candidates for such a role. An interesting suggestion was made in a recent study on a 117-year-old female whose longevity was associated with a significant shortening of telomeres (Santos-Pujol et al., 2025). The authors conclude that, in light of her good health, chromosomal attrition acted more like a chromosomal clock than a predictor of age-related diseases. This is a testable hypothesis that may benefit invertebrate studies.
With the expanding assortment of epigenomic modifications and their potential roles in various cellular and organismal contexts, it is becoming possible to consider other modifiers. One key area of focus is the modifications occurring at the histone level. Unlike DNA methylation, which is not universally present, histone and chromatin modifications are found in all eukaryotic organisms. Notably, the age-related remodelling of heterochromatin and the loss of chromatin associated with hypomethylation suggest a relationship between these two regulatory layers. Importantly, ageing-dependent remodelling of heterochromatin and chromatin loss, associated with hypomethylation, suggests a connection between these two regulatory layers (Ciccarone et al., 2018). A recent analysis of multiple tissues has shown that the dynamics of seven histone marks during human ageing yielded results comparable to those of DNA methylation age predictors (de Lima Camillo et al., 2025). The findings revealed a trend characterised by a loss of modifications linked to heterochromatin and an increase in marks associated with euchromatin, indicating an overall decline in epigenetic regulation with age. This area of research is expected to grow significantly within mammalian biomedical studies and may also provide valuable insights for similar inquiries in other lineages.
Other epigenomic mechanisms, such as RNA modifications, are starting to gain attention in invertebrate research (Zhang et al., 2015; Jiao and Palli, 2024). Their ubiquitous presence and roles in cellular mechanisms (Wang et al., 2022) suggest that their involvement in ageing processes could be leveraged to design RNA-based epigenetic clocks akin to DNA methylation (DNAm) epigenetic clocks. In this context, the somewhat unexpected role of the enzyme TET in demethylating N6-methyladenine in DNA and 5-methylcytosine in RNA in the fly D. melanogaster, which lacks a DNA methylation toolkit (Zhang et al., 2015), suggests that a more extensive repertoire of DNA and RNA modifications might be considered for quantifying ageing, even in species lacking DNA methylation.
However, various methods to quantify biological ageing may not necessarily measure the same thing and ultimately yield different results, suggesting that comparative analyses may pose a significant challenge (Belsky et al., 2018).
Ageing as an epigenetic, environmentally influenced process
In vertebrates, studies on convergent evolution have established that the genetic architecture of longevity-related genes is an important factor influencing the differing lifespans of closely related species with similar genomes. For instance, genetic diversity plays a role in the contrasting lifespans of two species of rougheye fish: the rockeye (Sebastes aleutianus), which lives for about 200 years, and its close relative, the blue rockfish (Sebastes mystinus), which typically lives for only 26 years (Kolora et al., 2021). These two species inhabit distinct environmental niches that present different survival challenges. The long-lived S. aleutianus resides in very deep, cold waters (−0.3°C–5.0°C) near the seabed, often within caves, while S. mystinus lives near the surface. The relative contributions of genetics and environment to prolonged longevity have been established for many organisms. For example, the protective environment of a eusocial insect colony contributes to the long lives of epigenetically generated reproductive queens. In humans, both genetics and environmental factors (the exposome) influence health, with the exposome shaping unique patterns of disease and mortality, independent of genetic factors (Argentieri et al., 2025). A computer model supports the evolutionary role of the environment in tuning lifespans. It shows that a limited lifespan can be detrimental to an individual in the short term but beneficial to their distant descendants. The model also predicts that the most beneficial lifespan varies with the environmental conditions (Werfel et al., 2015).
This interplay between contributing factors indicates that the epigenetic basis of longevity may have different implications across species (Mariani et al., 2010). Additionally, ageing exemplifies biological degeneracy, where different mechanisms can yield similar functional outcomes (Edelman and Gally, 2001; Maleszka et al., 2014). For example, in the ageing human brain, various cellular malfunctions can lead to similar symptoms known as dementia, and in species lacking a DNA methylation toolkit, alternative epigenomic layers are responsible for controlling genome-environment interactions (Mason et al., 2017; Maleszka et al., 2014). Conserved pathways, such as the insulin/IGF-1 signalling pathway and the mTOR node, regulate lifespan and can be studied in model organisms like yeast, nematodes, flies, and mice. However, it is crucial to approach studies at the epigenetic level and within the context of molecular clocks for ageing cautiously, interpreting results through a broader comparative framework. Furthermore, incorporating research on non-traditional and unusual species, while integrating both mechanistic and demographic studies, is essential (Cohen, 2018; Tian et al., 2017).
