 Kuerbanjiang Abuduxikuer
Kuerbanjiang Abuduxikuer Zhong-Die Li†
Zhong-Die Li†- Department of Hepatology, Children's Hospital of Fudan University, Shanghai, China
Familial glucocorticoid deficiency type 1 (FGD1) is an autosomal recessive disorder caused by mutations in the melanocortin 2 receptor (MC2R) gene, characterized by a low or undetectable serum cortisol level and a high adrenocorticotropic hormone (ACTH) level. Clinical manifestations include hypoglycemia, seizure, skin hyperpigmentation, hyperbilirubinemia, cholestasis, and a tall stature. Some dysmorphic features such as, a prominent forehead, hypertelorism, a broad nasal bridge, and small tapering fingers, have been reported. Children with FGD1 may have other isolated endocrine abnormalities. To date, no patient with FGD1 has been reported in mainland China. Here we report on a Chinese patient with FGD1 having a novel MC2R gene variant, a mild transverse palm crease, hypertelorism, and subtle/transient endocrine abnormalities relating to all three zones of the adrenal cortex and thyroid gland. We also reviewed cases with dysmorphic features or additional endocrine abnormalities.
Introduction
Familial glucocorticoid deficiency type 1 (FGD1) (OMIM #202200) is an autosomal recessive disorder caused by mutations in the melanocortin 2 receptor (MC2R) gene (OMIM *607397), characterized by a low or undetectable serum cortisol level and high adrenocorticotropic hormone (ACTH) level. Clinical manifestations include hypoglycemia, seizure, skin hyperpigmentation, hyperbilirubinemia, cholestasis, and a tall stature (1–6). Other dysmorphic features, such as a prominent forehead with hypertelorism (7), and a broad nasal bridge with small tapering fingers (8), have been reported. Children with FGD1 may have other isolated endocrine abnormalities (4, 6–17). Here we report on the first FGD1 patient from China with a novel MC2R gene variant, a mild transverse palm crease, hypertelorism, and subtle/transient endocrine abnormalities relating to all three zones of the adrenal cortex and thyroid gland.
Case Presentation
The female infant was born to healthy non-consanguineous parents (25-year-old father, and 22-year-old mother) after an uncomplicated first pregnancy and 40 weeks of gestation. A Cesarean section was performed due to a failed vaginal delivery, but the Apgar score until 15 min after birth and birth weight was normal (3,300 g). The patient was intubated after developing progressive tachypnea, moaning, and severe hypoglycemia (0.9 mmol/L). The patient developed hyperbilirubinemia that was unresponsive to phototherapy, but the parents took the baby home, for home care at the age of 9 days.
At the age of 1.4 months, the patient was admitted to a provincial hospital for jaundice, vomiting, afebrile seizures, and pneumonia. The lowest blood glucose level during the hospital stay was 1.4 mmol/L. The serum cortisol levels were extremely low (13.8–29.3 nmol/L, normal range 138–690 nmol/L) while adrenocorticotropic hormone levels were slightly lower or normal (6.0–18.5 pg/ml, normal range 6.4–40 pg/ml). A cortisol deficiency was diagnosed, but parents refused hormone replacement therapy. The patient was discharged after the pneumonia was resolved and blood glucose levels were stabilized.
At the age of 3.2 months, the patient was presented to our hospital for cholestasis without obvious symptoms of hypoglycemia, infection, alacrima, or achalasia. Repeated morning serum cortisol levels were extremely low (8.8–10.6 nmol/L, normal range 138–690 nmol/L), while ACTH was extremely elevated (1656.9–1911.8 pg/ml, normal range 6.4–40 pg/ml). Upon physical examination, significant jaundice, skin hyperpigmentation and slight hepatosplenomegaly (liver 2–2.5 cm below the right costal margin, and 2.5 cm below the xiphoid process; spleen 1.5–2.0 cm below the left costal margin) were observed. Slight dysmorphic features such as a transverse palmar crease in the right hand, a prominent forehead, hypertelorism (inner canthal distance greater than the palpebral fissure length) were noted. The palmar crease, and the changes in skin pigmentation are presented in Figure 1. Written informed consent was obtained from the parents for the publication of this case report and related images. Changes in body weight/length, complete blood count, procalcitonin, serum biochemistry, blood coagulation, and endocrine profiles throughout the disease course are provided in Table 1.
