 Toshiyuki Nishikido1,2
Toshiyuki Nishikido1,2 Kausik K. Ray1*
Kausik K. Ray1*- 1Imperial Centre for Cardiovascular Disease Prevention, Department of Primary Care and Public Health, School of Public Health, Imperial College London, London, United Kingdom
- 2Department of Cardiovascular medicine, Saga University, Saga, Japan
Low-density lipoprotein (LDL) is one of the principal risk factors for atherosclerosis. Circulating LDL particles can penetrate into the sub-endothelial space of arterial walls. These particles undergo oxidation and promote an inflammatory response, resulting in injury to the vascular endothelial wall. Persistent elevation of LDL-cholesterol (LDL-C) is linked to the progression of fatty streaks to lipid-rich plaque and thus atherosclerosis. LDL-C is a causal factor for atherosclerotic cardiovascular disease and lowering it is beneficial across a range of conditions associated with high risk of cardiovascular events. Therefore, all guidelines-recommended initiations of statin therapy for patients at high cardiovascular risk is irrespective of LDL-C. In addition, intensive LDL-C lowering therapy with statins has been demonstrated to result in a greater reduction of cardiovascular event risk in large clinical trials. However, many high-risk patients receiving statins fail to achieve the guideline-recommended reduction in LDL-C levels in routine clinical practice. Moreover, low levels of adherence and often high rates of discontinuation demand the need for further therapies. Ezetimibe has typically been used as a complement to statins when further LDL-C reduction is required. More recently, proprotein convertase subtilisin kexin 9 (PCSK9) has emerged as a novel therapeutic target for lowering LDL-C levels, with PCSK9 inhibitors offering greater reductions than feasible through the addition of ezetimibe. PCSK9 monoclonal antibodies have been shown to not only considerably lower LDL-C levels but also cardiovascular events. However, PCSK9 monoclonal antibodies require once- or twice-monthly subcutaneous injections. Further, their manufacturing process is expensive, increasing the cost of therapy. Therefore, several non-antibody treatments to inhibit PCSK9 function are being developed as alternative approaches to monoclonal antibodies. These include gene-silencing or editing technologies, such as antisense oligonucleotides, small interfering RNA, and the clustered regularly interspaced short palindromic repeats/Cas9 platform; small-molecule inhibitors; mimetic peptides; adnectins; and vaccination. In this review, we summarize the current knowledge base on the role of PCSK9 in lipid metabolism and an overview of non-antibody approaches for PCSK9 inhibition and their limitations. The subsequent development of alternative approaches to PCSK9 inhibition may give us more affordable and convenient therapeutic options for the management of high-risk patients.
Introduction
Low-density lipoprotein particles account for 90% or more of plasma apolipoprotein B-containing atherogenic lipoprotein particles, hence LDL cholesterol (LDL-C) accounts for the majority of circulating cholesterol carried by atherogenic particles. LDL particles are able to penetrate the endothelium of the artery intima, leading to its oxidative modification into highly atherogenic particles that subsequently induces an inflammatory response. The uptake of oxidized LDL by macrophages converts them to foam cells initiating the process of atherosclerosis within the fatty streak. Genetic, observational and trial data demonstrate that LDL-C is a causal factor for atherosclerotic cardiovascular disease (CVD) and that lowering it reduces clinical events (1). Therefore, the clinical benefit of lowering LDL-C is widely acknowledged, and current guidelines recommend lipid-lowering strategies principally with statins for individuals at high cardiovascular risk (2–4). Reduction in LDL-C levels with 3-hydroxy-3methylglutaryl coenzyme A reductase inhibitors (statins) results in a dose-dependent reduction in LDL-C and of CVD risk, proportional to the absolute magnitude of the reduction in LDL-C levels. The Cholesterol Treatment Trialists' meta-analyses of data from 170,000 participants in 26 randomized trials involving intensive statin therapy revealed that 1 mmol/L (~40 mg/dl) reduction in LDL-C levels resulted in a 10% relative reduction in all-cause mortality (Relative risk (RR) 0.90, 95% confidence interval (CI) 0.87–0.93) and 22% relative reduction of major vascular events (non-fatal myocardial infarction, coronary death, coronary revascularization, or stroke) (RR 0.78, 95% CI 0.76–0.80) (5). Furthermore, achieving very low levels of LDL-C has beneficial effects on CVD risk, according to the meta-analysis of eight randomized controlled statin trials (6). In a post-hoc analysis of the JUPITER trial, the participants attaining LDL-C levels below 50 mg/dl with Rosuvastatin 20 mg experienced the fewest CVD events without an increase of the incidence of adverse events (7, 8). The IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) demonstrated the incremental lowering of LDL-C levels by combining a non-statin drug with statin therapy (9). Among 18,144 patients who had experienced acute coronary syndromes, ezetimibe combined with statin therapy reduced the median time-adjusted average LDL-C level by 53.2 mg/dl after 1 year, and reduced the risk of a composite of cardiovascular death, major coronary event (non-fatal myocardial infarction, unstable angina, or coronary revascularization), or non-fatal stroke. These findings supported the notion that intensive LDL-C level reduction leads to improved outcome regardless of the lipid-modifying drug administered in combination with statins, particularly in high-risk patients (10). Observational data within the same study show that over a 7-year period those achieving the lowest LDL-C levels had the lowest risk and that such levels were safe. Life-long lowering of LDL-C levels resulting from genetic differences shows that the benefits of LDL-C lowering are cumulative as a genetically 13 mg/dl difference in LDL-C over 52 years offers the same reduction in risk as a 39 mg/dl over 5 years with statins. These data also suggest that that there may be benefits from early initiation of therapy (11, 12). Therefore, both the absolute magnitude of the reduction of LDL-C levels and the total duration of the period of low LDL-C levels should be considered when assessing the benefits of therapy.
Despite these findings, ~50% of patients treated with statins fail to achieve target the LDL-C levels recommended by the guidelines (13, 14). Moreover, 40% of the patients who receive high doses of statins do not achieve LDL-C levels below 70 mg/dl, even though patients with LDL-C levels below 50 mg/dl have a significantly lower risk of cardiovascular events than patients with LDL-C levels between 75 and 100 mg/dl. Hence, there is a large variation in the reduction in LDL-C levels in the general population. Risk factors do not exist in isolation, so risk factors such as diabetes mellitus, hypertension, abdominal obesity, smoking, etc., lead to higher absolute cardiovascular event risk when they occur together in a synergistic manner (15–17). This is highlighted further by the observation that individuals with established cardiovascular disease do not all have a 10-year risk of 20% but rather a wide variation in event rate. Part of this excess risk is because patients are not on optimal treatment and part of the need to reduce so called residual risk is to optimize control of all risk factors and achieve guideline-recommended treatment strategies (18). Even if guideline-based treatments were attained, there will be subsets of patients who by virtue of the risk factors they possess will require further LDL-C reduction or in whom LDL-C levels are high despite statins e.g., Familial hypercholesterolemia or who cannot tolerate therapies that are routinely available such as those with statin intolerance.
PCSK9 as a Promising Therapeutic Target
The LDL receptor (LDLR) on the liver surface controls plasma LDL levels by binding to circulating LDL, and the LDL/LDLR complex is internalized by clathrin-mediated endocytosis. In endosomes, at acidic pH, LDL is released and degraded, while the LDLR is recycled to the cell surface (19). LDLR expression is predominantly regulated at the transcriptional level by a negative feedback mechanism in the intracellular cholesterol pool. Proprotein convertase subtilisin kexin 9 (PCSK9) plays an important role in the regulation of LDL-C homeostasis. PCSK9 binds to the LDLR, and thus promotes the endosomal and lysosomal degradation of the PCSK9-LDLR complex in a non-enzymatic fashion, and prevents LDLR recycling to the hepatocyte membrane. This leads to a reduction in LDL uptake and an increase in circulating LDL levels (20–22) (Figure 1). The mutations in the PCSK9 gene were identified in French families with autosomal dominant hypercholesterolemia in 2003, which were not due to either LDLR or apoB (23, 24). Rare gain-of-function PCSK9 mutations promoted degradation of LDLR, resulting in elevated LDL-C levels and increased CVD risk. On the other hand, more frequent loss-of function PCSK9 mutations were found to be associated with lower LDL-C levels and a reduced risk of CVD without adverse consequences (25–28). These findings suggest that the inhibition of PCSK9 may comprise a safe and effective strategy for addressing hypercholesterolemia. Hence, PCSK9 inhibition has emerged as a promising therapeutic target for the management of LDL-C.
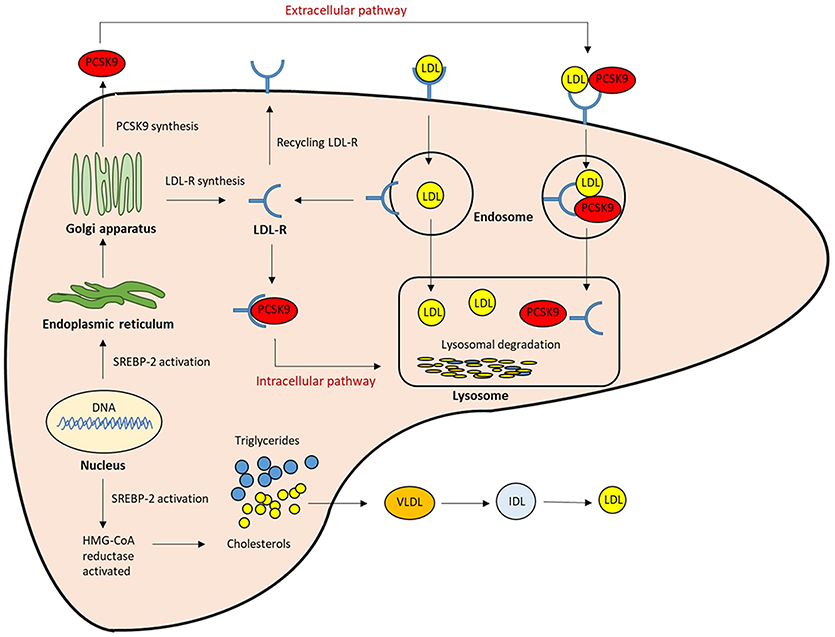
Figure 1. The role of PCSK9 in lipoprotein metabolism. The expression of LDLR and PCSK9 are regulated by SREBP-2. The PCSK9 secreted from the endoplasmic reticulum to the Golgi apparatus, and then from trans-Golgi network into the medium. It can be sorted directly to lysosomes as a complex with the LDLR (intracellular pathway), or secreted into the plasma and then internalized with the LDLR into clathrin-coated endosomes for lysosomal degradation (Extracellular pathway). Both mechanisms result in the reduction of LDLR, and then LDL clearance from the circulation is reduced.; LDL-R, LDL receptor; SREBP-2, Sterol regulatory element-binding protein-2; HMG-CoA, 3-hydroxy-3methylglutaryl coenzyme A.
Structure and Function of PCSK9
PCSK9 is the ninth member of the subtilisin family of kexin-like proprotein convertases. It is expressed predominantly in the liver and, to a lesser extent, in the small intestine, kidney, pancreas, and the central nervous system (29, 30). The human 22-kb PCSK9 gene is located at chromosome 1p32, and contains 12 exons and 11 introns. It encodes a 692-amino acid serine protease (31). PCSK9 expression is mainly regulated transcriptionally, by the sterol regulatory element-binding protein (SREBP)-2, a membrane-bound transcription factor that controls cellular lipid homeostasis (32). Among the three identified isoforms of SREBP, SREBP-2 regulates genes involved in cholesterol homeostasis, such as LDLR, PCSK9, and HMGCoAR, whereas SREBP-1c regulates genes involved in fatty acid synthesis (33). It has been shown that in vivo, SREBP-2 increases the sterol-dependent transcription of PCSK9, predominantly via binding the sterol-regulatory element (SRE) (34). Moreover, PCSK9 expression is also regulated by hepatocyte nuclear factor (HNF)-1α, which is an essential positive regulator of PCSK9 transcription. The HNF-1α-binding site is located between SRE and Sp1 site, as a tissue-specific cis-regulatory sequence of the PCSK9 promoter (35, 36).