Conclusion
There are no straightforward answers when it comes to the quantitative approaches to the biology of ageing, and research strategies should reflect this complexity. This difficulty is especially apparent when analysing the epigenetic machinery, which varies considerably between mammals and invertebrates. Significant challenges for invertebrate epigenetic clocks include computational methodology, particularly in areas such as interpretation, cell-type heterogeneity, and the adoption of emerging single-cell techniques. Technology is no longer a limiting factor, and with new developments emerging frequently, all epigenetic research will inevitably adapt to these changes. These new methodologies aim to establish guidelines for the rigorous development of interpretable epigenetic clocks at both cell-type and single-cell resolutions. Exploring additional ideas, predictions, and even speculations about epigenetic clocks in invertebrates would be valuable within the framework of modern theories of ageing and the concept of “genes for ageing.”
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
RM: Writing – review and editing, Writing – original draft, Resources, Conceptualization, Project administration.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amdam, G. V. (2011). Social context, stress, and plasticity of aging. Aging Cell 10 (1), 18–27. doi:10.1111/j.1474-9726.2010.00647.x
Argentieri, M. A., Amin, N., Nevado-Holgado, A. J., Sproviero, W., Collister, J. A., Keestra, S. M., et al. (2025). Integrating the environmental and genetic architectures of aging and mortality. Nat. Med. 31 (3), 1016–1025. doi:10.1038/s41591-024-03483-9
Ashby, R., Foret, S., Searle, I., and Maleszka, R. (2016). MicroRNAs in honey bee caste determination. Sci. Rep. 6, 18794. doi:10.1038/srep18794
Beira, J. V., and Paro, R. (2016). The legacy of drosophila imaginal discs. Chromosoma 125 (4), 573–592. doi:10.1007/s00412-016-0595-4
Bell, C. G., Lowe, R., Adams, P. D., Baccarelli, A. A., Beck, S., Bell, J. T., et al. (2019). DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20 (1), 249. doi:10.1186/s13059-019-1824-y
Belsky, D. W., Moffitt, T. E., Cohen, A. A., Corcoran, D. L., Levine, M. E., Prinz, J. A., et al. (2018). Eleven telomere, epigenetic clock, and biomarker-composite quantifications of biological aging: do they measure the same thing? Am. J. Epidemiol. 187 (6), 1220–1230. doi:10.1093/aje/kwx346
Bertile, F., Matallana-Surget, S., Tholey, A., Cristobal, S., and Armengaud, J. (2023). Diversifying the concept of model organisms in the age of -omics. Commun. Biol. 6 (1), 1062. doi:10.1038/s42003-023-05458-x
Bertucci-Richter, E. M., and Parrott, B. B. (2023). The rate of epigenetic drift scales with maximum lifespan across mammals. Nat. Commun. 14 (1), 7731. doi:10.1038/s41467-023-43417-6
Bewick, A. J., Vogel, K. J., Moore, A. J., and Schmitz, R. J. (2017). Evolution of DNA methylation across insects. Mol. Biol. Evol. 34 (3), 654–665. doi:10.1093/molbev/msw264
Blacher, P., Huggins, T. J., and Bourke, A. F. G. (2017). Evolution of ageing, costs of reproduction and the fecundity–longevity trade-off in eusocial insects. Proc. R. Soc. B Biol. Sci. 284 (1858), 20170380. doi:10.1098/rspb.2017.0380
Brink, K., Thomas, C., Jones, A., and Mallon, E. (2023). An epigenetic clock in an insect model system. bioRxiv.