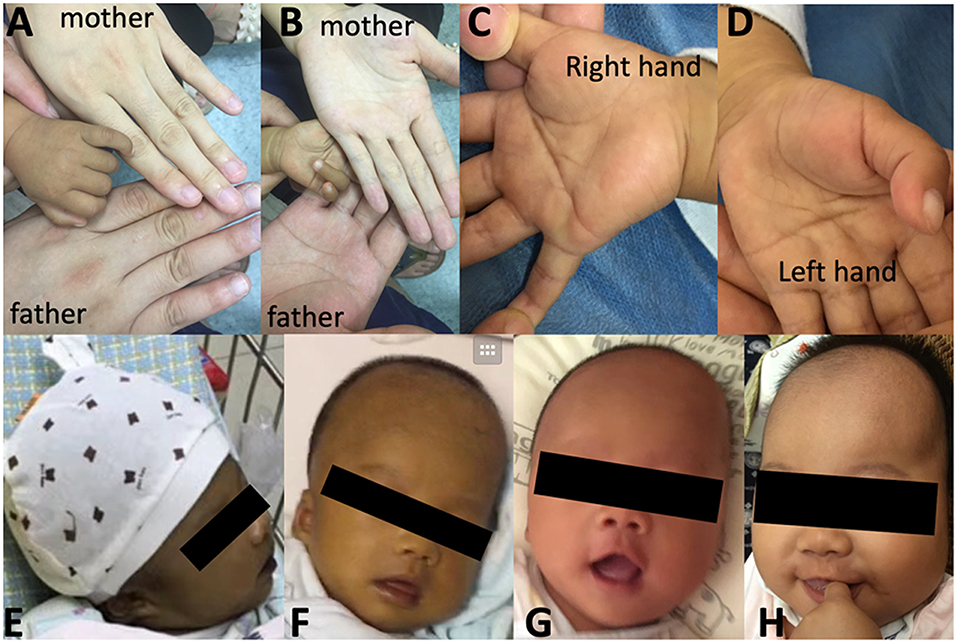
Figure 1. Skin pigmentation and palmar creases. Higher skin pigmentation of the hand compared to parents at 3.5 months (A,B), a slight transverse palmar crease on the right hand (C) but not on the left hand (D). Changes in skin pigmentation at 0.7 months (E), 1.6 months (F), 3 months (F), and 4.9 months (H, 1.5 months after hydrocortisone therapy and resolution of cholestasis). Written informed consent was obtained from the parents for the publication of personal images.
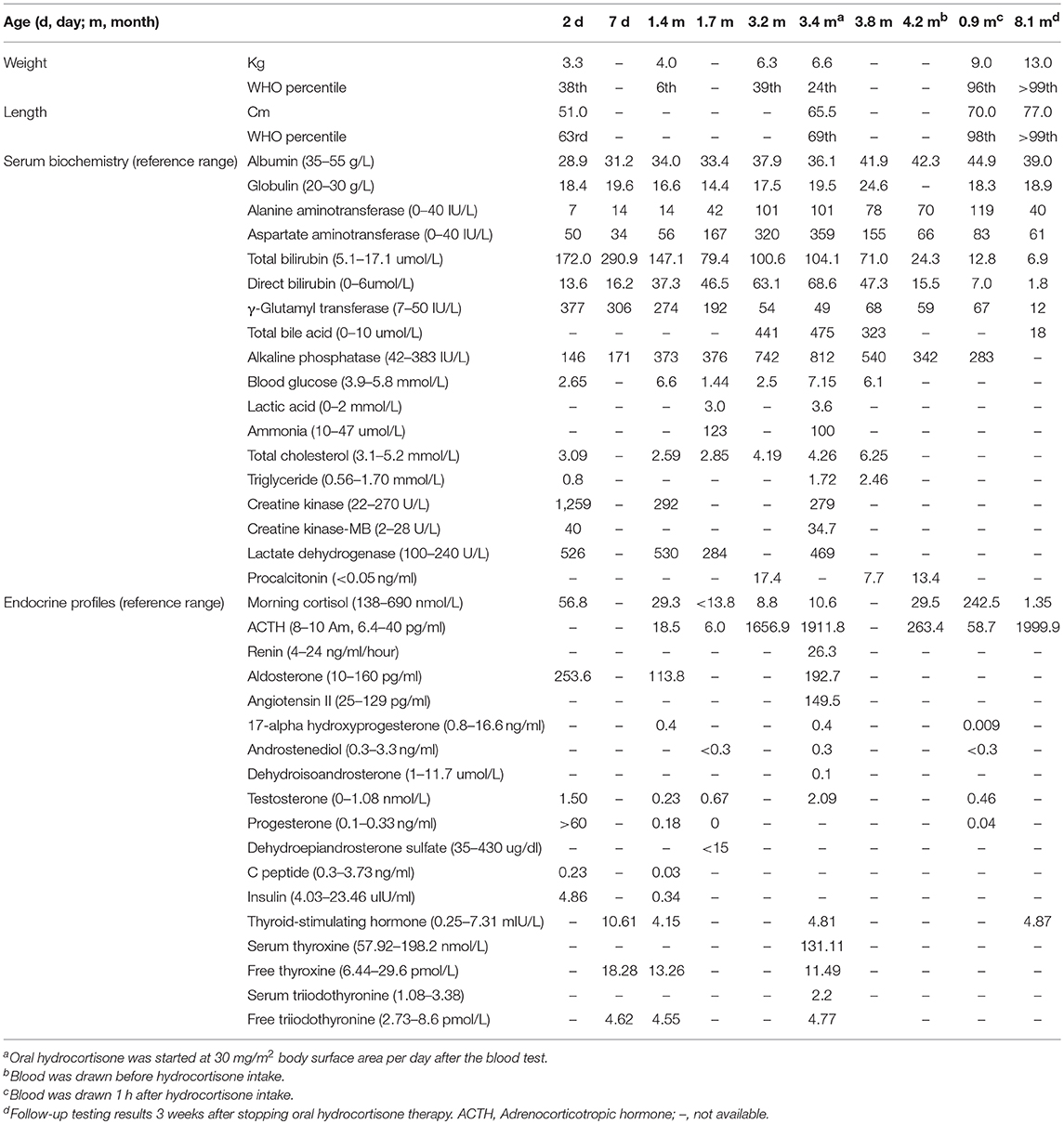
Table 1. Changes in body weight/length, complete blood count, procalcitonin, serum biochemistry, blood coagulation, and endocrine profiles.