PCSK9 is primarily synthesized as an inactive zymogen, proPCSK9 (74 kDa), composed of a single peptide with an N-terminal prodomain, a catalytic domain, and a cysteine- and histidine-rich C-terminal domain (21) (Figure 2). The C-terminal domain is composed of M1, M2, and M3 modules. M1 and M3 are associated with PCSK9 maturation and secretion, while M2 is required for intracellular LDLR degradation. In the endoplasmic reticulum (ER), the signal peptide of proPCSK9 is cleaved and the peptide undergoes co-translational autocatalytic cleavage at position Gln152 into mature PCSK9 (63 kDa). The cleaved prodomain (14 kDa) remains non-covalently bound to the catalytic domain. It subsequently acts as a chaperone to assist the proper folding of mature PCSK9, which is required for PCSK9 to exit from the ER. In addition, it blocks the catalytic site of mature PCSK9 enzymatically through the secretory pathway (37, 38). Thus, PCSK9 is secreted from the ER to the Golgi apparatus as an inactive mature PCSK9/prodomain complex, which prevents the binding of any other proteins or peptides to the catalytic sites and inhibits the protease activity of PCSK9 (32, 39–41). However, this complex can bind to specific target proteins and escort them to intracellular degradation compartments (42). The C-terminal domain of PCSK9 acts as a chaperone for PCSK9 mediated LDLR degradation (43–46). The LDLR ectodomain consists of the N-terminal ligand-binding domain and the epidermal growth factor (EGF) precursor homology domain (EGF-A, EGF-B, -propeller, and EGF-C domains). Thus, the PCSK9 catalytic domain directly binds to the first EGF-A domain of the LDLR on the cell surface (47, 48). On the surface of hepatocyte plasma membrane, the catalytic domain of secreted PCSK9 is internalized and the LDLR/PCSK9 complex enters the endosomal pathway. In the acidic compartment of the endosome, the affinity of PCSK9 toward LDLR is enhanced, while LDL is released from the LDLR. Thus, PCSK9 disrupts the recycling of LDLR to the cell membrane and acts to increase the plasma LDL levels. Furthermore, PCSK9 targets the LDLR for degradation not only via the extracellular pathway but also the intracellular pathway. PCSK9 also enhances intracellular LDLR degradation without recycling to the cell surface by direct intracellular trafficking from the trans-Golgi network to the late endosomes/lysosomes. This pathway requires clathrin light chains, and involves a different sorting mechanism than that of the extracellular degradation pathway (32, 49) (Figure 1).
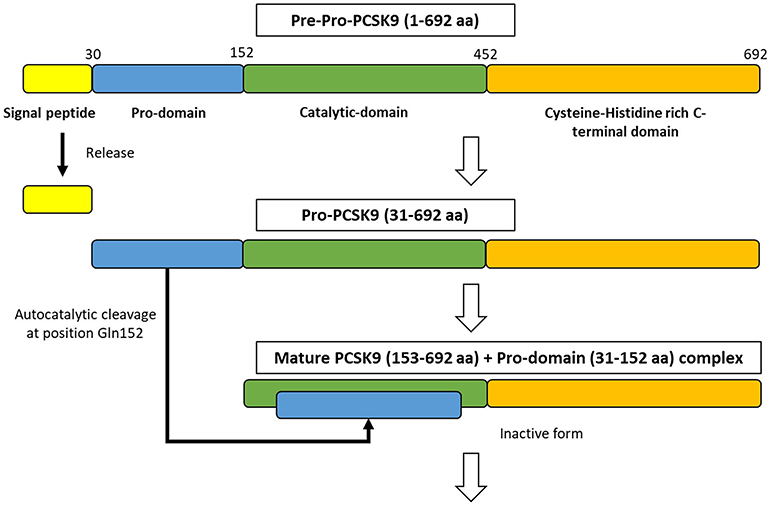
Figure 2. The synthesize of PCSK9 protein in the endoplasmic reticulum. PCSK9 comprises a single peptide (amino acid 1-30), a prodomain (amino acid 31-152), a catalytic domain (amino acid 153-452), and Cysteine-Histidine rich C-terminal domain (amino acid 453-692). In endoplasmic reticulum, proPCSK9 undergoes autocatalytic cleavage and the prodomain is separated from the mature PCSK9. The prodomain remains associated with the catalytic fragment, which inhibits the protease activity of the mature protein.; aa, amino acid.
PCSK9 Monoclonal Antibodies (MAbs)
The first approach to inhibit PCSK9 involved inhibiting the binding of PCSK9 to the LDLR (Figure 3). Two fully human anti-PCSK9 monoclonal antibodies (MAbs), alirocumab (SAR236553/REGN727 from Regeneron Pharmaceuticals/Sanofi) and evolocumab (AMG145 by Amgen), are currently approved for the treatment of hypercholesterolemia by the US Food and Drug Administration and European Medicines Agency. Anti-PCSK9 MAbs bind the catalytic domain of PCSK9, blocking extracellular interaction with the EGF-A domain of the LDLR by neutralizing PCSK9 (50). Subcutaneous injection of anti-PCSK9 MAbs provides a rapid and persistent reduction of LDL-C levels. Numerous trials and subsequent meta-analyses demonstrate that anti-PCSK9 MAbs reduce LDL-C levels by ~50–60% in patients with a diet-based therapy, alone or with various doses of stains without serious adverse events. Moreover, treatments involving PCSK9 MAbs have been shown to improve cardiovascular outcomes (51–55).
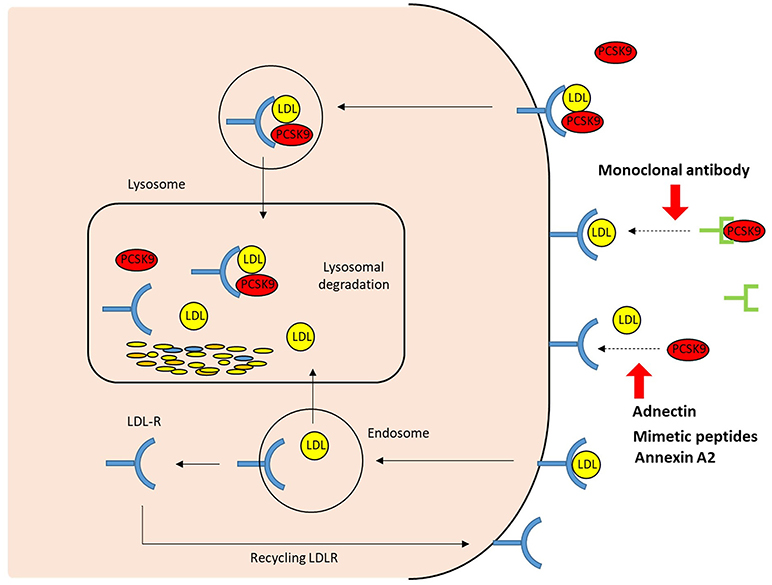
Figure 3. The extracellular mechanisms of PCSK9 inhibition. The strategies that target the binding between PCSK9 and LDLR extracellularly include PCSK9 monoclonal antibodies, adnectin (BMS-962476), mimetic peptides, and small molecules. By blocking PCSK9, these PCSK9 inhibitors increase LDLR recycling and LDL uptake; LDLR, LDL receptor.
Currently, two large-scale cardiovascular outcome trials have been conducted to evaluate the efficacy of the two PCSK9 MAbs mentioned above. The Further Cardiovascular Outcomes Research with PCSK9 inhibition in Subjects with Elevated Risk (FOURIER) study involving 27,564 high-risk patients with stable atherosclerotic cardiovascular disease is a large cardiovascular outcome clinical trial of the PCSK9 inhibitor evolocumab (56). The combination of evolocumab with statin therapy resulted in a substantial 59% reduction in LDL-C levels from a mean baseline value of 92–30 mg/dl, and a 15% relative reduction of cardiovascular events (cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization) after a median follow-up of 2.2 years.
The ODYSSEY OUTCOME trial enrolled 18,924 patients who had experienced a recent acute coronary syndrome and with elevated LDL-C levels despite high-intensity statin therapy (57). A reduction in LDL-C levels by 61% compared to placebo after one year was observed even though ~90% of patients were receiving high intensity statin therapy. Alirocumab reduced major adverse cardiovascular events (coronary heart disease death, non-fatal myocardial infarction, fatal or non-fatal ischemic stroke, and unstable angina requiring hospitalization) by 15% (hazard ratio: 0.85, 95% CI: 0.78–0.93) and all-cause death by 15% (hazard ratio: 0.85, 95% CI: 0.73–0.98). Taken together these two outcome trials demonstrate the effectiveness of PCSK9 inhibition in high risk populations with LDL-C > 70 mg/dl despite maximally tolerated/high intensity statin therapy.
There are issues which limit the widespread use of anti-PCSK9 MAbs. Anti-PCSK9 MAbs need to be administered by subcutaneous injection once or twice a month. The dosing frequency is low compared with other injectable treatments, such as insulin therapy. At the current dosing frequency, a high level of adherence over at least one year was reported for the self-injected anti-PCSK9 MAbs in a pooled analysis of six clinical trials of alirocumab (58). However, the use of injectable PCSK9 agent for essentially asymptomatic patients has several considerations; firstly maintaining the recommended schedule of injections over the long-term, and adequate refrigeration are required. Hence, it is unclear whether the adherence to these agents is any higher than that of daily administration of oral drugs in the real-world setting. In addition, PCSK9 inhibitors are not likely to be cost-effective for all patients given their current high price. It is estimated that the cost of PCSK9 inhibitors was ~$14,000 per person per year in the US and $5,000–7,000 per person per year in Europe (59–61). Specific high-risk sub-groups of patients with established vascular disease and in particular such patients with additionally a high baseline LDL-C would be expected to derive the greatest benefit and consequently these treatments are more cost-effective in such cases. Currently, the plan to cut the price of PCSK9 MAbs in the US have been announced by the pharmaceutical companies. Improving the cost effectiveness may provide an effective treatment option for a broader patient population. Whilst PCSK9 is a validated target for treatment there are several limitations to MAbs and recently, several approaches to inhibiting PCSK9 are under development as described below.
Non-antibody Approaches to PCSK9 Inhibition
Based on the biology of PCSK9 inhibition of PCSK9 expression, secretion from cells and interaction between PCSK9 and LDLR are considered to be effective and alternative approaches to MAbs (Figures 3, 4). The current therapeutic options involve gene editing or silencing technologies, small-molecule inhibitors, mimetic peptides, and vaccination (Table 1).
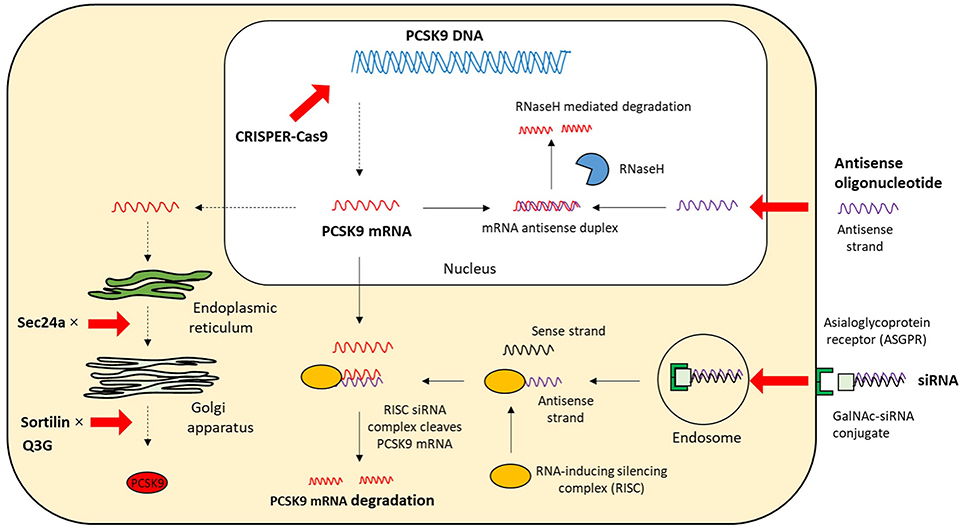
Figure 4. The intracellular mechanisms of PCSK9 inhibition. The strategies that target the expression and secretion of PCSK9 intracellularly include antisense oligonucleotide, siRNA, CRISPR/Cas9 gene editing, and small molecules; mRNA, messenger RNA; siRNA, small interfering RNA; CPRISPER-Cas9, Clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9; GalNAc, N-acetylgalactosamine; Q3G, quercetin-3-O-beta-glucoside; RNaseH, Ribonuclease H.
