 Zhengbo Tao1†
Zhengbo Tao1† Jinpeng Wang1†
Jinpeng Wang1† Kaicheng Wen1†
Kaicheng Wen1† Renqi Yao2
Renqi Yao2 Wacili Da1
Wacili Da1 Siming Zhou1
Siming Zhou1 Yan Meng1
Yan Meng1 Shui Qiu1
Shui Qiu1 Keda Yang1
Keda Yang1 Yue Zhu1
Yue Zhu1 Lin Tao1*
Lin Tao1*- 1Department of Orthopaedics, First Affiliated Hospital of China Medical University, Shenyang, China
- 2Department of Burn Surgery, Changhai Hospital, the Naval Medical University, Shanghai, China
Osteoporosis has become a worldwide disease characterized by a reduction in bone mineral density and the alteration of bone architecture leading to an increased risk of fragility fractures. And an increasing number of studies have indicated that osteoblasts undergo a large number of programmed death events by many different causes in osteoporosis and release NLRP3 and interleukin (e.g., inflammatory factors), which play pivotal roles in contributing to excessive differentiation of osteoclasts and result in exaggerated bone resorption. NLRP3 is activated during pyroptosis and processes the precursors of IL-1β and IL-18 into mature forms, which are released into the extracellular milieu accompanied by cell rupture. All of these compounds are the classical factors of pyroptosis. The cellular effects of pyroptosis are commonly observed in osteoporosis. Although many previous studies have focused on the pathogenesis of these inflammatory factors in osteoporosis, pyroptosis has not been previously evaluated. In this review, pyroptosis is proposed as a novel hypothesis of osteoporosis pathogenesis for the first time, thus providing a new direction for the treatment of osteoporosis in the future.
Background
Osteoporosis is a systemic bone metabolism disease with characteristics of microstructural deterioration of bone tissue and decreased bone density. Approximately 200 million people are suffering from osteoporosis, and the number is increasing every year. The risk of fractures is greatly increased because of the enhanced bone fragility, especially in the hip, vertebrae, and distal forearm. Approximately 8.9 million fractures are caused by osteoporosis, and it has become one of the main causes of death in elderly individuals (1). Bone is an active tissue that is continually resorbed and formed in a process known as remodeling, which is orchestrated by two main cell types: osteoclasts and osteoblasts. Osteoporosis develops when the rate of bone resorption exceeds that of bone formation, and it is caused by natural aging or other pathological conditions (2). Such conditions include low calcium intake (3), estrogen deficiency in postmenopausal women (4, 5), autoimmune diseases, such as rheumatoid arthritis (RA) (6), and the adverse reactions of some drugs, etc.
Over the last 60 years, two seminal clinical observations showing that the decline of ovarian function at menopause leads to a loss of bone mass and estrogen replacement prevents such loss (7, 8) have led to the development of the traditional estrogen-centric perspective of the pathogenesis of osteoporosis (9). In 1999, Eriksen EF et al. demonstrated that bone remodeling in early postmenopausal women is characterized by progressive osteoclastic hyperactivity via dramatic elevations in the number of basic multicellular units (10). However, due to advancements in our understanding of the aging process and the effects of aging and age-related oxidative stress (OS) on bones as well as the serious shortcomings of estrogen-based therapies, aging is presented as a pivotal determinant of the loss of strength and bone mass (9). The most enduring theory of aging proposed by Harman D in 1956 elucidates that OS resulting from an increase in intracellular ROS (reactive oxygen species) is the major determinant of aging and lifespan (11). A series of studies have also indicated that ROS can increase osteoclastogenesis by stimulating the receptor activators of the NF-κB ligand (RANKL) and TNF expression (12–14). As the research on inflammatory disease, including rheumatoid arthritis (6), psoriatic arthritis (15), and Crohn’s disease (16), continues, an array of evidence illustrates that inflammation also increases the loss of bone and risk of fractures, thus emphasizing its skeletal relevance (17, 18). Later in 2005, Weitzmann MN and Pacifici R further explained that certain stimuli of the body lead to an enhancement in the adaptive immune function and consequently upregulates the tumor necrosis factor α (TNF-α) produced by activated T cells, which promotes osteoclast formation and bone resorption (19). Obviously, in the process of osteoporosis, a large number of inflammatory factors and inflammatory signaling pathways are involved, which indicates that there is a certain mechanism between the occurrence of inflammation and osteoporosis that remains to be explored. Pyroptosis, a kind of programmed lytic cell death, is highly correlated to inflammation (20). Compared with apoptosis, pyroptosis, another type of programmed cell death that is non-lytic and usually immune-silent, mediates the development of many inflammatory diseases. Studies show that the cell phenotypes of pyroptosis are manifested in osteoporosis, although the connection between osteoporosis and pyroptosis has not been definitely reported until now. Therefore, this study aims to illuminate the action of pyroptosis on osteoporosis and propose it as a novel pathogenesis of osteoporosis.
Pyroptosis
Pyroptosis was first described in 1992 by Zychlinsky and colleagues (21) and is defined by the Nomenclature Committee on Cell Death (NCCD) as a programmed death mediated by gasdermin, which causes cells to swell until the cell membrane ruptures, causing the release of cell contents and activating a strong inflammatory response (22). Pyroptosis can be initiated by caspase-1 or caspase-11 and caspase-1 can be activated by a variety of inflammasomes, which are multi-protein signaling complexes. Once activated, inflammasomes assemble to form an intracellular macromolecular protein complex and convert the caspase-1 precursor to the active form (20, 23). Caspase-1 cleaves gasdermin D into the hydrophilic GSDMD-C-terminal domain and lipophilic GSDMD-N-terminal domain (24). The N-terminal domain is anchored on the cell membrane and polymerizes to compose a hollow annular oligomer at 10−20 nm in diameter (25–27), which disrupts the regular permeability barrier of the plasma membrane via breaking the concentration gradient of sodium and potassium, leading to an osmotic pressure change. When the number of pores exceeds the compensatory capacity of cells, water flows into the cells via an osmotic gradient, and then the cells swell, rupture, and die as a result (28). Furthermore, during this process, the IL-1β and IL-18 precursors are also cleaved into the mature form by caspase-1 (29, 30) and released through the gasdermin D pores or later during membrane rupture (31, 32). Moreover, additional immune cells will be recruited during this process to trigger an inflammatory response, which in turn induces pyroptosis (28).
In addition to caspase-1, caspase-11 can also mediate pyroptosis via the noncanonical pathway, it may directly recognize and bind with cytosolic lipopolysaccharide (LPS) with the priming of IFN-γ to initiate pyroptosis (33–37). Caspase-11 also acts on gasdermin D independently and produces an N-terminal fragment that oligomerizes and combines with the cell membrane to form pyroptotic pores (31, 38). However, Nobuhiko Kayagaki studied Casp11 gene-targeted mice and Casp11-/- mice and showed that caspase-11 is incapable of directly cleaving the IL-1 and IL-18 precursors, although is an essential effector of non-canonical caspase-1 activation and secretion of mature IL-1β and IL-18 (39, 40) (Figure 1 (30, 41)).