Genetic screening for abnormalities related to congenital adrenal hyperplasia (list of 44 genes are provided in Table 3), and multiplex ligation-dependent probe amplification (MLPA) analysis of the CYP21A2 gene were performed by a commercial genetic testing company (Customized target capture sequencing, http://www.mygenostics.com/ServiceTechnology.aspx?nid=263&pid=268). The result showed compound heterozygous variants in the melanocortin 2 receptor (MC2R) gene, but the result of the CYP21A2 gene MLPA analysis was negative for hot-spot mutations and copy number variants (Supplementary Figure 1). We conducted protein modeling with SWISS-model (https://www.swissmodel.expasy.org) using the most similar structure (5jtb.1.A, Adenosine receptor A2a), and polar contacts of wild-type and mutated amino acid residues were compared with Pymol software (https://pymol.org/2/). The c.433C>T/p.R145C was reported in the dbSNP152 (http://www.ncbi.nlm.nih.gov/snp/rs139218324), and gnomAD (http://gnomad-old.broadinstitute.org/variant/18-13885085-G-A), but not in the 1000 Genome Database (http://www.1000genomes.org/) and Exome Variant Server (http://evs.gs.washington.edu/EVS/). The c.712C>T/p.H238Y variant was not reported in the dbSNP152, gnomAD, 1000 Genome Database, and Exome Variant Server. The c.433C>T/p.R145C variant of maternal origin caused the change of arginine (polar, basic) at the amino acid position of 145 to cysteine (non-polar, neutral). R145 is a relatively conserved amino acid residue, and five out of eight in-silico prediction tools (Table 2) predicted this variant as pathogenic. This is a known disease-causing variant (HGMD CM116421, rs139218324), and reported to be associated with FGD1 in an adopted Chinese girl (18). Protein modeling showed no effect of R145C residue change on polar contact with V149. The c.712C>T/p.H238Y variant of paternal origin caused the change of amino acid residue histidine (polar, basic) at the position of 238 to tyrosine (polar, neutral). H238 is a strictly conserved residue, and all eight in-silico prediction tools predicted this variant as pathogenic (Table 2). Protein modeling showed that the H238Y mutation changed polar contact of the amino acid residue in the position of 238 with adjacent residues, and polar contact with N261 in the transmembrane domain (TMD) 7 was lost. Confirmation with Sanger sequencing, conservation status of amino acid residue that have been affected, protein modeling results, and in-silico pathogenicity prediction results for both MC2R variants are provided in Figure 2 and Table 2.
