 Simon McArthur* and Glenda E. Gillies
Simon McArthur* and Glenda E. Gillies
- Department of Medicine, Centre for Neuroscience, Imperial College London, London, UK
The nigrostriatal dopaminergic (NSDA) pathway degenerates in Parkinson’s disease (PD), which occurs with approximately twice the incidence in men than women. Studies of the influence of systemic estrogens in females suggest sex hormones contribute to these differences. In this review we analyze the evidence revealing great complexity in the response of the healthy and injured NSDA system to hormonal influences, and emphasize the importance of centrally generated estrogens. At physiological levels, circulating estrogen (in females) or estrogen precursors (testosterone in males, aromatized to estrogen centrally) have negligible effects on dopaminergic neuron survival in experimental PD, but can modify striatal dopamine levels via actions on the activity or adaptive responses of surviving cells. However, these effects are sexually dimorphic. In females, estradiol promotes adaptive responses in the partially injured NSDA pathway, preserving striatal dopamine, whereas in males gonadal steroids and exogenous estradiol have a negligible or even suppressive effect, effectively exacerbating dopamine loss. On balance, the different effects of gonadal factors in males and females contribute to sex differences in experimental PD. Fundamental sex differences in brain organization, including the sexually dimorphic networks regulating NSDA activity are likely to underpin these responses. In contrast, estrogen generated locally appears to preserve striatal dopamine in both sexes. The available data therefore highlight the need to understand the biological basis of sex-specific responses of the NSDA system to peripheral hormones, so as to realize the potential for sex-specific, hormone-based therapies in PD. Furthermore, they suggest that targeting central steroid generation could be equally effective in preserving striatal dopamine in both sexes. Clarification of the relative roles of peripheral and central sex steroid hormones is thus an important challenge for future studies.
Introduction
Sex steroid hormones produced by activation of the hypothalamo-pituitary–gonadal (HPG) axis have profound effects on brain development and function. In recent years estrogens have been a major focus of attention because of their emergent neuroprotective, neurotrophic, and psychotropic actions (Fink et al., 1996; McEwen and Alves, 1999; Gillies et al., 2004; Brann et al., 2007; Craig and Murphy, 2007). In all mammalian species studied, including humans, estrogens are considered the steroid hormones of the female sex due to their roles in regulating ovulation and reproductive behaviors. Not surprisingly, therefore, the vast majority of investigations into the roles of estrogens in the physiology and pathology of the brain have been conducted in females. These include studies of neurodegenerative disorders, such as Parkinson’s disease (PD) and Alzheimer’s disease, as well as schizophrenia and drug abuse, which show sex differences in their incidence, severity, and/or progression. However, there is a growing realization that estrogens are also important physiological regulators in males where they can be synthesized locally in many tissues either de novo or from steroid precursors, such as circulating testosterone (Sharpe, 1998; Jones et al., 2006). This includes the brain (Do Rego et al., 2009), where estrogens may act via their classical nuclear receptors, (ERα and ERβ), which are widely distributed in the brains of males as well as females, or via rapid membrane effects (Toran-Allerand, 2005; Balthazart and Ball, 2006; Brann et al., 2007; Micevych and Dominguez, 2009). This knowledge raises the possibility that the protective effects of estrogens and estrogen mimetics may be exploited for the therapy of brain disorders that affect men as well as women. However, if this potential is to be realized, we need to understand the relative contributions of systemically and centrally generated steroids to brain function under both normal and disrupted conditions. This review will focus on a system well-suited to addressing this issue, the nigrostriatal dopaminergic (NSDA) pathway that plays a central role in the fine control of sensorimotor function, and degenerates in PD (Klein and Schlossmacher, 2007). We shall consider the evidence that this pathway, whether intact or injured, exhibits sexual dimorphisms in its responsiveness to gonadal hormones, as well as innately in its physiology and pathology in many species. We shall also develop the view that in contrast to systemic sex steroids of gonadal origin, which have notable neuroprotective effects in female, but not male, rodents exposed to experimental PD, centrally generated sex steroids may be equally beneficial in both sexes.
Parkinson’s Disease: The Clinical Profile
Etiology and Sex Differences
Parkinson’s disease, the second most common neurodegenerative disorder of aging, affects approximately 1 in 500 people in the developed world, a figure set only to increase as the population ages (Pavon et al., 2010). Clinically, PD is characterized by a core set of symptoms, namely resting tremor, slowness of movement, and problems with balance and rigidity, which are commonly associated with a wide range of secondary symptoms including depression, anxiety, insomnia, and dementia (Weintraub et al., 2008). The major neuropathological lesion associated with the disease is the loss of dopaminergic input to the caudate nucleus and putamen (collectively termed the striatum), caused by the degeneration of dopaminergic cells in the substantia nigra pars compacta (SNc). These cells, the origin of the NSDA pathway, play a central role in the regulation of motor function, and their degeneration leads to the primary clinical symptoms of PD, although degeneration in other brain areas, including, for example, the noradrenergic cells of the locus coeruleus and the cholinergic neurons in the nucleus basalis, contribute to the secondary symptoms associated with the disease (Braak et al., 2003). Aside from about 3% of cases that are attributable to a single genetic event, in the majority of cases of PD the underlying cause(s) remain obscure (Klein and Schlossmacher, 2007). However, the cellular and molecular processes underlying the degeneration of dopaminergic cells have been extensively studied, and interactions between mitochondrial failure (Vives-Bauza et al., 2010), oxidative stress (Mattson, 2006), the formation of aggregated α-synuclein positive inclusions known as Lewy bodies (Auluck et al., 2010) and microglial inflammation (Halliday and Stevens, 2011) are thought to be responsible for neuronal death, although there remains some controversy as to which of these processes has primacy, and which are consequent upon the others. These findings support the now widely accepted theory that PD is a multi-factorial complex disease (probably better described as a clinical syndrome), with an etiology comprising an interaction between multiple genetic factors, the environment, and aging (Klein and Schlossmacher, 2007).
Being male appears high on the list of factors that contribute to the development of PD (Klein and Schlossmacher, 2007). Epidemiological studies have revealed a clear sex difference in the incidence (the number of new cases developed in a specific time-frame) of PD, with individual studies of male to female ratios for incidence rates reporting values ranging from 1.37 to 3.7 (Baldereschi et al., 2000; Swerdlow et al., 2001; Van Den Eeden et al., 2003; Wooten et al., 2004; Shulman and Bhat, 2006; Cantuti-Castelvetri et al., 2007; Haaxma et al., 2007; Taylor et al., 2007), with a large meta-analysis study concluding that the relative risk for men is 1.5 times greater than women (Elbaz et al., 2002). Thus, whilst approximately 3% of people aged over 65 present with PD, this figure breaks down by sex such that 2% of men, but only 1.3% of women, are affected during their lifetime (Elbaz et al., 2002). There is some controversy over the presence of sex differences in the age of onset of clinical PD symptoms, with some studies indicating that men present approximately 2 years earlier than women (Haaxma et al., 2007; Alves et al., 2009), although other work has not shown these differences (Baba et al., 2005).
These discrepancies may be instructive in their own right however, as studies that have found a difference tend to have examined younger patients, whilst those that report no significant sex differences in age at onset have focused more on patients that were older when they presented with the disease, possibly reflecting an interaction with menopause in females and a potential role for peripheral gonadal steroids. The literature also holds contradictions on sex differences in the rate of disease progression. On the one hand, increasing symptom severity was reported to be similar in males and females (Baba et al., 2005), whereas another study found a slower rate of decline of motor impairment in women, despite no evidence for a sex difference in concomitant neurochemical changes (Haaxma et al., 2007). To some extent, these contradictions may be explained by the findings that there are numerous sex differences in the clinical characteristics of PD, with females presenting with a milder PD phenotype, at least in the early clinical stages (Shulman and Bhat, 2006; Haaxma et al., 2007; Miller and Cronin-Golomb, 2010). For example, rigidity appears more frequently in men than women, whereas more women than men suffer dyskinesias and PD-associated depression; a sex-specific pattern is also emerging for disease-associated cognitive defects. The advent of new technologies to dissect the molecular neuropathology of PD may possibly provide a more objective analysis of underlying sexual dimorphisms. Indeed, comparison of gene expression profiles in laser dissected single SNc dopamine neurons in post-mortem brains of control subjects and idiopathic PD patients (late stage) indicate a clear impact of sex on both the normal and degenerating pathway: For example in the healthy brain genes involved in signal transduction and neuronal maturation were up-regulated in women, whereas an up-regulation of genes implicated in PD pathogenesis (alpha-synuclein, PINK-1) was seen in men relative to women; in PD brains changes in genes with protein kinase activity and genes associated with proteolysis and Wnt signaling predominate in women, whereas predominant gene changes in men were seen for those involved in protein- and copper-binding activities (Simunovic et al., 2010; Cantuti-Castelvetri et al., 2007). These differences, as well as indicating a basal sex bias, also suggest differences in the mechanism of the disease process. Thus, on balance, evidence from a variety of sources strongly supports the proposition that women are relatively protected from PD compared with men. They also highlight the fact that the goal of understanding PD pathogenesis and, hence, the development of improved treatments, requires a better knowledge of the basis for sex differences in PD. In the next section we shall argue for the key importance of sex steroids, which potently modify genetic and environmental influences.
Genes vs. Sex Hormones as Contributors to the Sex Bias in Parkinson’s Disease
Single nucleotide polymorphisms in several estrogen-related genes, including estrogen receptors and aromatase (estrogen synthase), have been positively associated with PD (Westberg et al., 2004; Hakansson et al., 2005; Chung et al., 2011; Palacios et al., 2011). On the basis that estrogens may be neuroprotective, this could have relevance for a sex bias, but understanding how single nucleotide polymorphisms influence disease risk remains a challenge (Klein and Schlossmacher, 2007). Moreover, as single gene effects account for so few PD cases, other factors must account for sex differences. On a more general level, it is increasingly appreciated that factors linked to the sex chromosomes could explain a certain degree of sex bias that is present in the great majority of common diseases (Kaminsky et al., 2006). Such effects could be attributable to a direct effect of Y chromosome genes or to differential expression of X chromosome genes due either to incomplete X chromosome silencing, or to sex differences in the genomic imprinting of X chromosome genes (Federman, 2006; van Nas et al., 2009). However, although pre-clinical evidence suggests that sex chromosome effects have a certain role to play in brain sexual differentiation (Federman, 2006; van Nas et al., 2009), their potential impact on disease susceptibility is not yet known. Other factors, which could act directly or interact with genetic factors, include life-style as a cause of sex differences in PD, with exposure to agrichemicals and head trauma being examples of factors which associate with a greater incidence in men (Wooten et al., 2004; Das et al., 2011).
Overwhelmingly however, sex hormones are the major factors that drive structural and functional sex differences in the normal brain, and are increasingly identified as key components in inducing differential susceptibility to complex disease, probably by inducing differences in the epigenetic regulation of autosomal genes (Kaminsky et al., 2006). By far the greatest degree of attention has focused upon the role of estrogen and its potential for neuroprotective actions, supported by several lines of epidemiological and clinical evidence (Dluzen and Horstink, 2003; Shulman and Bhat, 2006). For example, women who have undergone surgical removal of the ovaries present a significantly increased risk of developing PD (Benedetti et al., 2001; Ragonese et al., 2006). Circulating levels of estrogen also seem to be involved in modulating PD symptom severity in pre-menopausal women, as the disease burden has been shown to lessen during stages of the menstrual cycle characterized by high circulating estrogen, and to worsen at menses, when estrogen levels are lowest (Quinn and Marsden, 1986). In accord with these findings, there are numerous reports that estrogen-based hormone replacement therapy has positive effects on PD symptom burden (Saunders-Pullman et al., 1999; Tsang et al., 2000; Benedetti et al., 2001) and cessation of therapy has been associated with a marked deterioration in symptoms (Sandyk, 1989). Such therapies have also been associated with a decreased risk of developing PD (Currie et al., 2004; Liu and Dluzen, 2007), although this is not a universal finding (Popat et al., 2005), possibly because of the considerable variations in hormone replacement treatment regimes (Wise et al., 2005).