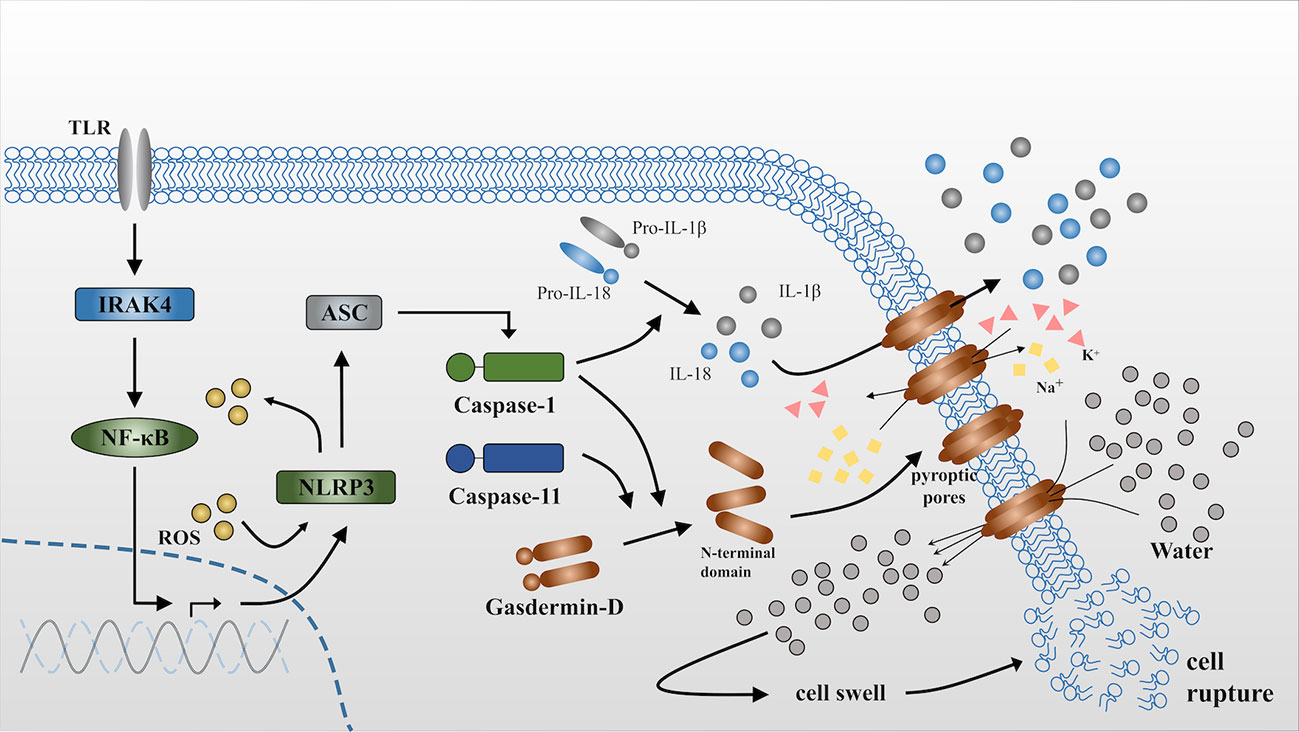
Figure 1 Release of IL-1β and IL-18 during pyroptosis. NLRP3 is activated via the NFκB-pathway when it receives stimulus from toll-like receptors (TLRs) (30). ASC is a bipartite molecule that bridges NLRP3 and caspase-1 precursors (41) and subsequently converts pro-caspase-1 into the activated form. Active caspase-1 can directly act on the precursors of IL-1β and IL-18 and cleave them into the mature form. By contrast, caspase-11 is itself the sensor for cytosolic lipopolysaccharide (LPS). Either caspase-1 or caspase-11 independently cleaves gasdermin D to produce N-terminal domains that anchor to the cell membrane and form pyroptotic pores. The influx of K+ and efflux of Na+ through the pyroptotic pores breaks the osmotic gradient of the cell and consequently, large quantities of water flow into the cell, resulting in cell swelling and rupture. IL-1β and IL-18 are released into the extracellular matrix through pyroptotic pores or accompanied by cell rupture. ROS can accelerate this process via triggering NLRP3 inflammasome formation as well as serving as effector molecules that will be demonstrated in the following.
Pyroptosis is a classical inflammatory reaction that induces T lymphocyte and macrophage activation. Nevertheless, excessive pyroptosis can lead to high levels of cell death, tissue injury, and organ failure, and even cause autoimmune inflammation or septic shock, resulting in irreversible damage to the body (42, 43). Numerous research has indicated that pyroptosis is closely connected with other diseases that cover almost all organs or tissues in the human body. For instance, pyroptosis is thought to be closely linked to various bacterial, viral, and fungal infections (44–46). Pyroptosis is also of great importance in the pathogenesis of many cardiovascular diseases, like atherosclerosis with the activation of NLRP3 (47, 48). Furthermore, Tan MS et al. reported that cerebral NLRP1 levels both in vitro and in vivo were upregulated, indicating the important role of NLRP1/caspase-1 signaling in the progression of Alzheimer’s disease (49). In addition, an array of evidence also suggests that pyroptosis performs a major part in the development of metabolic diseases and spontaneous inflammatory diseases (28). Moreover, IL-1 and IL-18 are undoubtedly important nodes of pyroptosis while NLRP3 is a key effector of pyroptosis.
Except for the diseases presented above, osteoporosis has not been definitely and directly connected with pyroptosis, although both phenomena have a close relationship with inflammation. Indeed, pyroptosis participates in the pathogenesis of osteoporosis mainly via activating NLRP3 and secreting mature IL-1β and IL-18, which will be demonstrated as follows.
IL-1
As mentioned above, T lymphocytes and macrophages are activated during pyroptosis. The activation of T cells and macrophages can induce the release of cytokines, including IL-1, IL-18, interferon-γ (IFN-γ), and TNF-α, etc. (50–52). Moreover, IL-1β is secreted out of the cell during pyroptosis. Therefore, a considerable elevation of IL-1 occurs during the process of pyroptosis, which upregulates osteoclastic formation in several ways. For example, early in 1989, Boyce BF et al. injected IL-1α once daily for three days into normal mice and indicated that IL-1 can stimulate bone formation and absorption systemically and that it has a profound long-term effect on bone remodeling (53). Later in 2007, it was demonstrated by Zwerina J et al. that IL-1 induces the expression of RANKL in mesenchymal stem cells (MSCs) and directly affects osteoclasts by enhancing the RANK expression. Further study by Aoki et al. in 2015 demonstrated that the overexpressed human IL-1α gene can cause osteopenia in hIL-1α Tg mice (54).
Effects of IL-1β on Bone Metabolism
There are 11 members in the IL-1 ligand family, and IL-1β has been regarded as the major therapeutic target for a variety of inflammatory conditions (55, 56). IL-1β binds to its receptor and initiates IL-1 signal transduction, which includes mitogen-activated protein kinase (MAPK) cascade and the NFκB-pathway (57, 58). These signaling pathways cause the activation of some transcription factors, such as c-Jun N-terminal kinase (JNK), AP-1, NF-κB, and p38 MAPK (55, 59).
It was first reported by S-M Dai et al. in 2004 that IL-1 stimulates osteoclastogenesis indirectly through the upregulation of the production of RANKL from rheumatoid arthritis synovial T cells (60). Binding IL-1β with its receptors on T lymphocytes, B lymphocytes, and macrophages promotes the production of RANKL (61), which combines with the RANK of osteoclast precursor cells and contributes to the differentiation and activation of osteoclasts (62, 63). For example, it was reported by Lee SK in 2006 that endogenous IL-1 enhances the response to RANKL of bone marrow cells, and this effect seems to be mediated by mechanisms associated with the enhancement of JNK activity (64). Moreover, IL-1β can activate osteoclasts by increasing the production of M-CSF (macrophage colony-stimulating factor) and also inhibit osteoclast apoptosis (65).
