 Ushashi C. Dadwal
Ushashi C. Dadwal Eric S. Chang
Eric S. Chang Uma Sankar
Uma Sankar- Department of Anatomy and Cell Biology, Indiana University School of Medicine, Indianapolis, IN, United States
The skeletal system is of paramount importance in advanced stage prostate cancer (PCa) as it is the preferred site of metastasis. Complex mechanisms are employed sequentially by PCa cells to home to and colonize the bone. Bone-resident PCa cells then recruit osteoblasts (OBs), osteoclasts (OCs), and macrophages within the niche into entities that promote cancer cell growth and survival. Since PCa is heavily reliant on androgens for growth and survival, androgen-deprivation therapy (ADT) is the standard of care for advanced disease. Although it significantly improves survival rates, ADT detrimentally affects bone health and significantly increases the risk of fractures. Moreover, whereas the majority patients with advanced PCa respond favorably to androgen deprivation, most experience a relapse of the disease to a hormone-refractory form within 1–2 years of ADT. The tumor adapts to surviving under low testosterone conditions by selecting for mutations in the androgen receptor (AR) that constitutively activate it. Thus, AR signaling remains active in PCa cells and aids in its survival under low levels of circulating androgens and additionally allows the cancer cells to manipulate the bone microenvironment to fuel its growth. Hence, AR and its downstream effectors are attractive targets for therapeutic interventions against PCa. Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2), was recently identified as a key downstream target of AR in coordinating PCa cell growth, survival, and migration. Additionally, this multifunctional serine/threonine protein kinase is a critical mediator of bone remodeling and macrophage function, thus emerging as an attractive therapeutic target downstream of AR in controlling metastatic PCa and preventing ADT-induced bone loss. Here, we discuss the role played by AR-CaMKK2 signaling axis in PCa survival, metabolism, cell growth, and migration as well as the cell-intrinsic roles of CaMKK2 in OBs, OCs, and macrophages within the bone microenvironment.
Introduction
Prostate cancer (PCa) is the second leading cause of cancer-related deaths in American men and accounts for 15% of all cancers diagnosed in men worldwide (1, 2). The American Cancer Society estimates that in 2018 alone, 164,690 men will be newly diagnosed with PCa and 29,430 men will die from it in the United States. Routine testing of prostate serum antigen (PSA) levels has resulted in early diagnosis and treatment of PCa. Consequently, men with early-stage PCa have a high, near 100%, 10-year rate of survival. Among patients with non-localized disease, however, about 40% develop metastases to distant sites such as bone, lymph nodes, lung, and brain and their 5-year survival rate drops dramatically to 30% (3). PCa displays a preferential tropism toward bone which is the primary site of metastasis in 80% of patients with advanced disease (4). Metastatic PCa becomes lodged in the bone marrow (BM)-rich axial skeleton, which provides the perfect “soil” for the disease to develop to an advanced form often termed “castrate-resistant PCa (CRPC)” as it is resistant to hormone-ablation.
Bone is an organ of utmost importance in PCa. Bone metastasis is often a leading cause of patient mortality in PCa (4). Once they “home” and colonize the bone, PCa cells disrupt the homeostatic balance between bone-forming osteoblasts (OBs) and bone-resorbing osteoclasts (OCs). Similar to breast cancer, PCa stimulates osteolysis. However, a unique feature of bone-lodged PCa cells is that they stimulate the OBs to produce weak woven bone instead of the strong lamellar bone that is normally synthesized. Such skeletal-related events (SREs) triggered by PCa in the bone culminate in pathological fractures, spinal cord compression, and sclerosis, detrimentally affecting the overall quality of life and survival rate among patients (5–8).
Prostate cancer cells express the androgen receptor (AR) and are heavily reliant on androgens for growth and survival. Hence, most patients with locally advanced or metastatic PCa receive androgen-deprivation therapy (ADT) as a gold standard treatment (9). Although it significantly improves survival rates, ADT detrimentally affects skeletal health, causing tremendous bone loss and rendering the patients at risk for fragility fractures (10). Therapies that inhibit bone resorption such as bisphosphonates prevent ADT-induced bone loss and may additionally delay bone colonization by the tumor by interfering with its ability to manipulate the bone microenvironment (11). PCa patients initially respond positively to ADT, though most experience a relapse of the cancer to a hormone-refractory form called CRPC, which occur when cancer cells adapt to growth under low androgen conditions by constitutively upregulating AR (12). Consequently, AR and its downstream effectors are attractive therapeutic targets to combat tumor growth in androgen-resistant PCa. Indeed, clinical studies indicate that AR inhibitors such as enzalutamide delay SREs and improve survival rates in patients (13–15). Still, novel therapies that preserve musculoskeletal heath while significantly inhibiting tumor growth are acutely needed in the clinic.
In this review, we will briefly discuss the steps involved in bone metastasis of PCa, the role of AR activation in the development of CRPC and skeletal complications associated with ADT. We will additionally discuss recent findings that identify Ca2+/calmodulin (CaM)-dependent protein kinase kinase 2 (CaMKK2), an AR-regulated gene with additional roles in bone remodeling and inflammation, as a novel therapeutic target to inhibit PCa growth and prevention of ADT-associated bone loss.
Bone Metastasis of PCa
Prostate cancer cells show an almost exclusive tropism for bone. Although the exact mechanisms that drive bone metastasis are unknown, it has been proposed that the BM microenvironment may provide the ideal condition for the PCa cells to thrive. The “seed and soil” hypothesis proposed by Steven Paget in 1889, wherein the “seeds” or tumor cells develop a tropism and metastasize to the “soil” or target organ that is well suited or “fertile ground” for its growth (16) still remains a guiding principle in understanding the role BM microenvironment plays in bone metastasis of PCa.
Metastasis of PCa to bone involves several steps including decreased cell adhesion, epithelial to mesenchymal transition (EMT), local migration, invasion, intravasation into the circulation, homing, and colonization of bone (17, 18). Cell–cell adhesion in normal prostate epithelium is maintained by integrins and tight junctions composed of cell adhesion molecules, such as selectins and cadherins. There are two main types of cadherins, E-cadherin and N-cadherin, expressed by epithelial cells and mesenchymal cells, respectively. During early transformation, prostate epithelial cells exhibit alterations in cell adhesion factors, including a downregulation of E-cadherin and an upregulation of N-cadherin, a process termed cadherin switching and a main feature in EMT. Decreased expression of integrins and Wnt-target protein β-catenin also play important roles in EMT (17–19). The next step is migration and it involves an upregulation of focal adhesion. During normal cell migration, focal adhesions formed on the leading edges of the cells are used as anchors by the cells to pull themselves forward over the extracellular matrix (ECM). Disassembly of focal adhesions on the rear edge of the cell enables the cell to move forward (20). This process involves the binding of focal adhesion kinases (FAKs) to integrins and their subsequent activation by the Src family of kinases, initiating signaling events including those involving Rho that regulate focal adhesion turnover and migration. Expression of FAK and Src as well as Rho activities are elevated in bone metastases and CRPC, indicating increased focal adhesion turnover and cell mobility.