The evidence discussed above supports the view that sex influences the incidence and nature of the degenerative processes in the NSDA pathway, and that sex hormones, specifically estrogens in females, play a key role. However, clinical studies do not permit investigations into the nature of estrogen’s actions or provide clues as to potential hormonal influences in men. Furthermore, they cannot distinguish between the effects of gonadally derived and centrally generated steroids, which are receiving increasing prominence as regulators of brain development, function, and behavior in the vertebrate brain (Do Rego et al., 2009). Significantly, recent evidence suggests that neurosteroids (that is, steroids synthesized in the CNS de novo or from peripherally derived precursors) are abundant in nigrostriatal regions in human post-mortem brains, with reports of altered levels in degenerative conditions, including PD (Bixo et al., 1997; di Michele et al., 2003; Luchetti et al., 2010). Therefore, pre-clinical experimental models of PD have an important role to play, and will be discussed in the following section.
Experimental Parkinson’s Disease
Animal Models
Many aspects of idiopathic PD are recreated in experimental animal models. The simplest of such models are produced by the use of the neurotoxins 6-hydroxydopamine (6-OHDA), typically administered directly to the NSDA pathway in rodents (Figure 1; Dluzen, 1997; Murray et al., 2003), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is given systemically in mice and non-human primates (Miller et al., 1998; Dluzen and Horstink, 2003), as well as centrally in non-human primates (Morissette and Di Paolo, 2009; Fox and Brotchie, 2010). Both toxins selectively kill dopaminergic cells through a combination of oxidative stress and the inhibition of mitochondrial respiration (Glinka et al., 1996; Blum et al., 2001; Simola et al., 2007). Methamphetamine has also been used to create nigral lesions in mice, although the similarities to parkinsonian degeneration have been less well characterized (Dluzen et al., 2002; Dluzen and Liu, 2008). Whilst such models are limited in that they cannot recreate the true (unknown) underlying cause of PD, they have proven of very great value in aiding our understanding of its symptoms and pathology, and have been essential in the development of therapeutic agents for the treatment of the condition (Lane and Dunnett, 2008; Fox and Brotchie, 2010; Potashkin et al., 2010). As such they are of great utility in studying the potential neuroprotective role of sex steroid hormones, and have provided significant insights into the interactions of estradiol with the degenerating nigrostriatal dopaminergic pathway.
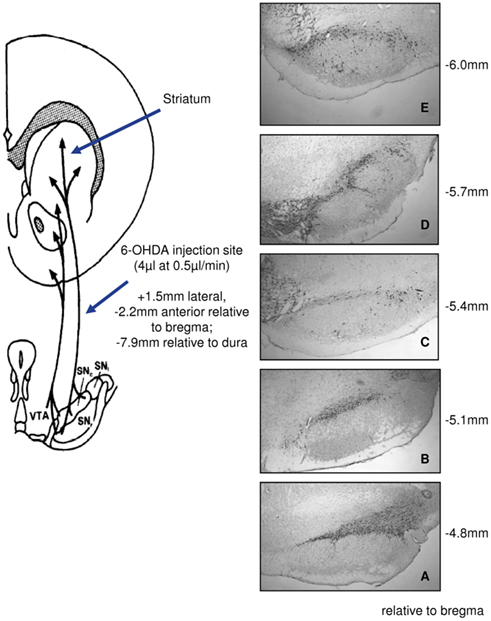
Figure 1. Experimental Parkinson’s disease created by unilateral infusions of 6-hydroxydopamine (6-OHDA). Schematic representation of placement of the lesions, under general anesthesia, into the left medial forebrain bundle (left panel) and immunocytochemical detection 1–2 weeks later of tyrosine hydroxylase, the rate-limiting enzyme in dopamine synthesis, to identify dopaminergic neurons through five levels (A–E) of the substantia nigra pars compacta (right panel).
Sex Differences in Experimental Parkinson’s Disease
The existence of sex differences in susceptibility to neurotoxic lesioning is widely reported in various animal models of PD, recapitulating the differences seen clinically. For example, clear sex differences are seen in the extent of both dopamine loss in the striatum and dopaminergic neuron loss in the SNc following administration of 6-hydroxydopamine (6-OHDA) to rats (Table 1; Figure 2 control, gonad-intact groups; Murray et al., 2003; Gillies et al., 2004). Moreover, the progressive loss of dopaminergic cells over a 5-week period post-lesioning was consistently greater in 6-OHDA-treated male rats, confirming a true sex difference rather a difference in the rate of neurodegeneration (Moroz et al., 2003). Similar sex differences in the extent of SNc and striatal lesions are seen in mice treated with MPTP or MA, with females being relatively protected (Miller et al., 1998; Dluzen, 2000; Yu and Liao, 2000; Liu and Dluzen, 2007). These differences are present following partial lesions (<60%) of the nigrostriatal pathway using sub-maximal doses of 6-OHDA, but with larger lesions (70–90%) sex differences are no longer apparent (Gillies et al., 2004; Gillies and McArthur, 2010b). Together, these pre-clinical findings corroborate the clinical studies suggesting that females may be more able to resist the onset and/or progression of the neurodegenerative lesions. However, they also demonstrate that the degree of intrinsic neuroprotection in the female brain can be over-ridden once neurodegeneration reaches a certain extent. Additionally, these studies demonstrate that the use of an experimental regime of sub-maximal lesions in the NSDA pathway, considered to be a model of pre-clinical/early stage PD (Schwarting and Huston, 1996), has potential for investigating the factors that provide the female advantage, hence revealing novel therapeutic approaches to delay disease onset and/or progression.

Table 1. Sex differences in experimental PD.
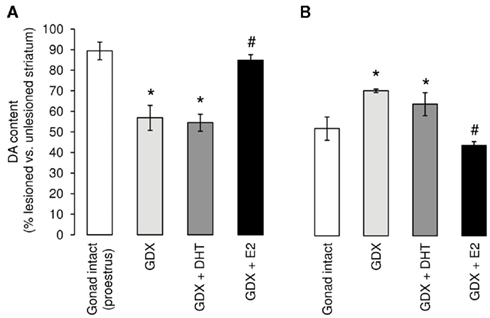
Figure 2. Manipulation of the hormone environment exerts sex-specific effects on striatal lesions induced by 6-hydroxydopamine (6-OHDA). Male and female rats underwent bilateral gonadectomy (GDX) or sham operation (Gonad-intact) followed immediately, while still under anesthesia, by subcutaneous implantation of a pellet, containing either estradiol (replaced to proestrus levels), 5α-dihydrotestosterone (to mimic physiological androgen levels), or a placebo pellet (full details in Murray et al., 2003). Approximately 1 week later all animals received a unilateral infusion of 6-hydroxydopamine (6-OHDA; 1 μg) into the left medial forebrain bundle and, after killing 2 weeks later, brains were processed to assess lesion size. Results represent the percentage striatal dopamine (DA) remaining in the lesioned (left) side compared with the contralateral, unlesioned (right) side. Sham-operated animals (Gonad-intact, □) are taken as controls. (A) Following ovariectomy ( ) levels of DA in the female striatum were significantly reduced compared with their sham-operated counterparts. This effect was unaltered by treatment with the non-aromatizable androgen, dihydrotestosterone (DHT,
) levels of DA in the female striatum were significantly reduced compared with their sham-operated counterparts. This effect was unaltered by treatment with the non-aromatizable androgen, dihydrotestosterone (DHT,  ), but was reversed by estradiol (E2, ■). (B) Following orchidectomy (), levels of DA in the male striatum were significantly greater than their sham-operated counterparts (□). This effects was unaffected by treatment with DHT (), but was reversed by estradiol (E2, ■). *p < 0.05 Vs. gonad-intact; #p < 0.05 vs. GDX. (Modified from Murray et al., 2003).
), but was reversed by estradiol (E2, ■). (B) Following orchidectomy (), levels of DA in the male striatum were significantly greater than their sham-operated counterparts (□). This effects was unaffected by treatment with DHT (), but was reversed by estradiol (E2, ■). *p < 0.05 Vs. gonad-intact; #p < 0.05 vs. GDX. (Modified from Murray et al., 2003).
Impact of Peripheral Sex Steroid Hormones on Striatal Lesion Size
The reduction in the levels of dopamine and its metabolites in the striatum are widely used as indicators of the magnitude of destruction (size of lesion) induced in rodent models of PD. Despite the greater male susceptibility to PD, the vast majority of studies have focused on the neuroprotective effects of estrogens in female rodents and, to a lesser extent, female non-human primates. However, convergent evidence indicates that gonadal factors play a different role in males (Gillies et al., 2004; McArthur et al., 2007; Gillies and McArthur, 2010a), which merits separate consideration of the sexes.
Females
The loss of striatal dopamine in the 6-OHDA-partially lesioned nigrostriatal system is far greater after ovariectomy than in gonad-intact female rats (Murray et al., 2003) an effect which can be reversed by replacement of estradiol to physiological levels prior to lesioning (Figure 2A; Dluzen, 1997; Murray et al., 2003; Ferraz et al., 2008). Estradiol treatment of ovariectomized animals also reduces striatal dopamine depletion induced by MPTP in mice (Miller et al., 1998; Dluzen and Horstink, 2003) and non-human primates (Morissette and Di Paolo, 2009), or by MA in mice (Yu and Liao, 2000). Equally, female rats lesioned with any of these three toxins at proestrus, when estrogen levels are maximal, exhibited significantly smaller losses of striatal dopamine than similar animals lesioned at diestrus, the period of the estrus cycle when estrogen levels are at their nadir (Yu and Liao, 2000; Datla et al., 2003; Dluzen and Horstink, 2003). However, just as the female advantage in experimental PD is lost as lesion size approaches or exceeds approximately 60%, so is the neuroprotective effect of estradiol lost with larger lesions (Cordellini et al., 2011). Moreover, replacement of estradiol to high, supraphysiological levels may fail to protect or even worsen lesion size (Ramirez et al., 2003; Bourque et al., 2009; Cordellini et al., 2011). These studies demonstrate that the levels of circulating estradiol prevailing at the time of injury are critical and that physiological levels, whether endogenous or administered exogenously, have protective capacity in the partially injured female NSDA system. They therefore support the prevailing view that sex differences in PD could be attributable largely to the protective role of gonadal factors, namely estradiol.
Males
Unlike in females, gonadectomy significantly reduced striatal lesion size in male rats exposed to sub-maximal doses of 6-OHDA (Figure 2B). Although some studies in mice report that castration had no significant effect on neurotoxin-induced striatal dopamine loss, (Dluzen et al., 1994; Yu and Wagner, 1994; Gao and Dluzen, 2001), others found that testosterone treatment of either intact or gonadectomized CD-1 male mice worsened striatal dopamine loss in animals with mild/moderate (40%) lesions (Dluzen et al., 1994; Lewis and Dluzen, 2008). On balance, these findings suggest that gonadally derived testosterone has detrimental effects in experimental PD, which appears to be a cross-species phenomenon. However, with more extensive lesions (70%) testosterone treatment caused no further loss of striatal dopamine (Dluzen et al., 1994). Thus, although the effects of gonadally derived hormones are opposite in males and females, in both sexes hormonal influences appear to have a significant effect only in experimental models mimicking the pre-clinical/early phases of the degenerative process. Despite the general finding that estradiol provides universal protection against dopamine loss in experimental PD in female mice and rats, whether estrogens are protective in males is more equivocal, possibly because of differences in species, strain, neurotoxin, and hormone treatment regimes employed in various studies. For example, in the highly sensitive C56Bl/6 mouse strain, estradiol may protect male mice against MPTP-induce striatal lesions (Dluzen, 1996; Ekue et al., 2002), whereas it lacked any protective capacity against MA toxicity (Gao and Dluzen, 2001; Yu et al., 2002). Moreover, in the 6-OHDA rat model, treatment of castrated males with physiological levels of estradiol exacerbated lesion size, thereby reversing the effects of gonadectomy, whereas the non-aromatizable androgen, dihydrotestosterone (DHT), had no effect on striatal dopamine loss and failed to reverse the effects of gonadectomy (Figure 2; Murray et al., 2003). These findings suggest that any influences of circulating testosterone to promote degeneration in the male NSDA system operate only after its conversion to estradiol by aromatase enzymes, which are prevalent in the striatum (Kuppers et al., 2000).