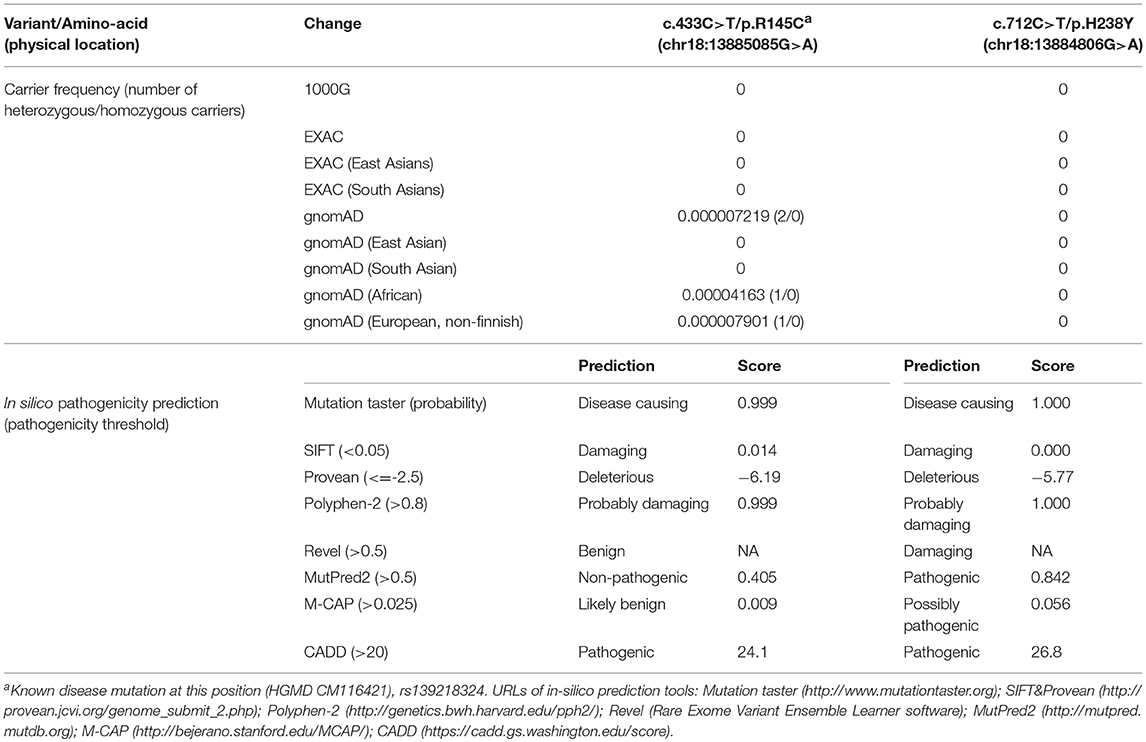
Table 2. Carrier frequency and in-silico pathogenicity prediction results of MC2R gene variants.

Table 3. Diagnostic evaluation.
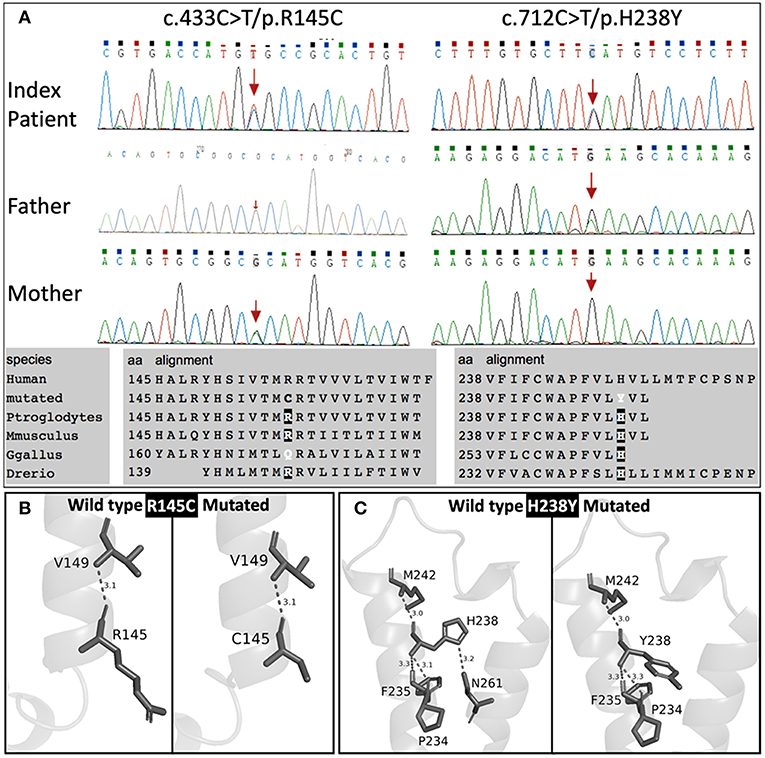
Figure 2. Sanger sequencing results with conservation status of amino acid residues (A), and protein modulation (B,C).
Extensive etiologic evaluations from birth until the last follow-up (4.9 months) are provided in Table 3. After ruling out other causes of hypoglycemia, cholestasis, and adrenal deficiency, a diagnosis of FGD1 was made. Oral hydrocortisone was started at a dose of 30 mg/m2 body surface area (divided into three doses) at the age of 3.4 months. Cholestasis was resolved at 4.9 months, skin hyperpigmentation was improved, and no further episodes of hypoglycemia occurred. Morning serum cortisol levels 1 h after hydrocortisone intake was normal, while ACTH levels returned to near normal levels. However, parents decided to stop the medication at the age of 7.4-months, and serum cortisol/ACTH levels returned to extreme levels at the age of 8.1-months (Table 1).
Discussion
ACTH unresponsiveness was first described by Shepard et al. (19) in 1959, melanocortin receptors were cloned in 1992 (20), and the first FGD1 caused by the MC2R gene mutation was reported by Clark et al. (2).