Once the transformed prostate epithelial cells gain the ability to migrate, they need to dissociate from the ECM, which is composed of the basement membrane and connective tissue. Prostate epithelial cells that have undergone mesenchymal transition have the ability to secrete proteases such as matrix metalloproteases and serine proteinases, such as urokinase-type plasminogen activator and PSA, which partially degrade the ECM, allowing the cells to disseminate, invade the surrounding tissue and intravasate into blood vessels (17, 20, 21). Homing to the target organ is only possible if the PCa cells survive in the circulation, and they achieve this by attaching to the vascular endothelium. PCa cells have been shown to interact with BM endothelial cells (BMECs) with high affinity through a mechanism involving E-selectin receptor on PCa cells and E-selectin on BMECs and integrins such as αVβ3, αVβ1, and α3β1 (18). Additionally, CD44 on PCa cells binds to vascular cell adhesion molecule 1 on BMECs in a process that is enhanced by IL-17 and IGF1 in circulation. The subsequent homing of PCa cells to bone is facilitated by multiple chemokine-mediated mechanisms. For instance, BM stromal cells and OBs in the bone secrete C–X–C motif chemokine ligand 12 (CXCL12) or stromal derived factor-1 (SDF1) whereas PCa cells express its receptor CXCR4. CXCL12/SDF–CXCR4 interaction allows PCa cells to home to the bone, adapting a similar mechanism as the one utilized by hematopoietic stem cells (22). Additionally, CXCL12/SDF1 from OBs activates the expression of the adhesion molecule αVβ3 integrin on PCa cells that further contribute to their homing to the BM. Further, the expression of yet another chemokine ligand CXCL16 allows PCa cells to recruit and convert CXCR6-expressing mesenchymal stem cells into cancer-associated fibroblasts that also secrete high levels of CXCL12/SDF1. Finally, recent reports provide evidence for microRNA (miR)-containing exosomes from PCa cells arriving early in the BM to enable the modification of the bone microenvironment to favor cancer cell homing to the bone (23–25).
Colonization of the bone by PCa is aided by their ability to (a) attach to the bone matrix and (b) manipulate the BM microenvironment into favoring their growth and survival. PCa express two key integrins αVβ3 and α2β1, which allow the cells to attach to the bone matrix and migrate along the surface to identify suitable “niches” for their outgrowth. PCa cells preferentially home to OB-rich niches within the bone, allowing physical contact between these two cell types, facilitated in part by adhesion molecules such as cadherin-11 expressed on both OBs and malignant PCa cells (26, 27). Interestingly, physical contact between PCa and OBs appear to disrupt the normal order of matters within the bone. Kimura et al. noted that in the presence of PCa cells, the bone-resident OBs which usually line neatly along the collagen matrix become disorganized and that this anisotropy requires cell–cell contact (28). Unlike other solid tumor malignancies which are mostly osteolytic, bone-metastatic PCa is primarily an osteoblastic disease driven in part by the ability of PCa cells to perform “osteomimicry” wherein they adopt the genetic and phenotypic characteristics of OBs (29). OB growth and differentiation are governed by complex signaling pathways, such as Wnts, bone morphogenic proteins (BMPs), insulin growth factor (IGF), and transforming growth factor β (TGF-β) (30, 31). In contrast, OC differentiation is regulated by receptor activator of NF-κB ligand (RANKL), osteoprotegerin, parathyroid hormone, and TGF-β. Differentiated OBs secrete these factors, but many are also released from the bone matrix by OCs themselves during bone resorption. Interestingly, bone-lodged PCa cells produce many of the same factors that stimulate the proliferation and differentiation of OBs and OCs (17, 30). In addition to producing factors that favor bone cell differentiation, PCa cells also induce other cell types to transdifferentiate into OBs (32). Recently, Lin et al. reported an endothelial cell-to-OB conversion as one of the mechanisms underlying osteoblastic bone disease in PCa (33). These authors showed that PCa induces the overexpression of BMP4 in BMECs driving their transdifferentiation to OBs (33). Recent studies from multiple myeloma highlight the importance of osteocytes, the most abundant bone cells, in tumor-bone interactions (34). Although studies have indicated a role for osteocytes in PCa (35), more research is needed to fully comprehend the contribution of these cells to bone metastasis by PCa. Taken together, these studies suggest that cancer cells disrupt the homeostatic mechanisms within the BM and hijack the normal paracrine and autocrine mechanisms regulating normal bone remodeling to create a “vicious cycle” that ultimately favors PCa colonization and growth within the bone (Table 1).
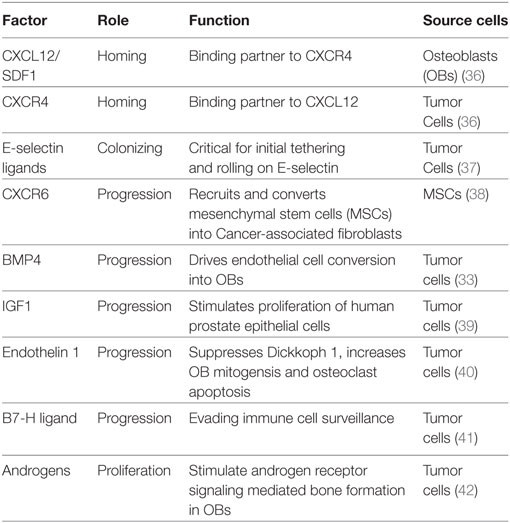
Table 1. Growth factors involved in aiding skeletal metastasis of prostate cancer.
Androgens, AR, Bone, and ADT
Since the original description by Charles Huggins in 1942 of the heavy dependence of PCa on androgens and the benefits of orchiectomy in PCa patients, androgens, and AR have remained the main therapeutic targets in PCa treatment (43–46). In men, Leydig cells of the testis produce about 90% of the circulating androgens or testosterone and the adrenal cortex produces the remaining 10% (47). Testosterone diffuses into the prostate epithelial cells where it is converted into dihydrotestosterone (DHT) by the enzyme 5α-reductase (47, 48). DHT binds to AR, a member of the nuclear hormone receptor family of transcription factors. Upon ligand binding, AR translocates to the nucleus, undergoes homodimerization and binds to androgen response elements (ARE) within the promoters of AR-target genes such as PSA. AR then recruits cofactors and initiates the transcription of target genes that regulate proliferation, metabolism, and survival of PCa cells (45, 49, 50).
The goal of ADT is to starve the tumor cells of androgens by drastically diminishing their circulating amount (<5% of normal range). This is achieved by blocking testosterone production surgically via castration or chemically by treating patients with luteinizing hormone releasing hormone agonists or first generation antiandrogen drugs, such as flutamide, nilutamide, and bicalutamide, that competitively block DHT binding to AR (51). Testosterone is converted into estradiol, the primary male estrogen via aromatization and it binds to the estrogen receptor α (ERα) present on both OBs and OCs. OBs express both AR and ERα, whereas OCs express only ERα. These receptors promote OB survival, numbers, and activity, while ERα inhibits OC differentiation. Moreover, the combined action of these two nuclear receptors stimulate periosteal apposition and lengthening of the epiphyseal growth plate in men while maintaining their cortical and trabecular bone. The continued periosteal growth during adult life in men partially offsets age-related increase in endosteal bone loss (7, 10). All these processes are affected by ADT as it suppresses not only androgens but also estradiol, resulting in the abrogation of the stimulatory effect of androgens on OBs and the inhibitory effect of estradiol on OCs. This triggers increased bone turnover in patients on ADT, resulting in a significantly high rate of bone loss at 4.6% per year, which exceeds the annual bone loss in aging men and postmenopausal women (10). The maximum bone loss occurs during the first year of therapy, ranging from 1.5 to 4%, depending on the skeletal location examined (10). Thus, ADT renders these men, who are often older and possess lower bone mass to begin with, four times more likely to develop osteoporosis. This enhances their risk of fragility fractures and in turn, their mortality risk (7).
Nitrogen-containing bisphosphonates, such as alendronate, risedronate, and zoledronic acid, as well as denosumab, a monoclonal antibody to RANKL are all FDA-approved to treat osteoporosis in PCa patients on ADT. Selective ER modulators such as raloxifene and Toremifene have also been shown to preserve bone in clinical trials with PCa patients undergoing ADT (7, 15, 52–56). Moreover, second-generation antiandrogens, such as abiraterone and enzalutamide, as well as radiotherapies such as Radium-223 have shown to suppress tumor growth and delay SREs (13, 14, 57–60). Teriparatide, though FDA-approved, is not recommended for PCa patients at risk for bone metastasis. A list of current therapies and novel compounds in clinical trials in the treatment of bone-metastatic PCa are detailed in Tables 2 and 3.