The preceding observations support the view that detrimental effects of gonadal factors in the male NSDA system, as well as protective effects of ovarian factors in females compound to create sex differences in striatal susceptibility to injury. Furthermore, they reveal that estrogen therapy has opposite effects on striatal dopamine loss in males (increased) and females (reduced). Together, these pre-clinical findings complement the clinical evidence (discussed above) for sexual dimorphisms in the mechanisms underlying NSDA degeneration and highlight the potential for centrally generated as well as systemic sex steroid hormones to critically influence these processes. Furthermore, they reveal that the reported clinical benefits of estrogens in the female sex may not simply translate to the opposite sex.
Impact of Peripheral Sex Steroid Hormones on SNc Lesion Size
It is implicit in studies of experimental PD that the extent of striatal dopamine loss equates with the extent of dopaminergic neuron loss in the SNc, as corroborated by the data in Table 1, showing similar sex differences in lesion size at both cell body and terminal levels. Therefore, the majority of studies measure only striatal dopamine levels. However, despite substantial evidence that systemic hormonal status affects striatal dopamine loss (discussed above), gonadectomy, and replacement with estradiol or DHT did not affect the loss dopaminergic neurons in males (McArthur et al., 2007) or females (Ferraz et al., 2003, 2008; Moroz et al., 2003; McArthur et al., 2007) exposed to 6-OHDA. Moreover, dopaminergic cell numbers in the normal SNc, which are significantly greater in males compared with females (see legend Table 1; Murray et al., 2003; Gillies et al., 2004; Dewing et al., 2006; McArthur et al., 2007), were not affected by gonadectomy, with or without hormone replacement, in either sex. These studies suggest that the inherent sex differences in dopaminergic cell numbers, as well as sex differences in dopaminergic cell loss in experimental PD, are not hormone-dependent, and that hormonal influences on striatal dopamine depletion occur independently of dopaminergic cell survival.
Sex Hormone Influences on Adaptation vs. Survival: Physiological and Supraphysiological Effects
The evidence discussed thus far demonstrates a dissociation of the effects of physiological levels of circulating estradiol at the level of the cell body and nerve terminals in the partially injured NSDA system in both males and females, although these effects are sexually dimorphic. We therefore need to ask how systemic estradiol can preserve striatal dopamine levels in the NSDA pathway after injury in females, but not males, without affecting dopaminergic neuron numbers in either sex.
Available evidence supports the view that hormonal influences on the activity of the surviving neurons rather than on neuron survival may be one explanation for these observations (McArthur et al., 2007; Gillies and McArthur, 2010a,b). In both clinical and experimental PD the surviving neurons and associated basal ganglia circuitry are known to have a remarkable capacity for compensation, and this is thought to account for the finding that overt motor symptoms may not be apparent until around 80% of striatal dopamine and 60% of SNc perikarya are lost (Castaneda et al., 1990; Song and Haber, 2000; Bezard et al., 2001, 2003; Bassilana et al., 2005). The adaptive mechanisms include increased tyrosine hydroxylase expression leading to synthesis and release of dopamine, maintaining striatal functionality. Notably, circulating estradiol (endogenous and exogenous) promotes all these parameters (McDermott et al., 1994; Pasqualini et al., 1995; Becker, 1999; Ohtani et al., 2001; Serova et al., 2004; Dluzen, 2005), as well as striatal dopamine turnover (McArthur et al., 2007) in females, but not males. Proestrus levels of estradiol are also far more effective in the female compared with the male striatum in suppressing the density of the dopamine transporter (McArthur et al., 2007), which is both a critical regulator of dopaminergic neuron dynamics and a gateway for the entry of neurotoxins into dopaminergic neurons. Additionally, estradiol replacement promotes behavioral recovery after 6-OHDA lesions in female rodents, but fails to do so in males (Tamas et al., 2005). Hence, a combination of physiological levels of estradiol in the systemic circulation with a mild/moderate injury to the NSDA would render females, but not males, more able to preserve striatal dopamine levels and adapt to injury, without changing cell numbers.
Less evidence is available to explain the origins of the hormone-independent sex differences in dopaminergic cell numbers in the intact (males > females) and injured SNc (males < females; Murray et al., 2003; Dewing et al., 2006; McArthur et al., 2007). Of potential relevance is the discovery that a proportion of dopaminergic neurons within the murine SNc co-express the Sry gene (the sex determining region of the Y chromosome; Dewing et al., 2006), which was previously thought to be relevant solely to ensuring testicular differentiation in the embryo. Transient suppression of this gene in the adult mouse brain not only reduced the numbers of dopaminergic neurons in the male SNc to those seen in the normal female SNc, but also compromised fine motor control, emphasizing the relevance of this co-expression for NSDA functionality (Dewing et al., 2006). However, the impact of Sry on dopaminergic neuron survival in the injured SNc remains to be determined.
Despite the foregoing discussion, it must be acknowledged that under certain circumstances estrogens have been reported to protect against the neurotoxin-induced loss of nigral dopaminergic cells (DeGiorgio et al., 2002; Quesada and Micevych, 2004). Generally, however, this requires high, pharmacological doses which are known to activate various non-specific processes that are common to neuroprotection against many types of injury throughout the brain, including anti-oxidant activity and effects on genes involved in regulating apoptosis (Green and Simpkins, 2000; Hurn and Macrae, 2000; Garcia-Segura et al., 2001; Behl, 2002; Murray et al., 2003; Dewing et al., 2006; Brann et al., 2007; McArthur et al., 2007; Wise et al., 2009). Therefore, we have proposed that neuroprotection afforded by supraphysiological levels of estradiol in the systemic circulation is not specific to the degenerating NSDA system, whereas levels within the physiological range uniquely modify NSDA activity rather than nigral cell survival (Gillies and McArthur, 2010b).
Impact of Centrally Generated Sex Steroids in Experimental PD
Incontrovertible evidence now shows that in addition to responding to sex steroid hormones, the brain itself is a steroidogenic organ, synthesizing neurosteroids (estrogens, progestogens, and androgens) de novo from cholesterol or after metabolism of precursors, which may derive from peripheral sources (Do Rego et al., 2009). Accordingly, the complex series of enzymes required for steroid biosynthesis are all found in the brain, with discrete distributions in neurons and glia, which are thought to act co-operatively in the production of a range of steroids to influence brain development, physiology, and pathology (Mensah-Nyagan et al., 1999; Zwain and Yen, 1999; Pelletier, 2010). The most important of these regarding the potential neuromodulatory and neuroprotective actions of estrogen is the enzyme aromatase (cytochrome P450 19A1, EC 1.14.14.1; Azcoitia et al., 2010, 2011; Boon et al., 2010), which catalyzes the synthesis of various isoforms of estrogens, including estradiol, estrone, 2-hydroxy estradiol, and the 17α- and 17β- isomers (Saldanha et al., 2009). Importantly, these enzymes are present in both male and female brains, although with some sex differences in distribution (reviewed by Balthazart in this issue). In particular, concentrations of estrogen in male brains have been estimated as being at least six times greater than in the plasma (Mukai et al., 2006), and may potentially be even greater at local sites due to the concentration of the hormone at the synapse (Balthazart et al., 2002).
In the normal brain it is thought that aromatase expression occurs primarily in neurons, where it may have a role in modulating neurotransmission (Balthazart and Ball, 2006). Aromatase activity, and hence the production of estrogens, has a further important role in the response to neural injury, as seen in a variety of experimental models (Garcia-Segura, 2008). For example, studies of hippocampal excitotoxicity revealed significantly greater neuronal loss in the absence of effective aromatase activity, whether through pharmacological or genetic ablation (Azcoitia et al., 2001). Moreover, up-regulation of aromatase expression, principally in astrocytes, has been reported following a diverse range of neural injuries, indicating a protective role in male and female brains (Azcoitia et al., 2003, 2005, 2010; Carswell et al., 2005; Garcia-Ovejero et al., 2005; Garcia-Segura, 2008). A role for aromatase in the NSDA system has received less attention, but the enzyme is present in the undamaged striatum throughout life (Kuppers and Beyer, 1998; Kipp et al., 2006). Evidence suggests that it plays an important role in nigrostriatal development as one feature characterizing the aromatase knock-out mouse is a dramatic decrease in SNc dopaminergic neuron number and striatal dopaminergic fiber innervation when compared with wild-type littermates (Morale et al., 2008). A functional link between aromatase and the nigrostriatal dopaminergic system is also suggested by the finding that administration of the aromatase inhibitor anastrozole to the otherwise uninjured nigrostriatal pathway significantly enhanced striatal dopamine content (McArthur et al., 2007). Furthermore, aromatase appears to play a protective role following neurotoxic lesioning of the nigrostriatal system (Figure 3). Infusion of anastrozole into the medial forebrain bundle of male rats prior to administering 6-OHDA caused a significant potentiation of dopamine loss in the degenerating striatum (McArthur et al., 2007). Studies using the aromatase knock-out mouse also suggest that central aromatase activity protects females against neurotoxin-induced striatal dopamine depletion (Morale et al., 2008). Interestingly, anastrozole failed to influence the loss of SNc dopaminergic neurons in the partially damaged NSDA pathway (McArthur et al., 2007). Together, these observations support the view that in males centrally generated endogenous estrogens primarily promote plastic/adaptive compensatory mechanisms within the surviving dopaminergic neurons to maintain striatal dopamine content, without affecting cell survival, in a manner similar to that described above for systemic estrogens in females (Figure 2, see Impact of Peripheral Sex Steroid Hormones on Striatal Lesion Size and Impact of Peripheral Sex Steroid Hormones on SNc Lesion Size). Additionally, these findings reveal that in males estradiol can have opposite effects on striatal susceptibility to injury depending on its central or systemic origin to reduce or worsen damage, respectively.
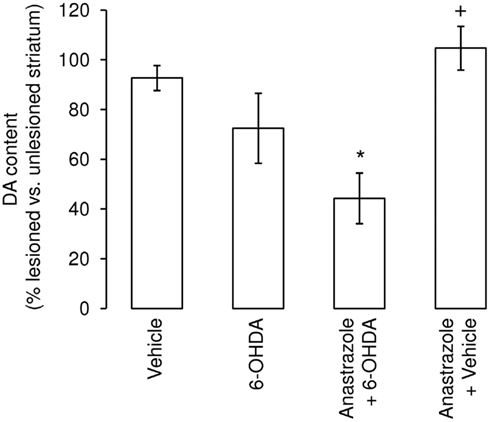
Figure 3. Inhibition of striatal aromatase activity augments the lesion size following administration of 6-hydroxydopamine (6-OHDA). Administration of the selective aromatase inhibitor anastrozole (7 μg) to the left medial forebrain bundle of male rats concurrently with 1 μg 6-hydroxydopamine significantly increased the degree of striatal dopamine loss seen 1 week later. *p < 0.05 Vs. vehicle-treated group; +p < 0.05 vs. anastrozole +6-OHDA group. (Modified from McArthur et al., 2007).