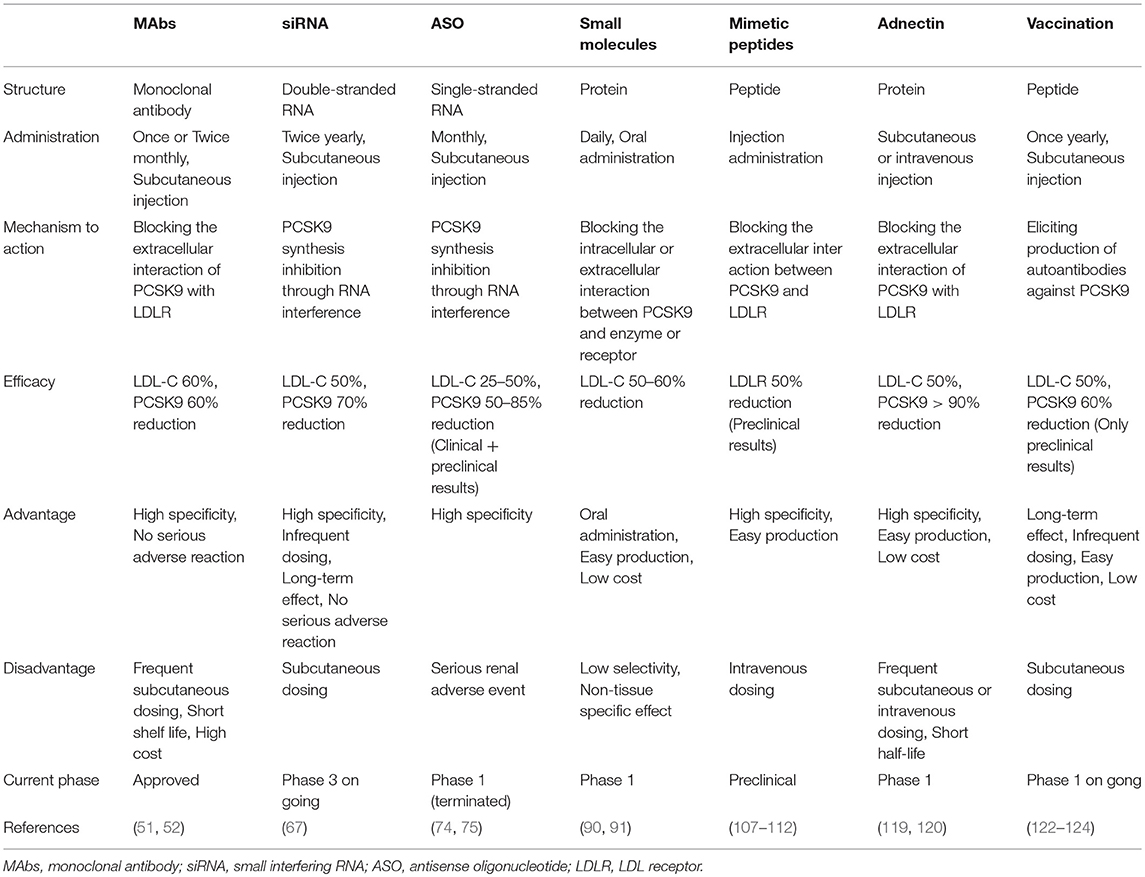
Table 1. The comparison among different approaches to PCSK9 inhibition.
Interfering With PCSK9 Expression
Small Interfering RNA (siRNA)
RNA interference is an endogenous post-transcriptional mechanism for regulating gene expression in almost any type of cell. It is specifically mediated by small interfering RNA (siRNA). siRNAs are double-stranded RNA molecules (~20–25 bp long), consisting of two short-stranded RNA molecules, a passenger sense strand, and a guide (anti-sense) strand that is complementary to the target mRNA. The siRNA duplex is separated by an enzyme called DICER and the guide anti-sense strand is loaded onto the RNA-induced silencing complex, which leads to sequence-specific gene silencing by catalytic cleavage and degradation of the targeted mRNA (62, 63). The single antisense strand/ RISC complex is highly stable hence multiple mRNA transcripts can be catalytically degraded.
siRNA molecules can be synthesized relatively easily and must be modified to prevent degradation by endonucleases. ALN-PCS, a PCSK9-specific siRNA formulated in a lipid nanoparticle, was developed as a PCSK9 inhibitor. This new approach resulted in a rapid and durable reduction of PCSK9 mRNA and LDL-C levels. Healthy volunteers received ALN-PCS by intravenous infusion for 60 min. The highest dose of ALN-PCS reduced PCSK9 mRNA and protein levels by 70% and LDL-C levels by 40% after 3 days. The formulation of ALN-PCS was subsequently improved to limit the dose and volume of drug injected (64). Inclisiran (ALN-PCSsc: ALN-60,212 by Alnylam/The Medicines Company) is a fully chemically modified stabilized PCSK9-specific siRNA. Inclisiran is composed of one 2′-deoxy, eleven 2′-fluoro-, and thirty-two 2′-O- methyl-modified nucleotides, and a triantennary N-acetylgalactosamine (GalNAc) that is conjugated to the 3′-end of the passenger strand. The asialoglycoprotein receptor expressed highly on the hepatocyte surface specifically recognizes the GalNAc (65), which enables a rapid uptake of inclisiran with high specificity, only by the liver, after a subcutaneous low-volume injection. The plasma levels of inclisiran thus fall to undetectable levels within 24 h (66). This mechanism effectively prevents any off-target effects because PCSK9 is also present in extrahepatic tissues.
In phase II of the placebo-controlled, double-blind, randomized trial (ORION-1 trial), the efficacy, safety, and tolerability of inclisiran were evaluated in patients who had a history of atherogenic CVD or high atherosclerotic CVD-risk with elevated LDL-C levels despite having received the maximum tolerated doses of lipid-lowering therapy (67, 68). Patients who received two doses of inclisiran (300 mg per dose) on days 1 and 90 exhibited mean percent reductions of the PCSK9 and LDL-C levels of 69.1 and 52.6%, respectively, on day 180. Furthermore, no significant serious adverse events induced by inclisiran were apparent, and thus it was deemed to be relatively safe and well-tolerated during the study. These observations suggest that the administration of inclisiran by the healthcare professionals once or twice a year might result in potent and long-lasting lowering of LDL-C levels. This may improve treatment adherence and individual variability in response to the treatment. However, the mechanism of the long-lasting effect remains unknown and, hence, the effect of inclisiran should be investigated in a pharmacokinetic study. Further, safety monitoring over a long period of time in further clinical studies is also needed. Currently, there are some ongoing phase III trials (ORION 4, 5, 9, 10, and 11) for a large number of patients with the risk of atherosclerotic CVD or familial hypercholesterolemia. ORION-4 trial will evaluate the effect of inclisiran on cardiovascular outcomes in approximately 15,000 participants with pre-existing atherosclerotic CVD.
Antisense Oligonucleotides (ASOs)
ASOs, which consist of short, single-stranded nucleotides (12–25 bp long), interfere with gene expression by binding to the target mRNA directly, via Watson-Crick base paring, within the nucleus or cytoplasm. The majority of currently developed ASO drugs rely on an RNase H-dependent cleavage to degrade mRNA in a DNA-RNA hybrid (69). The activity of RNase H-dependent ASO is different between nucleus and cytoplasm due to some factors such as specificity and concentration of RNase H protein (70, 71). ASOs against PCSK9 mRNA inhibit protein synthesis to specifically reduce intracellular and extracellular PCSK9 levels.
Second-generation ASOs targeting PCSK9 mRNA (ISIS 394814/BMS 844421 by Bristol Myers Squibb/ISIS Pharmaceutical), with an affinity-enhancing modification of a nucleic acid-building block called, 2′-O-methoxyethyl RNA, were initially explored in preclinical studies. It was demonstrated that the administration of high doses of 2-O-methoxyethyl RNA ASOs lowered PCSK9 mRNA levels by 92% and increased LDLR protein levels 2-fold, which resulted in a reduction of LDL-C levels by 38% in vivo (72). However, the development of these ASOs was terminated because of insufficient binding affinity. Subsequently, shorter ASOs (SPC4061/SPC5001 by Santaris Pharma A/S) relying on locked nucleic acid technology is being developed instead of second-generation ASOs. These ASOs targeting PCSK9 have a stable conformation, and higher binding affinity and specificity for PCSK9 mRNA, inducing rapid, sustained, and potent effects compared with longer oligonucleotide. Early studies suggest that PCSK9 mRNA levels are reduced by 60% within 24 h post-injection, and the effect was sustained for over 16 days (73). In non-human primates, the hepatic PCSK9 mRNA levels and plasma LDL-C levels decreased by 85 and 50%, respectively (74). Meanwhile, subcutaneous administration of SPC5001 lowered the PCSK9 mRNA and LDL-C levels in a dose-dependent manner, by ~50 and 25%, respectively, in healthy human volunteers. The drug also decreased apolipoprotein B and increased apolipoprotein A1 levels (75). Nevertheless, transient renal tubular toxicity (one subject experienced an onset of acute tubular necrosis) and injection site reactions were also observed, and further clinical development of SPC4061/SPC5001 was terminated (75, 76). The exact cause of acute kidney also injury remains uncertain. Hence, understanding the molecular mechanism of renal toxicity and a strategy for sensitive detection of renal injury are important in the future development of this oligonucleotide drug. As another example, an anti-PCSK9 antisense oligonucleotide modified by a bridged nucleic acid is being developed; in early studies it exhibited high potency and low toxicity. In mice fed an atherogenic diet and administered this compound twice weekly for 6 weeks, a dose-dependent reduction of PCSK9 mRNA and LDL-C levels was observed (77).
The technology of antisense oligonucleotide synthesis is progressing by developing various structural modifications of the phosphate backbone, sugar moieties, and targeting ligand (78). These allow low frequency of administration and low individual variations compared with other small-molecule drugs. However, these modifications might cause potential off-target effects or adverse effects (79). It is important that ASOs accurately match the target mRNA, and are specifically delivered to the hepatocytes to reduce off target gene toxicity or effects in other tissues which may be undesirable.
Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/CRISPR-Associated Protein 9 (Cas9)
CRISPR/(Cas9) (by Academic project/AstraZeneca) is a useful genome-editing tool that allows a quick destruction or editing of a target gene. This system is more accurate, faster, and cheaper than other existing genome-editing tools. It consists of Cas9 endonuclease and single guide RNA (sgRNA), which is ~100 bp long. A 20-nt sequence at the 5′-end of sgRNA recognizes and hybridizes with a complementary sequence of target DNA site located directly next to a protospacer-adjacent motif sequence (80). The Cas9-sgRNA–mediated DNA cleavage generates a double-strand break that triggers DNA repair machinery by non-homologous end-joining or homology-directed repair (81). Non-homologous end-joining is error-prone and induces disruptive insertions and/or deletions at the target site, while homology-directed repair can use an exogenous DNA repair template to perform precise knock-in of a desired alteration of the genomic DNA (82, 83). The CRISPR/Cas9 system has been shown to effectively disrupt the PCSK9 gene in mouse liver, resulting in significantly reduced plasma PCSK9 protein levels and increased hepatic LDLR levels. The plasma cholesterol levels were reduced by 35–40%, which was consistent with the 35–52% reduction observed in PCSK9 knockout mice compared with wild-type mice (84–86). Non-sense mutations resulting in loss-of-function of PCSK9 are associated with significant reduction of both LDL-C levels and coronary heart disease risk, with no adverse clinical consequences (28). This implies that PCSK9 genome editing technique might be a novel strategy that may be used to permanently alter the human genome for the prevention of coronary heart disease in patients with hypercholesterolemia.