Hypothyroidism had been reported in an FGD1 patient with compound heterozygous L46fs/V49M mutation. The TSH level of 13.9 mIU/L at 3-months of age was normalized after a week of L-thyroxine therapy and remained normal when the medicine was stopped after 3 months (6). Our patient had hypothyroidism (TSH 10.61 mIU/L) during the neonatal period, but the repeated TSH levels without hormone replacement therapy at the age of 1.4 months was normal. Partial or complete deficiency of sex hormones such as 17-alpha hydroxyprogesterone, androstenediol, dehydroisoandrosterone, testosterone, progesterone, and dehydroepiandrosterone sulfate (DHES) had been reported in several FGD1 patients (4, 7, 9, 10, 13–15, 17). Besides low levels of androstenediol, DHES, and 17-alpha-hydroxyprogesterone, our patient also had lower levels of dehydroisoandrosterone and progesterone, as well as higher levels of testosterone. Although FGD1 patients may experience delayed development of pubic hair, other sexual characteristics did not seem to be affected. Slight abnormalities in renin or aldosterone levels have been reported (7, 8, 11, 13, 14), but angiotensin II levels have never been reported to be elevated in FGD1 patients. Renin, aldosterone, and angiotensin II levels were slightly elevated in our patient without any abnormalities in serum electrolytes, blood pressure, and kidney function. Patients with severe or homozygous truncating mutations in the MC2R gene, mild disturbances in renin–angiotensin–aldosterone axis may need temporary replacement of mineralocorticoid but did not cause long-term mineralocorticoid deficiency after stopping the intervention (11, 12).
A tall stature in the presence of normal growth hormone levels is one of the features of FGD1 (7, 8, 10, 14, 15, 17) accompanied by some dysmorphic features (such as hypertelorism, relative frontal prominence, epicanthic folds, large head circumference, and small tapering fingers) (7, 8). In vitro studies indicated that excess levels of ACTH increases chondrocyte precursors leading to chondrogenic phenotypes (21), and low levels of cortisol may fail to inhibit the synthesis of insulin-like growth factor-binding protein-5 (22). A single transverse palmar crease was associated with 107 genes and 172 disease entities (http://www.geneontology.org/formats/oboInOwl#id: HP:0007598), but not with the MC2R gene or FGD1. Since none of the transverse palmar associated genes were screened in this patient, we cannot rule out the possibility that variants in other genes may have caused this phenotype. Our patient had a normal body weight and length percentiles until hydrocortisone treatment, but both weight and length percentiles exceeded the 95th percentile 1.5 months post-treatment. This might be due to inadequate suppression and a prolonged effect of elevated ACTH. A tall stature is believed to return to normal after continuous treatment with hydrocortisone. However, growth parameters should be monitored in patients with FGD1, and growth hormone levels together with bone maturity should be evaluated when necessary. We also observed slight dysmorphic features, such as a prominent forehead, and hypertelorism, but no epicanthic folds were observed in our patient. Other endocrine abnormalities, and dysmorphic features in previously reported cases are summarized in Table 4.