Furthermore, IL-1 is also regarded as a crucial mediator of TNF-mediated osteogenesis (66, 67). TNF and IL-1 are initially involved in different signaling pathways, which are fused with the activation of NF-κB and the stimulation of the MAPK system. Therefore, the crosstalk effect of the two cytokines provides an effective signal to osteoclastogenesis, inhibits osteoblastic function, and regulates the lifespan of skeletal cells (19). For example, Shi Wei et al. reported that IL-1 regulates the osteoclastogenic effect of TNF-α by directly stimulating the differentiation of osteoclast precursors and enhancing the RANKL expression of stromal cell (68). Later in 2007, Zwerina J et al. crossed arthritic human TNF-transgenic (hTNF-tg) mice with IL-1α and β-deficient (IL-1-/-) mice and observed that compared with hTNFtg mice, osteoclast formation and bone erosion were greatly reduced in IL-1-/-hTNFtg mice, suggesting that IL-1 must be an indispensable mediator of TNF-induced osteoclastic formation, and this finding was further reinforced by K Polzer et al. with similar methods in 2015 (69). Moreover, several researchers have proved that TNF-α and IL-1β have a potent anti-apoptotic effect in osteoclasts. In 1999, Eijiro Jimi et al. confirmed that IL-1 induced the viability and multinucleation of osteoclasts and stimulated their pit-forming activity due to the ability to activate NF-κB (70). The prolongation of the osteoclastic lifespan may represent a crucial contribution toward accelerated bone resorption (19, 71).
On the other hand, IL-1 also has an impact on osteoblasts. IL-1α can induce the viability of decreased MC3T3-E1 cells and inhibit osteoblast differentiation through the JNK and p38 MAPK pathways (72). Zhang YZ et al. performed the knockdown of TLR4 in osteoblasts and reduced the levels of IL-1, TNF-α, and IL-6, and subsequently, the cell viability improved because of the inhibition of the inflammatory pathway (73). Conversely, in an osteoporosis model, rats subjected to an ovariectomy showed remarkably higher levels of IL-1, IL-6, and TNF-α in serum compared with control rats and the differentiation of MSCs was blocked as well (74). Obviously, osteoblasts had lost their abilities in an inflammatory environment.
Effects of IL-1β on Dendritic Cells
Immune cells may have the same ability to differentiate into osteoclasts. Dendritic cells (DCs), which are regarded as professional antigen-presenting cells capable of efficiently activating naive T lymphocytes, are able to differentiate into osteoclasts in vitro at the early development stage (75). Indeed, immature dendritic cells can be induced into osteoclasts with the assistance of M-CSF and RANKL (76). DCs contribute to osteoclastogenesis indirectly and their major ability is activating naive T cells, which can then produce RANKL and stimulate osteoclasts differentiation (77, 78). Interestingly, it was suggested by Akagawa that DC-derived osteoclastic formation is dramatically faster and more efficient in terms of fusion rate than the classically described monocyte-derived osteoclastic formation pathway (79). The notion of the transdifferentiation of DCs was further reinforced in 2007 by Carole Speziani, who showed that murine IL-1β and TNF-α can increase bone resorption and DC fusion into osteoclasts when M-CSF and RANKL are present and compared with TNF-α, IL-1β had a higher efficiency in promoting bone resorption (80).
IL-18
As an IL-1 cytokine superfamily member, IL-18 is also secreted out of the cell via the processing of caspase-1 (28, 56). After combining with its receptor, IL-18 shares signaling pathways with IL-1 receptors and toll-like receptors (TLRs) and activates many transcription factors, like NF-κB, AP-1, and MAPK (81, 82). In 1999, Gracie et al. showed that the mRNA and protein of IL-18 are abundant in the joints of RA patients and proved that IL-18 can stimulate the significant production of IFN-γ, GM-CSF, and TNF-α (83). Indeed, IL-18 plays a key role in the increased production of TNF-α and IL-1β (84). For example, Joosten LA et al. reported that the significant suppression of IL-1β and TNF-α levels emerged after the blockade of endogenous IL-18. The role of IL-1β and TNF-α in osteoclastic formation has been described above. Therefore, IL-18 can facilitate the differentiation of osteoclasts via several pathways, including inducing the secretion of interferon -γ (IFN-γ) as well as acting on T lymphocytes.
IL-18 Induces the Secretion of IFN-γ
IL-18 is characterized as an IFN-γ inducing factor because of its ability to augment the production of IFN-γ by natural killer cells and activated T cells in the presence of IL-12 (85, 86). For example, primary data derived from gene knockout experiments by Yoshimoto T show that IL-18 might favor the Th1 response and subsequently induce IFN-γ production in vivo (87). IL-18 from CD8α+ dendritic cells can stimulate the IFN-γ secretion of memory CD4+ T cells in the spleen which activate the NLRC4 inflammasome against S. Typhimurium (88).
Initially, IFN-γ was described as the anti-osteoclastogenic factor because of its potent inhibitory effect for osteoclastogenesis in vitro (51). This notion reinforced that IFN-γ is a bone resorption inhibitor when Vermeire K et al. found that silencing of IFN-γR–/– signaling would result in a severer collagen-induced arthritis and faster bone resorption (89). However, it is also observed that IFN-γ can promote bone resorption and lead to bone loss in various conditions. For example, it was demonstrated by Baker PJ that a lack of IFN-γ in mice led to decreased bone loss (90). Furthermore, as a potent antigen presentation inducer, IFN-γ is also an inducer of T cell activation. When IFN-γ levels are increased in vivo, activated T cells secrete pro-osteoclastogenic factors, offsetting the anti-osteoclastogenic influence of IFN-γ (19). The controversy was eventually solved later in 2007 by Yuhao Gao et al., who showed that IFN-γ had both indirect pro-osteoclastogenic and direct anti-osteoclastogenic effects in vivo while the net balance of the two opposing forces is biased towards bone resorption under inflammatory conditions (91). Therefore, IL-18 promotes osteoclastogenesis via increasing the production of IFN-γ under the inflammatory condition caused by pyroptosis.
IL-18 Acts on T Lymphocytes
IL-18 can also directly act on T lymphocytes and subsequently facilitate bone resorption. For example, an observation by S-M Dai showed that IL-18 indirectly stimulated osteoclastic formation through the upregulation of the production of RANKL from T cells, which is as effective as IL-1β but less potent than TNF-α (60). Moreover, IL-18 is also involved in driving the response of TH17 cells to induce IL-17 production (92, 93), and IL-17 can directly induce human monocytes to differentiate into osteoclasts in the presence of TNF-α and RANKL (94). Similar to IL-1β, IL-17 can also increase murine DC development into osteoclasts, which likely occurs via the induction of IL-1β or TNF-α (80) (Figure 2).