However, the genome-editing technology entails ethical, legal, and safety issues that need to be resolved before it can be used as a general widely accepted therapeutic approach in human gene (87). The major limitation of CRISPR-Cas9 editing is more frequent off-target mutation than the intended mutation. The targeting specificity of Cas9 is regulated by a connection between the guide sequence of sgRNA and the protospacer-adjacent motif next to the target sequence in the genome. However, off-target cleavage is observed on a DNA sequence that harbors several mismatches with the distal protospacer-adjacent motif in the sgRNA-guiding sequence (88). It is important to detect potential off-target sites and optimize the CRISPR/Cas9 system to reduce the off-target activity without sacrificing the efficiency of on-target cleavage and minimize off-target mutations. Currently, CRISPR-Cas9 technology is widely adapted in various fields as a promising genome-editing tool that is easy to use and characterized by good cost-effectiveness. The long-term benefit and safety in human subjects need to be assessed by designing sgRNA with higher specify than currently attainable.
Small Molecules
Cholesteryl ester transfer protein (CETP) under normal physiological conditions transfers equimolar amount of triglycerides to high-density lipoprotein, while it promotes the transport of cholesteryl ester from high-density lipoprotein to apolipoprotein B-containing lipoproteins, such as very low-density lipoprotein, intermediate-density lipoprotein, and LDL (89). Hence, CETP inhibitor pharmacologically increases high-density lipoprotein-cholesterol levels and decreases LDL-C levels. A new CETP inhibitor, called K-312 (by Kowa), reduces PCSK9 expression in hepatocytes by decreasing the active forms of SREBP-1 and SREBP-2 that regulate PCSK9 promoter activity (90). In vivo, cholesterol-fed white rabbits were administered K-312 for 2 weeks which resulted in a 63% reduction of PCSK9 mRNA levels in the liver, reduced LDL-C levels (vehicle vs. K-312: 272.5 ± 42.2 mg/dl vs. 193.0 ± 36.5 mg/dl), and increased high-density lipoprotein-cholesterol levels (vehicle vs. K-312: 29.9 ± 3.7 mg/dl vs. 93.3 ± 14.7 mg/dl). Even though the plasma CETP activity was suppressed by K-312, the suppression of PCSK9 mRNA expression by K-312 was observed after siRNA silencing of CETP in vitro. Therefore, K-312 can decrease not only LDL-C levels but also PCSK9 expression via a mechanism independent of CETP inhibition. In addition, K-312 treatment has been shown to attenuate the progression of atherosclerosis. Hence, this compound may serve as a new effective therapy for hypercholesterolemia and possibly prevention of CVD.
PF-06446846 (by Pfizer) is an orally active small molecule, which directly and selectively inhibits translation of PCSK9 by stalling the 80 S ribosome in the proximity of codon 34. Its activity is dependent on the amino acid sequence of the nascent chain within the ribosome exit tunnel. Orally-administered PF-06446846 reduces plasma PCSK9 and LDL-C levels without liver toxicity symptoms in vivo (91). This promising approach had the potential of becoming a potent therapeutic option inhibiting PCSK9 synthesis by oral dosing; however, its development was discontinued for competitive reasons (92).
Berberine (BBR) is a natural cholesterol-lowering compound. It is an isoquinoline plant alkaloid (2,3-methylenedioxy-9,10-dimethoxyprotoberberine chloride) isolated from Huanglian (Coptis chinensis), and exerts different pharmacological and therapeutic effects on carbohydrate and lipid metabolism, diabetes mellitus, the immune system, endothelial function, and cardiovascular system, etc. (93). Previous studies indicated that BBR up-regulates the expression of LDLR-coding gene independent of intracellular cholesterol levels and down-regulates PCSK9 transcription. It has been demonstrated that oral administration of BBR reduced plasma levels of total cholesterol by 29%, triglycerides by 35%, and LDL-C by 25% in 32 patients with hypercholesterolemia (94). PCSK9 synthesis is controlled at transcriptional level by SREBPs and HNF-1α, which work cooperatively with SREBP-2 to transactivate the PCSK9 promoter (35). BBR simultaneously decreases HNF-1α and SREBP-2 levels, leading to a strong suppression of PCSK9 transactivation. Further, in vitro, BBR reduces PCSK9 mRNA levels in a time- and dose-dependent manner (95). This mechanism may negatively affect LDL-C metabolism because SREBPs are also important for increasing LDLR transcription; however, BBR was found to elevate LDLR expression independent of SREBPs via a post-transcriptional mechanism that stabilizes the LDLR mRNA in vivo (94). Nevertheless, further studies into the precise mechanism of BBR inhibition of PCSK9 are needed, before BBR may progress to the next stage of drug development as a new therapeutic molecule for lowering LDL-C levels.
Oleanolic acid is a pentacyclic triterpenoid that is common in nature, and widely distributed in plants and medicinal herbs as a free acid or as a saponin aglycone (96). It exerts various beneficial effects, such as anti-cancer, hepatoprotective, hypolipidemic (97), anti-oxidative, anti-inflammatory (98, 99), and endothelial-protective effects (100). Previous reports demonstrate that oleanolic acid treatment reduces total serum cholesterol, LDL-C, triglyceride, and free fatty acid levels in mice. Moreover, oleanolic acid reduced PCSK9 protein and mRNA levels in a time- and dose-dependent manner in vitro (39). However, the mechanism of PCSK9 inhibition and the utility as a PCSK9 inhibitor remain unknown.
All small-molecule inhibitors have the advantage of oral administration and lower production cost than MAbs. However, the achievement of oral delivery will require the maintenance of stability and potency to avoid proteolytic degradation in the gastrointestinal tract. In addition, broad blood distribution due to oral administration may increase the possibility of harmful side effects because of off-target interactions, since it is less specific than MAbs.
Blocking the Function of PCSK9
Small Molecules
Oral administration of small-molecule inhibitors specific to PCSK9 appears to constitute a promising therapeutic strategy to disrupt the interaction of PCSK9 and LDLR. The small-molecule inhibitors can be produced easily and inexpensively. However, this approach remains challenging because it is difficult to design small molecules that specifically bind to the site of interaction between the catalytic subunit of PCSK9 and the EGF-A domain of LDLR because of the fairly large size of the flat interface (47, 101). In addition, small molecules have generally poor stability, selectivity, and potency. Small molecule-inhibitors of PCSK9 might be less specific than PCSK9 MAbs which increase the potential of adverse drug reactions and allosteric effects on different intracellular functions of PCSK9.
In spite of the above, DS-9001a (by Daiichi Sankyo/Pieris Pharmaceuticals), a small biologic molecule (~22 kDa) which requires subcutaneous administration, was recently created. DS-9001a is composed of an albumin-binding domain fused with an artificial lipocalin mutein (ABD-fused anticalin protein). The lipocalin architecture provides a high structural plasticity of the binding site, and enables potent and specific recognition of the target protein (102). DS-9001a strongly inhibits the binding of PCK9 to LDLR and attenuates LDLR degradation. A single intravenous injection of DS-9001a resulted in a reduction of LDL-C levels by ~62% and this effect was sustained for up to 21 days in the cynomolgus monkey (103). It can be manufactured using a bacterial expression system, which is associated with a low production cost. In addition, the small size and high solubility of DS-9001a enable the administration of increased drug concentrations at lower volumes than those of MAbs (104). A single ascending-dose study of DS9001a performed its safety and tolerability in healthy volunteers. Further, pharmacokinetic/pharmacodynamic analysis elucidated a dose-dependent reduction of free PCSK9 and LDL-C levels. Effective half-life of DS-9001a was ~12.5 days (105). Therefore, DS-9001a may constitute a potent therapeutic option, as an alternative to anti-PCSK9 MAb therapy.
Mimetic Peptides
Mimetic peptides are small amino acid stretches designed to biologically mimic the peptic structure of a target protein. Several peptides that mimic the EGF-A or EGF-AB binding domains of LDLR have been developed as competitive PCSK9 inhibitors, which bind to the catalytic domain of PCSK9, and limit the interaction between PCSK9 and LDLR. Peptides have emerged as therapeutic alternatives that bridge the gap between small molecules and large antibodies (106). A synthetic LDLR EGF-A domain peptide has been found to inhibit PCSK9-mediated degradation of LDLR and maintain LDL uptake in a dose-dependent manner (44, 107). Furthermore, recently a truncated 26 amino acid EGF-A analog, which is a smaller peptide-based inhibitor of PCSK9/LDLR binding, has been synthesized (108). This exhibits an enhanced binding affinity to PCSK9, promote LDLR recycling and lowers LDL-C levels. In addition, an LDL-R mimetic with the gain-of-function H306Y substitution which increases the affinity of the receptor for PCSK9 is in preclinical development as other peptide candidates (109). Pep2-8 is also the small peptide that mimics the secondary structural elements of the EGF-A domain, hindering the interaction between PCSK9 and the LDLR (110). Alternatively, approaches involving the catalytic domain, prodomain, or C-terminal domain of PCSK9 can be employed. It has been demonstrated that mimetics derived from PCSK9 fragments reduce PCSK9-mediated degradation of LDLR in vivo (111, 112).
Annexin A2 (AnxA2) is an extracellular endogenous antagonist that interacts with the C-terminal domain of PCSK9, and has received attention as a natural inhibitor of PCSK9 binding to LDLR (113, 114). High levels of AnxA2 are present in the lung, pancreas, colon, and adrenal tissues, while low levels of this antagonist are present in the liver, kidney, and spleen. The repeat-one domain of AnxA2 can specifically bind to the C-terminal domain of PCSK9 at the cell surface and inhibit PCSK9-mediated degradation of the LDLR. In AnxA2 knock-out mice, there was an ~2-fold increase in the circulating PCSK9 levels and an ~1.4-fold increase in LDL-C levels was observed, in addition to a reduction of LDLR protein levels by ~50% in extrahepatic tissues. On the other hand, an adenoviral overexpression of AnxA2 in the liver results in an increase of LDLR protein levels, mostly in extrahepatic tissues (114). Furthermore, a peptide mimicking AnxA2 repeat-one domain directly inhibited the interaction between PCSK9 and LDLR (115). Thus, the extrahepatic physiological role of AnxA2 in the regulation of PCSK9 enhances the degradation of LDLR. A small peptide mimicking AnxA2 may constitute a potential approach for PCSK9 inhibition.
Adnectin
Adnectin is a synthetic protein based on the 10th type III domain of human fibronectin. Its variable loops can be designed to efficiently introduce a surface that binds therapeutically relevant targets with high affinity and specificity (116). It is small and compact, with no sequence homology to immunoglobulins, although it possesses a β-sheet–fold structure with diversified loops analogous to the antibody variable regions (117, 118). Adnectin BMS-962476 (by Bristol-Myers Squibb/Adnexus) is a PCSK9-targeting polypeptide conjugated with polyethylene glycol to enhance its pharmacokinetic profile, which binds human PCSK9 with a subnanomolar affinity. Adnectin hinders the interaction between extracellular PCSK9 and the EGF-A domain of LDLR as a potential alternative to MAbs, and prevents PCSK9-induced degradation of the LDLR. Its production is easier and less expensive than that of antibodies since the molecule can be produced using bacterial expression systems. A preclinical trial revealed that BMS-962476 (5 mg/kg) rapidly reduces free PCSK9 levels by more than 99%, while increasing 6-fold the total PCSK9 levels in the cynomolgus monkey. Hence, LDL-C levels were reduced by up to 55% within 48 h, with the effects persisting for up to 3 weeks (119). In the first study involving humans, a single ascending regimen subcutaneous or intravenous dose of BMS-962476 resulted in a reduction of free PCSK9 levels by more than 90% without serious adverse events; LDL-C levels were reduced by up to 48% at maximum dose between day 4 and 14 (120). These observations suggested that BMS-962476 is a well-tolerated and effective agent without notable safety issues at an early phase of development. However, its effect should be followed over a longer time period in a large number of subjects. Nevertheless, BMS-962476 is a promising therapeutic drug and an alternative to MAbs against circulating PCSK9 through which to achieve significant lowering of LDL-C levels.