Brink, K., Thomas, C. L., Jones, A., Chan, T. W., and Mallon, E. B. (2024). Exploring the ageing methylome in the model insect, Nasonia vitripennis. BMC Genomics 25 (1), 305. doi:10.1186/s12864-024-10211-7
Ciccarone, F., Tagliatesta, S., Caiafa, P., and Zampieri, M. (2018). DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech. Ageing Dev. 174, 3–17. doi:10.1016/j.mad.2017.12.002
Cohen, A. A. (2018). Aging across the tree of life: the importance of a comparative perspective for the use of animal models in aging. Biochim. Biophys. Acta Mol. Basis Dis. 1864 (9 Pt A), 2680–2689. doi:10.1016/j.bbadis.2017.05.028
Cramer, J. M., Pohlmann, D., Gomez, F., Mark, L., Kornegay, B., Hall, C., et al. (2017). Methylation specific targeting of a chromatin remodeling complex from sponges to humans. Sci. Rep. 7 (1), 40674. doi:10.1038/srep40674
Dabe, E. C., Sanford, R. S., Kohn, A. B., Bobkova, Y., and Moroz, L. L. (2015). DNA methylation in basal metazoans: insights from ctenophores. Integr. Comp. Biol. 55 (6), 1096–1110. doi:10.1093/icb/icv086
de Lima Camillo, L. P., Asif, M. H., Horvath, S., Larschan, E., and Singh, R. (2025). Histone mark age of human tissues and cell types. Sci. Adv. 11 (1), eadk9373. doi:10.1126/sciadv.adk9373
Dickman, M. J., Kucharski, R., Maleszka, R., and Hurd, P. J. (2013). Extensive histone post-translational modification in honey bees. Insect Biochem. Mol. Biol. 43 (2), 125–137. doi:10.1016/j.ibmb.2012.11.003
Edelman, G. M., and Gally, J. A. (2001). Degeneracy and complexity in biological systems. Proc. Natl. Acad. Sci. U. S. A. 98 (24), 13763–13768. doi:10.1073/pnas.231499798
Elango, N., Hunt, B. G., Goodisman, M. A. D., and Yi, S. V. (2009). DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, apis mellifera. Proc. Natl. Acad. Sci. U. S. A. 106 (27), 11206–11211. doi:10.1073/pnas.0900301106
Engelhardt, J., Scheer, O., Stadler, P. F., and Prohaska, S. J. (2022). Evolution of DNA methylation across ecdysozoa. J. Mol. Evol. 90 (1), 56–72. doi:10.1007/s00239-021-10042-0
Fairfield, E. A., Richardson, D. S., Daniels, C. L., Butler, C. L., Bell, E., and Taylor, M. I. (2021). Ageing European lobsters (homarus gammarus) using DNA methylation of evolutionarily conserved ribosomal DNA. Evol. Appl. 14 (9), 2305–2318. doi:10.1111/eva.13296
Field, A. E., Robertson, N. A., Wang, T., Havas, A., Ideker, T., and Adams, P. D. (2018). DNA methylation clocks in aging: categories, causes, and consequences. Mol. Cell 71 (6), 882–895. doi:10.1016/j.molcel.2018.08.008
Flores, K., Wolschin, F., Corneveaux, J. J., Allen, A. N., Huentelman, M. J., and Amdam, G. V. (2012). Genome-wide association between DNA methylation and alternative splicing in an invertebrate. BMC Genomics 13, 480. doi:10.1186/1471-2164-13-480
Foret, S., Kucharski, R., Pellegrini, M., Feng, S. H., Jacobsen, S. E., Robinson, G. E., et al. (2012). DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc. Natl. Acad. Sci. U. S. A. 109 (13), 4968–4973. doi:10.1073/pnas.1202392109
Hardison, R. C. (2016). A guide to translation of research results from model organisms to human. Genome Biol. 17 (1), 161. doi:10.1186/s13059-016-1026-9
Hearn, J., Plenderleith, F., and Little, T. J. (2021). DNA methylation differs extensively between strains of the same geographical origin and changes with age in Daphnia magna. Epigenetics and Chromatin 14 (1), 4. doi:10.1186/s13072-020-00379-z