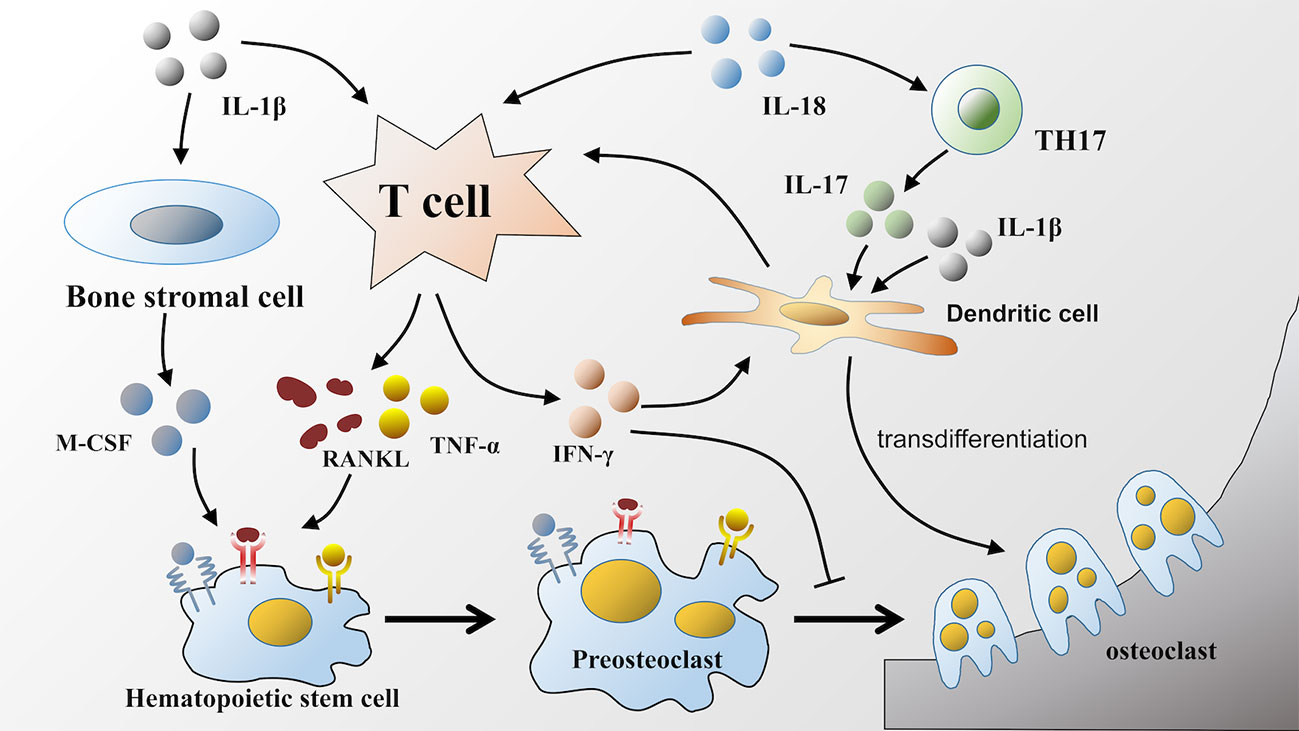
Figure 2 Effects of IL-1β and IL-18 on osteoclastic formation. IL-1β and IL-18 released during pyroptosis facilitate osteoclastic formation in various ways. IL-1β and IL-18 can both act on T lymphocytes to upregulate the production of RANKL. Furthermore, IL-1β also participates in TNF-α mediated osteoclastogenesis, as well as facilitates the production of M-CSF by bone stromal cells. RANKL, TNF-α, and M-CSF bind to corresponding receptors respectively on hematopoietic stem cells to promote its differentiation into osteoclasts. Unlike IL-1β, IL-18 can increase the production of IFN-γ by T lymphocytes, which has dual effects on osteoclastic differentiation. Moreover, IL-18 can also upregulate the production of IL-17. Either IL-17 or IL-1β can induce the transdifferentiation of dendritic cells into osteoclasts.
NLRP3
Inflammatory responses must be induced in macrophages before inflammasomes are activated, and chronic inflammatory responses represent a primary risk factor for inflammatory or autoimmune diseases (95). As demonstrated above, IL-1β and IL-18 are known to produce a number of effects in osteoclastogenesis; therefore, excessive activation of NLRP3 can affect osteoclastic formation by processing IL-1β and IL-18 precursors. Moreover, there are two signals required for the activation of the NLRP3 inflammasome: the NF-κB-dependent pathway and an agonist to induce oligomerization (96). The NF-κB-dependent pathway is of great significance in osteoclastic differentiation. Therefore, excessive activation of inflammasomes would play a pivotal role in the occurrence and development of inflammatory/autoimmune diseases.
Effects of NLRP3 on the Release of IL-1β and IL-18
NLRP3 is capable of promoting progressive and debilitating arthritis characterized by granulocytic infiltration, elevated cytokines, bone erosion, and osteoporosis in humanized mice. For example, John N. Snouwaert et al. demonstrated that IL-1β was elevated in the samples from NLRP3 humanized animals but IL-18 and IL-6 levels were significantly higher. Moreover, kyphosis as well as imageological changes to the hindlimbs were observed in mutant mice with advancing age, which is consistent with osteoporosis (97). Moreover, Jenko B et al. showed that at the genetic level, the NLRP3 inflammasome components expressed were significantly higher in patients with RA compared to controls and the NF-κB signaling pathway was upregulated as a result, which also facilitates the formation of osteoclasts (98, 99). Another study also investigated the regulation of NLRP3 inflammasomes in the release of IL-1β. Vande Walle et al. indicated that the rheumatoid arthritis susceptibility gene A20 was lacking, increasing the expression of pro-IL-1β and NLRP3 genes, which led to caspase-1 activation mediated by the NLRP3 inflammasome, IL-1β secretion, and the occurrence of pyroptosis, although a deficiency of NLRP3 greatly suppressed this process (100).
Interplay of NLRP3 With ROS
ROS are produced by some known NLRP3 inflammasome activators, and they are critical triggers for the formation and activation of the NLRP3 inflammasome (101). In 2007, Cruz CM et al. discovered that inhibiting NADPH oxidase-derived ROS can prevent the activation of caspase-1 induced by ATP and the secretion of IL-1β in alveolar macrophages (102), and they initially proposed the hypothesis of ROS involvement in the formation and activation of the NLRP3 inflammasome. In the following year, this hypothesis was further substantiated by Dostert C et al., who significantly knocked down the p22phox subunit of NADPH oxidase and found that the release of IL-1β was significantly suppressed (103). However, apart from serving as a triggering factor to activate NLRP3, ROS have also been demonstrated to be effector molecules that result in pathological processes (104). When NLRP3 inflammasomes are activated by various stimuli, the local inflammatory response occurs with the production of ROS and cytokines (105). If ROS derived from NLRP3 activation are significantly generated relative to eliminated during this process, then intracellular and extracellular oxidative stress occurs and subsequently contributes to an array of pathophysiological processes and respective diseases. For example, recent studies have also indicated that ROS are important components in the pathogenesis of osteoporosis via upregulating the differentiation of osteoclasts and facilitating the resorption of bone (106, 107). Therefore, as is demonstrated at the beginning of this review, excessive production of ROS due to the aging process activates NLRP3 inflammasomes, which in turn produce more ROS and aggravate the condition of high amounts of ROS. Under the circumstances, excessive bone resorption emerges due to the overactivation of osteoclasts by ROS, which eventually results in osteoporosis.
Effects of NLRP3 on Osteogenesis
NLRP3 directly inhibits osteogenesis by affecting osteoblasts and plays a core role in osteoporosis induced by inflammation. Early in 2008, McCall SH et al. proved that osteoblasts expressed NLRP3 which mediated bacterially induced cell death and participated in bone loss during inflammation (103). NLRP3 is considered essential in many bacterial infections caused by osteoporosis (100, 108–110). When NLRP3 inflammasomes increase, the caspase-1 pathway is activated and the expression levels of IL-1 and IL-18 are upregulated, resulting in the death of osteoblasts (111). Although not explicitly stated, this finding is evidence of pyroptosis in osteoblasts. In addition, in estrogen-deficient osteoporosis, direct bacterial infection was not observed but NLRP3 levels still rose, the viability of osteoblasts was significantly increased, and osteoporosis was also relieved when NLRP3 was inhibited or inactivated (112, 113).