PCSK9 Vaccination
As an alternative strategy of long-term PCSK9 inhibition, a peptide-based anti-PCSK9 vaccination approach has been developed. PCSK9 vaccine stimulates the immune system to generate high-affinity PCSK9-specific antibodies, and then blocks the ability of PCSK9 to bind to the LDLR. Even though active vaccination exerts the same therapeutic effect as passive administration of PCSK9 MAbs, the effect can be achieved with fewer injections, at a lower dose, and potentially in a cost-effective manner, without the possibility of inducing a drug-neutralizing immune response. Induction of an antibody response against PCSK9 is limited by B-cell tolerance; however, efficient activation of self-reactive B-cells is provoked by the presence of foreign T-helper epitopes or antigen multivalency (121), which induce particularly robust, high-titer, and long-lasting autoantibody response against foreign antigens. A peptide-based anti-PCSK9 vaccine called AT04A consists of short peptides (by Affiris), mimicking fragments of a mature human PCSK9 protein, conjugated to a foreign carrier protein (Keyhole limpet hemocyanin) that provides T-helper cell epitopes (122). The amino acid sequence of mimotope-peptides used for vaccination is different from the native peptide sequence to be targeted, but these peptides are still able to induce a highly PCSK9-specific antibody response. These peptides are recognized as foreign by the immune system because they do not share sequence identity with other human proteins, and offer a potent means of inducing specific antibodies to self-antigens. For instance, the total cholesterol and LDL-C levels were reduced by up to 30 and 50% in anti-PCSK9 immunized mice, respectively. The atherosclerosis was reduced by 64% in blood vessels with this approach. Moreover, the vaccine-generated anti-PCSK9 antibody had a half-life of ~4 months and resulted in sustained significant reductions in cholesterol levels for at least 1 year in mice. Booster vaccination with the same dose a year after the initial immunization effectively reactivated the anti-PCSK9 antibody response, revealing the potential of a yearly re-boosting immunization (123).
One approach for inducing a strong antibody response against self-antigens involves the displaying of self-antigen in a highly dense, repetitive format on the surface of virus-like particles (VLP). VLPs are formed by a self-assembly of the viral structure protein without a viral nucleic acid, which is used to produce multivalent vaccines. PCSK9-dysplaying VLPs elicit high-titer peptide-specific PCSK9-reactive response in mouse. A VLP-based vaccine targeting PCSK9 synergistically has been shown to reduce LDL-C levels by 30–40% in macaques treated with statins (124).
These findings suggest that PCSK9 vaccination could be a promising strategy for sustained reduction of LDL-C levels. However, longer-term safety, in addition to efficacy, is an important concern for its successful development. It is possible that active immunization would cause antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity with non-specific cell destruction (125). A previous study demonstrated that PCSK9 vaccine is well-tolerated in short- and long-term experiments, and that immunization does not induce any apparent side effects (123). Further clinical studies are necessary to identify the optimal immunization scheme and to evaluate long-term safety. Accordingly, phase I clinical trials (AT04A and AT06A) are currently ongoing to test that.
Inhibition of PCSK9 Secretion
PCSK9 is synthesized and transported from the ER to the Golgi apparatus, and is subsequently secreted into circulation. A loss-of -function mutation in the prodomain, results in an inhibition of the secretion of PCSK9 because protein trafficking is disrupted (25). Inhibition of pathways which are major mediators of PCSK9 secretion may prevent PCSK9 from reaching the cell surface, which may in turn be used for preserving LDLR expression in the liver and subsequent reduction of plasma LDL-C levels.
Small Molecules
Sortilin is encoded by the gene SORT1, which is a high-affinity sorting receptor of PCSK9 and thus influences cholesterol levels. It predominately co-localizes with PCSK9 in the trans-Golgi network. Sortilin facilitates cellular secretion of PCSK9 in the late secretory pathway from the trans-Golgi network to the plasma membrane (126). The strongest interaction between sortilin and PCSK9 occurs at a pH 6.5, whereas it is completely lost at pH 5.5. Thereby, sortilin binds PCSK9 in the trans-Golgi network (pH ~6.5) and releases it in secretary vesicles (pH ~5.5). Increased intracellular PCSK9 levels and reduced circulating PCSK9 levels occur in sortilin-deficient mice. These effects increase LDLR levels in the liver and reduce plasma LDL-C levels. On the other hand, overexpression of human sortilin in the liver increases circulating PCSK9 levels and reduces hepatic LDLR levels, thus increasing plasma LDL-C levels. Moreover, a significant positive correlation between sortilin and PCSK9 is observed in human cohort studies. In healthy individuals, a significant positive correlation between sortilin and PCSK9 was noted, suggesting that sortilin expression affects circulating PCSK9 levels (126, 127). In addition, sortilin is associated with not only dysregulated lipoprotein metabolism but also the development of type 2 diabetes mellitus and progression of atherosclerosis. These findings suggest that sortilin might be a potential therapeutic target to reduce atherogenic risk. However, sortilin is essential for proper neuronal functionality on the nervous system. Therefore, caution is needed when considering the use of sortilin as a therapeutic tool (128, 129).
Sec24a, a protein which is incorporated into the coat protein complex II-coated vesicles, is required for the transport of PCSK9 from the ER to the Golgi apparatus. In Sec24a-deficient mice, reduced plasma PCSK9 levels and elevated hepatic PCSK9 levels are observed, which lead to an increase in LDLR expression on the liver surface. In addition, overexpression of Sec24a promotes the secretion of PCSK9 (130, 131). The pharmacological inhibition of Sec24a function may be a useful alternative potential approach to inhibit PCSK9 secretion. However, it is worth noting that it is important to identify the presence of other proteins that require Sec24a for their secretion from the ER.
SRT3025 is a third-generation synthetic Sirtuin-1 (SIRT1)-activating compound, currently under investigation as an allosteric activator of SIRT1 (by GSK/Sirtis). SIRT1, a member of the sirtuin family of NAD ± dependent deacetylases, contributes to the regulation of body energy homeostasis. It is broadly expressed in many tissues including adipose tissue, liver, pancreas and skeletal muscle. Therefore, SIRT1 plays also an essential role in the regulation of hepatic lipid metabolism (132). In vitro SRT3025 attenuates PCSK9 secretion from hepatocytes and reduces plasma PCSK9 levels in vivo, and thus increases LDLR expression in the hepatocytes. This is accompanied by a significant reduction of plasma LDL-C levels and atherosclerosis (133). Administration of SIRT1 to elderly individuals was safe, and serum cholesterol, LDL-C, and triglyceride decreased (134). Employing a pharmacological SIRT1 activator such as SRT3025 might offer a potential anti-atherosclerotic strategy.
Quercetin is a phytochemical which occurs predominantly as quercetin-3-O-beta-glucoside (Q3G) in a broad range of fruits and vegetables. Oral administration of Q3G was shown to have cholesterol lowering and insulin lowering properties (135, 136). Q3G has been found to reduce PCSK9 secretion and to increase LDLR expression, leading to increased LDL-C uptake and reduction in plasma LDL-C levels. Moreover, it was reported that Q3G does not affect SEC24A expression, but significantly inhibits intracellular sortilin level (by 50%) and its m-RNA level (by 40%) (137). However, another report suggested that Q3G does not affect PCSK9 expression in mice fed a low-cholesterol diet; in mice fed a high-cholesterol diet, reduction of circulating PCSK9, and a corresponding increase in cellular PCSK9 and LDLR levels was observed (136). This might be caused by a corrective activity of Q3G, in that Q3G reverses the LDLR/PCSK9 ratio in the liver. These observations suggest that Q3G supplementation on the background of a high-cholesterol diet results in regulation of PCSK9 expression and secretion, by inhibiting sortilin expression. This results in LDL-C clearance by increasing hepatic LDLR, and reversing hypercholesterolemia caused by the diet. Further studies on its effects on lipid and glucose metabolism may offer a chance of developing a therapy effective in high-risk patients with dyslipidemia and diabetes mellitus.
PCSK9 sequence variation resulting from the PCSK9-Gln152/His substitution was identified in a French-Canadian family, which results in restricted proteolysis and secretion through separate mechanisms through separate mechanisms. It was reported that the loss-of-function PCSK9-Gln152/His variant is associated with reduced circulating PCSK9 and LDL-C levels (by 79 and 48%, respectively). In addition, the Gln152/His amino acid substitution reduces the ability of proPCSK9 to undergo autocatalytic cleavage and subsequent secretion in vitro (138, 139). These findings support the notion that inhibition of autocatalytic processing in the ER may be a novel strategy to specifically alter PCSK9 secretion.
Future Perspectives and Controversies
Statins are the first-line drug treatment for hypercholesterolemia; however, even the maximally tolerated dose of statin, alone or in combination with ezetimibe, is not sufficient for attaining the LDL-C goals in patients with the highest risk and most extreme levels of cholesterol. Therefore, PCSK9 inhibitors have attracted a lot of attention as a potent option for regulating LDL-C levels. PCSK9 expression is regulated by a SREBP-2 transcription factor, which induces the expression of genes involved in cholesterol synthesis and uptake, according to the intracellular cholesterol levels (34). Statins induce SREBP-2 activity by inhibiting the 3-hydroxy-3methylglutaryl coenzyme A reductase. This leads to both a reduction of endogenous cholesterol synthesis and an increase in LDLR expression (Figure 1). However, statins also enhance PCSK9 expression by activating SREBP-2, which attenuates the reduction of plasma LDL-C levels by degrading LDLR. This mechanism at least in part explains the observation that doubling of a statin dose only results in an additional 6% reduction of LDL-C levels. This suggests that adding PCSK9 inhibitors to a statin therapy may result in a beneficial synergistic effect (140). It was reported that as many as 20% of patients who received statin therapy exhibited statin-associated muscle symptoms in a routine care setting. Statin intolerance greatly contributes to the burden of CVD together with patient related factors such as medication discontinuation and low adherence (141, 142). Therefore, inhibiting PCSK9 may be a useful alternative approach in high-risk patients with statin intolerance.
Each approach to PCSK9 inhibition has distinct advantages and disadvantages (Table 1). Even though the use of anti-PCSK9 MAbs can lead to a remarkable reduction in LDL-C levels and an acceptable risk of adverse events, their inconvenience on account of 12 or 24 yearly subcutaneous injections and high manufacturing costs are major impediments to practical use. On the other hand, the new approaches to PCSK9 inhibition may offer the possibility to improve on the durability of effect, route of administration, adherence, storage, and cost effectiveness, even though none of these have been approved. For example, small molecules can be administered orally without adverse events at the injection site, including pain. Thus, they might be broadly more acceptable to patients. In addition, the productions of these inhibitors are easier and cheaper than those of anti-PCSK9 MAbs. Thus, oral administration in combination with statins may be an attractive strategy. One advantage of siRNA and vaccination over anti-PCSK9 MAbs is the infrequency of administrations (once or twice a year vs. 12–24 injection per year), which not only offers the potential of significant and consistent reduction of LDL-C levels, but also improvement in adherence and, subsequently, low inter-patient variability in response to treatment due to non-compliance. The manufacturing cost of inclisiran is assumed to be lower than that of anti-PCSK9 MAbs and the same as that of small molecules because of the relatively easy manufacturing process and stability of dried oligonucleotides at room temperature (66). PCSK9 vaccination is also likely to be cheaper than anti-PCSK9 MAbs and easy to apply to large populations in most countries (125). These promising approaches to PCSK9 inhibition are expected to provide potential benefits beyond those of anti-PCSK9 MAbs.
Anti-PCSK9 MAbs inhibit only the extracellular PCSK9 activity but, on the other hand, some approaches focused on how PCSK9 expression and secretion affect intracellular PCSK9 activity. For example, inclisiran reduces the intracellular and extracellular PCSK9 levels by inhibiting PCSK9 synthesis in the hepatocytes. However, the magnitude of the effect of inclisiran on all lipid and lipoprotein levels is broadly similar to that of anti-PCSK9 MAbs (67, 68). This suggests that the inhibition of intracellular PCSK9 may not contribute meaningfully to the regulation of PCSK9-mediated LDLR degradation, which is dependent on the PCSK9 in the circulation. Nevertheless, inclisiran has a long-lasting and durable effect of LDL-C reduction compared to anti-PCSK9 MAbs after a single injection and this may be associated more with the effect of altering intracellular PCSK9 metabolism or quite simply mean that the inclisiran RISC complex is highly stable and catalytic and hence can degrade multiple transcripts. Since the function and mechanism of intracellular PCSK9 inhibition remain unclear, additional lipoprotein kinetic studies would help to reveal the effect of the inhibition of intracellular pathway on lipid metabolism. In addition, relevant kinetic studies of other approaches including the inhibition of secretion will be helpful to understand the potential role of PCSK9 in the intracellular pathway.