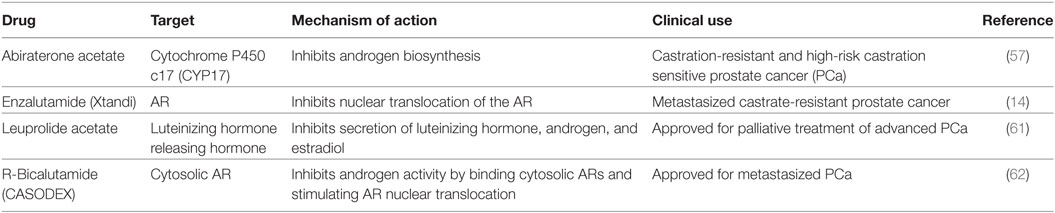
Table 2. Androgen receptor (AR) targeted therapies—FDA-approved drugs in clinic.
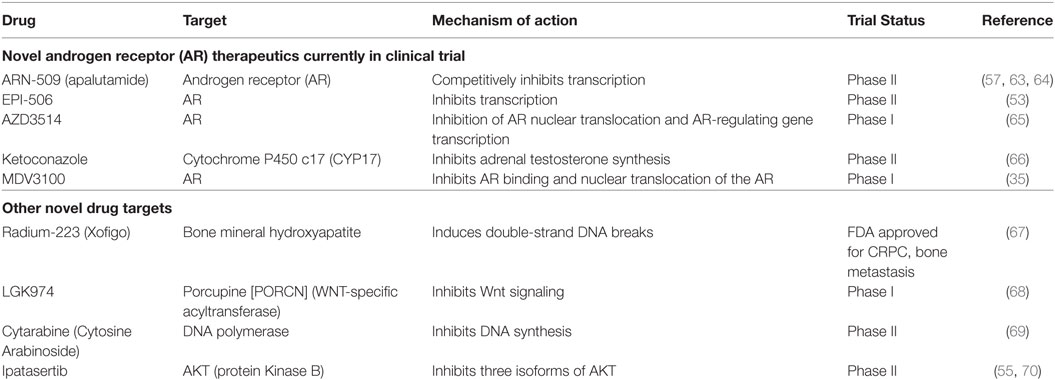
Table 3. Novel therapies against castrate-resistant prostate cancer (CRPC) currently in trials.
AR Activation in CRPC
Androgen-deprivation therapy results in diminished tumor burden in about 90% of patients with advanced PCa. However, over time, the cancer cells adapt undergo selection to proliferate and survive under low levels of circulating androgens by upregulating AR and becoming unresponsive to ADT. The disease at this stage is termed CRPC (17, 71). AR is the main driver of CPRC development, while a minority of metastatic PCa are associated with the loss of p53, PTEN, or Rb (13, 72). The main mechanisms for AR reactivation in CRPC include amplification leading to overexpression, activating mutations, structural gene alterations, expression of constitutively active variants, mutations in the AR that confer broader ligand specificity to the receptor, upregulation of co-regulators, increased expression of steroidogenic enzymes, as well as upregulation of cross-talk signal transduction pathways such as interleukin 6, STAT3, Src, and IGF that can activate AR in a ligand-independent manner (71). These mechanisms have been extensively reviewed elsewhere (21, 72–74). Gain-of-function AR splicing variants (AR-Vs) often lack portions of the ligand-binding domain (LBD) but possess constitutive transcriptional activity even in the absence of androgens. The most well-characterized among these is AR-V7 whose expression has been shown to increase in response to ADT and has been shown to confer resistance to drugs, such as abiraterone and enzalutamide that either block androgen synthesis or antagonize AR (75). AR-V7 was identified as the most frequently occurring variant in patients with CRPC and its expression correlates with increased disease recurrence (76–78). AR-V7 expression is associated with the upregulation of some AR-target genes relevant for proliferation and survival, such as UBE2C, CCNA2, C-MYC, AKT1, EDN2, and ETS2 (71, 78).
Mechanisms that enable CRPCs to activate AR and continually acquire resistance to therapies underscore the importance of gaining a comprehensive understanding of the downstream effectors of AR signaling that play crucial roles in cancer progression, as they could serve as druggable targets in the treatment of CRPC. Several studies have attempted to identify AR-regulated genes by focusing on genome-wide AR-binding sites on cell lines and clinical samples, or by examining temporal regulation of androgen stimulation in one or more PCa cell lines such as LNCaP (harbors an LBD mutation of AR), VCaP (contains AR gene amplification) or C4-2B (a CRPC cell line) (66, 79–85). These studies have identified several AR-target genes with functions in gene transcription (NKX3.1, FOX family), growth stimulation (IGF1R), cell cycle regulation (CDK6, UBE2C), signaling (MEK5, FKBP5), autophagy (ATG4B, ULK1, TFEB), non-coding RNA (miR-21, miR141), glycolysis (GLUT1), and central metabolism [MTOR, Ca2+/CaM-dependent protein kinase kinase 2 (CaMKK2)]. Among these, CaMKK2 has emerged as an attractive therapeutic candidate in PCa as it is a direct target of AR, containing AREs on its promoter and is consistently overexpressed in clinical CRPC samples as well as AR-positive PCa cell lines (86, 87).
CaMKK2: A Molecular Hub Directed by AR in PCa Cells
Intracellular Ca2+ is a universal second messenger that regulates diverse cellular processes. Transient variations in intracellular Ca2+ are immediately sensed by the ubiquitous high-affinity intracellular Ca2+ receptor, CaM. This initiates a cascade of Ca2+/CaM-mediated signaling events that culminate in changes to key cellular events such as proliferation, differentiation, survival, and metabolism (88). In particular, Ca2+/CaM complexes bind to and activate CaM kinases (CaMKs), which are a family of multifunctional Ser/Thr protein kinases that includes CaMKK1, CaMKK2, CaMKI, CaMKII, and CaMKIV. The upstream kinases, CaMKKs 1 and 2, are activated through Ca2+/CaM binding and intramolecular phosphorylation. Binding of Ca2+/CaM allows the activation loop in CaMKs to unravel and expose a critical threonine residue that becomes phosphorylated by the two upstream CaMKKs, resulting in their full activation, triggering the formation of a CaMK signaling cascade that is regulated by Ca2+/CaM at multiple levels (89–91). Interestingly, unlike CaMKK1, which is solely dependent on Ca2+/CaM for activity, CaMKK2 possesses considerable autonomous activity in the absence of Ca2+/CaM. This autonomous activity is regulated by phosphorylation by Ca2+/CaM-independent kinases such as glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (CDK5) (92, 93). As it is not dependent on rapid fluxes in intracellular Ca2+ for basal activity, CaMKK2 is capable of responding to other stimuli of longer duration and phosphorylating novel substrates outside of the CaMK cascade. Indeed, CaMKK2 (not CaMKK1) directly phosphorylates and activates adenosine monophosphate activated protein kinase (AMPK), a heterotrimeric kinase that co-ordinates cellular energy balance, autophagy, cell proliferation, and cytoskeletal organization (94, 95). The CaMKK2–AMPK pathway plays key roles in the regulation of hypothalamic feeding behavior, hepatic gluconeogenesis, adipocyte differentiation, and macroautophagy (94, 96–98). Recent studies indicate roles for CaMKK2 in hepatic cancer, macrophage-mediated inflammation, and bone remodeling through non-AMPK-mediated mechanisms (99–103).