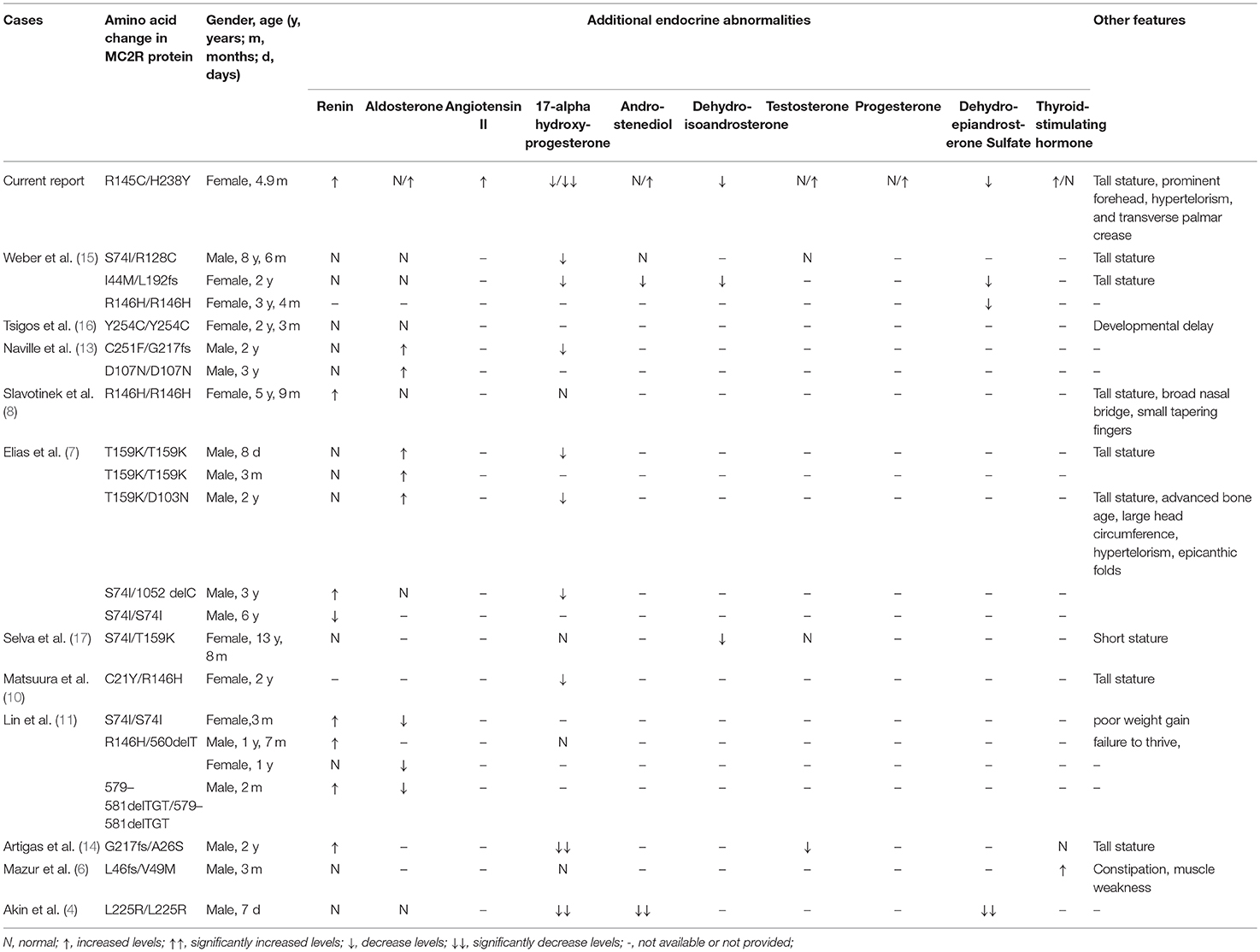
Table 4. Other endocrine abnormalities and dysmorphic features in published cases.
The underlying mechanism of the MC2R gene mutations causing FGD1 is ACTH resistance, either due to the trafficking failure of the receptor from the endoplasmic reticulum to the cell surface, or due to ineffective binding to ACTH (5). according to the last published review in 2018 (23), a total of 28 missense mutations, three non-sense mutations, and eight small insertion/deletions in the MC2R gene were reported in the literature. Most naturally occurring or site-directed mutants cause defective trafficking of the MC2R protein toward the cell membrane while others may lead to defective binding with ACTH (Figure 3). R145C found in our patient was previously reported in an adopted Chinese FGD1 child (18) and a known disease-causing variant in HGMD (CM116421), but no functional study was conducted to evaluate its effect in protein function. Located in the transmembrane domain 4 (TMD4), R145C is adjacent to R146H, which is also a naturally occurring disease causing mutant that causes decreased binding to ACTH and defective membrane trafficking. Site directed mutagenesis of another adjacent residue (T147D) resulted in a trafficking defect, but T147A did not affect protein trafficking toward the cell membrane (24). TMD4 plays a critical role in the activation of the rainbow trout melanocortin-2 receptor (25). R145C may have caused abnormal MC2R protein function by affecting receptor localization in the cell membrane, or activation of the receptor itself. H238Y (located in the TMD6) in our patient is a novel variant in a highly conserved residue not only among different species, but also among other melanocortin receptors. An in vitro study of site directed mutagenesis at the same amino acid residue (H238A) caused a 1.4-fold decrease in membrane trafficking of the MC2R protein from Golgi apparatus toward the cell membrane. H238 residue is also a component of the proposed ACTH binding site consisting of E80, D104, D107, F168, F178, F235, H238, and F258 (26). These findings suggest that the H238Y variant may affect cortisol production by affecting the MC2R localization and ACTH binding. Further functional studies are needed to prove variants found in our patient affect the MC2R protein function.
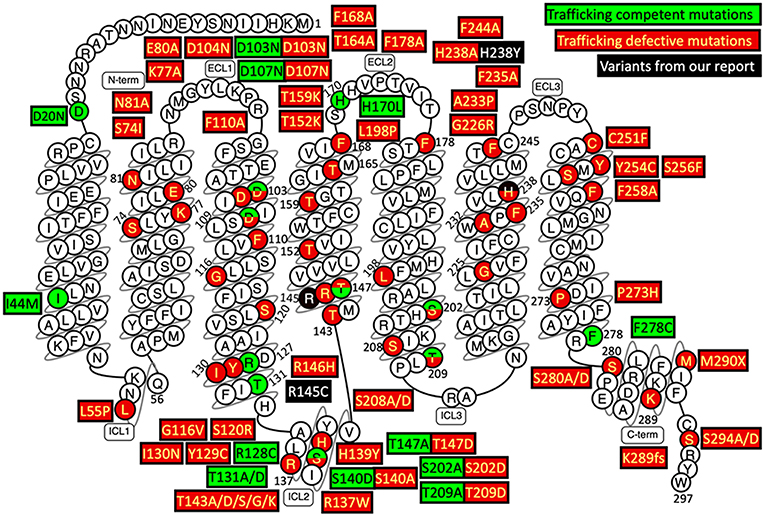
Figure 3. Naturally occurring and site-directed MC2R gene mutants their effect on protein function. This snake diagram of MC2R protein was obtained from G protein-coupled receptors database (GPCRdb, http://gpcrdb.org/family/001_002_014_002/).