New therapeutic strategies with novel mechanisms of action are being developed, which appear to be reasonably effective. However, the successful development entirely depends on their safety (143, 144) (Table 2). The efficacy of PCSK9 MAbs and loss-of-function mutation of PCSK9 revealed that very low LDL-C levels had no major safety concern (28, 145, 146). Nevertheless, PCSK9 has other possible physiological roles in the inflammatory response, glucose metabolism, and the nervous system beyond lipid metabolism. Several studies reported that it causes insulin intolerance, progression of lesion inflammation, and neurogenesis or neuronal apoptosis (147). EBBINGHAUS (Evaluating PCSK9 Binding antiBody Influence oN cognitive in HeAlth cardiovascular Risk Subjects) evaluated the possible adverse effects of evolocumab on cognition in 1974 patients from the FOURIER trial, but found no evidence of statistically significant adverse effects on cognition (148). In contrast, meta-analysis of data from more than 10,000 patients receiving PCSK9 MAbs raised the possibility through adverse event reporting of an increased risk of cognitive impairment, which was unrelated to achieved LDL-C levels (149–151). The new therapeutics to inhibit PCSK9 may be associated with unexpected off-target effects within the same or different organs and the long-term safety of new mechanisms of action and technologies is still unknown. Therefore, further studies are required to elucidate the global physiological function of PCSK9; improve our understanding of the mechanisms by which each approach reduces PCSK9; and long-term assessment of safety. Should the safety be proven, these will pave the way for additional potent therapeutic options for the reduction of LDL-C.
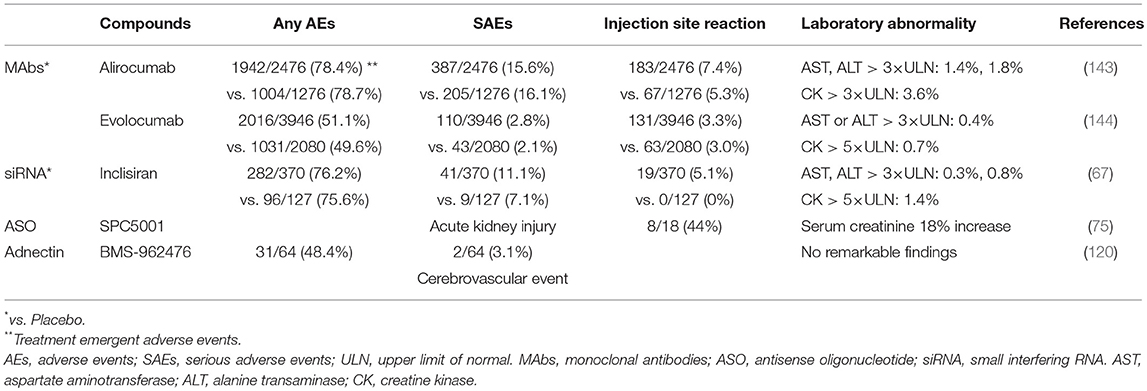
Table 2. The adverse effects in clinical trials.
Author Contributions
TN searched the literature and wrote the manuscript. KR provided guidance and edited the manuscript.
Conflict of Interest Statement
KR has received personal fees (data safety monitoring board) from AbbVie, Inc.; consultant fees/honoraria from Aegerion, Algorithm, Amgen, AstraZeneca, Boehringer Ingelheim, Cerenis, Eli Lilly and Company, Ionis Pharmaceuticals, Kowa, Medicines Company, MSD, Novartis, Pfizer, Regeneron Pharmaceuticals, Inc., Resverlogix, Sanofi, and Takeda; and research grants from Kowa, Pfizer, and Regeneron Pharmaceuticals, Inc. CD, MB-Bobanovic. TN was supported by a grant from the Uehara Memorial Foundation.
Abbreviations
AnxA2, annexin A2; ASO, antisense oligonucleotide; BBR, berberine; CETP, cholesteryl ester transfer protein; Cas9, CRIPSR-associated protein 9; CRISPR, clustered regularly interspaced short palindromic repeat; CVD, cardiovascular disease; EGF, epidermal growth factor; ER, endoplasmic reticulum; HNF, hepatocyte nuclear factor; LDL, low-density lipoprotein; LDL-C, LDL cholesterol; LDLR, LDL receptor; MAb, monoclonal antibody; PCSK9, proprotein convertase subtilisin kexin 9; Q3G, quercetin-3-O-beta-glucoside; sgRNA, single guide RNA; siRNA, small-interfering RNA; Sirt1, Sirtuin-1; SRE, sterol regulatory element; SREBP, sterol regulatory element-binding protein; VLP, virus-like particle.
References
1. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. (2017) 38:2459–72. doi: 10.1093/eurheartj/ehx144
2. Piepoli MF, Hoes AW, Agewall S, Albus C, Brotons C, Catapano AL, et al. 2016 European guidelines on cardiovascular disease prevention in clinical practice: the sixth joint task force of the European society of cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the european association for cardiovascular prevention & rehabilitation (EACPR). Eur Heart J. (2016) 37:2315–81. doi: 10.1093/eurheartj/ehw106
3. Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults. J Am Coll Cardiol. (2014) 63:2889–934. doi: 10.1016/j.jacc.2013.11.002
4. Jacobson TA, Ito MK, Maki KC, Orringer CE, Bays HE, Jones PH, et al. National lipid association recommendations for patient-centered management of dyslipidemia: part 1–full report. J Clin Lipidol. (2015) 9:129–69. doi: 10.1016/j.jacl.2015.02.003
5. Cholesterol Treatment Trialists C, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet (2010) 376:1670–81. doi: 10.1016/S0140-6736(10)61350-5
6. Boekholdt SM, Hovingh GK, Mora S, Arsenault BJ, Amarenco P, Pedersen TR, et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J Am Coll Cardiol. (2014) 64:485–94. doi: 10.1016/j.jacc.2014.02.615
7. Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol < 50 mg/dl with rosuvastatin. The jupiter trial (justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin). J Am Coll Cardiol. (2011) 57:1666–75. doi: 10.1016/j.jacc.2010.09.082
8. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the jupiter trial. Lancet (2009) 373:1175–82. doi: 10.1016/S0140-6736(09)60447-5
9. Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. (2015) 372:2387–97. doi: 10.1056/NEJMoa1410489
10. Giugliano RP, Wiviott SD, Blazing MA, De Ferrari GM, Park JG, Murphy SA, et al. Long-term safety and efficacy of achieving very low levels of low-density lipoprotein cholesterol: a prespecified analysis of the improve-it trial. JAMA Cardiol. (2017) 2:547–55. doi: 10.1001/jamacardio.2017.0083
11. Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, et al. Effect of long–term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a mendelian randomization analysis. J Am Coll Cardiol. (2012) 60:2631–9. doi: 10.1016/j.jacc.2012.09.017
12. Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med. (2016) 375:2144–53. doi: 10.1056/NEJMoa1604304
13. Wong ND, Chuang J, Zhao Y, Rosenblit PD. Residual dyslipidemia according to low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B among statin-treated US adults: National Health and Nutrition Examination Survey 2009-2010. J Clin Lipidol. (2015) 9:525–32. doi: 10.1016/j.jacl.2015.05.003
14. Gitt AK, Drexel H, Feely J, Ferrieres J, Gonzalez-Juanatey JR, Thomsen KK, et al. Persistent lipid abnormalities in statin-treated patients and predictors of LDL-cholesterol goal achievement in clinical practice in Europe and Canada. Eur J Prev Cardiol. (2012) 19:221–30. doi: 10.1177/1741826711400545
15. D'Agostino RB Sr, Grundy S, Sullivan LM, Wilson P, Group CHDRP. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA (2001) 286:180–7. doi: 10.1001/jama.286.2.180
16. Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet (2004) 364:937–52. doi: 10.1016/S0140-6736(04)17018-9
17. Jackson R, Lawes CM, Bennett DA, Milne RJ, Rodgers A. Treatment with drugs to lower blood pressure and blood cholesterol based on an individual's absolute cardiovascular risk. Lancet (2005) 365:434–41. doi: 10.1016/S0140-6736(05)17833-7
18. Kaasenbrood L, Boekholdt SM, van der Graaf Y, Ray KK, Peters RJ, et al. Distribution of Estimated 10-year risk of recurrent vascular events and residual risk in a secondary prevention population. Circulation (2017) 134:1419–29. doi: 10.1161/CIRCULATIONAHA.116.021314
19. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science (1986) 232:34–47.
20. Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci USA. (2004) 101:7100–5. doi: 10.1073/pnas.0402133101
21. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. (2004) 279:48865–75. doi: 10.1074/jbc.M409699200
22. Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. (2004) 279:50630–8. doi: 10.1074/jbc.M410077200
23. Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. (2003) 34:154–6. doi: 10.1038/ng1161
24. Abifadel M, Guerin M, Benjannet S, Rabes JP, Le Goff W, Julia Z, et al. Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis (2012) 223:394–400. doi: 10.1016/j.atherosclerosis.2012.04.006
25. Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. (2006) 79:514–23. doi: 10.1086/507488
26. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. (2005) 37:161–5. doi: 10.1038/ng1509
27. Kent ST, Rosenson RS, Avery CL, Chen YI, Correa A, Cummings SR, et al. PCSK9 loss-of-function variants, low-density lipoprotein cholesterol, and risk of coronary heart disease and stroke: data from 9 studies of blacks and whites. Circ Cardiovasc Genet. (2017) 10:e001632. doi: 10.1161/CIRCGENETICS.116.001632
28. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. (2006) 354:1264–72. doi: 10.1056/NEJMoa054013
29. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. (2003) 100:928–33. doi: 10.1073/pnas.0335507100
30. Zaid A, Roubtsova A, Essalmani R, Marcinkiewicz J, Chamberland A, Hamelin J, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology (2008) 48:646–54. doi: 10.1002/hep.22354
31. Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. (2012) 11:367–83. doi: 10.1038/nrd3699
32. Burke AC, Dron JS, Hegele RA, Huff MW. PCSK9: regulation and target for drug development for dyslipidemia. Annu Rev Pharmacol Toxicol. (2017) 57:223–44. doi: 10.1146/annurev-pharmtox-010716-104944
33. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI15593
34. Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. (2008) 49:399–409. doi: 10.1194/jlr.M700443-JLR200
35. Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem. (2009) 284:28885–95. doi: 10.1074/jbc.M109.052407
36. Dong B, Li H, Singh AB, Cao A, Liu J. Inhibition of PCSK9 transcription by berberine involves down-regulation of hepatic HNF1alpha protein expression through the ubiquitin-proteasome degradation pathway. J Biol Chem. (2015) 290:4047–58. doi: 10.1074/jbc.M114.597229
37. Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. (2007) 14:413–9. doi: 10.1038/nsmb1235
38. Piper DE, Jackson S, Liu Q, Romanow WG, Shetterly S, Thibault ST. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure (2007) 15:545–52. doi: 10.1016/j.str.2007.04.004
39. He NY, Li Q, Wu CY, Ren Z, Gao Y, Pan LH, et al. Lowering serum lipids via PCSK9-targeting drugs: current advances and future perspectives. Acta Pharmacol Sin. (2017) 38:301–11. doi: 10.1038/aps.2016.134
40. Wierod L, Cameron J, Strom TB, Leren TP. Studies of the autoinhibitory segment comprising residues 31-60 of the prodomain of PCSK9: Possible implications for the mechanism underlying gain-of-function mutations. Mol Genet Metab Rep. (2016) 9:86–93. doi: 10.1016/j.ymgmr.2016.11.003
41. Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA. (2008) 105:1820–5. doi: 10.1073/pnas.0712064105
42. Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. (2014) 114:1022–36. doi: 10.1161/CIRCRESAHA.114.301621
43. Zhang DW, Garuti R, Tang WJ, Cohen JC, Hobbs HH. Structural requirements for PCSK9- mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci USA. (2008) 105:13045–50. doi: 10.1073/pnas.0806312105
44. Tveten K, Holla OL, Cameron J, Strom TB, Berge KE, Laerdahl JK, et al. Interaction between the ligand-binding domain of the LDL receptor and the C-terminal domain of PCSK9 is required for PCSK9 to remain bound to the LDL receptor during endosomal acidification. Hum Mol Genet. (2012) 21:1402–9. doi: 10.1093/hmg/ddr578