CaMKK2 is increasingly being considered a hub of signaling mechanisms that regulate PCa cell metabolism, proliferation and migration downstream of AR (104). Frigo et al. identified the presence of an AR-binding region located 2.3-kb upstream of the CaMKK2 transcriptional start site and reported the recruitment of AR to this region in an androgen-dependent manner (87). These authors also found that the knockdown of CaMKK2 or its pharmacological inhibition using a selective inhibitor STO-609 or inhibition of the CaMKK2-target protein AMPK abrogates PCa cell migration and invasion (68, 87, 105). Overexpression of CaMKK2 alone was sufficient to induce AMPK phosphorylation and facilitate PCa cell migration, implying that androgens promote PCa cell migration through an AR-CaMKK2-AMPK signaling axis (87). Massie et al. integrated genome-wide AR-binding transcript profiling with an analysis of androgen-stimulated recruitment of the transcriptional machinery to a core set of AR-binding sites and identified CaMKK2 to be consistently enriched in PCa clinical cohorts, in a pattern similar to that of the established PCa marker AMACR (86). Similar to previous reports (87), these authors also observed AR recruitment to CaMKK2 promoter in both androgen-dependent and CRPC cell lines and an early upregulation of the CaMKK2 transcripts and protein within 4 and 12 h of androgen stimulation, respectively, indicating direct AR regulation (86). Subsequent functional studies identified CaMKK2 as a key effector of AR in stimulating glycolysis through its activation of AMPK and phosphofructokinase (PFK), which in turn drives anabolism and PCa cell proliferation (86). Of note, the AR-CaMKK2–AMPK–PFK axis does not affect cellular biosynthesis through mTOR in PCa, indicating its primary role in regulating glucose uptake and lactate production. In vivo inhibition of CaMKK2 using STO-609 resulted in a significant reduction in the growth of C4-2B xenografts in nude mice, and this treatment was additive with AR inhibition achieved via castration (86). It should be noted that CaMKK2 inhibition by itself did not affect the size of the normal prostate or its epithelium in nude mice, and the CaMKK2−/− mice do not possess any prostate anomalies or fertility deficits (86, 87). Thus, the inhibition of CaMKK2, rather than AR itself, may offer greater selective advantage over PCa at all stages.
Karacosta et al. examined PCa in clinical samples and found strong CaMKK2 immunoreactivity in the epithelium of malignant glands, compared to extremely low expression in the adjacent normal epithelium (106). Moreover, CaMKK2 staining intensity increased with the Gleason score of the tumors, and the staining pattern shifted from predominantly cytoplasmic to perinuclear and nuclear (106). CaMKK2 intensity increased with tumor progression in a transgenic adenocarcinoma of the mouse prostate (TRAMP) mouse model of PCa, and its expression was higher in castration-resistant tumor xenografts than androgen-responsive ones. These authors also observed upregulation of CaMKK2 as well as its nuclear translocation in LNCaP following DHT treatment and a reversal with androgen withdrawal. Further, silencing of CaMKK2 using small interfering RNA elicited G1 arrest of LNCaP cells, reducing their proliferation, along with lowering the levels of PSA as well as AR-regulated cell cycle proteins such as cyclin D1 and hyperphosphorylated Rb (106). Karacosta et al. proposed a novel positive feedback loop in the PCa in which CaMKK2 is induced by AR, and it in turn stabilizes AR to promote its transcriptional activity and cell cycle progression (106). In a recent follow-up study, these authors confirmed the higher nuclear expression of CaMKK2 in CRPC C4-2 cells, and showed that this occurs due to the association of CaMKK2 with nuclear pore complexes through its direct interaction with nucleoporin 62 (NUP62) (107). These authors showed that silencing NUP62 reduces the growth and viability of C4-2 cells, and provided evidence for the recruitment of NUP62, CaMKK2, and AR complexes to the AR-binding regions in the promoters of target genes such as PSA, suggesting a novel CaMKK2-NUP62 mechanism of AR transcriptional regulation in advanced PCa (107).
Similar to the aforementioned studies, Shima et al. performed genome-wide analysis of a small set of clinical samples and found a sixfold higher CaMKK2 expression in PCa compared to normal prostate (108). However, in contrast to previous studies (82, 87, 106, 107), these authors provide evidence for an inhibitory role for CaMKK2 to AR signaling and hypothesize that while CaMKK2 supports growth of tumors in early PCa, it inhibits excessive proliferation in CRPC (108). Whereas additional studies are warranted to validate these intriguing findings and hypotheses, the consensus emerging from all of these studies is that CaMKK2 is a key effector of AR signaling in PCa cells, regulating cell cycle by stabilizing AR, cell migration through AMPK signaling, and glycolysis by activating the AMPK–PFK pathway. AR is essential for PCa cell viability, proliferation, invasion, and bone metastasis, and the tumor cells are under constant selective pressure to maintain AR signaling, especially under the conditions of low testosterone such as ADT (86). Therefore, targeting downstream effectors such as CaMKK2 would be an effective approach to abrogate AR signaling in metastatic PCa.
CaMKK2 in Bone Microenvironment
CaMKK2 and Bone Cells
Prostate cancer recruits OBs and OCs within the bone microenvironment and transforms them into entities that support tumor growth (30). Studies discussed above show that CaMKK2 is expressed in PCa cells where it acts as a molecular hub downstream of AR in regulating tumor cell growth. CaMKK2 is expressed by OBs and OCs and plays important cell-intrinsic roles in these cells (99). During homeostatic conditions, CaMKK2 stimulates OC differentiation by activating phosphorylated form of cyclic adenosine monophosphate (cAMP) response element binding protein (pCREB) and its transcriptional target, nuclear factor of activated T cells c1 in a CaMKIV-dependent manner. Hence, inhibition or deletion of CaMKK2 inhibits OCs. On the other hand, CaMKK2 inhibits OB differentiation by inhibiting cAMP-protein kinase A (PKA) signaling under normal conditions. Therefore, the inhibition or absence of CaMKK2 relieves this inhibition and results in the stimulation of OB differentiation (99). Mice null for CaMKK2 possess higher bone mass along with significantly more OBs and fewer multinuclear OCs. Inhibition of CaMKK2 promotes bone fracture healing, and confers protection from ovariectomy and age-related osteoporosis (99, 100, 102). Taken together, these studies reveal profound roles for CaMKK2 in the two main bone cell types that interact with PCa in the bone microenvironment.
CaMKK2 and Macrophages
Immune cells, such as macrophages and lymphocytes, are also part of the bone–tumor microenvironment and play important roles in tumor growth and bone metastasis (109). For example, chronic inflammation sustained by macrophage activation plays a pivotal role in the regulation of tumor microenvironment in many solid tumors (104). Chronic inflammatory conditions existing within the tumor recruit myeloid cells and induce their differentiation into tumor-associated macrophages, the infiltration of which negatively correlates with prognosis in advanced PCa. Recently, Roca et al. reported that macrophage-driven efferocytosis accelerates CXCL5-mediated inflammation and PCa growth within the bone (110). Among immune cells, CaMKK2 is selectively expressed in macrophages and its ablation impairs their ability to spread, phagocytose, and produce inflammatory cytokines and chemokines in response to lipopolysaccharides (101). CaMKK2 regulates metabolic responses and cytokine release in response toll-like receptor/integrin stimulation in macrophages. Indeed, Camkk2−/− mice are resistant to irritants that lead to systemic inflammation (101). Thus, CaMKK2 plays roles in multiple cell types, including OBs, OCs, and macrophages, that form the PCa microenvironment in the bone. AR is expressed in OBs and macrophages, and it plays an indirect role in OCs through ERα. However, whether CaMKK2 plays a role downstream of AR in OBs and macrophages is unknown. Nevertheless, we hypothesize that AR signaling in PCa cells uses CaMKK2 as a downstream hub regulating several molecular mechanisms in OBs, OCs, and macrophages to manipulate the BM niche to the benefit of the cancer cells (Figure 1).