Differential Effects of Peripheral and Central Sex Steroid Hormones
Further studies are required to solve this puzzle, but it seems likely that there are different modes of action for centrally generated and systemic steroids (Prange-Kiel and Rune, 2006). Hence, we hypothesize that aromatase activity in the area of discrete neuronal injury will be responsible for the local production of estradiol which acts directly on the nigral dopaminergic neurons to activate protective, compensatory mechanisms in both males and females (Table 2). In contrast, gonadal or exogenous steroids administered peripherally can freely access most brain regions, including networks interacting with and regulating the NSDA system. These systems are known to be sexually differentiated and may therefore exert indirect, sex-specific influences on its activity and, hence, capacity to adapt to injury. In males the net effect would be an increased striatal vulnerability to 6-OHDA which over-rides any beneficial effects of locally synthesized estrogens (including those derived from circulating testosterone), whereas in females the net effect is protective. In support of this, numerous examples where systemic estrogen promotes NSDA activity in females but not males were given in Section “Impact of Peripheral Sex Steroid Hormones on SNc Lesion Size.” Although this could be due to direct effects on dopaminergic neurons, estrogen-sensitive sexually dimorphic input pathways could be targeted. For example, noradrenergic transmission from the locus coeruleus exerts positive influences on neurotransmission and adaptive responses in the injured NSDA system (Marien et al., 2004), but tyrosine hydroxylase expression in this pathway is up-regulated by circulating estradiol in female mice, but down-regulated by testosterone (after conversion to estradiol) in male mice (Thanky et al., 2002). The striatal medium spiny neurons, which provide critical GABAergic interneuron regulation of the basal ganglia, are also sexually differentiated targets for estradiol, which suppresses their negative influence on dopaminergic terminals (increasing release) in females, but not males (Hu et al., 2004, 2006; Schultz et al., 2009). Other sexually differentiated, estrogen-sensitive networks that could modify adaptations in the injured NSDA system include the mesocortical dopaminergic pathways (Kritzer and Creutz, 2008) and the serotonergic system of the dorsal raphe (Klink et al., 2002). Whatever the precise mechanisms involved, together, these observations highlight brain aromatase as an attractive therapeutic target in both sexes.

Table 2. Summary of potential modes of action of peripheral and central sex steroid hormones on the nigrostriatal dopaminergic (NSDA) system in male and female brain.
A question remains as to the cellular source of aromatase activity, and hence the locally derived estrogen, in the lesioned striatum. Aromatase expression has been identified within developing striatal neurons (Kuppers and Beyer, 1998), but there is a lack of information on its presence in the injured adult NSDA system. However, the balance of evidence from other forms of neuronal injury, including penetrative trauma, excitotoxicity, and hypoxia/ischemia, would favor its up-regulation in reactive glia, especially astrocytes (Garcia-Segura et al., 1999; Wynne and Saldanha, 2004; Carswell et al., 2005; Garcia-Ovejero et al., 2005; Azcoitia et al., 2010) which are considered the most active steroidogenic cells in the rat brain (Zwain and Yen, 1999).
Glia as Producers of and Targets for Estrogen
Injury to the brain, whether by trauma, ischemia, infection, or degenerative diseases, such as Parkinson’s and Alzheimer’s, triggers an immune response (Lucas et al., 2006; Klegeris et al., 2007). Although this differs in important respects from the peripheral immune response in ways that yet need to be fully defined, brain innate immunity shares the same purpose of host-defense which, if excessively or chronically activated can become damaging to the host and exacerbate on-going disease processes (Rogers et al., 2007). Two types of glial cells, the astrocytes, and microglia, play critical, active, and co-operative roles in mounting and regulating brain immune responses (Farina et al., 2007) and, depending on the phase of the response (activation vs. resolution) and nature of the stimulus, exhibit both pro-inflammatory (potentially harmful) and anti-inflammatory (protective) features (Minghetti, 2005; Lucas et al., 2006). Thus augmentation of glial protective and repair processes as well as suppression of their neurotoxic potential are attractive novel therapeutic strategies. Notably, steroid hormones, especially estrogens, have been credited with the powerful ability to regulate the balance of astrocytic and microglial activity toward a favorable pro- vs. anti-inflammatory status (Garcia-Ovejero et al., 2005; Marchetti et al., 2005; Block et al., 2007; Tapia-Gonzalez et al., 2008; Vegeto et al., 2008; Arevalo et al., 2011). Moreover, just as peripheral immune functions are sexually dimorphic in humans and animals, regulated to a large degree by sex hormones (Azevedo et al., 2001), evidence is emerging for sex differences in the neuroimmune response (Santos-Galindo et al., 2011). Thus, knowledge of hormonal influences on glia is essential for the effective design of new treatments as well as our understanding of sexual dimorphisms in CNS disorders. Importantly, endogenous regulation of glial reactivity by sex steroids may come both from peripheral, gonadally derived sources as well as the CNS glia themselves.
Astrocytes
Classically, astrocytes, the most abundant type of cell within the brain parenchyma, have been thought of as merely providing structural and metabolic support to neurons. Over the past decade, however, it has steadily become clear that they perform many other functions, such as the regulation of and participation in neurotransmitter release and synaptic function (Halassa and Haydon, 2010). Increasing evidence suggests that they may also play an important role in regulating the neuronal response to injury (Barres, 2008), with reactive astrogliosis being a feature of many CNS disorders, representing context-dependent influences that have the potential to be both beneficial or detrimental (Hamby and Sofroniew, 2010). The regulatory mechanisms are complex and not fully understood, but it is particularly relevant to the present discussion that astrocytes can both produce estrogens (as discussed above, see Differential Effects of Peripheral and Central Sex Steroid Hormones; Zwain and Yen, 1999) and respond to this sex steroid hormone.
Up-regulation of astrocyte aromatase activity at sites of injury, and the associated increase in local synthesis of estradiol, may trigger a variety of protective mechanisms in vulnerable neurons that are considered general, non-specific actions of estrogens. These include modification of apoptotic pathways and anti-oxidant activity, that have been extensively reviewed elsewhere (Green and Simpkins, 2000; Garcia-Segura et al., 2001; Behl, 2002; Brann et al., 2007; Garcia-Segura, 2008; Nilsen, 2008; Saldanha et al., 2009). In addition to being a likely local source of aromatase/estrogens in the damaged brain, astrocytes could also contribute to sex differences in damage/repair processes. For example, one study, using primary astrocyte cultures derived from the neonatal rat cortex, showed that females have a greater capacity for aromatization and estradiol production compared with males; it remains to be seen whether this effect is region-specific and/or age-dependent. Sex differences in neurosteroid levels have, however, been reported in various areas of the normal and injured adult rat brain (Cosimo Melcangi and Garcia-Segura, 2010; Pesaresi et al., 2010), and neurosteroid levels are changed in affected regions in several models of neurodegeneration, including the striatum in the 6-OHDA-lesioned rat model of PD (Melcangi et al., 2010). Together, these findings suggest that differing capacities for constitutive and/or injury-induced neurosteroid production could contribute to sex differences in the incidence and/or manifestation of CNS pathologies.
As well as targeting neurons, astrocyte-derived estrogens also exert important effects on glia, namely astrocytes themselves and microglia, the macrophage-like, innate immune cells of the brain. The evidence for constitutive astrocyte expression of ERs is contradictory (Azcoitia et al., 1999; Blurton-Jones and Tuszynski, 2001; Milner et al., 2001, 2005), but it appears that a key part of the neural response to various types of injury seen in rodents and primates includes an up-regulation of ERα as well as aromatase within astrocytes (Garcia-Segura et al., 1999; Blurton-Jones and Tuszynski, 2001; Carswell et al., 2005; Garcia-Ovejero et al., 2005). Although ER expression in the astrocytes of the damaged NSDA pathway has not been specifically studied, under normal conditions ERα is present at low levels in the striatum and SNc, but not in dopaminergic cells, whereas ERβ was not detected in the striatum (Shughrue, 2004), but was found to be present in 40% of nigral dopaminergic neurons as well as in 30% of astrocytes in the female rat (Quesada et al., 2007). Importantly, in addition to their neuroprotective and neurotrophic activities, astrocytes may also release factors such as pro-inflammatory cytokines, chemokines, and reactive oxygen/nitrogen species in response to injury which, although necessary for local resistance and repair mechanisms, if present in excess for prolonged periods can become neurotoxic and are thought to contribute to the progression of neurodegenerative disorders such as PD (Marchetti et al., 2005; Morale et al., 2006; Yokoyama et al., 2011). Thus, the orchestrated up-regulation of both aromatase and ERs in reactive astrocytes may serve as a negative feedback regulator of the astrocytic response, thereby representing an important component of the brain’s response to injury. In direct support for this view, in vitro studies have demonstrated that estradiol treatment reduces the production of cytokines and reactive oxygen species by astrocytes from many brain regions, including the midbrain, following inflammatory challenge (Kipp et al., 2007; Cerciat et al., 2010; Santos-Galindo et al., 2011). Furthermore, in vivo studies using the MPTP-treated mouse model of PD, showed that circulating estradiol (endogenous or exogenous) can down-regulate injury-induced astrogliosis and astrocytic production of inducible nitric oxide synthetase, which correlated with its protective effects (Morale et al., 2006; Tripanichkul et al., 2006). Although not in the context of PD, a recent study has provided an elegant demonstration that astrocytes are, indeed, the mediators of estrogenic neuroprotection by showing that conditional deletion of ERα from astrocytes, but not from neurons, prevented the neuroprotective effects of systemic ERα ligands in murine experimental autoimmune encephalomyelitis (Spence et al., 2011).
Microglia
Microglia are critical components of the neuroimmune, host-defense response to injury- and disease-induced neurodegeneration. They constantly monitor the brain microenvironment for signs of tissue damage or infection, whereupon they rapidly become activated and co-ordinate the brain’s immune response (Nimmerjahn et al., 2005). This involves production of a host of pro-inflammatory and pro-oxidant mediators which, if the neuroinflammatory response is not curtailed, will exacerbate rather than protect against neural damage, leading to a detrimental neurological outcome (Hirsch et al., 1998; Block et al., 2007; Rogers et al., 2007; Vegeto et al., 2008). Their significance in PD is underscored by the presence of high numbers of microglia in close proximity to degenerating neurons of post-mortem parkinsonian brains (McGeer et al., 1988; Banati et al., 1998; Hirsch et al., 1998; Orr et al., 2005; Hamza et al., 2010; Halliday and Stevens, 2011). These microglia express high levels of pro-oxidant enzymes including inducible nitric oxide synthase (Hunot et al., 1996), NADPH oxidase, and cyclo-oxygenase (Knott et al., 2000), thereby representing an important source of the reactive oxygen species known to contribute to oxidative stress in the dopaminergic neurons of the parkinsonian SNc (Shadrina et al., 2010). Although the steroidogenic capacity of microglia has received relatively little attention compared with that of astrocytes, they do express the machinery for steroidogenesis (Garcia-Ovejero et al., 2005), including steroid converting enzymes that would enable them to produce estrogenic compounds from precursors (Gottfried-Blackmore et al., 2008), and they also express ERs (Vegeto et al., 2003; Baker et al., 2004; Sierra et al., 2008). Together, these observations suggest that microglia contribute to the neurodegenerative process in PD and could be targets for and mediators of the neuroprotective effects of estrogens (Garcia-Ovejero et al., 2005; Vegeto et al., 2008). In support of this, studies using either primary mesencephalic neuronal cultures or dopaminergic cell lines (Liu et al., 2005; McArthur et al., 2010) have shown that microglia activated by treatment with bacterial lipopolysaccharide (LPS) can cause the death of dopaminergic neurons through the release of toxic factors such as reactive oxygen/nitrogen species and TNFα, an effect that can be prevented by co-treatment with estradiol (Liu et al., 2005). Similar effects have been reported in vivo, where administration of estradiol to ovariectomized mice treated with MPTP significantly suppressed microglial production of iNOS and nitric oxide, limiting striatal dopamine loss (Morale et al., 2006).