45. Nassoury N, Blasiole DA, Tebon Oler A, Benjannet S, Hamelin J, Poupon V, et al. The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic (2007) 8:718–32. doi: 10.1111/j.1600-0854.2007.00562.x
46. Poirier S, Hamouda HA, Villeneuve L, Demers A1, Mayer G. Trafficking dynamics of PCSK9-induced LDLR degradation: focus on human PCSK9 mutations and C-terminal domain. PLoS ONE (2016) 11:e0157230. doi: 10.1371/journal.pone.0157230
47. Lo Surdo P, Bottomley MJ, Calzetta A, Settembre EC, Cirillo A, Pandit S, et al. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. (2011) 12:1300–5. doi: 10.1038/embor.2011.205
48. Norata GD, Tibolla G, Catapano AL. PCSK9 inhibition for the treatment of hypercholesterolemia: promises and emerging challenges. Vascul Pharmacol. (2014) 62:103–11. doi: 10.1016/j.vph.2014.05.011
49. Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem. (2009) 284:28856–64. doi: 10.1074/jbc.M109.037085
50. Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci USA. (2009) 106:9820–5. doi: 10.1073/pnas.0903849106
51. Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. (2015) 372:1489–99. doi: 10.1056/NEJMoa1501031
52. Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. (2015) 372:1500–9. doi: 10.1056/NEJMoa1500858
53. Navarese EP, Kolodziejczak M, Schulze V, Gurbel PA, Tantry U, Lin Y, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med. (2015) 163:40–51. doi: 10.7326/M14-2957
54. Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev. (2017) 4:CD011748. doi: 10.1002/14651858.CD011748.pub2
55. Ray KK, Ginsberg HN, Davidson MH, Pordy R, Bessac L, Minini P, et al. Reductions in atherogenic lipids and major cardiovascular events: a pooled analysis of 10 ODYSSEY trials comparing alirocumab with control. Circulation (2016) 134:1931–43. doi: 10.1161/CIRCULATIONAHA.116.024604
56. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, De Ferrari GM, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. (2017) 376:1713–22. doi: 10.1056/NEJMoa1615664
57. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. (2018) 379:2097–107. doi: 10.1056/NEJMoa1801174
58. Farnier M, Colhoun HM, Sasiela WJ, Edelberg JM, Asset G, Robinson JG. Long-term treatment adherence to the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab in 6 ODYSSEY Phase III clinical studies with treatment duration of 1 to 2 years. J Clin Lipidol. (2017) 11:986–97. doi: 10.1016/j.jacl.2017.05.016
59. Arrieta A, Page TF, Veledar E, Nasir K. Economic evaluation of PCSK9 inhibitors in reducing cardiovascular risk from health system and private payer perspectives. PLoS ONE (2017) 12:e0169761. doi: 10.1371/journal.pone.0169761
60. Whayne TF. Outcomes, access, and cost issues involving PCSK9 inhibitors to lower LDL-cholesterol. Drugs (2018) 78:287–91. doi: 10.1007/s40265-018-0867-9
61. Gulizia MM, Colivicchi F, Ricciardi G, Giampaoli S, Maggioni AP, Averna M, et al. ANMCO/ISS/AMD/ANCE/ARCA/FADOI/GICR-IACPR/SICI-GISE/SIBioC/SIC/SICOA/SID/SIF/SIMEU /SIMG/SIMI/SISA joint consensus document on cholesterol and cardiovascular risk: diagnostic-therapeutic pathway in Italy. Eur Heart J Suppl. (2017) 19:D3–54. doi: 10.1093/eurheartj/sux029
62. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
63. Bernards R. Exploring the uses of RNAi–gene knockdown and the Nobel Prize. N Engl J Med. (2006) 355:2391–3. doi: 10.1056/NEJMp068242
64. Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet (2014) 383:60–8. doi: 10.1016/S0140-6736(13)61914-5
65. Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. (2014) 136:16958–61. doi: 10.1021/ja505986a
66. Khvorova A. Oligonucleotide therapeutics - a new class of cholesterol-lowering drugs. N Engl J Med. (2017) 376:4–7. doi: 10.1056/NEJMp1614154
67. Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med. (2017) 376:1430–40. doi: 10.1056/NEJMoa1615758
68. Ray KK, Stoekenbroek RM, Kallend D, Leiter LA, Landmesser U, Wright RS, et al. Effect of an siRNA therapeutic targeting PCSK9 on atherogenic lipoproteins: pre-specified secondary end points in ORION 1. Circulation (2018) 138:1304–16. doi: 10.1161/CIRCULATIONAHA.118.034710
69. Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. (2010) 50:259–93. doi: 10.1146/annurev.pharmtox.010909.105654
70. Vickers TA, Crooke ST. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. (2015) 43:8955–63. doi: 10.1093/nar/gkv920
71. Liang XH, Sun H, Nichols JG, Crooke ST. RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol Ther. (2017) 25:2075–92. doi: 10.1016/j.ymthe.2017.06.002
72. Graham MJ, Lemonidis KM, Whipple CP, Subramaniam A, Monia BP, Crooke ST, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. (2007) 48:763–7. doi: 10.1194/jlr.C600025-JLR200
73. Gupta N, Fisker N, Asselin MC, Lindholm M, Rosenbohm C, Orum H, et al. A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS ONE (2010) 5:e10682. doi: 10.1371/journal.pone.0010682
74. Lindholm MW, Elmen J, Fisker N, Hansen HF, Persson R, Moller MR, et al. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol Ther. (2012) 20:376–81. doi: 10.1038/mt.2011.260
75. van Poelgeest EP, Hodges MR, Moerland M, Tessier Y, Levin AA, Persson R, et al. Antisense-mediated reduction of proprotein convertase subtilisin/kexin type 9 (PCSK9): a first-in-human randomized, placebo-controlled trial. Br J Clin Pharmacol. (2015) 80:1350–61. doi: 10.1111/bcp.12738
76. van Poelgeest EP, Swart RM, Betjes MG, Moerland M, Weening JJ, Tessier Y, et al. Acute kidney injury during therapy with an antisense oligonucleotide directed against PCSK9. Am J Kidney Dis. (2013) 62:796–800. doi: 10.1053/j.ajkd.2013.02.359
77. Yamamoto T, Harada-Shiba M, Nakatani M, Wada S, Yasuhara H, Narukawa K, et al. Cholesterol-lowering action of BNA-based antisense oligonucleotides targeting PCSK9 in atherogenic diet-induced hypercholesterolemic mice. Mol Ther Nucleic Acids (2012) 1:e22. doi: 10.1038/mtna.2012.16
78. Wierzbicki AS, Viljoen A. Anti-sense oligonucleotide therapies for the treatment of hyperlipidaemia. Expert Opin Biol Ther. (2016) 16:1125–34. doi: 10.1080/14712598.2016.1196182
79. Nordestgaard BG, Nicholls SJ, Langsted A, Ray KK, Tybjærg-Hansen A. Advances in lipid-lowering therapy through gene-silencing technologies. Nat Rev Cardiol. (2018) 15:261–72. doi: 10.1038/nrcardio.2018.3
80. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337:816–21. doi: 10.1126/science.1225829
81. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science (2014) 346:1258096. doi: 10.1126/science.1258096
82. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. (2013) 8:2281–308. doi: 10.1038/nprot.2013.143
83. Wang X, Raghavan A, Chen T, Qiao L, Zhang Y, Ding Q, et al. CRISPR-Cas9 targeting of PCSK9 in human hepatocytes in vivo-brief report. Arterioscler Thromb Vasc Biol. (2016) 36:783–6. doi: 10.1161/ATVBAHA.116.307227
84. Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. (2014) 115:488–92. doi: 10.1161/CIRCRESAHA.115.304351
85. Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature (2015) 520:186–91. doi: 10.1038/nature14299
86. Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci USA. (2005) 102:5374–9. doi: 10.1073/pnas.0501652102
87. Lanphier E, Urnov F, Haecker SE, Werner M, Smolenski J. Don't edit the human germ line. Nature (2015) 519:410–1. doi: 10.1038/519410a
88. Zhang Y, Ge X, Yang F, Zhang L, Zheng J, Tan X, et al. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci Rep. (2014) 4:5405. doi: 10.1038/srep05405
89. Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, et al. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med. (1990) 323:1234–8. doi: 10.1056/NEJM199011013231803
90. Miyosawa K, Watanabe Y, Murakami K, Murakami T, Shibata H, Iwashita M, et al. New CETP inhibitor K-312 reduces PCSK9 expression: a potential effect on LDL cholesterol metabolism. Am J Physiol Endocrinol Metab. (2015) 309:E177–90. doi: 10.1152/ajpendo.00528.2014
91. Lintner NG, McClure KF, Petersen D, Londregan AT, Piotrowski DW, Wei L, et al. Selective stalling of human translation through small-molecule engagement of the ribosome nascent chain. PLoS Biol. (2017) 15:e2001882. doi: 10.1371/journal.pbio.2001882
92. Mullard A. Nine paths to PCSK9 inhibition. Nat Rev Drug Discov. (2017) 16:299–301. doi: 10.1038/nrd.2017.83
93. Imanshahidi M, Hosseinzadeh H. Pharmacological and therapeutic effects of Berberis vulgaris and its active constituent, berberine. Phytother Res. (2008) 22:999–1012. doi: 10.1002/ptr.2399
94. Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. (2004) 10:1344–51. doi: 10.1038/nm1135
95. Cameron J, Ranheim T, Kulseth MA, Leren TP, Berge KE. Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis (2008) 201:266–73. doi: 10.1016/j.atherosclerosis.2008.02.004
96. Sultana N, Ata A. Oleanolic acid and related derivatives as medicinally important compounds. J Enzyme Inhib Med Chem. (2008) 23:739–56. doi: 10.1080/14756360701633187
97. Chen S, Wen X, Zhang W, Wang C, Liu J, Liu C. Hypolipidemic effect of oleanolic acid is mediated by the miR-98-5p/PGC-1beta axis in high-fat diet-induced hyperlipidemic mice. FASEB J. (2017) 31:1085–96. doi: 10.1096/fj.201601022R
98. Tsai SJ, Yin MC. Antioxidative and anti-inflammatory protection of oleanolic acid and ursolic acid in PC12 cells. J Food Sci. (2008) 73:H174–8. doi: 10.1111/j.1750-3841.2008.00864.x
99. Wang X, Liu R, Zhang W, Zhang X, Liao N, Wang Z, et al. Oleanolic acid improves hepatic insulin resistance via antioxidant, hypolipidemic and anti-inflammatory effects. Mol Cell Endocrinol. (2013) 376:70–80. doi: 10.1016/j.mce.2013.06.014
100. Rodriguez-Rodriguez R, Stankevicius E, Herrera MD, Ostergaard L, Andersen MR, Ruiz-Gutierrez V, et al. Oleanolic acid induces relaxation and calcium-independent release of endothelium-derived nitric oxide. Br J Pharmacol. (2008) 155:535–46. doi: 10.1038/bjp.2008.289
101. Banerjee Y, Santos RD, Al-Rasadi K, Rizzo M. Targeting PCSK9 for therapeutic gains: Have we addressed all the concerns? Atherosclerosis (2016) 248:62–75. doi: 10.1016/j.atherosclerosis.2016.02.018
102. Rothe C, Skerra A. Anticalin((R)) proteins as therapeutic agents in human diseases. BioDrugs (2018) 32:233–43. doi: 10.1007/s40259-018-0278-1
103. Masuda Y, Yamaguchi S, Suzuki C, Aburatani T, Nagano Y, Miyauchi R, et al. Generation and characterization of a novel small biologic alternative to proprotein convertase subtilisin/kexin type 9 (PCSK9) antibodies, DS-9001a, albumin binding domain-fused anticalin protein. J Pharmacol Exp Ther. (2018) 365:368–78. doi: 10.1124/jpet.117.246652
104. Gebauer M, Skerra A. Anticalins small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. (2012) 503:157–88. doi: 10.1016/B978-0-12-396962-0.00007-0
105. Kato M, He L, McGuire K, Dishy V, Zamora CA. A Randomized, Placebo Controlled, Single Ascending Dose Study to Assess the Safety, PK and PD of DS-9001a, a Novel Small Biologic PCSK9 Inhibitor, in Healthy Subjects. ASCPT meeting. Orland, CA (2018).
106. Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. (2013) 81:136–47. doi: 10.1111/cbdd.12055
107. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. (2007) 282:18602–12. doi: 10.1074/jbc.M702027200
108. Schroeder CI, Swedberg JE, Withka JM, Rosengren KJ, Akcan M, Clayton DJ, et al. Design and synthesis of truncated EGF-A peptides that restore LDL-R recycling in the presence of PCSK9 in vitro. Chem Biol. (2014) 21:284–94. doi: 10.1016/j.chembiol.2013.11.014
109. McNutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. (2009) 284:10561–70. doi: 10.1074/jbc.M808802200
110. Zhang Y, Eigenbrot C, Zhou L, Shia S, Li W, Quan C, et al. Identification of a small peptide that inhibits PCSK9 protein binding to the low density lipoprotein receptor. J Biol Chem. (2014) 289:942–55. doi: 10.1074/jbc.M113.514067
111. Du F, Hui Y, Zhang M, Linton MF, Fazio S, Fan D. Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J Biol Chem. (2011) 286:43054–61. doi: 10.1074/jbc.M111.273474
112. Alghamdi RH, O'Reilly P, Lu C, Gomes J, Lagace TA, Basak A. LDL-R promoting activity of peptides derived from human PCSK9 catalytic domain (153-421): design, synthesis and biochemical evaluation. Eur J Med Chem. (2015) 92:890–907. doi: 10.1016/j.ejmech.2015.01.022
113. Mayer G, Poirier S, Seidah NG. Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J Biol Chem. (2008) 283:31791–801. doi: 10.1074/jbc.M805971200
114. Seidah NG, Poirier S, Denis M, Parker R, Miao B, Mapelli C, et al. Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PLoS ONE (2012) 7:e41865. doi: 10.1371/journal.pone.0041865
115. Ly K, Saavedra YG, Canuel M, Routhier S, Desjardins R, Hamelin J, et al. Annexin A2 reduces PCSK9 protein levels via a translational mechanism and interacts with the M1 and M2 domains of PCSK9. J Biol Chem. (2014) 289:17732–46. doi: 10.1074/jbc.M113.541094
116. Walker RG, Thompson TB. Fibronectin-based scaffold domain proteins that bind myostatin: a patent evaluation of WO2014043344. Expert Opin Ther Pat. (2015) 25:619–24. doi: 10.1517/13543776.2015.1007954
117. Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. (2011) 24:3–9. doi: 10.1093/protein/gzq097
118. Ramamurthy V, Krystek SR Jr, Bush A, Wei A, Emanuel SL, Das Gupta R, et al. Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure (2012) 20:259–69. doi: 10.1016/j.str.2011.11.016
119. Mitchell T, Chao G, Sitkoff D, Lo F, Monshizadegan H, Meyers D, et al. Pharmacologic profile of the Adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J Pharmacol Exp Ther. (2014) 350:412–24. doi: 10.1124/jpet.114.214221
120. Stein EA, Kasichayanula S, Turner T, Kranz T, Arumugam U, et al. 2014. Ldl cholesterol reduction with Bms-962476, an adnectin inhibitor of Pcsk9: results of a single ascending dose study. J Am Coll Cardiol. 63:A1372. doi: 10.1016/s0735-1097(14)61372-3
121. Chackerian B, Remaley A. Vaccine strategies for lowering LDL by immunization against proprotein convertase subtilisin/kexin type 9. Curr Opin Lipidol. (2016) 27:345–50. doi: 10.1097/MOL.0000000000000312
122. Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur Heart J. (2017) 38:2499–507. doi: 10.1093/eurheartj/ehx260
123. Galabova G, Brunner S, Winsauer G, Juno C, Wanko B, Mairhofer A, et al. Peptide-based anti-PCSK9 vaccines - an approach for long-term LDLc management. PLoS ONE (2014) 9:e114469. doi: 10.1371/journal.pone.0114469
124. Crossey E, Amar MJA, Sampson M, Peabody J, Schiller JT, Chackerian B, et al. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine (2015) 33:5747–55. doi: 10.1016/j.vaccine.2015.09.044
125. Civeira F, Jarauta E. Vaccine against PCSK9: the natural strategy from passive to active immunization for the prevention of atherosclerosis. J Thorac Dis. (2017) 9:4291–4. doi: 10.21037/jtd.2017.10.18
126. Gustafsen C, Kjolby M, Nyegaard M, Mattheisen M, Lundhede J, Buttenschon H, et al. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab. (2014) 19:310–8. doi: 10.1016/j.cmet.2013.12.006
127. Hu D, Yang Y, Peng DQ. Increased sortilin and its independent effect on circulating proprotein convertase subtilisin/kexin type 9 (PCSK9) in statin-naive patients with coronary artery disease. Int J Cardiol. (2017) 227:61–5. doi: 10.1016/j.ijcard.2016.11.064
128. Goettsch C, Kjolby M, Aikawa E. Sortilin and its multiple roles in cardiovascular and metabolic diseases. Arterioscler Thromb Vasc Biol. (2018) 38:19–25. doi: 10.1161/ATVBAHA.117.310292
129. Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS. Sortilin is essential for proNGF-induced neuronal cell death. Nature (2004) 427:843–8. doi: 10.1038/nature02319
130. Debose-Boyd RA, Horton JD. Opening up new fronts in the fight against cholesterol. Elife (2013) 2:e00663. doi: 10.7554/eLife.00663
131. Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, et al. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife (2013) 2:e00444. doi: 10.7554/eLife.00444
132. Ye X, Li M, Hou T, Gao T, Zhu WG, Yang Y. Sirtuins in glucose and lipid metabolism. Oncotarget (2017) 8:1845–59. doi: 10.18632/oncotarget.12157
133. Miranda MX, van Tits LJ, Lohmann C, Arsiwala T, Winnik S, Tailleux A, et al. The Sirt1 activator SRT3025 provides atheroprotection in Apoe-/- mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur Heart J. (2015) 36:51–9. doi: 10.1093/eurheartj/ehu095
134. Libri V, Brown AP, Gambarota G, Haddad J, Shields GS, Dawes H, et al. A pilot randomized, placebo controlled, double blind phase I trial of the novel SIRT1 activator SRT2104 in elderly volunteers. PLoS ONE (2012) 7:e51395. doi: 10.1371/journal.pone.0051395
135. Jung CH, Cho I, Ahn J, Jeon TI, Ha TY. Quercetin reduces high-fat diet-induced fat accumulation in the liver by regulating lipid metabolism genes. Phytother Res. (2013) 27:139–43. doi: 10.1002/ptr.4687
136. Mbikay M, Mayne J, Sirois F, Fedoryak O, Raymond A, Noad J, et al. Mice fed a high-cholesterol diet supplemented with quercetin-3-glucoside show attenuated hyperlipidemia and hyperinsulinemia associated with differential regulation of PCSK9 and LDLR in their liver and pancreas. Mol Nutr Food Res. (2018) 62:e1700729. doi: 10.1002/mnfr.201700729
137. Mbikay M, Sirois F, Simoes S, Mayne J, Chretien M. Quercetin-3-glucoside increases low-density lipoprotein receptor (LDLR) expression, attenuates proprotein convertase subtilisin/kexin 9 (PCSK9) secretion, and stimulates LDL uptake by Huh7 human hepatocytes in culture. FEBS Open Biol. (2014) 4:755–62. doi: 10.1016/j.fob.2014.08.003
138. Mayne J, Dewpura T, Raymond A, Bernier L, Cousins M, Ooi TC, et al. Novel loss-of-function PCSK9 variant is associated with low plasma LDL cholesterol in a French-Canadian family and with impaired processing and secretion in cell culture. Clin Chem. (2011) 57:1415–23. doi: 10.1373/clinchem.2011.165191
139. Chorba JS, Shokat KM. The proprotein convertase subtilisin/kexin type 9 (PCSK9) active site and cleavage sequence differentially regulate protein secretion from proteolysis. J Biol Chem. (2014) 289:29030–43. doi: 10.1074/jbc.M114.594861
140. Lambert G, Sjouke B, Choque B, Kastelein JJ, Hovingh GK. The PCSK9 decade. J Lipid Res. (2012) 53:2515–24. doi: 10.1194/jlr.R026658
141. Zhang H, Plutzky J, Skentzos S, Morrison F, Mar P, Shubina M, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. (2013) 158:526–34. doi: 10.7326/0003-4819-158-7-201304020-00004
142. Serban MC, Colantonio LD, Manthripragada AD, Monda KL, Bittner VA, Banach M, et al. Statin intolerance and risk of coronary heart events and all-cause mortality following myocardial infarction. J Am Coll Cardiol. (2017) 69:1386–95. doi: 10.1016/j.jacc.2016.12.036
143. Jones PH, Bays HE, Chaudhari U, Pordy R, Lorenzato C, Miller K, et al. Safety of alirocumab (A PCSK9 monoclonal antibody) from 14 randomized trials. Am J Cardiol. (2016) 118:1805–11. doi: 10.1016/j.amjcard.2016.08.072
144. Toth PP, Descamps O, Genest J, Sattar N, Preiss D, Dent R, et al. Pooled safety analysis of evolocumab in over 6000 patients from double-blind and open-label extension studies. Circulation (2017) 135:1819–31. doi: 10.1161/CIRCULATIONAHA.116.025233
145. Giugliano RP, Keech A, Murphy SA, Huber K, Tokgozoglu SL, Lewis BS, et al. Clinical efficacy and safety of evolocumab in high-risk patients receiving a statin: secondary analysis of patients with low LDL cholesterol levels and in those already receiving a maximal-potency statin in a randomized clinical trial. JAMA Cardiol. (2017) 2:1385–91. doi: 10.1001/jamacardio.2017.3944
146. Robinson JG, Rosenson RS, Farnier M, Chaudhari U, Sasiela WJ, Merlet L, et al. Safety of very low low-density lipoprotein cholesterol levels with alirocumab: pooled data from randomized trials. J Am Coll Cardiol. (2017) 69:471–82. doi: 10.1016/j.jacc.2016.11.037
147. Norata GD, Tavori H, Pirillo A, Fazio S, Catapano AL. Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc Res. (2016) 112:429–42. doi: 10.1093/cvr/cvw194
148. Giugliano RP, Mach F, Zavitz K, Kurtz C, Im K, Kanevsky E, et al. Cognitive function in a randomized trial of evolocumab. N Engl J Med. (2017) 377:633–43. doi: 10.1056/NEJMoa1701131
149. Swiger KJ, Martin SS. PCSK9 inhibitors and neurocognitive adverse events: exploring the FDA directive and a proposal for N-of-1 trials. Drug Saf. (2015) 38:519–26. doi: 10.1007/s40264-015-0296-6
150. Khan AR, Bavishi C, Riaz H, Farid TA, Khan S, Atlas M, et al. Increased risk of adverse neurocognitive outcomes with proprotein convertase subtilisin-kexin type 9 inhibitors. Circ Cardiovasc Qual Outcomes (2017) 10:e003153. doi: 10.1161/CIRCOUTCOMES.116.003153
151. Lipinski MJ, Benedetto U, Escarcega RO, Biondi-Zoccai G, Lhermusier T, Baker NC, et al. The impact of proprotein convertase subtilisin-kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta-analysis. Eur Heart J. (2016) 37:536–45. doi: 10.1093/eurheartj/ehv563
Keywords: PCKS9 inhibition, LDL cholesterol, statin, monoclonal antibody, adverse effect
Citation: Nishikido T and Ray KK (2019) Non-antibody Approaches to Proprotein Convertase Subtilisin Kexin 9 Inhibition: siRNA, Antisense Oligonucleotides, Adnectins, Vaccination, and New Attempts at Small-Molecule Inhibitors Based on New Discoveries. Front. Cardiovasc. Med. 5:199. doi: 10.3389/fcvm.2018.00199
Received: 27 September 2018; Accepted: 28 December 2018;
Published: 29 January 2019.
Edited by:
Nathan D. Wong, University of California, Irvine, United StatesReviewed by:
Matti Sakari Jauhiainen, Minerva Foundation Institute for Medical Research, FinlandChristoph Sinning, Universitäres Herzzentrum Hamburg GmbH (UHZ), Germany
Copyright © 2019 Nishikido and Ray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kausik K. Ray, k.ray@imperial.ac.uk