Conclusions and Perspectives
Under inflammatory conditions, a series of corresponding molecules recognized by inflammasomes activate caspase-1 and cleave gasdermin D into a reactive N-terminal fragment with the participation of caspase-11. The N-terminal fragments anchor to the cell membrane and lead to cell swelling and death via the formation of pyroptotic pores. Precursors of IL-1β and IL-18 are processed by caspase-1 during pyroptosis and secreted into the extracellular milieu along with cell debris, which aggravates the inflammatory condition. IL-1β and IL-18 can directly or indirectly facilitate the differentiation of hematopoietic stem cells and preosteoclasts into mature osteoclasts, which leads to the excessive formation of osteoclasts. The formation of osteoclasts exceeds that of osteoblasts, which leads to superfluous bone resorption and an imbalance of bone remodeling, which subsequently results in osteoporosis. Moreover, the stimulation of pyroptosis on osteoclastogenesis may be more obvious in age-related osteoporosis due to the dual action of ROS as a triggering factor and an effector of NLRP3 inflammasomes. Therefore, we speculate that pyroptosis is the main pathogenesis of osteoporosis (detailed hypothesis in Figure 3). When the body is exposed to external stimuli, such as bacterial infection, estrogen deficiency, or aging signals, many inflammatory pathways are activated, IL-1 and IL-18 secretion of inflammatory factors increase, and NLRP3 inflammasomes amplify this signal. Pyroptosis occurs in osteoblasts, which further activate osteoclasts and leads to the aggravation of bone loss. The current clinical drugs used to treat osteoporosis are mainly classic anti-osteoclast drugs, like bisphosphonates or expensive osteogenic teriparatide. Besides, denosumab and romosozumab show promise in the treatment of osteoporosis according to recent research (114). Our study highlights the potential of essential components during pyroptosis, such as NLRP3, caspase-1, or the N-terminal of gasdermin D, as drug targets. However, the detailed activation and action mechanisms are not thoroughly understood, which has hampered the discovery and development of novel therapeutics against these targets. Here, we summarized some important factors involved in pyroptosis and their crosstalk pathways related to osteoporosis (Table 1) (108, 109, 113, 115–118) and some cytokines involved in the pathogenesis of inflammation-induced osteoporosis (Figure 4). Some of the pro-inflammatory cytokines, such as IL-7, IL-12, IL-23, and IL-17, as well as INF-γ, have been shown to possess dual, osteoclastogenic, and anti-osteoclastogenic properties. It seems that their net effect depends on the specific pathophysiological condition of the bone in which they are being studied in in vivo, while in experiments in vitro it depends on the developmental stage of osteoclasts. Among them, IFN-γ can suppress osteoclastogenesis via directly targeting osteoclast precursors, but indirectly promote osteoclast formation and bone resorption by stimulating antigen-dependent T cell activation and production of the osteoclastogenic factors RANKL and TNF-γ. Under the conditions of estrogen deficiency, infection, and inflammation, the net effect of these two opposing properties is biased toward bone resorption (91). And the dual effects of IL-12 can be regarded as the opposing properties of IFN-γ because it serves as a potent inducer of IFN-γ (119). The effect of IL17 is concentration-dependent, which means low concentrations of IL-17 can promote bone resorption while high concentrations of IL-17 can restrain the differentiation of osteoclasts (120, 121). Hence, further investigations into the action of additional components present in pyroptosis on the activation and proliferation of osteoclasts and osteoblasts are of great significance in the pathogenesis of osteoporosis as well as the development of therapeutic strategies.
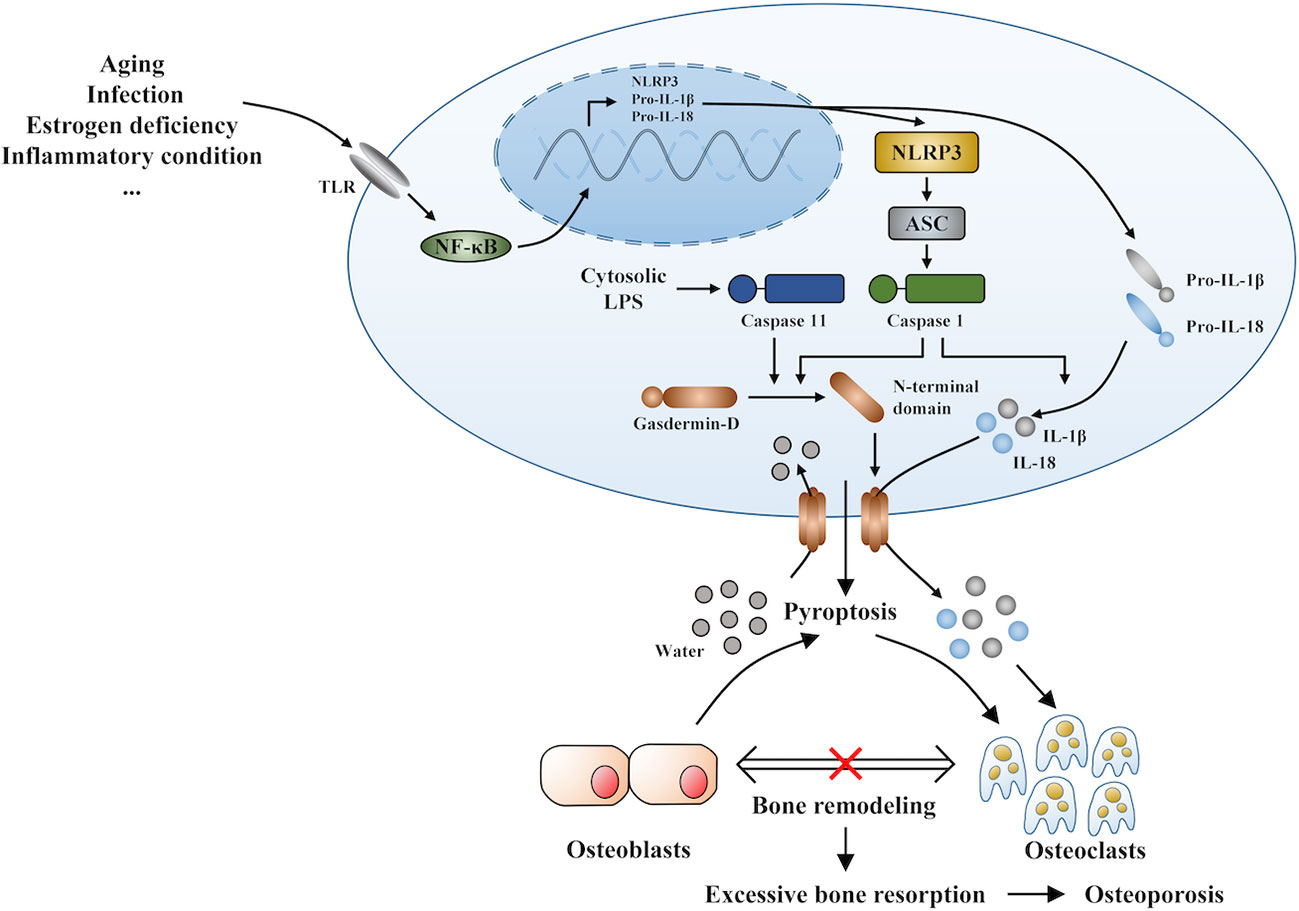
Figure 3 The novel hypothesis of pyroptosis as an osteoporosis pathogenesis.