Despite the principal research focus on suppressing the potentially harmful phenotypes induced by reactive gliosis in neuronal injury, glia, and microglia in particular, perform the critically protective role of removing dead or dying cells before they can enter secondary necrosis and become pro-inflammatory foci (Neumann et al., 2009). This removal of apoptotic cells by phagocytosis is an important component of inflammatory resolution, as their engulfment induces anti-inflammatory microglial activation and actively contributes to repair and the restoration of homeostasis. In order to investigate this phenomenon we have developed an in vitro system comprising a microglial cell line and a dopaminergic neuronal cell line in which apoptosis can be induced by treatment with the selective dopaminergic neurotoxin, 6-OHDA (McArthur et al., 2010). In this model, apoptotic, neuron-like cells are phagocytosed by the microglia, a process enhanced in a dose-dependent manner by estradiol (Figure 4), suggesting that promotion of microglial phagocytosis is yet another pathway by which estrogens may serve as neuroprotective agents in PD.
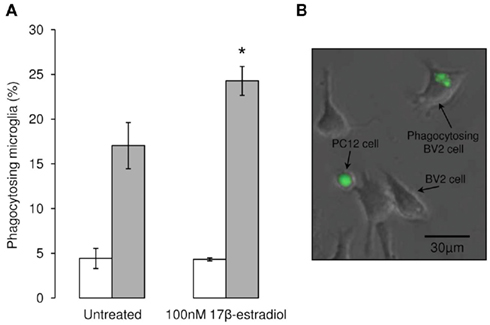
Figure 4. Estradiol enhances microglial phagocytosis. We used our established in vitro model of co-cultures of the murine microglial cell line BV2 and the rat neuron-like cell line PC12 to investigate phagocytosis, for full details see (McArthur et al., 2010), to study the effects of estradiol treatment upon the removal of apoptotic neurons. (A) Treatment of BV2 microglia for 16 h with 100 nM 17β-estradiol had no effect upon the phagocytosis of vehicle-treated, non-apoptotic PC12 cells (□), but significantly increased the number of microglia actively phagocytosing apoptotic, 6-OHDA-treated PC12 cells () when compared with untreated control microglia, *p < 0.05 vs. untreated microglia. (B) Typical example of BV2 microglia phagocytosing CMFDA-labeled, green fluorescent PC12 cells after 6-hydroxydopamine treatment.
Further work is needed to finally provide definitive evidence as to whether peripherally or centrally generated steroids are the prime mediators in regulating glial activity. Of note, several aspects of the neuroprotective actions of estrogens are achieved only with doses of estradiol that have been termed “pharmacological” because they exceed the normal circulating levels in females and, certainly, in males. These include estrogen’s ability to promote non-inflammatory phagocytosis of apoptotic neuron-like cells (see above, Figure 4 and McArthur, Vohra, Solito and Gillies, unpublished observations) as well as its anti-oxidant and anti-neuroinflammatory actions (Green and Simpkins, 2000; Liu et al., 2005; Nilsen, 2008). However, our recent knowledge that brain injury may induce an increase both in the central production of estrogens, as well as glial sensitivity to estrogens, highlights the likelihood that circulating “physiological” levels may well be exceeded in the brain where much higher levels may be achieved (Mukai et al., 2006). Thus it is tempting to speculate that local production of estrogens via glial aromatase may represent the principal mode whereby estrogen exerts its neuroprotective effects, probably through regulation of the pro- and anti-inflammatory balance of astrocytic and microglial reactivity, with only a limited contribution from gonadal hormones. Our evidence that central administration of an aromatase inhibitor can exacerbate striatal damage in experimental PD would support this view (see Impact of Centrally Generated Sex Steroids in Experimental PD, Figure 3; McArthur et al., 2007). However, other studies suggest that a gonadal source of estrogens in females is important for regulating microglial reactivity in animal models of inflammation (Vegeto et al., 2006). Equally, a gonadal source of an estrogen precursor (testosterone) in males, which is subsequently aromatized in the brain, appears to be important for neuroprotection against excitotoxic injury (Azcoitia et al., 2001). It therefore remains an open question as to the relative importance of centrally and peripherally generated estrogens.
Concluding Remarks
Parkinson’s disease is already one of the most prevalent neurodegenerative disorders, second only to Alzheimer’s disease, and its incidence and prevalence are expected to rise markedly in the near future (Pavon et al., 2010). However, despite many decades of research, options to prevent or delay disease onset and/or progression are unmet therapeutic needs, and dopamine replacement therapies to alleviate motor symptoms have remained the principal therapeutic option for almost half a century (Meissner et al., 2011). A wealth of evidence now documents notable sex differences in the epidemiology, clinical features, molecular pathology, and treatment outcomes in PD, with women being relatively protected from, and having a more benign pre-clinical phase of the disease compared with men (Haaxma et al., 2007; Pavon et al., 2010). Knowledge of factors underlying sex differences in the pathophysiology of PD will therefore offer considerable insight into improving diagnosis and treatments, which need to be tailored to the specific needs of men and women. In this review we have focused on the precise contributions of sex steroid hormones, and estrogens in particular. Much attention has focused on the ostensibly neuroprotective effects of estrogen in experimental models of PD, but the term “neuroprotection” has been used broadly and often describes a relative sparing of striatal dopamine rather than a defined limitation of neuronal death. Therefore, we have developed the view that any protective capacity offered by endogenous sex steroid hormones in PD centers on their ability to promote adaptive compensatory mechanisms in the partially injured neuronal network, with potential to influence onset or progression of disease. However, inherent sexual dimorphisms in the NSDA-related circuitry, which may themselves be largely attributable to the sex-differentiating influences of gonadal steroids during development, represent biological factors that will also affect male/female differences in vulnerability to CNS disorders and responses to sex hormones (Gillies and McArthur, 2010a). Together, these factors account for sex-specific responses to peripheral sex hormones in experimental PD, which are protective in females, but not in males. Importantly, however, it appears that aromatase activity local to the damaged NSDA has a more restricted effect on the neural network, allowing centrally generated estrogen to protect against striatal dopamine loss in males as well as females. These observations leave us with two challenging questions which urgently need to be answered: precisely what is the nature of the sexually dimorphic neural circuitry which underlies sex differences in responsiveness of the healthy and injured NSDA system, and what are the relative contributions of estrogens/estrogen precursors derived from the gonads and neural cells (especially astrocytes)? Although in its infancy, a better understanding of such “neuroendocrine” mechanisms in the NSDA offers considerable potential for the development of novel, sex-specific, hormone-based treatments in PD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alves, G., Muller, B., Herlofson, K., Hogenesch, I., Telstad, W., Aarsland, D., Tysnes, O. B., and Larsen, J. P. (2009). Incidence of Parkinson’s disease in Norway: the Norwegian ParkWest study. J. Neurol. Neurosurg. Psychiatr. 80, 851–857.
Arevalo, M. A., Diz-Chaves, Y., Santos-Galindo, M., Bellini, M. J., and Garcia-Segura, L. M. (2011). Selective oestrogen receptor modulators decrease the inflammatory response of glial cells. J. Neuroendocrinol. doi: 10.1111/j.1365-2826.2011.02156.x. [Epub ahead of print]..
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 26, 211–233.
Azcoitia, I., Santos-Galindo, M., Arevalo, M. A., and Garcia-Segura, L. M. (2010). Role of astroglia in the neuroplastic and neuroprotective actions of estradiol. Eur. J. Neurosci. 32, 1995–2002.
Azcoitia, I., Sierra, A., and Garcia-Segura, L. M. (1999). Localization of estrogen receptor beta-immunoreactivity in astrocytes of the adult rat brain. Glia 26, 260–267.
Azcoitia, I., Sierra, A., Veiga, S., and Garcia-Segura, L. M. (2003). Aromatase expression by reactive astroglia is neuroprotective. Ann. N. Y. Acad. Sci. 1007, 298–305.
Azcoitia, I., Sierra, A., Veiga, S., and Garcia-Segura, L. M. (2005). Brain steroidogenesis: emerging therapeutic strategies to prevent neurodegeneration. J. Neural Transm. 112, 171–176.
Azcoitia, I., Sierra, A., Veiga, S., Honda, S., Harada, N., and Garcia-Segura, L. M. (2001). Brain aromatase is neuroprotective. J. Neurobiol. 47, 318–329.
Azcoitia, I., Yague, J. G., and Garcia-Segura, L. M. (2011). Estradiol synthesis within the human brain. Neuroscience 191, 139–147.
Azevedo, R. B., Lacava, Z. G., Miyasaka, C. K., Chaves, S. B., and Curi, R. (2001). Regulation of antioxidant enzyme activities in male and female rat macrophages by sex steroids. Braz. J. Med. Biol. Res. 34, 683–687.
Baba, Y., Putzke, J. D., Whaley, N. R., Wszolek, Z. K., and Uitti, R. J. (2005). Gender and the Parkinson’s disease phenotype. J. Neurol. 252, 1201–1205.
Baker, A. E., Brautigam, V. M., and Watters, J. J. (2004). Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor beta. Endocrinology 145, 5021–5032.
Baldereschi, M., Di Carlo, A., Rocca, W. A., Vanni, P., Maggi, S., Perissinotto, E., Grigoletto, F., Amaducci, L., and Inzitari, D. (2000). Parkinson’s disease and parkinsonism in a longitudinal study: two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 55, 1358–1363.
Balthazart, J., Baillien, M., and Ball, G. F. (2002). Interactions between aromatase (estrogen synthase) and dopamine in the control of male sexual behavior in quail. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 132, 37–55.
Balthazart, J., and Ball, G. F. (2006). Is brain estradiol a hormone or a neurotransmitter? Trends Neurosci. 29, 241–249.
Banati, R. B., Daniel, S. E., and Blunt, S. B. (1998). Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 13, 221–227.
Barres, B. A. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440.
Bassilana, F., Mace, N., Li, Q., Stutzmann, J. M., Gross, C. E., Pradier, L., Benavides, J., Menager, J., and Bezard, E. (2005). Unraveling substantia nigra sequential gene expression in a progressive MPTP-lesioned macaque model of Parkinson’s disease. Neurobiol. Dis. 20, 93–103.
Becker, J. B. (1999). Gender differences in dopaminergic function in striatum and nucleus accumbens. Pharmacol. Biochem. Behav. 64, 803–812.
Benedetti, M. D., Maraganore, D. M., Bower, J. H., Mcdonnell, S. K., Peterson, B. J., Ahlskog, J. E., Schaid, D. J., and Rocca, W. A. (2001). Hysterectomy, menopause, and estrogen use preceding Parkinson’s disease: an exploratory case-control study. Mov. Disord. 16, 830–837.
Bezard, E., Dovero, S., Prunier, C., Ravenscroft, P., Chalon, S., Guilloteau, D., Crossman, A. R., Bioulac, B., Brotchie, J. M., and Gross, C. E. (2001). Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J. Neurosci. 21, 6853–6861.
Bezard, E., Gross, C. E., and Brotchie, J. M. (2003). Presymptomatic compensation in Parkinson’s disease is not dopamine-mediated. Trends Neurosci. 26, 215–221.
Bixo, M., Andersson, A., Winblad, B., Purdy, R. H., and Backstrom, T. (1997). Progesterone, 5alpha-pregnane-3,20-dione and 3alpha-hydroxy-5alpha-pregnane-20-one in specific regions of the human female brain in different endocrine states. Brain Res. 764, 173–178.
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69.
Blum, D., Torch, S., Lambeng, N., Nissou, M.-F., Benabid, A.-L., Sadoul, R., and Verna, J.-M. (2001). Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 65, 135–172.