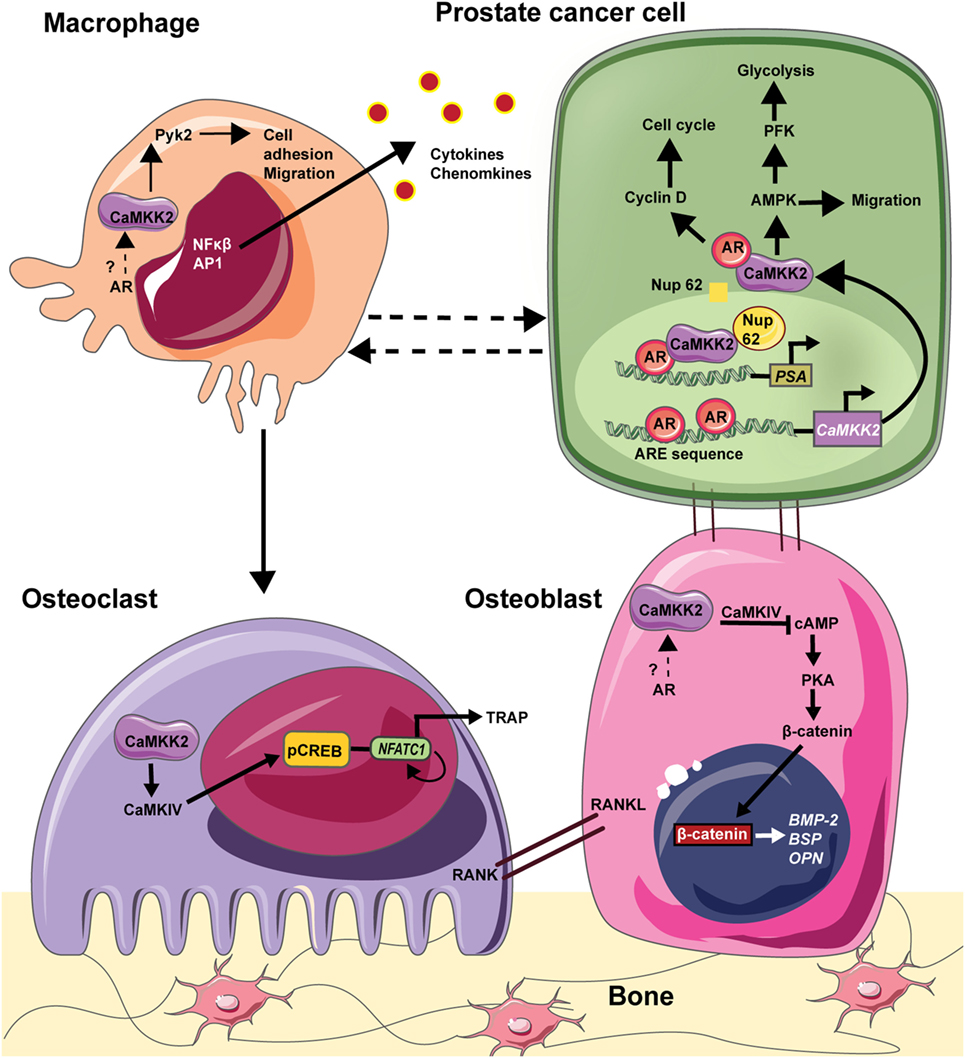
Figure 1. CAMKK2 as a molecular hub downstream in the bone–prostate cancer (PCa) microenvironment. In PCa cells, the androgen receptor (AR) binds to androgen response element (ARE) on CaMKK2 promoter which is situated upstream of the transcriptional start site. Thus, CaMKK2 is a direct transcriptional target of AR and its expression is highly elevated in metastatic PCa. Once transcribed and translated, CaMKK2 binds to AR initiating a positive feedback loop to stimulate AR transcriptional activity in the activation of AR-dependent genes that regulate cell cycle progression such as cyclin D. Additionally, CaMKK2 through its activation of AMPK regulates PCa cell migration. CaMKK2-AMPK signaling pathway also regulates cellular glycolysis via the activation of phosphofructokinase (PFK). This drives PCa cell anabolism and in turn promotes cell proliferation and tumor growth. Furthermore, in CRPCs, CaMKK2 binds to nucleoporin 62 (NUP62) to enter the nucleus, where it along with AR and NUP62 are recruited to the ARE in the promoters of downstream targets such as prostate serum antigen (PSA). PCa cells that metastasize to the bone physically interacts with OBs to alter their organization and function. Although both AR and CaMKK2 are expressed in OBs, whether CaMKK2 operates downstream of AR in these cells is not known. In OBs, CaMKK2 signaling inhibits cyclic adenosine monophosphate (cAMP) production and protein kinase A (PKA) activation. PKA is an important regulator of OB differentiation. Thus, the inhibition of CaMKK2 would relieve this inhibition of PKA signaling and OB differentiation. In osteoclasts (OCs), CaMKK2 signaling through CaMKIV-pCREB activates nuclear factor of activated T cells c1 (NFATc1), which is the master regulator of OC differentiation. In macrophages, CaMKK2 regulates cytoskeletal rearrangement via its regulation of Pyk2. Moreover, CaMKK2-CaMK1 signaling regulates cytokine/chemokine production by macrophages. Thus, CaMKK2 is a key component of AR signaling in PCa cells and additionally regulates multiple cell types that constitute the tumor microenvironment within the bone.
Perspectives and Concluding Remarks
Complex mechanisms employed by PCa cells allow it to home and thrive in bone, their preferred site of metastasis. Once lodged in the bone, the cancer cells recruit OBs, OCs, and macrophages within the skeletal niche to become entities that secrete growth factors and chemokines that allow the PCa cells to proliferate and survive even under low circulating testosterone conditions such as following ADT. AR signaling remains critical for PCa cell survival even under ADT and this creates selective pressure for the generation of AR gene mutations that facilitate the constitutive activation of the AR signaling cascade. Thus, AR and its downstream effectors are attractive therapeutic targets against bone-metastatic PCa.
The CaMKK2-AMPK signaling pathway operates downstream of AR to mediate PCa cell cycle, metabolism, migration, and invasion. CaMKK2 inhibition interferes with the growth and survival of bone-lodged PCa, and will presumably interfere with its ability to secrete factors that modify OBs into cancer-promoting entities. Similar to PCa, AR signaling plays an active pro-survival role in OBs. However, whether it operates upstream of CaMKK2 in OBs is unclear. Various signaling pathways, including cAMP-PKA, CDK5, and GSK3, have been implicated as upstream regulators of CaMKK2 in other cell types. In addition to AR-binding elements, CaMKK2 promoter also contains consensus-binding sites for several transcription factors including runt-related transcription factor 2 (RUNX2), the master regulator of OB differentiation. In macrophages, CaMKK2 is activated by toll-like receptors, Gq-coupled receptors, and voltage-gated Ca2+ channels on the plasma membrane (101). Although monocytes express AR, its role in the regulation of CaMKK2 in these cells is unclear. Nevertheless, we can conclude from the studies discussed above that the AR-CaMKK2 signaling axis acts as a molecular hub promoting PCa survival and in turn its ability to manipulate the bone microenvironment. Cell-intrinsic roles of CaMKK2 in OBs, OCs, and macrophages may aid in this process, ultimately enhancing the malignancy, SREs, and bone fragility.
In addition to the studies reviewed herein, CaMKK2 inhibition or genetic ablation has been shown to protect against diet-induced glucose intolerance, insulin resistance, diabetes, hepatocellular carcinoma, and non-alcoholic high fat liver disease [reviewed in Ref. (111)]. In case of PCa, CaMKK2 emerges as an attractive and druggable target downstream of AR that when inhibited, abrogates tumor growth, inhibits macrophage-mediated inflammation, and improves bone health. Future studies will provide a comprehensive understanding of the precise molecular mechanisms by which CaMKK2 regulates PCa cells as well as how AR-CaMKK2 signaling in these cells affects CaMKK2 function in bone cells and macrophages that constitute the bone microenvironment. Nonetheless, highly selective small molecule inhibitors of CaMKK2 should be developed as potent “dual-hit” therapeutic interventions to abrogate bone-metastatic PCa growth while preventing ADT-associated bone loss. Together with improving bone mass and strength in PCa patients, who are often elderly, CaMKK2 inhibition would offer the best odds for long-term disease-free survival.
Author Contributions
UD and US contributed to conception, design, and writing of the manuscript. EC contributed to the literature search and drafted tables for manuscript. US critically reviewed the drafts of the manuscript and wrote the final version. UD and US read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Servier Medical art for the use of their image bank. This work was supported by grants from the American Cancer Society (RSG-13-301-01) and National Institutes of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) R01 AR068332 to US.