In conclusion, we reported on the first child with FGD1 from mainland China, carrying a novel MC2R variant, and reviewed previously reported cases with additional features.
Informed Consent
Written informed consent was obtained from the parents of the participant for the publication of this case report.
Ethics Statement
Ethical Committee of Children's Hospital of Fudan University approved the study.
Author Contributions
J-SW designed the report and approved the final submission. KA and Z-DL collected data. KA analyzed relevant information and conduced protein modeling. Z-DL analyzed the next generation sequencing data. J-SW, KA, Z-DL, X-BX, Y-CL, and JZ, clinically managed the patient. Both KA and Z-DL wrote the manuscript, contributed equally for this manuscript, and will be first co-authors.
Funding
This study was supported by grants (No. 81873543 and No. 81570468) from the National Natural Science Foundation of China, awarded to J-SW.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2019.00359/full#supplementary-material
Supplementary Figure 1. CYP21A2 gene MLPA analysis was negative for hot-spot mutations and copy number variants.
References
1. Chung TT, Chan LF, Metherell LA, Clark AJ. Phenotypic characteristics of familial glucocorticoid deficiency (FGD) type 1 and 2. Clin Endocrinol. (2010) 72:589–94. doi: 10.1111/j.1365-2265.2009.03663.x
2. Clark AJ, McLoughlin L, Grossman A. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet. (1993) 341:461–2. doi: 10.1016/0140-6736(93)90208-X
3. Tsigos C, Arai K, Hung W, Chrousos GP. Hereditary isolated glucocorticoid deficiency is associated with abnormalities of the adrenocorticotropin receptor gene. J Clin Invest. (1993) 92:2458–61. doi: 10.1172/JCI116853
4. Akin MA, Akin L, Coban D, Ozturk MA, Bircan R, Kurtoglu S. A novel mutation in the MC2R gene causing familial glucocorticoid deficiency type 1. Neonatology. (2011) 100:277–81. doi: 10.1159/000323913
5. Chung TT, Webb TR, Chan LF, Cooray SN, Metherell LA, King PJ, et al. The majority of adrenocorticotropin receptor (melanocortin 2 receptor) mutations found in familial glucocorticoid deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J Clin Endocrinol Metab. (2008) 93:4948–54. doi: 10.1210/jc.2008-1744
6. Mazur A, Koehler K, Schuelke M, Skunde M, Ostanski M, Huebner A. Familial glucocorticoid deficiency type 1 due to a novel compound heterozygous MC2R mutation. Horm Res. (2008) 69:363–8. doi: 10.1159/000117393
7. Elias LL, Huebner A, Metherell LA, Canas A, Warne GL, Bitti ML, et al. Tall stature in familial glucocorticoid deficiency. Clin Endocrinol. (2000) 53:423–30. doi: 10.1046/j.1365-2265.2000.01122.x
8. Slavotinek AM, Hurst JA, Dunger D, Wilkie AO. ACTH receptor mutation in a girl with familial glucocorticoid deficiency. Clin Genet. (1998) 53:57–62. doi: 10.1034/j.1399-0004.1998.531530112.x
9. Weber A, Clark AJ, Perry LA, Honour JW, Savage MO. Diminished adrenal androgen secretion in familial glucocorticoid deficiency implicates a significant role for ACTH in the induction of adrenarche. Clin Endocrinol. (1997) 46:431–7. doi: 10.1046/j.1365-2265.1997.1580969.x
10. Matsuura H, Shiohara M, Yamano M, Kurata K, Arai F, Koike K. Novel compound heterozygous mutation of the MC2R gene in a patient with familial glucocorticoid deficiency. J Pediatr Endocrinol Metab. (2006) 19:1167–70. doi: 10.1515/JPEM.2006.19.9.1167
11. Lin L, Hindmarsh PC, Metherell LA, Alzyoud M, Al-Ali M, Brain CE, et al. Severe loss-of-function mutations in the adrenocorticotropin receptor (ACTHR, MC2R) can be found in patients diagnosed with salt-losing adrenal hypoplasia. Clin Endocrinol. (2007) 66:205–10. doi: 10.1111/j.1365-2265.2006.02709.x
12. Chan LF, Metherell LA, Krude H, Ball C, O'Riordan SM, Costigan C, et al. Homozygous nonsense and frameshift mutations of the ACTH receptor in children with familial glucocorticoid deficiency (FGD) are not associated with long-term mineralocorticoid deficiency. Clin Endocrinol. (2009) 71:171–5. doi: 10.1111/j.1365-2265.2008.03511.x