Blurton-Jones, M., and Tuszynski, M. H. (2001). Reactive astrocytes express estrogen receptors in the injured primate brain. J. Comp. Neurol. 433, 115–123.
Boon, W. C., Chow, J. D., and Simpson, E. R. (2010). The multiple roles of estrogens and the enzyme aromatase. Prog. Brain Res. 181, 209–232.
Bourque, M., Dluzen, D. E., and Di Paolo, T. (2009). Neuroprotective actions of sex steroids in Parkinson’s disease. Front. Neuroendocrinol. 30, 142–157.
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211.
Brann, D. W., Dhandapani, K., Wakade, C., Mahesh, V. B., and Khan, M. M. (2007). Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids 72, 381–405.
Cantuti-Castelvetri, I., Keller-Mcgandy, C., Bouzou, B., Asteris, G., Clark, T. W., Frosch, M. P., and Standaert, D. G. (2007). Effects of gender on nigral gene expression and Parkinson disease. Neurobiol. Dis. 26, 606–614.
Carswell, H. V., Dominiczak, A. F., Garcia-Segura, L. M., Harada, N., Hutchison, J. B., and Macrae, I. M. (2005). Brain aromatase expression after experimental stroke: topography and time course. J. Steroid Biochem. Mol. Biol. 96, 89–91.
Castaneda, E., Whishaw, I. Q., and Robinson, T. E. (1990). Changes in striatal dopamine neurotransmission assessed with microdialysis following recovery from a bilateral 6-OHDA lesion: variation as a function of lesion size. J. Neurosci. 10, 1847–1854.
Cerciat, M., Unkila, M., Garcia-Segura, L. M., and Arevalo, M. A. (2010). Selective estrogen receptor modulators decrease the production of interleukin-6 and interferon-gamma-inducible protein-10 by astrocytes exposed to inflammatory challenge in vitro. Glia 58, 93–102.
Chung, S. J., Armasu, S. M., Biernacka, J. M., Lesnick, T. G., Rider, D. N., Cunningham, J. M., and Maraganore, D. M. (2011). Variants in estrogen-related genes and risk of Parkinson’s disease. Mov. Disord. 26, 1234–1242.
Cordellini, M. F., Piazzetta, G., Pinto, K. C., Delattre, A. M., Matheussi, F., Carolino, R. O., Szawka, R. E., Anselmo-Franci, J. A., and Ferraz, A. C. (2011). Effect of different doses of estrogen on the nigrostriatal dopaminergic system in two 6-hydroxydopamine-induced lesion models of Parkinson’s disease. Neurochem. Res. 36, 955–961.
Cosimo Melcangi, R., and Garcia-Segura, L. M. (2010). Sex-specific therapeutic strategies based on neuroactive steroids: in search for innovative tools for neuroprotection. Horm. Behav. 57, 2–11.
Craig, M. C., and Murphy, D. G. (2007). Estrogen: effects on normal brain function and neuropsychiatric disorders. Climacteric 10(Suppl. 2), 97–104.
Currie, L. J., Harrison, M. B., Trugman, J. M., Bennett, J. P., and Wooten, G. F. (2004). Postmenopausal estrogen use affects risk for Parkinson disease. Arch. Neurol. 61, 886–888.
Das, K., Ghosh, M., Nag, C., Nandy, S. P., Banerjee, M., Datta, M., Devi, G., and Chaterjee, G. (2011). Role of familial, environmental and occupational factors in the development of Parkinson’s disease. Neurodegener. Dis. 8, 345–351.
Datla, K. P., Murray, H. E., Pillai, A. V., Gillies, G. E., and Dexter, D. T. (2003). Differences in dopaminergic neuroprotective effects of estrogen during estrous cycle. Neuroreport 14, 47–50.
DeGiorgio, L. A., Attardi, B., Shimizu, Y., Ogata, M., and Volpe, B. T. (2002). 17 beta-estradiol treatment retards excitotoxic delayed degeneration in substantia nigra reticulata neurons. Brain Res. 936, 15–20.
Dewing, P., Chiang, C. W., Sinchak, K., Sim, H., Fernagut, P. O., Kelly, S., Chesselet, M. F., Micevych, P. E., Albrecht, K. H., Harley, V. R., and Vilain, E. (2006). Direct regulation of adult brain function by the male-specific factor SRY. Curr. Biol. 16, 415–420.
di Michele, F., Longone, P., Romeo, E., Lucchetti, S., Brusa, L., Pierantozzi, M., Bassi, A., Bernardi, G., and Stanzione, P. (2003). Decreased plasma and cerebrospinal fluid content of neuroactive steroids in Parkinson’s disease. Neurol. Sci. 24, 172–173.
Dluzen, D. (1997). Estrogen decreases corpus striatal neurotoxicity in response to 6- hydroxydopamine. Brain Res. 767, 340–344.
Dluzen, D., and Horstink, M. (2003). Estrogen as neuroprotectant of nigrostriatal dopaminergic system: laboratory and clinical studies. Endocrine 21, 67–75.
Dluzen, D., Jain, R., and Liu, B. (1994). Modulatory effects of testosterone on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J. Neurochem. 62, 94–101.
Dluzen, D. E. (1996). Effects of testosterone upon MPTP-induced neurotoxicity of the nigrostriatal dopaminergic system of C57/B1 mice. Brain Res. 715, 113–118.
Dluzen, D. E. (2000). Neuroprotective effects of estrogen upon the nigrostriatal dopaminergic system. J. Neurocytol. 29, 387–399.
Dluzen, D. E. (2005). Unconventional effects of estrogen uncovered. Trends Pharmacol. Sci. 26, 485–487.
Dluzen, D. E., Anderson, L. I., and Pilati, C. F. (2002). Methamphetamine-gonadal steroid hormonal interactions: effects upon acute toxicity and striatal dopamine concentrations. Neurotoxicol. Teratol. 24, 267–273.
Dluzen, D. E., and Liu, B. (2008). Gender differences in methamphetamine use and responses: a review. Gend. Med. 5, 24–35.
Do Rego, J. L., Seong, J. Y., Burel, D., Leprince, J., Luu-the, V., Tsutsui, K., Tonon, M. C., Pelletier, G., and Vaudry, H. (2009). Neurosteroid biosynthesis: enzymatic pathways and neuroendocrine regulation by neurotransmitters and neuropeptides. Front. Neuroendocrinol. 30, 259–301.
Ekue, A., Boulanger, J. F., Morissette, M., and Di Paolo, T. (2002). Lack of effect of testosterone and dihydrotestosterone compared to 17beta-oestradiol in 1-methyl-4-phenyl-1,2,3,6, tetrahydropy-ridine-mice. J. Neuroendocrinol. 14, 731–736.
Elbaz, A., Bower, J. H., Maraganore, D. M., Mcdonnell, S. K., Peterson, B. J., Ahlskog, J. E., Schaid, D. J., and Rocca, W. A. (2002). Risk tables for parkinsonism and Parkinson’s disease. J. Clin. Epidemiol. 55, 25–31.
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145.
Ferraz, A. C., Matheussi, F., Szawka, R. E., Rizelio, V., Delattre, A. M., Rigon, P., Hermel Edo, E., Xavier, L. L., Achaval, M., and Anselmo-Franci, J. A. (2008). Evaluation of estrogen neuroprotective effect on nigrostriatal dopaminergic neurons following 6-hydroxydopamine injection into the substantia nigra pars compacta or the medial forebrain bundle. Neurochem. Res. 33, 1238–1246.
Ferraz, A. C., Xavier, L. L., Hernandes, S., Sulzbach, M., Viola, G. G., Anselmo-Franci, J. A., Achaval, M., and Da Cunha, C. (2003). Failure of estrogen to protect the substantia nigra pars compacta of female rats from lesion induced by 6-hydroxydopamine. Brain Res. 986, 200–205.
Fink, G., Sumner, B. E., Rosie, R., Grace, O., and Quinn, J. P. (1996). Estrogen control of central neurotransmission: effect on mood, mental state, and memory. Cell. Mol. Neurobiol. 16, 325–344.
Fox, S. H., and Brotchie, J. M. (2010). The MPTP-lesioned non-human primate models of Parkinson’s disease. Past, present, and future. Prog. Brain Res. 184, 133–157.
Gao, X., and Dluzen, D. E. (2001). The effect of testosterone upon methamphetamine neurotoxicity of the nigrostriatal dopaminergic system. Brain Res. 892, 63–69.
Garcia-Ovejero, D., Azcoitia, I., Doncarlos, L. L., Melcangi, R. C., and Garcia-Segura, L. M. (2005). Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Res. Brain Res. Rev. 48, 273–286.
Garcia-Segura, L. M. (2008). Aromatase in the brain: not just for reproduction anymore. J. Neuroendocrinol. 20, 705–712.
Garcia-Segura, L. M., Azcoitia, I., and Doncarlos, L. L. (2001). Neuroprotection by estradiol. Prog. Neurobiol. 63, 29–60.
Garcia-Segura, L. M., Wozniak, A., Azcoitia, I., Rodriguez, J. R., Hutchison, R. E., and Hutchison, J. B. (1999). Aromatase expression by astrocytes after brain injury: implications for local estrogen formation in brain repair. Neuroscience 89, 567–578.
Gillies, G. E., and McArthur, S. (2010a). Estrogen actions in the brain and the basis for differential action in men and women: a case for sex-specific medicines. Pharmacol. Rev. 62, 155–198.
Gillies, G. E., and McArthur, S. (2010b). Independent influences of sex steroids of systemic and central origin in a rat model of Parkinson’s disease: a contribution to sex-specific neuroprotection by estrogens. Horm. Behav. 57, 23–34.
Gillies, G. E., Murray, H. E., Dexter, D., and Mcarthur, S. (2004). Sex dimorphisms in the neuroprotective effects of estrogen in an animal model of Parkinson’s disease. Pharmacol. Biochem. Behav. 78, 513–522.
Glinka, Y., Tipton, K. F., and Youdim, M. B. (1996). Nature of inhibition of mitochondrial respiratory complex I by 6-Hydroxydopamine. J. Neurochem. 66, 2004–2010.
Gottfried-Blackmore, A., Sierra, A., Jellinck, P. H., Mcewen, B. S., and Bulloch, K. (2008). Brain microglia express steroid-converting enzymes in the mouse. J. Steroid Biochem. Mol. Biol. 109, 96–107.
Green, P. S., and Simpkins, J. W. (2000). Neuroprotective effects of estrogens: potential mechanisms of action. Int. J. Dev. Neurosci. 18, 347–358.
Haaxma, C. A., Bloem, B. R., Borm, G. F., Oyen, W. J., Leenders, K. L., Eshuis, S., Booij, J., Dluzen, D. E., and Horstink, M. W. (2007). Gender differences in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr. 78, 819–824.
Hakansson, A., Westberg, L., Nilsson, S., Buervenich, S., Carmine, A., Holmberg, B., Sydow, O., Olson, L., Johnels, B., Eriksson, E., and Nissbrandt, H. (2005). Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 133B, 88–92.
Halassa, M. M., and Haydon, P. G. (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 72, 335–355.
Halliday, G. M., and Stevens, C. H. (2011). Glia: initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 26, 6–17.
Hamby, M. E., and Sofroniew, M. V. (2010). Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7, 494–506.
Hamza, T. H., Zabetian, C. P., Tenesa, A., Laederach, A., Montimurro, J., Yearout, D., Kay, D. M., Doheny, K. F., Paschall, J., Pugh, E., Kusel, V. I., Collura, R., Roberts, J., Griffith, A., Samii, A., Scott, W. K., Nutt, J., Factor, S. A., and Payami, H. (2010). Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 42, 781–785.