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer (2010) 127(12):2893–917. doi:10.1002/ijc.25516
2. Pernar CH, Ebot EM, Wilson KM, Mucci LA. The epidemiology of prostate cancer. Cold Spring Harb Perspect Med (2018). doi:10.1101/cshperspect.a030361
3. Noone AM, Howlader N, Krapcho M, Miller D, Brest A, Yu M, et al. editors. SEER Cancer Statistics Review, 1975–2015. Bethesda, MD: National Cancer Institute (2017). Available from: https://seer.cancer.gov/csr/1975_2015/ (Accessed: April, 2018).
4. Bubendorf L, Schöpfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol (2000) 31(5):578–83. doi:10.1053/hp.2000.6698
5. Saad F, Gleason DM, Murray R, Tchekmedyian S, Venner P, Lacombe L, et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst (2004) 96(11):879–82. doi:10.1093/jnci/djh235
6. Saad F, Ivanescu C, Phung D, Loriot Y, Abhyankar S, Beer TM, et al. Skeletal-related events significantly impact health-related quality of life in metastatic castration-resistant prostate cancer: data from PREVAIL and AFFIRM trials. Prostate Cancer Prostatic Dis (2017) 20(1):110–6. doi:10.1038/pcan.2016.62
7. Bienz M, Saad F. Androgen-deprivation therapy and bone loss in prostate cancer patients: a clinical review. Bonekey Rep (2015) 4:716. doi:10.1038/bonekey.2015.85
8. Oefelein MG, Ricchiuti V, Conrad W, Resnick MI. Skeletal fractures negatively correlate with overall survival in men with prostate cancer. J Urol (2002) 168(3):1005–7. doi:10.1097/00005392-200209000-00024
9. Cooperberg MR, Grossfeld GD, Lubeck DP, Carroll PR. National practice patterns and time trends in androgen ablation for localized prostate cancer. J Natl Cancer Inst (2003) 95(13):981–9. doi:10.1093/jnci/95.13.981
10. Cianferotti L, Bertoldo F, Carini M, Kanis JA, Lapini A, Longo N, et al. The prevention of fragility fractures in patients with non-metastatic prostate cancer: a position statement by the international osteoporosis foundation. Oncotarget (2017) 8(43):75646–63. doi:10.18632/oncotarget.17980
11. Nagao K, Matsuyama H, Nozawa M, Hara I, Nishioka T, Komura T, et al. Zoledronic acid combined with androgen-deprivation therapy may prolong time to castration-resistant prostate cancer in hormone-naïve metastatic prostate cancer patients – a propensity scoring approach. Asian J Urol (2016) 3(1):33–8. doi:10.1016/j.ajur.2015.10.003
12. Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol (2004) 164(1):217–27. doi:10.1016/S0002-9440(10)63112-4
13. Logothetis C, Morris MJ, Den R, Coleman RE. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastasis Rev (2018) 37(1):189–96. doi:10.1007/s10555-017-9719-4
14. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med (2012) 367(13):1187–97. doi:10.1056/NEJMoa1207506
15. Tombal B, Borre M, Rathenborg P, Werbrouck P, Van Poppel H, Heidenreich A, et al. Long-term antitumor activity and safety of enzalutamide monotherapy in hormone naive prostate cancer: 3-year open label followup results. J Urol (2018) 199(2):459–64. doi:10.1016/j.juro.2017.08.103
16. Paget S. The distribution of secondary growths in cancer of the breast. Lancet (1889) 1:571–3. doi:10.1016/S0140-6736(00)49915-0
17. Jin JK, Dayyani F, Gallick GE. Steps in prostate cancer progression that lead to bone metastasis. Int J Cancer (2011) 128(11):2545–61. doi:10.1002/ijc.26024
18. Park SH, Keller ET, Shiozawa Y. Bone marrow microenvironment as a regulator and therapeutic target for prostate cancer bone metastasis. Calcif Tissue Int (2018) 102(2):152–62. doi:10.1007/s00223-017-0350-8
19. Yokoyama NN, Shao S, Hoang BH, Mercola D, Zi X. Wnt signaling in castration-resistant prostate cancer: implications for therapy. Am J Clin Exp Urol (2014) 2(1):27–44.
20. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer (2014) 14:598. doi:10.1038/nrc3792
21. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene (2013) 32(49):5501–11. doi:10.1038/onc.2013.206
22. Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res (2002) 62(6):1832–7.
23. Hannafon BN, Ding WQ. Intercellular communication by exosome-derived microRNAs in cancer. Int J Mol Sci (2013) 14(7):14240–69. doi:10.3390/ijms140714240
24. Ye Y, Li SL, Ma YY, Diao YJ, Yang L, Su MQ, et al. Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Oncotarget (2017) 8(55):94834–49. doi:10.18632/oncotarget.22014
25. Kumar S, Nag A, Mandal CC. A comprehensive review on miR-200c, a promising cancer biomarker with therapeutic potential. Curr Drug Targets (2015) 16(12):1381–403. doi:10.2174/1389450116666150325231419
26. Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res (2008) 6(8):1259–67. doi:10.1158/1541-7786.MCR-08-0077
27. Lee YC, Bilen MA, Yu G, Lin SC, Huang CF, Ortiz A, et al. Inhibition of cell adhesion by a cadherin-11 antibody thwarts bone metastasis. Mol Cancer Res (2013) 11(11):1401–11. doi:10.1158/1541-7786.MCR-13-0108
28. Kimura Y, Matsugaki A, Sekita A, Nakano T. Alteration of osteoblast arrangement via direct attack by cancer cells: new insights into bone metastasis. Sci Rep (2017) 7:44824. doi:10.1038/srep44824
29. Rucci N, Teti A. Osteomimicry: how the seed grows in the soil. Calcif Tissue Int (2018) 102(2):131–40. doi:10.1007/s00223-017-0365-1
30. Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer (2016) 16:373. doi:10.1038/nrc.2016.44
31. Lee Y, Schwarz E, Davies M, Jo M, Gates J, Wu J, et al. Differences in the cytokine profiles associated with prostate cancer cell induced osteoblastic and osteolytic lesions in bone. J Orthop Res (2003) 21(1):62–72. doi:10.1016/S0736-0266(02)00095-5
32. Peng J, Kang Y. The bony side of endothelial cells in prostate cancer. Dev Cell (2017) 41(5):451–2. doi:10.1016/j.devcel.2017.05.015
33. Lin SC, Lee YC, Yu G, Cheng CJ, Zhou X, Chu K, et al. Endothelial-to-osteoblast conversion generates osteoblastic metastasis of prostate cancer. Dev Cell (2017) 41(5):467–80.e3. doi:10.1016/j.devcel.2017.05.005
34. Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, et al. Bidirectional notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res (2016) 76(5):1089–100. doi:10.1158/0008-5472.CAN-15-1703
35. Sottnik JL, Dai J, Zhang H, Campbell B, Keller ET. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res (2015) 75(11):2151–8. doi:10.1158/0008-5472.CAN-14-2493
36. Sun YX, Schneider A, Jung Y, Wang J, Dai J, Wang J, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res (2005) 20(2):318–29. doi:10.1359/JBMR.041109
37. Dimitroff CJ, Lechpammer M, Long-Woodward D, Kutok JL. Rolling of human bone-metastatic prostate tumor cells on human bone marrow endothelium under shear flow is mediated by E-selectin. Cancer Res (2004) 64(15):5261–9. doi:10.1158/0008-5472.CAN-04-0691
38. Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun (2013) 4:1795. doi:10.1038/ncomms2766
39. Wolk A, Mantzoros CS, Andersson SO, Bergström R, Signorello LB, Lagiou P, et al. Insulin-like growth factor 1 and prostate cancer risk: a population-based, case-control study. J Natl Cancer Inst (1998) 90(12):911–5. doi:10.1093/jnci/90.12.911
40. Schmidt LJ, Tindall DJ. Steroid 5 α-reductase inhibitors targeting BPH and prostate cancer. J Steroid Biochem Mol Biol (2011) 125(1):32–8. doi:10.1016/j.jsbmb.2010.09.003
41. Chavin G, Sheinin Y, Crispen PL, Boorjian SA, Roth TJ, Rangel L, et al. Expression of immunosuppresive B7-H3 ligand by hormone-treated prostate cancer tumors and metastases. Clin Cancer Res (2009) 15(6):2174–80. doi:10.1158/1078-0432.CCR-08-2262
42. Chiang C, Chiu M, Moore AJ, Anderson PH, Ghasem-Zadeh A, McManus JF, et al. Mineralization and bone resorption are regulated by the androgen receptor in male mice. J Bone Miner Res (2009) 24(4):621–31. doi:10.1359/jbmr.081217
43. Huggins C. Effect of orchiectomy and irradiation on cancer of the prostate. Ann Surg (1942) 115(6):1192–200. doi:10.1097/00000658-194206000-00030
44. Benoist GE, van der Meulen E, van Oort IM, Beumer JH, Somford DM, Schalken JA, et al. Development and validation of a bioanalytical method to quantitate enzalutamide and its active metabolite N-desmethylenzalutamide in human plasma: application to clinical management of metastatic castration-resistant prostate cancer patients. Ther Drug Monit (2018) 40(2):222–9. doi:10.1097/FTD.0000000000000484
45. Schweizer MT, Yu EY. AR-signaling in human malignancies: prostate cancer and beyond. Cancers (Basel) (2017) 9(1):E7. doi:10.3390/cancers9010007
46. Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med (2018) 378(15):1408–18. doi:10.1056/NEJMoa1715546
47. Allan CA, McLachlan RI. Chapter 139 – androgen deficiency disorders A2. 7th ed. In: Larry JJ, Groot LJD, de Kretser DM, Giudice LC, Grossman AB, Melmed S, et al., editors. Endocrinology: Adult and Pediatric. Philadelphia: W.B. Saunders (2016). p. 2394–420.e13.