13. Naville D, Barjhoux L, Jaillard C, Faury D, Despert F, Esteva B, et al. Demonstration by transfection studies that mutations in the adrenocorticotropin receptor gene are one cause of the hereditary syndrome of glucocorticoid deficiency. J Clin Endocrinol Metab. (1996) 81:1442–8. doi: 10.1210/jcem.81.4.8636348
14. Artigas RA, Gonzalez A, Riquelme E, Carvajal CA, Cattani A, Martínez-Aguayo A, et al. A novel adrenocorticotropin receptor mutation alters its structure and function, causing familial glucocorticoid deficiency. J Clin Endocrinol Metab. (2008) 93:3097–105. doi: 10.1210/jc.2008-0048
15. Weber A, Toppari J, Harvey RD, Klann RC, Shaw NJ, Ricker AT, et al. Adrenocorticotropin receptor gene mutations in familial glucocorticoid deficiency: relationships with clinical features in four families. J Clin Endocrinol Metab. (1995) 80:65–71. doi: 10.1210/jcem.80.1.7829641
16. Tsigos C, Arai K, Latronico AC, DiGeorge AM, Rapaport R, Chrousos GP. A novel mutation of the adrenocorticotropin receptor (ACTH-R) gene in a family with the syndrome of isolated glucocorticoid deficiency, but no ACTH-R abnormalities in two families with the triple A syndrome. J Clin Endocrinol Metab. (1995) 80:2186–9. doi: 10.1210/jcem.80.7.7608277
17. Selva KA, LaFranchi SH, Boston B. A novel presentation of familial glucocorticoid deficiency (FGD) and current literature review. J Pediatr Endocrinol Metab. (2004) 17:85–92. doi: 10.1515/JPEM.2004.17.1.85
18. Aza-Carmona M, Barreda-Bonis AC, Guerrero-Fernández J, González-Casado I, Gracia R, Heath KE. Familial glucocorticoid deficiency due to compound heterozygosity of two novel MC2R mutations. J Pediatr Endocrinol Metab. (2011) 24:395–7. doi: 10.1515/jpem.2011.024
19. Shepard TH, Landing BH, Mason DG. Familial Addison's disease; case reports of two sisters with corticoid deficiency unassociated with hypoaldosteronism. AMA J Dis Child. (1959) 97:154–62. doi: 10.1001/archpedi.1959.02070010156002
20. Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. (1992) 257:1248–51. doi: 10.1126/science.1325670
21. Evans JF, Niu QT, Canas JA, Shen CL, Aloia JF, Yeh JK. ACTH enhances chondrogenesis in multipotential progenitor cells and matrix production in chondrocytes. Bone. (2004) 35:96–107. doi: 10.1016/j.bone.2004.03.015
22. Gabbitas B, Pash JM, Delany AM, Canalis E. Cortisol inhibits the synthesis of insulin-like growth factor-binding protein-5 in bone cell cultures by transcriptional mechanisms. J Biol Chem. (1996) 271:9033–8. doi: 10.1074/jbc.271.15.9033
23. Novoselova TV, Chan LF, Clark AJL. Pathophysiology of melanocortin receptors and their accessory proteins. Best Pract Res Clin Endocrinol Metab. (2018) 32:93–106. doi: 10.1016/j.beem.2018.02.002
24. Fridmanis D, Roga A, Klovins J. ACTH Receptor (MC2R) Specificity: what do we know about underlying molecular mechanisms? Front Endocrinol. (2017) 8:13. doi: 10.3389/fendo.2017.00013
25. Liang L, Davis PV, Dores MR, Dores RM. The melanocortin-2 receptor of the rainbow trout: identifying a role for critical positions in transmembrane domain 4, extracellular loop 2, and transmembrane domain 5 in the activation of rainbow trout MC2R. Gen Comp Endocrinol. (2018) 257:161–7. doi: 10.1016/j.ygcen.2017.05.003
Keywords: melanocortin 2 receptor (MC2R), familial glucocorticoid deficiency (FGD) type 1, cholestasis, skin hyperpigmentation, hypoglycemia, cortisol, adrenocorticotropic hormone (ACTH), linear overgrowth
Citation: Abuduxikuer K, Li Z-D, Xie X-B, Li Y-C, Zhao J and Wang J-S (2019) Novel Melanocortin 2 Receptor Variant in a Chinese Infant With Familial Glucocorticoid Deficiency Type 1, Case Report and Review of Literature. Front. Endocrinol. 10:359. doi: 10.3389/fendo.2019.00359
Received: 01 March 2019; Accepted: 21 May 2019;
Published: 06 June 2019.
Edited by:
Eli Hershkovitz, Soroka Medical Center, IsraelReviewed by:
Louise Metherell, Queen Mary University of London, United KingdomAdrian Clark, St. George's, University of London, United Kingdom
Copyright © 2019 Abuduxikuer, Li, Xie, Li, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-She Wang, jshwang@shmu.edu.cn
†These authors have contributed equally to this work