Hirsch, E. C., Hunot, S., Damier, P., and Faucheux, B. (1998). Glial cells and inflammation in Parkinson’s disease: a role in neurodegeneration? Ann. Neurol. 44, S115–S120.
Hu, M., Crombag, H. S., Robinson, T. E., and Becker, J. B. (2004). Biological basis of sex differences in the propensity to self-administer cocaine. Neuropsychopharmacology 29, 81–85.
Hu, M., Watson, C. J., Kennedy, R. T., and Becker, J. B. (2006). Estradiol attenuates the K+-induced increase in extracellular GABA in rat striatum. Synapse 59, 122–124.
Hunot, S., Boissiere, F., Faucheux, B., Brugg, B., Mouatt-Prigent, A., Agid, Y., and Hirsch, E. C. (1996). Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 72, 355–363.
Hurn, P. D., and Macrae, I. M. (2000). Estrogen as a neuroprotectant in stroke. J. Cereb. Blood Flow Metab. 20, 631–652.
Jones, M. E., Chin Boon, W., Proietto, J., and Simpson, E. R. (2006). Of mice and men: the evolving phenotype of aromatase deficiency. Trends Endocrinol. Metab. 17, 55–64.
Kaminsky, Z., Wang, S. C., and Petronis, A. (2006). Complex disease, gender and epigenetics. Ann. Med. 38, 530–544.
Kipp, M., Karakaya, S., Johann, S., Kampmann, E., Mey, J., and Beyer, C. (2007). Oestrogen and progesterone reduce lipopolysaccharide-induced expression of tumour necrosis factor-alpha and interleukin-18 in midbrain astrocytes. J. Neuroendocrinol. 19, 819–822.
Kipp, M., Karakaya, S., Pawlak, J., Araujo-Wright, G., Arnold, S., and Beyer, C. (2006). Estrogen and the development and protection of nigrostriatal dopaminergic neurons: concerted action of a multitude of signals, protective molecules, and growth factors. Front. Neuroendocrinol. 27, 376–390.
Klegeris, A., Mcgeer, E. G., and Mcgeer, P. L. (2007). Therapeutic approaches to inflammation in neurodegenerative disease. Curr. Opin. Neurol. 20, 351–357.
Klein, C., and Schlossmacher, M. G. (2007). Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology 69, 2093–2104.
Klink, R., Robichaud, M., and Debonnel, G. (2002). Gender and gonadal status modulation of dorsal raphe nucleus serotonergic neurons. Part II. Regulatory mechanisms. Neuropharmacology 43, 1129–1138.
Knott, C., Stern, G., and Wilkin, G. P. (2000). Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 16, 724–739.
Kritzer, M. F., and Creutz, L. M. (2008). Region and sex differences in constituent dopamine neurons and immunoreactivity for intracellular estrogen and androgen receptors in mesocortical projections in rats. J. Neurosci. 28, 9525–9535.
Kuppers, E., and Beyer, C. (1998). Expression of aromatase in the embryonic and postnatal mouse striatum. Brain Res. Mol. Brain Res. 63, 184–188.
Kuppers, E., Ivanova, T., Karolczak, M., and Beyer, C. (2000). Estrogen: a multifunctional messenger to nigrostriatal dopaminergic neurons. J. Neurocytol. 29, 375–385.
Lane, E., and Dunnett, S. (2008). Animal models of Parkinson’s disease and L-dopa induced dyskinesia: how close are we to the clinic? Psychopharmacology (Berl.) 199, 303–312.
Lewis, C., and Dluzen, D. E. (2008). Testosterone enhances dopamine depletion by methamphetamine in male, but not female, mice. Neurosci. Lett. 448, 130–133.
Liu, B., and Dluzen, D. E. (2007). Oestrogen and nigrostriatal dopaminergic neurodegeneration: animal models and clinical reports of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 34, 555–565.
Liu, X., Fan, X. L., Zhao, Y., Luo, G. R., Li, X. P., Li, R., and Le, W. D. (2005). Estrogen provides neuroprotection against activated microglia-induced dopaminergic neuronal injury through both estrogen receptor-alpha and estrogen receptor-beta in microglia. J. Neurosci. Res. 81, 653–665.
Lucas, S. M., Rothwell, N. J., and Gibson, R. M. (2006). The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147(Suppl. 1), S232–S240.
Luchetti, S., Bossers, K., Frajese, G. V., and Swaab, D. F. (2010). Neurosteroid biosynthetic pathway changes in substantia nigra and caudate nucleus in Parkinson’s disease. Brain Pathol. 20, 945–951.
Marchetti, B., Serra, P. A., L’episcopo, F., Tirolo, C., Caniglia, S., Testa, N., Cioni, S., Gennuso, F., Rocchitta, G., Desole, M. S., Mazzarino, M. C., Miele, E., and Morale, M. C. (2005). Hormones are key actors in gene x environment interactions programming the vulnerability to Parkinson’s disease: glia as a common final pathway. Ann. N. Y. Acad. Sci. 1057, 296–318.
Marien, M. R., Colpaert, F. C., and Rosenquist, A. C. (2004). Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res. Brain Res. Rev. 45, 38–78.
Mattson, M. P. (2006). Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid. Redox Signal. 8, 1997–2006.
McArthur, S., Cristante, E., Paterno, M., Christian, H., Roncaroli, F., Gillies, G. E., and Solito, E. (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 185, 6317–6328.
McArthur, S., Murray, H. E., Dhankot, A., Dexter, D. T., and Gillies, G. E. (2007). Striatal susceptibility to a dopaminergic neurotoxin is independent of sex hormone effects on cell survival and DAT expression but is exacerbated by central aromatase inhibition. J. Neurochem. 100, 678–692.
McDermott, J. L., Liu, B., and Dluzen, D. E. (1994). Sex differences and effects of estrogen on dopamine and DOPAC release from the striatum of male and female CD-1 mice. Exp. Neurol. 125, 306–311.
McEwen, B. S., and Alves, S. E. (1999). Estrogen actions in the central nervous system. Endocr. Rev. 20, 279–307.
McGeer, P. L., Itagaki, S., Boyes, B. E., and Mcgeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291.
Meissner, W. G., Frasier, M., Gasser, T., Goetz, C. G., Lozano, A., Piccini, P., Obeso, J. A., Rascol, O., Schapira, A., Voon, V., Weiner, D. M., Tison, F., and Bezard, E. (2011). Priorities in Parkinson’s disease research. Nat. Rev. Drug Discov. 10, 377–393.
Melcangi, R. C., Caruso, D., Levandis, G., Abbiati, F., Armentero, M. T., and Blandini, F. (2010). Modifications of neuroactive steroid levels in an experimental model of nigrostriatal degeneration: potential relevance to the pathophysiology of Parkinson’s disease. J. Mol. Neurosci. doi: 10.1007/s12031-011-9570-y. [Epub ahead of print].
Mensah-Nyagan, A. G., Do-Rego, J. L., Beaujean, D., Luu-the, V., Pelletier, G., and Vaudry, H. (1999). Neurosteroids: expression of steroidogenic enzymes and regulation of steroid biosynthesis in the central nervous system. Pharmacol. Rev. 51, 63–81.
Micevych, P., and Dominguez, R. (2009). Membrane estradiol signaling in the brain. Front. Neuroendocrinol. 30, 315–327.
Miller, D. B., Ali, S. F., O’callaghan, J. P., and Laws, S. C. (1998). The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann. N. Y. Acad. Sci. 844, 153–165.
Miller, I. N., and Cronin-Golomb, A. (2010). Gender differences in Parkinson’s disease: clinical characteristics and cognition. Mov. Disord. 25, 2695–2703.
Milner, T. A., Ayoola, K., Drake, C. T., Herrick, S. P., Tabori, N. E., Mcewen, B. S., Warrier, S., and Alves, S. E. (2005). Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J. Comp. Neurol. 491, 81–95.
Milner, T. A., Mcewen, B. S., Hayashi, S., Li, C. J., Reagan, L. P., and Alves, S. E. (2001). Ultrastructural evidence that hippocampal alpha estrogen receptors are located at extranuclear sites. J. Comp. Neurol. 429, 355–371.
Minghetti, L. (2005). Role of inflammation in neurodegenerative diseases. Curr. Opin. Neurol. 18, 315–321.
Morale, M. C., L’episcopo, F., Tirolo, C., Giaquinta, G., Caniglia, S., Testa, N., Arcieri, P., Serra, P. A., Lupo, G., Alberghina, M., Harada, N., Honda, S., Panzica, G. C., and Marchetti, B. (2008). Loss of aromatase cytochrome P450 function as a risk factor for Parkinson’s disease? Brain Res. Rev. 57, 431–443.
Morale, M. C., Serra, P. A., L’episcopo, F., Tirolo, C., Caniglia, S., Testa, N., Gennuso, F., Giaquinta, G., Rocchitta, G., Desole, M. S., Miele, E., and Marchetti, B. (2006). Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 138, 869–878.
Morissette, M., and Di Paolo, T. (2009). Effect of estradiol on striatal dopamine activity of female hemiparkinsonian monkeys. J. Neurosci. Res. 87, 1634–1644.
Moroz, I. A., Rajabi, H., Rodaros, D., and Stewart, J. (2003). Effects of sex and hormonal status on astrocytic basic fibroblast growth factor-2 and tyrosine hydroxylase immunoreactivity after medial forebrain bundle 6-hydroxydopamine lesions of the midbrain dopamine neurons. Neuroscience 118, 463–476.
Mukai, H., Takata, N., Ishii, H. T., Tanabe, N., Hojo, Y., Furukawa, A., Kimoto, T., and Kawato, S. (2006). Hippocampal synthesis of estrogens and androgens which are paracrine modulators of synaptic plasticity: synaptocrinology. Neuroscience 138, 757–764.
Murray, H. E., Pillai, A. V., Mcarthur, S. R., Razvi, N., Datla, K. P., Dexter, D. T., and Gillies, G. E. (2003). Dose- and sex-dependent effects of the neurotoxin 6-hydroxydopamine on the nigrostriatal dopaminergic pathway of adult rats: differential actions of estrogen in males and females. Neuroscience 116, 213–222.
Neumann, H., Kotter, M. R., and Franklin, R. J. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–295.
Nilsen, J. (2008). Estradiol and neurodegenerative oxidative stress. Front. Neuroendocrinol. 29, 463–475.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Ohtani, H., Nomoto, M., and Douchi, T. (2001). Chronic estrogen treatment replaces striatal dopaminergic function in ovariectomized rats. Brain Res. 900, 163–168.
Orr, C. F., Rowe, D. B., Mizuno, Y., Mori, H., and Halliday, G. M. (2005). A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 128, 2665–2674.
Palacios, N., Weisskopf, M., Simon, K., Gao, X., Schwarzschild, M., and Ascherio, A. (2011). Polymorphisms of caffeine metabolism and estrogen receptor genes and risk of Parkinson’s disease in men and women. Parkinsonism Relat. Disord. 16, 370–375.
Pasqualini, C., Olivier, V., Guibert, B., Frain, O., and Leviel, V. (1995). Acute stimulatory effect of estradiol on striatal dopamine synthesis. J. Neurochem. 65, 1651–1657.
Pavon, J. M., Whitson, H. E., and Okun, M. S. (2010). Parkinson’s disease in women: a call for improved clinical studies and for comparative effectiveness research. Maturitas 65, 352–358.
Pelletier, G. (2010). Steroidogenic enzymes in the brain: morphological aspects. Prog. Brain Res. 181, 193–207.
Pesaresi, M., Maschi, O., Giatti, S., Garcia-Segura, L. M., Caruso, D., and Melcangi, R. C. (2010). Sex differences in neuroactive steroid levels in the nervous system of diabetic and non-diabetic rats. Horm. Behav. 57, 46–55.