48. Schmidt LJ, Tindall DJ. Steroid 5 α-reductase inhibitors targeting BPH and prostate cancer. J Steroid Biochem Mol Biol (2011) 125(1):32–8. doi:10.1016/j.jsbmb.2010.09.003
49. Robinson-Rechavi M, Escriva Garcia H, Laudet V. The nuclear receptor superfamily. J Cell Sci (2003) 116(Pt 4):585–6. doi:10.1242/jcs.00247
50. Takayama K-I, Inoue S. Transcriptional network of androgen receptor in prostate cancer progression. Int J Urol (2013) 20(8):756–68. doi:10.1111/iju.12146
51. Labrie F, Cusan L, Gomez J, Luu-The V, Candas B, Bélanger A, et al. Major impact of hormonal therapy in localized prostate cancer – death can already be an exception. J Steroid Biochem Mol Biol (2004) 92(5):327–44. doi:10.1016/j.jsbmb.2004.10.011
52. Smith MR, Fallon MA, Lee H, Finkelstein JS. Raloxifene to prevent gonadotropin-releasing hormone agonist-induced bone loss in men with prostate cancer: a randomized controlled trial. J Clin Endocrinol Metab (2004) 89(8):3841–6. doi:10.1210/jc.2003-032058
53. Smith MR, Morton RA, Barnette KG, Sieber PR, Malkowicz SB, Rodriguez D, et al. Toremifene to reduce fracture risk in men receiving androgen deprivation therapy for prostate cancer. J Urol (2013) 189(1 Suppl):S45–50. doi:10.1016/j.juro.2012.11.016
54. Smith MR, Morton RA, Barnette KG, Sieber PR, Malkowicz SB, Rodriguez D, et al. Toremifene to reduce fracture risk in men receiving androgen deprivation therapy for prostate cancer. J Urol (2010) 184(4):1316–21. doi:10.1016/j.juro.2010.06.022
55. Smith MR, Malkowicz SB, Brawer MK, Hancock ML, Morton RA, Steiner MS. Toremifene decreases vertebral fractures in men younger than 80 years receiving androgen deprivation therapy for prostate cancer. J Urol (2011) 186(6):2239–44. doi:10.1016/j.juro.2011.07.090
56. Smith MR, Coleman RE, Klotz L, Pittman K, Milecki P, Ng S, et al. Denosumab for the prevention of skeletal complications in metastatic castration-resistant prostate cancer: comparison of skeletal-related events and symptomatic skeletal events. Ann Oncol (2015) 26(2):368–74. doi:10.1093/annonc/mdu519
57. James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med (2017) 377(4):338–51. doi:10.1056/NEJMoa1702900
58. Suominen MI, Fagerlund KM, Rissanen JP, Konkol YM, Morko JP, Peng Z, et al. Radium-223 inhibits osseous prostate cancer growth by dual targeting of cancer cells and bone microenvironment in mouse models. Clin Cancer Res (2017) 23(15):4335–46. doi:10.1158/1078-0432.CCR-16-2955
59. Parimi S, Tsang E, Alexander A, Mckenzie M, Bachand F, Sunderland K, et al. A population-based study of the use of radium 223 in metastatic castration-resistant prostate cancer: factors associated with treatment completion. Can Urol Assoc J (2017) 11(10):350–5. doi:10.5489/cuaj.4415
60. Sartor O, Vogelzang NJ, Sweeney C, Fernandez DC, Almeida F, Iagaru A, et al. Radium-223 safety, efficacy, and concurrent use with abiraterone or enzalutamide: first U.S. experience from an expanded access program. Oncologist (2018) 23(2):193–202. doi:10.1634/theoncologist.2017-0413
61. Huntjens KM, Kosar S, van Geel TACM, Geusens PP, Willems P, Kessels A, et al. Risk of subsequent fracture and mortality within 5 years after a non-vertebral fracture. Osteoporos Int (2010) 21(12):2075–82. doi:10.1007/s00198-010-1178-5
62. Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem (2002) 277(29):26321–6. doi:10.1074/jbc.M203310200
63. Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res (2012) 72(6):1494–503. doi:10.1158/0008-5472.CAN-11-3948
64. Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, Hager JH, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol (2013) 31(28):3525–30. doi:10.1200/JCO.2013.50.1684
65. James GD, Symeonides SN, Marshall J, Young J, Clack G. Continual reassessment method for dose escalation clinical trials in oncology: a comparison of prior skeleton approaches using AZD3514 data. BMC Cancer (2016) 16:703. doi:10.1186/s12885-016-2702-6
66. Blessing AM, Rajapakshe K, Reddy Bollu L, Shi Y, White MA, Pham AH, et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy (2017) 13(3):506–21. doi:10.1080/15548627.2016.1268300
67. Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, Fosså SD, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med (2013) 369(3):213–23. doi:10.1056/NEJMoa1213755
68. Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem (2002) 277(18):15813–8. doi:10.1074/jbc.M201075200
69. Dhani NC, Emmenegger U, Adams L, Jongstra J, Tannock IF, Sridhar SS, et al. Phase II study of cytarabine in men with docetaxel-refractory, castration-resistant prostate cancer with evaluation of TMPRSS2-ERG and SPINK1 as serum biomarkers. BJU Int (2012) 110(6):840–5. doi:10.1111/j.1464-410X.2011.10922.x
70. Kim SB, Dent R, Im SA, Espié M, Blau S, Tan AR, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol (2017) 18(10):1360–72. doi:10.1016/S1470-2045(17)30450-3
71. Jernberg E, Bergh A, Wikstrom P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr Connect (2017) 6(8):R146–61. doi:10.1530/EC-17-0118
72. Wadosky KM, Koochekpour S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget (2016) 7(39):64447–70. doi:10.18632/oncotarget.10901
73. Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clin Cancer Res (2009) 15(10):3251–5. doi:10.1158/1078-0432.CCR-08-1171
74. Antonarakis E. AR signaling in human malignancies: prostate cancer and beyond. Cancers (2018) 10(1):22. doi:10.3390/cancers10010022
75. Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, et al. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res (2014) 20(6):1590–600. doi:10.1158/1078-0432.CCR-13-1863
76. De Laere B, van Dam PJ, Whitington T, Mayrhofer M, Diaz EH, Van den Eynden G, et al. Comprehensive profiling of the androgen receptor in liquid biopsies from castration-resistant prostate cancer reveals novel intra-AR structural variation and splice variant expression patterns. Eur Urol (2017) 72(2):192–200. doi:10.1016/j.eururo.2017.01.011
77. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res (2009) 69(6):2305–13. doi:10.1158/0008-5472.CAN-08-3795
78. Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One (2011) 6(4):e19059. doi:10.1371/journal.pone.0019059
79. Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell (2009) 138(2):245–56. doi:10.1016/j.cell.2009.04.056