Heinze, J., and Giehr, J. (2021). The plasticity of lifespan in social insects. Philosophical Trans. R. Soc. B Biol. Sci. 376 (1823), 20190734. doi:10.1098/rstb.2019.0734
Ivasyk, I., Olivos-Cisneros, L., Valdés-Rodríguez, S., Droual, M., Jang, H., Schmitz, R. J., et al. (2023). DNMT1 mutant ants develop normally but have disrupted oogenesis. Nat. Commun. 14 (1), 2201. doi:10.1038/s41467-023-37945-4
Jiang, Y., Hu, J., Li, Y., Tang, X., Peng, X., Xie, L., et al. (2025). Comprehensive genomic analysis reveals novel transposable element-derived MicroRNA regulating caste differentiation in honeybees. Mol. Biol. Evol. 42 (4). doi:10.1093/molbev/msaf074
Jiao, Y., and Palli, S. R. (2024). RNA modifications in insects. Front. Insect Sci. 4, 1448766. doi:10.3389/finsc.2024.1448766
Jones, O. R., Scheuerlein, A., Salguero-Gómez, R., Camarda, C. G., Schaible, R., Casper, B. B., et al. (2014). Diversity of ageing across the tree of life. Nature 505 (7482), 169–173. doi:10.1038/nature12789
Keller, L., and Jemielity, S. (2006). Social insects as a model to study the molecular basis of ageing. Exp. Gerontol. 41 (6), 553–556. doi:10.1016/j.exger.2006.04.002
Kolora, S. R. R., Owens, G. L., Vazquez, J. M., Stubbs, A., Chatla, K., Jainese, C., et al. (2021). Origins and evolution of extreme life span in Pacific Ocean rockfishes. Sci. (New York, NY) 374 (6569), 842–847. doi:10.1126/science.abg5332
Koncevičius, K., Nair, A., Šveikauskaitė, A., Šeštokaitė, A., Kazlauskaitė, A., Dulskas, A., et al. (2024). Epigenetic age oscillates during the day. Aging Cell 23 (7), e14170. doi:10.1111/acel.14170
Kramer, B. H., van Doorn, G. S., Weissing, F. J., and Pen, I. (2016). Lifespan divergence between social insect castes: challenges and opportunities for evolutionary theories of aging. Curr. Opin. Insect Sci. 16, 76–80. doi:10.1016/j.cois.2016.05.012
Kucharski, R., Ellis, N., Jurkowski, T. P., Hurd, P. J., and Maleszka, R. (2023). The PWWP domain and the evolution of unique DNA methylation toolkits in Hymenoptera. iScience 26 (11), 108193. doi:10.1016/j.isci.2023.108193
Kucharski, R., Maleszka, J., Foret, S., and Maleszka, R. (2008). Nutritional control of reproductive status in honeybees via DNA methylation. Sci. (New York, NY) 319 (5871), 1827–1830. doi:10.1126/science.1153069
Kucharski, R., and Maleszka, R. (2020). Exploring DNA methylation diversity in the honey bee brain by ultra-deep amplicon sequencing. Epigenomes 4 (2), 10. doi:10.3390/epigenomes4020010
Lewis, S. H., Ross, L., Bain, S. A., Pahita, E., Smith, S. A., Cordaux, R., et al. (2020). Widespread conservation and lineage-specific diversification of genome-wide DNA methylation patterns across arthropods. PLoS Genet. 16 (6), e1008864. doi:10.1371/journal.pgen.1008864
Li-Byarlay, H., Li, Y., Stroud, H., Feng, S., Newman, T. C., Kaneda, M., et al. (2013). RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc. Natl. Acad. Sci. U. S. A. 110 (31), 12750–12755. doi:10.1073/pnas.1310735110
Liu, R., Morselli, M., Yampolsky, L. Y., Peshkin, L., and Pellegrini, M. (2025). Genome-wide DNA methylation patterns in Daphnia magna are not significantly associated with age. Epigenetics Chromatin 18 (1), 17. doi:10.1186/s13072-025-00580-y
Maleszka, R. (2024). Reminiscences on the honeybee genome project and the rise of epigenetic concepts in insect science. Insect Mol. Biol. 33 (5), 444–456. doi:10.1111/imb.12888