Popat, R. A., Van Den Eeden, S. K., Tanner, C. M., Mcguire, V., Bernstein, A. L., Bloch, D. A., Leimpeter, A., and Nelson, L. M. (2005). Effect of reproductive factors and postmenopausal hormone use on the risk of Parkinson disease. Neurology 65, 383–390.
Potashkin, J. A., Blume, S. R., and Runkle, N. K. (2010). Limitations of animal models of Parkinson’s disease. Parkinsons Dis. 2011, 658083.
Prange-Kiel, J., and Rune, G. M. (2006). Direct and indirect effects of estrogen on rat hippocampus. Neuroscience 138, 765–772.
Quesada, A., and Micevych, P. E. (2004). Estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxdopamine lesions. J. Neurosci. Res. 75, 107–116.
Quesada, A., Romeo, H. E., and Micevych, P. (2007). Distribution and localization patterns of estrogen receptor-beta and insulin-like growth factor-1 receptors in neurons and glial cells of the female rat substantia nigra: localization of ERbeta and IGF-1R in substantia nigra. J. Comp. Neurol. 503, 198–208.
Quinn, N. P., and Marsden, C. D. (1986). Menstrual-related fluctuations in Parkinson’s disease. Mov. Disord. 1, 85–87.
Ragonese, P., D’amelio, M., and Savettieri, G. (2006). Implications for estrogens in Parkinson’s disease: an epidemiological approach. Ann. N. Y. Acad. Sci. 1089, 373–382.
Ramirez, A. D., Liu, X., and Menniti, F. S. (2003). Repeated estradiol treatment prevents MPTP-induced dopamine depletion in male mice. Neuroendocrinology 77, 223–231.
Rogers, J., Mastroeni, D., Leonard, B., Joyce, J., and Grover, A. (2007). Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int. Rev. Neurobiol. 82, 235–246.
Saldanha, C. J., Duncan, K. A., and Walters, B. J. (2009). Neuroprotective actions of brain aromatase. Front. Neuroendocrinol. 30, 106–118.
Sandyk, R. (1989). Estrogens and the pathophysiology of Parkinson’s disease. Int. J. Neurosci. 45, 119–122.
Santos-Galindo, M., Acaz-Fonseca, E., Bellini, M. J., and Garcia-Segura, L. M. (2011). Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol. Sex Differ. 2, 7.
Saunders-Pullman, R., Gordon-Elliott, J., Parides, M., Fahn, S., Saunders, H. R., and Bressman, S. (1999). The effect of estrogen replacement on early Parkinson’s disease. Neurology 52, 1417–1421.
Schultz, K. N., Von Esenwein, S. A., Hu, M., Bennett, A. L., Kennedy, R. T., Musatov, S., Toran-Allerand, C. D., Kaplitt, M. G., Young, L. J., and Becker, J. B. (2009). Viral vector-mediated overexpression of estrogen receptor-alpha in striatum enhances the estradiol-induced motor activity in female rats and estradiol-modulated GABA release. J. Neurosci. 29, 1897–1903.
Schwarting, R. K., and Huston, J. P. (1996). The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog. Neurobiol. 50, 275–331.
Serova, L. I., Maharjan, S., Huang, A., Sun, D., Kaley, G., and Sabban, E. L. (2004). Response of tyrosine hydroxylase and GTP cyclohydrolase I gene expression to estrogen in brain catecholaminergic regions varies with mode of administration. Brain Res. 1015, 1–8.
Shadrina, M. I., Slominsky, P. A., and Limborska, S. A. (2010). Molecular mechanisms of pathogenesis of Parkinson’s disease. Int. Rev. Cell Mol. Biol. 281, 229–266.
Shughrue, P. J. (2004). Estrogen attenuates the MPTP-induced loss of dopamine neurons from the mouse SNc despite a lack of estrogen receptors (ERalpha and ERbeta). Exp. Neurol. 190, 468–477.
Shulman, L. M., and Bhat, V. (2006). Gender disparities in Parkinson’s disease. Expert Rev. Neurother. 6, 407–416.
Sierra, A., Gottfried-Blackmore, A., Milner, T. A., Mcewen, B. S., and Bulloch, K. (2008). Steroid hormone receptor expression and function in microglia. Glia 56, 659–674.
Simola, N., Morelli, M., and Carta, A. R. (2007). The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 11, 151–167.
Simunovic, F., Yi, M., Wang, Y., Stephens, R., and Sonntag, K. C. (2010). Evidence for gender-specific transcriptional profiles of nigral dopamine neurons in Parkinson disease. PLoS ONE 5, e8856. doi: 10.1371/journal.pone.0008856
Song, D. D., and Haber, S. N. (2000). Striatal responses to partial dopaminergic lesion: evidence for compensatory sprouting. J. Neurosci. 20, 5102–5114.
Spence, R. D., Hamby, M. E., Umeda, E., Itoh, N., Du, S., Wisdom, A. J., Cao, Y., Bondar, G., Lam, J., Ao, Y., Sandoval, F., Suriany, S., Sofroniew, M. V., and Voskuhl, R. R. (2011). Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proc. Natl. Acad. Sci. U.S.A. 108, 8867–8872.
Swerdlow, R. H., Parkerjr, W. D., Currie, L. J., Bennettjr, J. P., Harrison, M. B., Trugman, J. M., and Wooten, G. F. (2001). Gender ratio differences between Parkinson’s disease patients and their affected relatives. Parkinsonism Relat. Disord. 7, 129–133.
Tamas, A., Lubics, A., Szalontay, L., Lengvari, I., and Reglodi, D. (2005). Age and gender differences in behavioral and morphological outcome after 6-hydroxydopamine-induced lesion of the substantia nigra in rats. Behav. Brain Res. 158, 221–229.
Tapia-Gonzalez, S., Carrero, P., Pernia, O., Garcia-Segura, L. M., and Diz-Chaves, Y. (2008). Selective oestrogen receptor (ER) modulators reduce microglia reactivity in vivo after peripheral inflammation: potential role of microglial ERs. J. Endocrinol. 198, 219–230.
Taylor, K. S., Cook, J. A., and Counsell, C. E. (2007). Heterogeneity in male to female risk for Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr. 78, 905–906.
Thanky, N. R., Son, J. H., and Herbison, A. E. (2002). Sex differences in the regulation of tyrosine hydroxylase gene transcription by estrogen in the locus coeruleus of TH9-LacZ transgenic mice. Brain Res. Mol. Brain Res. 104, 220–226.
Toran-Allerand, C. D. (2005). Estrogen and the brain: beyond ER-alpha, ER-beta, and 17beta-estradiol. Ann. N. Y. Acad. Sci. 1052, 136–144.
Tripanichkul, W., Sripanichkulchai, K., and Finkelstein, D. I. (2006). Estrogen down-regulates glial activation in male mice following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication. Brain Res. 1084, 28–37.
Tsang, K. L., Ho, S. L., and Lo, S. K. (2000). Estrogen improves motor disability in Parkinsonian postmenopausal women with motor fluctuations. Neurology 54, 2292–2298.
Van Den Eeden, S. K., Tanner, C. M., Bernstein, A. L., Fross, R. D., Leimpeter, A., Bloch, D. A., and Nelson, L. M. (2003). Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 157, 1015–1022.
van Nas, A., Guhathakurta, D., Wang, S. S., Yehya, N., Horvath, S., Zhang, B., Ingram-Drake, L., Chaudhuri, G., Schadt, E. E., Drake, T. A., Arnold, A. P., and Lusis, A. J. (2009). Elucidating the role of gonadal hormones in sexually dimorphic gene coexpression networks. Endocrinology 150, 1235–1249.
Vegeto, E., Belcredito, S., Etteri, S., Ghisletti, S., Brusadelli, A., Meda, C., Krust, A., Dupont, S., Ciana, P., Chambon, P., and Maggi, A. (2003). Estrogen receptor-alpha mediates the brain antiinflammatory activity of estradiol. Proc. Natl. Acad. Sci. U.S.A. 100, 9614–9619.
Vegeto, E., Belcredito, S., Ghisletti, S., Meda, C., Etteri, S., and Maggi, A. (2006). The endogenous estrogen status regulates microglia reactivity in animal models of neuroinflammation. Endocrinology 147, 2263–2272.
Vegeto, E., Benedusi, V., and Maggi, A. (2008). Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front. Neuroendocrinol. 29, 507–519.
Vives-Bauza, C., Tocilescu, M., Devries, R. L., Alessi, D. M., Jackson-Lewis, V., and Przedborski, S. (2010). Control of mitochondrial integrity in Parkinson’s disease. Prog. Brain Res. 183, 99–113.
Weintraub, D., Comella, C. L., and Horn, S. (2008). Parkinson’s disease – Part 1: pathophysiology, symptoms, burden, diagnosis, and assessment. Am. J. Manag. Care 14, S40–S48.
Westberg, L., Hakansson, A., Melke, J., Shahabi, H. N., Nilsson, S., Buervenich, S., Carmine, A., Ahlberg, J., Grundell, M. B., Schulhof, B., Klingborg, K., Holmberg, B., Sydow, O., Olson, L., Johnels, E. B., Eriksson, E., and Nissbrandt, H. (2004). Association between the estrogen receptor beta gene and age of onset of Parkinson’s disease. Psychoneuroendocrinology 29, 993–998.
Wise, P. M., Dubal, D. B., Rau, S. W., Brown, C. M., and Suzuki, S. (2005). Are estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the Women’s health initiative. Endocr. Rev. 26, 308–312.
Wise, P. M., Suzuki, S., and Brown, C. M. (2009). Estradiol: a hormone with diverse and contradictory neuroprotective actions. Dialogues Clin. Neurosci. 11, 297–303.
Wooten, G. F., Currie, L. J., Bovbjerg, V. E., Lee, J. K., and Patrie, J. (2004). Are men at greater risk for Parkinson’s disease than women? J. Neurol. Neurosurg. Psychiatry 75, 637–639.
Wynne, R. D., and Saldanha, C. J. (2004). Glial aromatization decreases neural injury in the zebra finch (Taeniopygia guttata): influence on apoptosis. J. Neuroendocrinol. 16, 676–683.
Yokoyama, H., Uchida, H., Kuroiwa, H., Kasahara, J., and Araki, T. (2011). Role of glial cells in neurotoxin-induced animal models of Parkinson’s disease. Neurol. Sci. 32, 1–7.
Yu, L., Kuo, Y., Cherng, C. G., Chen, H. H., and Hsu, C. H. (2002). Ovarian hormones do not attenuate methamphetamine-induced dopaminergic neurotoxicity in mice gonadectomized at 4 weeks postpartum. Neuroendocrinology 75, 282–287.
Yu, L., and Liao, P. C. (2000). Sexual differences and estrous cycle in methamphetamine-induced dopamine and serotonin depletions in the striatum of mice. J. Neural Transm. 107, 419–427.
Yu, Y. L., and Wagner, G. C. (1994). Influence of gonadal hormones on sexual differences in sensitivity to methamphetamine-induced neurotoxicity. J. Neural Transm. Park. Dis. Dement. Sect. 8, 215–221.
Keywords: nigrostriatal pathway, estrogen, Parkinson’s disease, central vs. gonadal steroids, sex
Citation: McArthur S and Gillies GE (2011) Peripheral vs. central sex steroid hormones in experimental parkinson’s disease. Front. Endocrin. 2:82. doi: 10.3389/fendo.2011.00082
Received: 05 October 2011;
Accepted: 10 November 2011;
Published online: 30 November 2011.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Rafael Vazquez-Martinez, University of Cordoba, SpainDean Dluzen, Northeast Ohio Medical University, USA
Copyright: © 2011 McArthur and Gillies. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Simon McArthur, Department of Medicine, Centre for Neuroscience, Imperial College London, London SW7 2AZ, UK. e-mail:c2ltb24ubWNhcnRodXJAaW1wZXJpYWwuYWMudWs=