80. Velasco AM, Gillis KA, Li Y, Brown EL, Sadler TM, Achilleos M, et al. Identification and validation of novel androgen-regulated genes in prostate cancer. Endocrinology (2004) 145(8):3913–24. doi:10.1210/en.2004-0311
81. Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell (2005) 8(5):393–406. doi:10.1016/j.ccr.2005.10.001
82. Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, Neal DE, et al. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep (2007) 8(9):871–8. doi:10.1038/sj.embor.7401046
83. Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A (2002) 99(18):11890–5. doi:10.1073/pnas.182376299
84. Sadeghi M, Ranjbar B, Ganjalikhany MR, Khan FM, Schmitz U, Wolkenhauer O, et al. MicroRNA and transcription factor gene regulatory network analysis reveals key regulatory elements associated with prostate cancer progression. PLoS One (2016) 11(12):e0168760. doi:10.1371/journal.pone.0168760
85. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (2004) 6(1):1–6. doi:10.1016/S1476-5586(04)80047-2
86. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J (2011) 30(13):2719–33. doi:10.1038/emboj.2011.158
87. Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, et al. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res (2011) 71(2):528–37. doi:10.1158/0008-5472.CAN-10-2581
88. Lu KP, Means AR. Regulation of the cell cycle by calcium and calmodulin. Endocr Rev (1993) 14(1):40–58. doi:10.1210/edrv-14-1-40
89. Colomer J, Means AR. Physiological roles of the Ca2+/CaM-dependent protein kinase cascade in health and disease. Subcell Biochem (2007) 45:169–214. doi:10.1007/978-1-4020-6191-2_7
90. Hook SS, Means AR. Ca2+/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol (2001) 41(1):471–505. doi:10.1146/annurev.pharmtox.41.1.471
91. Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem (2012) 287(38):31658–65. doi:10.1074/jbc.R112.356485
92. Green MF, Scott JW, Steel R, Oakhill JS, Kemp BE, Means AR. Ca2+/calmodulin-dependent protein kinase kinase {beta} is regulated by multisite phosphorylation. J Biol Chem (2011) 286(32):28066–79. doi:10.1074/jbc.M111.251504
93. Tokumitsu H, Hatano N, Fujimoto T, Yurimoto S, Kobayashi R. Generation of autonomous activity of Ca(2+)/calmodulin-dependent protein kinase kinase beta by autophosphorylation. Biochemistry (2011) 50(38):8193–201. doi:10.1021/bi201005g
94. Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab (2008) 7(5):377–88. doi:10.1016/j.cmet.2008.02.011
95. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol (2012) 13(4):251–62. doi:10.1038/nrm3311
96. Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell (2007) 25(2):193–205. doi:10.1016/j.molcel.2006.12.009
97. Anderson KA, Lin F, Ribar TJ, Stevens RD, Muehlbauer MJ, Newgard CB, et al. Deletion of CaMKK2 from the liver lowers blood glucose and improves whole-body glucose tolerance in the mouse. Mol Endocrinol (2012) 26(2):281–91. doi:10.1210/me.2011-1299
98. Lin F, Ribar TJ, Means AR. The Ca2+/calmodulin-dependent protein kinase kinase, CaMKK2, inhibits preadipocyte differentiation. Endocrinology (2011) 152(10):3668–79. doi:10.1210/en.2011-1107
99. Cary RL, Waddell S, Racioppi L, Long F, Novack DV, Voor MJ, et al. Inhibition of Ca(2)(+)/calmodulin-dependent protein kinase kinase 2 stimulates osteoblast formation and inhibits osteoclast differentiation. J Bone Miner Res (2013) 28(7):1599–610. doi:10.1002/jbmr.1890
100. Pritchard ZJ, Cary RL, Yang C, Novack DV, Voor MJ, Sankar U. Inhibition of CaMKK2 reverses age-associated decline in bone mass. Bone (2015) 75:120–7. doi:10.1016/j.bone.2015.01.021
101. Racioppi L, Noeldner PK, Lin F, Arvai S, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2 regulates macrophage-mediated inflammatory responses. J Biol Chem (2012) 287(14):11579–91. doi:10.1074/jbc.M111.336032
102. Williams JN, Kambrath AV, Patel RB, Kang KS, Mével E, Li Y, et al. Inhibition of CaMKK2 enhances fracture healing by stimulating Indian hedgehog signaling and accelerating endochondral ossification. J Bone Miner Res (2018) 33(5):930–44. doi:10.1002/jbmr.3379
103. Lin F, Marcelo KL, Rajapakshe K, Coarfa C, Dean A, Wilganowski N, et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology (2015) 62(2):505–20. doi:10.1002/hep.27832
104. Racioppi L. CaMKK2: a novel target for shaping the androgen-regulated tumor ecosystem. Trends Mol Med (2013) 19(2):83–8. doi:10.1016/j.molmed.2012.12.004
105. York B, Li F, Lin F, Marcelo KL, Mao J, Dean A, et al. Pharmacological inhibition of CaMKK2 with the selective antagonist STO-609 regresses NAFLD. Sci Rep (2017) 7(1):11793. doi:10.1038/s41598-017-12139-3
106. Karacosta LG, Foster BA, Azabdaftari G, Feliciano DM, Edelman AM. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J Biol Chem (2012) 287(29):24832–43. doi:10.1074/jbc.M112.370783
107. Karacosta LG, Kuroski LA, Hofmann WA, Azabdaftari G, Mastri M, Gocher AM, et al. Nucleoporin 62 and Ca(2+)/calmodulin dependent kinase kinase 2 regulate androgen receptor activity in castrate resistant prostate cancer cells. Prostate (2016) 76(3):294–306. doi:10.1002/pros.23121
108. Shima T, Mizokami A, Miyagi T, Kawai K, Izumi K, Kumaki M, et al. Down-regulation of calcium/calmodulin-dependent protein kinase kinase 2 by androgen deprivation induces castration-resistant prostate cancer. Prostate (2012) 72(16):1789–801. doi:10.1002/pros.22533
109. Owen KL, Parker BS. Beyond the vicious cycle: the role of innate osteoimmunity, automimicry and tumor-inherent changes in dictating bone metastasis. Mol Immunol (2017). doi:10.1016/j.molimm.2017.11.023
110. Roca H, Jones JD, Purica MC, Weidner S, Koh AJ, Kuo R, et al. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J Clin Invest (2018) 128(1):248–66. doi:10.1172/JCI92466
Keywords: castrate-resistant prostate cancer, androgen-deprivation therapy, CAMKK2, bone–tumor microenvironment, treatment induced bone loss
Citation: Dadwal UC, Chang ES and Sankar U (2018) Androgen Receptor-CaMKK2 Axis in Prostate Cancer and Bone Microenvironment. Front. Endocrinol. 9:335. doi: 10.3389/fendo.2018.00335
Received: 17 February 2018; Accepted: 31 May 2018;
Published: 18 June 2018
Edited by:
Chandi C. Mandal, Central University of Rajasthan, IndiaReviewed by:
Michael Koutsilieris, National and Kapodistrian University of Athens, GreeceJawed Akhtar Siddiqui, University of Nebraska Medical Center, United States
Copyright: © 2018 Dadwal, Chang and Sankar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uma Sankar, usankar@iupui.edu