Maleszka, R., de Couet, H. G., and Miklos, G. L. G. (1998). Data transferability from model organisms to human beings: insights from the functional genomics of the flightless region of drosophila. Proc. Natl. Acad. Sci. U. S. A. 95 (7), 3731–3736. doi:10.1073/pnas.95.7.3731
Maleszka, R., and Kucharski, R. (2022). Without mechanisms, theories and models in insect epigenetics remain a black box. Trends Genet. 38 (11), 1108–1111. doi:10.1016/j.tig.2022.05.004
Maleszka, R., Mason, P. H., and Barron, A. B. (2014). Epigenomics and the concept of degeneracy in biological systems. Brief. Funct. Genomics 13 (3), 191–202. doi:10.1093/bfgp/elt050
Mariani, F., Pérez-Barahona, A., and Raffin, N. (2010). Life expectancy and the environment. J. Econ. Dyn. Control 34 (4), 798–815. doi:10.1016/j.jedc.2009.11.007
Mason, P. H., Maleszka, R., and Dominguez, D. J. (2017). Another stage of development: biological degeneracy and the study of bodily ageing. Mech. Ageing Dev. 163, 46–51. doi:10.1016/j.mad.2016.12.007
May, R. M. (1988). How many species are there on Earth? Sci. (New York, NY) 241 (4872), 1441–1449. doi:10.1126/science.241.4872.1441
Miklos, G. L., and Maleszka, R. (2000). Deus ex genomix. Nat. Neurosci. 3 (5), 424–425. doi:10.1038/74786
Miklos, G. L. G., and Maleszka, R. (2011). Epigenomic communication systems in humans and honey bees: from molecules to behavior. Hormones Behav. 59 (3), 399–406. doi:10.1016/j.yhbeh.2010.05.016
Omholt, S. W., and Amdam, G. V. (2004). Epigenetic regulation of aging in honeybee workers. Sci. Aging Knowl. Environ. 2004 (26), pe28. doi:10.1126/sageke.2004.26.pe28
Promislow, D. E. L., Flatt, T., and Bonduriansky, R. (2022). The biology of aging in insects: from drosophila to other insects and back. Annu. Rev. Entomol. 67, 83–103. doi:10.1146/annurev-ento-061621-064341
Regev, A., Lamb, M. J., and Jablonka, E. (1998). The role of DNA methylation in invertebrates: developmental regulation or genome defense? Mol. Biol. Evol. 15 (7), 880–891. doi:10.1093/oxfordjournals.molbev.a025992
Santos-Pujol, E., Noguera-Castells, A., Casado-Pelaez, M., García-Prieto, C. A., Vasallo, C., Campillo-Marcos, I., et al. (2025). The multiomics blueprint of extreme human lifespan. bioRxiv.
Sarkies, P. (2022). Encyclopaedia of eukaryotic DNA methylation: from patterns to mechanisms and functions. Biochem. Soc. Trans. 50 (3), 1179–1190. doi:10.1042/BST20210725
Seale, K., Teschendorff, A., Reiner, A. P., Voisin, S., and Eynon, N. (2024). A comprehensive map of the aging blood methylome in humans. Genome Biol. 25 (1), 240. doi:10.1186/s13059-024-03381-w
Setola, V., and Roth, B. L. (2003). Why mice are neither miniature humans nor small rats: a cautionary tale involving 5-hydroxytryptamine-6 serotonin receptor species variants. Mol. Pharmacol. 64 (6), 1277–1278. doi:10.1124/mol.64.6.1277
Teschendorff, A. E., and Horvath, S. (2025). Epigenetic ageing clocks: statistical methods and emerging computational challenges. Nat. Rev. Genet. 26, 350–368. doi:10.1038/s41576-024-00807-w
Tian, X., Seluanov, A., and Gorbunova, V. (2017). Molecular mechanisms determining lifespan in Short- and long-lived species. Trends Endocrinol. Metab. 28 (10), 722–734. doi:10.1016/j.tem.2017.07.004
Wang, K., Liu, H., Hu, Q., Wang, L., Liu, J., Zheng, Z., et al. (2022). Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 7 (1), 374. doi:10.1038/s41392-022-01211-8
Wang, Y., Jorda, M., Jones, P. L., Maleszka, R., Ling, X., Robertson, H. M., et al. (2006). Functional CpG methylation system in a social insect. Sci. (New York, NY) 314 (5799), 645–647. doi:10.1126/science.1135213
Wedd, L., Kucharski, R., and Maleszka, R. (2022). DNA methylation in honey bees and the unresolved questions in insect methylomics. Adv. Exp. Med. Biol. 1389, 159–176. doi:10.1007/978-3-031-11454-0_7
Werfel, J., Ingber, D. E., and Bar-Yam, Y. (2015). Programed death is favored by natural selection in spatial systems. Phys. Rev. Lett. 114 (23), 238103. doi:10.1103/PhysRevLett.114.238103
Wojciechowski, M., Lowe, R., Maleszka, J., Conn, D., Maleszka, R., and Hurd, P. J. (2018). Phenotypically distinct female castes in honey bees are defined by alternative chromatin states during larval development. Genome Res. 28 (10), 1532–1542. doi:10.1101/gr.236497.118
Wojciechowski, M., Rafalski, D., Kucharski, R., Misztal, K., Maleszka, J., Bochtler, M., et al. (2014). Insights into DNA hydroxymethylation in the honeybee from in-depth analyses of TET dioxygenase. Open Biol. 4 (8), 140110. doi:10.1098/rsob.140110
Xu, G., Lyu, H., Yi, Y., Peng, Y., Feng, Q., Song, Q., et al. (2021). Intragenic DNA methylation regulates insect gene expression and reproduction through the MBD/Tip60 complex. iScience 24 (2), 102040. doi:10.1016/j.isci.2021.102040
Yang, J.-H., Hayano, M., Griffin, P. T., Amorim, J. A., Bonkowski, M. S., Apostolides, J. K., et al. (2023). Loss of epigenetic information as a cause of mammalian aging. Cell 186 (2), 305–26.e27. doi:10.1016/j.cell.2022.12.027
Yin, Z., Zhu, M., Davidson, E. H., Bottjer, D. J., Zhao, F., and Tafforeau, P. (2015). Sponge grade body fossil with cellular resolution dating 60 Myr before the Cambrian. Proc. Natl. Acad. Sci. 112 (12), E1453–E1460. doi:10.1073/pnas.1414577112
Ying, H., Hayward, D. C., Klimovich, A., Bosch, T. C. G., Baldassarre, L., Neeman, T., et al. (2022). The role of DNA methylation in genome defense in Cnidaria and other invertebrates. Mol. Biol. Evol. 39 (2). doi:10.1093/molbev/msac018
Ying, K., Liu, H., Tarkhov, A. E., Sadler, M. C., Lu, A. T., Moqri, M., et al. (2024). Causality-enriched epigenetic age uncouples damage and adaptation. Nat. Aging 4 (2), 231–246. doi:10.1038/s43587-023-00557-0
Yu, G., Wu, Q., Gao, Y., Chen, M., and Yang, M. (2019). The epigenetics of aging in invertebrates. Int. J. Mol. Sci. 20 (18), 4535. doi:10.3390/ijms20184535
Zhang, G., Huang, H., Liu, D., Cheng, Y., Liu, X., Zhang, W., et al. (2015). N6-methyladenine DNA modification in drosophila. Cell 161 (4), 893–906. doi:10.1016/j.cell.2015.04.018
Zhang, Q., Vallerga, C. L., Walker, R. M., Lin, T., Henders, A. K., Montgomery, G. W., et al. (2019). Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 11 (1), 54. doi:10.1186/s13073-019-0667-1
Keywords: epigenomics, PWWP domain, social insect, honey bee, epigenetic diversity, ageing
Citation: Maleszka R (2025) Is the concept of mammalian epigenetic clocks universal and applicable to invertebrates?. Front. Genet. 16:1633921. doi: 10.3389/fgene.2025.1633921
Received: 23 May 2025; Accepted: 25 July 2025;
Published: 08 August 2025.
Edited by:
Osman A. El-Maarri, University of Bonn, GermanyReviewed by:
Michele Zampieri, Sapienza University of Rome, ItalyCopyright © 2025 Maleszka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryszard Maleszka, cnlzemFyZC5tYWxlc3prYUBhbnUuZWR1LmF1