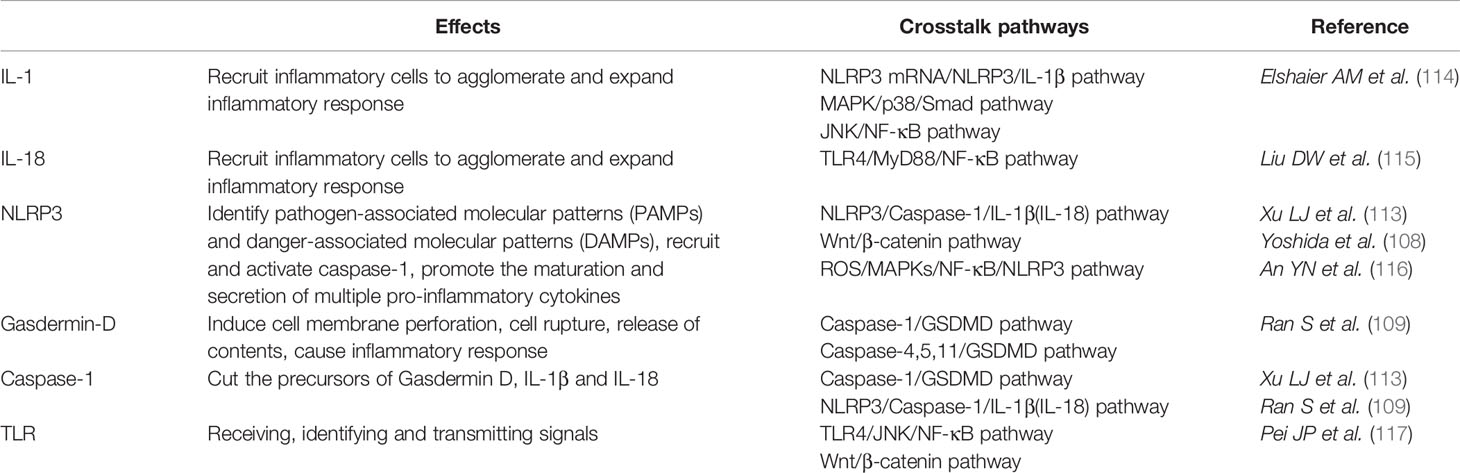
Table 1 Some important factors involved in pyroptosis and their crosstalk pathways.
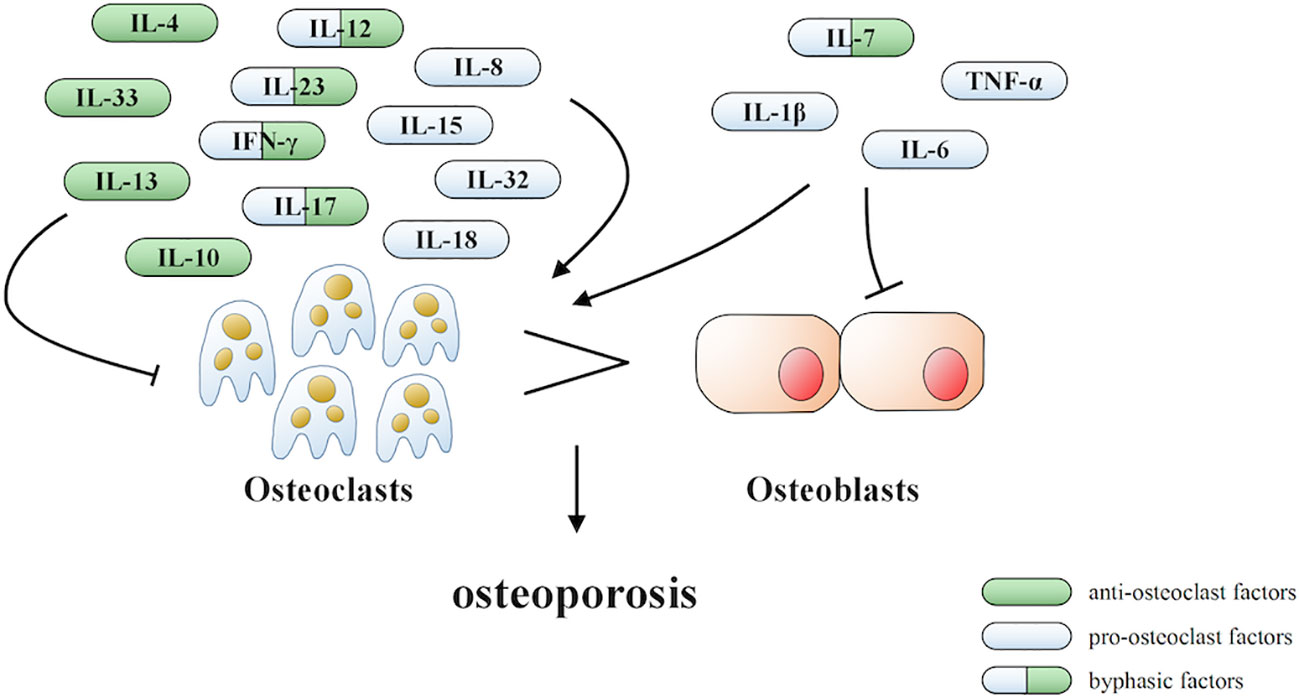
Figure 4 Cytokines involved in the pathogenesis of inflammation-induced osteoporosis.
Author Contributions
ZT and JW made substantial contributions to the conception and design of the work. RY, WD, SZ, YM, SQ, and KY performed the acquisition and analysis of literature. ZT, JW, and KW drafted the work. All the authors approved the version to be published. LT and YZ are responsible for this study. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
IL-1, interleukin-1; IL-18, interleukin-18; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain containing 3; GSDMD, gasdermin D; ROS, reactive oxygen species; DC, dendritic cells; RANK, receptor activator of NF-κB; RANKL, receptor activator of NF-κB ligands; M-CSF, macrophage colony-stimulating factor; MSCs, mesenchymal stem cells; RA, rheumatoid arthritis.
References
1. Genant HK, Cooper C, Poor G, Reid I, Ehrlich G, Kanis J, et al. Interim report and recommendations of the World Health Organization Task-Force for Osteoporosis. Osteoporosis Int (1999) 10:259–64. doi: 10.1007/s001980050224
2. Weitzmann MN, Ofotokun I. Physiological and pathophysiological bone turnover - role of the immune system. Nat Rev Endocrinol (2016) 12:518–32. doi: 10.1038/nrendo.2016.91
3. Johnston CC, Miller JZ, Slemenda CW, Reister TK, Hui S, Christian JC, et al. Calcium supplementation and increases in bone mineral density in children. N Engl J Med (1992) 327:82–7. doi: 10.1056/NEJM199207093270204
4. Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest (2006) 116:1186–94. doi: 10.1172/JCI28550
5. Ravn P, Cizza G, Bjarnason NH, Thompson D, Daley M, Wasnich RD, et al. Low body mass index is an important risk factor for low bone mass and increased bone loss in early postmenopausal women. Early Postmenopausal Intervention Cohort (EPIC) study group. J Bone Min. Res (1999) 14:1622–7. doi: 10.1359/jbmr.1999.14.9.1622
6. van Staa TP, Geusens P, Bijlsma JW, Leufkens HG, Cooper C. Clinical assessment of the long-term risk of fracture in patients with rheumatoid arthritis. Arthritis Rheum (2006) 54:3104–12. doi: 10.1002/art.22117
7. Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev (2000) 21:115–37. doi: 10.1210/er.21.2.115
8. Riggs BL, Khosla S, Melton LJ. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev (2002) 23:279–302. doi: 10.1210/edrv.23.3.0465
9. Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev (2010) 31:266–300. doi: 10.1210/er.2009-0024
10. Eriksen EF, Langdahl B, Vesterby A, Rungby J, Kassem M. Hormone replacement therapy prevents osteoclastic hyperactivity: A histomorphometric study in early postmenopausal women. J Bone Min Res (1999) 14:1217–21. doi: 10.1359/jbmr.1999.14.7.1217
11. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol (1956) 11:298–300. doi: 10.1093/geronj/11.3.298
12. Bai XC, Lu D, Liu AL, Zhang ZM, Li XM, Zou ZP, et al. Reactive oxygen species stimulates receptor activator of NF-kappaB ligand expression in osteoblast. J Biol Chem (2005) 280:17497–506. doi: 10.1074/jbc.M409332200
13. Lean JM, Jagger CJ, Kirstein B, Fuller K, Chambers TJ. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology (2005) 146:728–35. doi: 10.1210/en.2004-1021
14. Agidigbi TS, Kim C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int J Mol Sci (2019) 20(14):3576. doi: 10.3390/ijms20143576
15. Kocijan R, Englbrecht M, Haschka J, Simon D, Kleyer A, Finzel S, et al. Quantitative and Qualitative Changes of Bone in Psoriasis and Psoriatic Arthritis Patients. J Bone Min Res (2015) 30:1775–83. doi: 10.1002/jbmr.2521
16. Haschka J, Hirschmann S, Kleyer A, Englbrecht M, Faustini F, Simon D, et al. High-resolution Quantitative Computed Tomography Demonstrates Structural Defects in Cortical and Trabecular Bone in IBD Patients. J Crohn’s Colitis (2016) 10:532–40. doi: 10.1093/ecco-jcc/jjw012
17. Schett G, Kiechl S, Weger S, Pederiva A, Mayr A, Petrangeli M, et al. High-sensitivity C-reactive protein and risk of nontraumatic fractures in the Bruneck study. Arch Internal Med (2006) 166:2495–501. doi: 10.1001/archinte.166.22.2495
18. Pasco JA, Kotowicz MA, Henry MJ, Nicholson GC, Spilsbury HJ, Box JD, et al. High-sensitivity C-reactive protein and fracture risk in elderly women. JAMA (2006) 296:1353–5. doi: 10.1001/jama.296.11.1353
19. Weitzmann MN, Pacifici R. The role of T lymphocytes in bone metabolism. Immunol Rev (2005) 208:154–68. doi: 10.1111/j.0105-2896.2005.00324.x
20. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev (2015) 265:130–42. doi: 10.1111/imr.12287
21. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature (1992) 358:167–9. doi: 10.1038/358167a0
22. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-018-0102-y
23. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
24. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
25. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
26. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35:1766–78. doi: 10.15252/embj.201694696
27. Liang H, Liu Y. Gasdermins pore cell membrane to pyroptosis. Sci China Life Sci (2016) 59:1090–2. doi: 10.1007/s11427-016-0243-4
28. Gong W, Shi Y, Ren J. Research progresses of molecular mechanism of pyroptosis and its related diseases. Immunobiology (2019) 3:151884. doi: 10.1016/j.imbio.2019.11.019
29. Edgeworth JD, Spencer J, Phalipon A, Griffin GE, Sansonetti PJ. Cytotoxicity and interleukin-1beta processing following Shigella flexneri infection of human monocyte-derived dendritic cells. Eur J Immunol (2002) 32:1464–71. doi: 10.1002/1521-4141(200205)32:5<1464::AID-IMMU1464>3.0.CO;2-G
30. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov (2018) 17:588–606. doi: 10.1038/nrd.2018.97
31. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol (2017) 17:151–64. doi: 10.1038/nri.2016.147
32. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
33. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science (2013) 341:1250–3. doi: 10.1126/science.1240988
34. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
35. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187—+. doi: 10.1038/nature13683
36. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart, et al. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
37. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science (2013) 339:975–8. doi: 10.1126/science.1230751
38. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Dueber EC. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
39. Garrison JB, Correa RG, Gerlic M, Yip KW, Krieg A, Tamble CM, et al. ARTS and Siah Collaborate in a Pathway for XIAP Degradation. Mol Cell (2011) 41:107–16. doi: 10.1016/j.molcel.2010.12.002
40. Agostini L, Martinon F, Burns K, Mcdermott MF, Tschopp J. NALP3 Forms an IL-1??-Processing Inflammasome with Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity (2004) 20:319–25. doi: 10.1016/S1074-7613(04)00046-9
41. Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
42. Langdahl BL. New treatments of osteoporosis. Osteoporosis Sarcopenia (2015) 1:4–21. doi: 10.1016/j.afos.2015.07.007
43. Poli G, Brancorsini S, Cochetti G, Barillaro F, Mearini E. Expression of inflammasome-related genes in bladder cancer and their association with cytokeratin 20 messenger RNA. Urol Oncol Semin Orig Invest (2015) 33:505.e501–7. doi: 10.1016/j.urolonc.2015.07.012
44. Zheng Z, Wei C, Guan K, Yuan Y, He X. Bacterial E3 Ubiquitin Ligase IpaH4.5 of Shigella flexneri Targets TBK1 To Dampen the Host Antibacterial Response. J Immunol (2015) 196:1199. doi: 10.4049/jimmunol.1501045
45. Banerjee D, Chakraborty B, Chakraborty B. Anthrax: Where Margins are Merging between Emerging Threats and Bioterrorism. Indian J Dermatol (2017) 62:456–8. doi: 10.4103/ijd.IJD_378_17
46. Eklund D, Welin A, Andersson H, Verma D, Soderkvist P, Stendahl O, et al. Human Gene Variants Linked to Enhanced NLRP3 Activity Limit Intramacrophage Growth of Mycobacterium tuberculosis. J Infect Dis (2014) 209:749–53. doi: 10.1093/infdis/jit572
47. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol (2011) 51(4):600–6. doi: 10.1016/j.yjmcc.2010.10.033
48. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 466:652–2. doi: 10.1038/nature09316
49. Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, et al. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis (2014) 5(8):e1382. doi: 10.1038/cddis.2014.348
50. Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med (2000) 343:37–49. doi: 10.1056/NEJM200007063430107
51. Takayanagi H, Ogasawara K, Hida S, Chiba T, Taniguchi T. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature (2000) 408:600–5. doi: 10.1038/35046102
52. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2016) 79:541. doi: 10.1146/annurev-physiol-022516-034339
53. Boyce BF, Aufdemorte TB, Garrett IR, Yates AJ, Mundy GR. Effects of interleukin-1 on bone turnover in normal mice. Endocrinology (1989) 125:1142–50. doi: 10.1210/endo-125-3-1142
54. Aoki Y, Ichimura S, Kikuchi T, Yoshihara Y, Fujikawa K. Overexpression of the human interleukin 1α gene causes osteopenia in mice. J Rheumatol (2005) 32:320–4.
55. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
56. Ge Y, Huang M, Yao YM. Recent advances in the biology of IL-1 family cytokines and their potential roles in development of sepsis. Cytokine Growth Factor Rev (2019) 45:24–34. doi: 10.1016/j.cytogfr.2018.12.004
57. Schaeffer Hans J, Weber Michael J. Mitogen-Activated Protein Kinases: Specific Messages from Ubiquitous Messengers. Mol Cell Biol (1999) 19:2435–44. doi: 10.1128/MCB.19.4.2435
58. Renard P, Raes M. The proinflammatory transcription factor NF??B: A potential target for novel therapeutical strategies. Cell Biol Toxicol (1999) 15:341–4. doi: 10.1023/A:1007652414175
59. Dinarello CA. Overview of the interleukin-1 family of ligands and receptors. Semin Immunol (2013) 25:389–93. doi: 10.1016/j.smim.2013.10.001
60. Dai SM, Nishioka K, Yudoh K. Interleukin (IL) 18 stimulates osteoclast formation through synovial T cells in rheumatoid arthritis: comparison with IL1 beta and tumour necrosis factor alpha. Ann Rheumatic Dis (2004) 63:1379–86. doi: 10.1136/ard.2003.018481
61. Nakamura I, Jimi E. Regulation of osteoclast differentiation and function by interleukin-1. Vitam Horm (2006) 74:357–70. doi: 10.1016/S0083-6729(06)74015-8
62. Lu X, Gilbert L, He X, Rubin J, Nanes MS. Transcriptional regulation of the osterix (Osx, Sp7) promoter by tumor necrosis factor identifies disparate effects of mitogen-activated protein kinase and NF kappa B pathways. J Biol Chem (2006) 281(10):6297–306. doi: 10.1074/jbc.M507804200
63. Ono T, Nakashima T. Recent advances in osteoclast biology. Histochem Cell Biol (2018) 149:325–41. doi: 10.1007/s00418-018-1636-2
64. Lee SK, Gardner AE, Kalinowski JF, Jastrzebski SL, Lorenzo JA. RANKL-stimulated osteoclast-like cell formation in vitro is partially dependent on endogenous interleukin-1 production. Bone (2006) 38:0–685. doi: 10.1016/j.bone.2005.10.011
65. Sampaio Lacativa PG, Fleiuss de Farias ML. Osteoporosis and inflammation. Arq Bras Endocrinol Metabol (2010) 54:123–32. doi: 10.1590/S0004-27302010000200007
66. Kim JH, Jin HM, Kim K, Song I, Youn BU, Matsuo K, et al. The Mechanism of Osteoclast Differentiation Induced by IL-1. J Immunol (2009) 183:1862–70. doi: 10.4049/jimmunol.0803007
67. Zwerina J, Redlich K, Polzer K, Joosten L, Krönke G, Distler J, et al. TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci U S A (2007) 104:11742–7. doi: 10.1073/pnas.0610812104
68. Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest (2005) 115:282–90. doi: 10.1172/JCI200523394
69. Polzer K, Joosten L, Gasser J, Distler JH, Ruiz G, Baum W, et al. Interleukin-1 is essential for systemic inflammatory bone loss. Ann Rheumatic Dis (2010) 69:284–90. doi: 10.1136/ard.2008.104786
70. Jimi E, Akiyama S, Tsurukai T, Okahashi N, Kobayashi K, Udagawa N, et al. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J Immunol (1999) 163:434–42.
71. Lacey DL, Tan HL, Lu J, Kaufman S, Polverino AJ. Osteoprotegerin Ligand Modulates Murine Osteoclast Survival in Vitro and in Vivo. Am J Pathol (2000) 157:435–48. doi: 10.1016/S0002-9440(10)64556-7
72. Guo C, Yang X-G, Wang F, Ma X-Y. IL-1α induces apoptosis and inhibits the osteoblast differentiation of MC3T3-E1 cells through the JNK and p38 MAPK pathways. Int J Mol Med (2016) 38:319–27. doi: 10.3892/ijmm.2016.2606
73. Zhang Y, Shen X, Cheng L, Chen R, Zhao F, Zhong S, et al. Toll-like receptor 4 knockout protects against diabetic-induced imbalance of bone metabolism via autophagic suppression. Mol Immunol (2020) 117:12–9. doi: 10.1016/j.molimm.2019.10.025
74. Feng C, Xiao L, Yu JC, Li DY, Tang TY, Liao W, et al. Simvastatin promotes osteogenic differentiation of mesenchymal stem cells in rat model of osteoporosis through BMP-2/Smads signaling pathway. Eur Rev Med Pharmacol Sci (2020) 24:434–43. doi: 10.26355/eurrev_202001_19943
75. Alnaeeli M, Penninger JM, Teng Y-TA. Immune Interactions with CD4+ T Cells Promote the Development of Functional Osteoclasts from Murine CD11c+ Dendritic Cells. J Immunol (2006) 177:3314–26. doi: 10.4049/jimmunol.177.5.3314
76. Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdincombe C, et al. Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood (2004) 104(13):4029. doi: 10.1182/blood-2004-01-0041
77. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Galibert LA. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature (1997) 390:175–9. doi: 10.1038/36593
78. Takahashi N, Udagawa N, Suda T. A New Member of Tumor Necrosis Factor Ligand Family, ODF/OPGL/TRANCE/RANKL, Regulates Osteoclast Differentiation and Function. Biochem Biophys Res Commun (1999) 256(3):449–55. doi: 10.1006/bbrc.1999.0252
79. Akagawa KS, Takasuka N, Nozaki Y, Komuro I, Azuma M, Ueda M, et al. Generation of CD1+RelB+ dendritic cells and tartrate-resistant acid phosphatase-positive osteoclast-like multinucleated giant cells from human monocytes. Blood (1996) 88:4029–39. doi: 10.1182/blood.V88.10.4029.bloodjournal88104029
80. Speziani C, Rivollier A, Gallois A, Coury F, Mazzorana M, Azocar O, et al. Murine dendritic cell transdifferentiation into osteoclasts is differentially regulated by innate and adaptive cytokines. Eur J Immunol (2007) 37:747–57. doi: 10.1002/eji.200636534
81. Reddy P. Interleukin-18: recent advances. Curr Opin Hematol (2004) 11(6):405–10. doi: 10.1097/01.moh.0000141926.95319.42
82. Cheung H, Chen N-J, Cao Z, Ono N, Ohashi PS, Yeh W-C. Accessory Protein-Like Is Essential for IL-18-Mediated Signaling. J Immunol (2005) 174:5351–7. doi: 10.4049/jimmunol.174.9.5351
83. Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest (1999) 104(10):1393–401. doi: 10.1172/JCI7317
84. Joosten LAB, Van De Loo FA, Lubberts E, Helsen MMA, Van Den Berg WB. An IFN- -Independent Proinflammatory Role of IL-18 in Murine Streptococcal Cell Wall Arthritis. J Immunol (2001) 165:6553–8. doi: 10.4049/jimmunol.165.11.6553
85. Okamura H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, et al. Cloning Of A New Cytokine That Induces Ifn-Gamma Production By T Cells. Nature (1995) 378:88–91. doi: 10.1038/378088a0
86. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol (2010) 10:117–0. doi: 10.1038/nri2691
87. Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity (1998) 8(3):383–90. doi: 10.1016/s1074-7613(00)80543-9
88. Kupz A, Guarda G, Gebhardt T, Sander LE, Short KR, Diavatopoulos DA, et al. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8+ T cells. Nat Immunol (2012) 13:162–9. doi: 10.1038/ni.2195
89. Vermeire K, Heremans H, Vandeputte M, Huang S, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol (1997) 158(11):5507–13.
90. Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC. CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun (1999) 67(6):2804–9. doi: 10.1128/IAI.67.6.2804-2809.1999
91. Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, et al. IFN-γ stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest (2007) 117:122–32. doi: 10.1172/JCI30074
92. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: An Effector CD4 T Cell Lineage with Regulatory T Cell Ties. Immunity (2006) 24(6):677–88. doi: 10.1016/j.immuni.2006.06.002
93. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
94. Yago T, Nanke Y, Ichikawa N, Kobashigawa T, Mogi M, Kamatani N, et al. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-α antibody: A novel mechanism of osteoclastogenesis by IL-17. J Cell Biochem (2009) 108(4):947–55. doi: 10.1002/jcb.22326
95. Yi YS. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Kor J Physiol Pharmacol (2018) 22:1–15. doi: 10.4196/kjpp.2018.22.1.1
96. von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol (2013) 31:73–106. doi: 10.1146/annurev-immunol-032712-095944
97. Snouwaert JN, Nguyen M, Repenning PW, Dye R, Livingston EW, Kovarova M, et al. An NLRP3 Mutation Causes Arthropathy and Osteoporosis in Humanized Mice. Cell Rep (2016) 17:3077–88. doi: 10.1016/j.celrep.2016.11.052
98. Jenko B, Praprotnik S, Tomšic M, Dolžan V. NLRP3 and Polymorphisms Influence Higher Disease Activity in Rheumatoid Arthritis. J Med Biochem (2016) 35:319–23. doi: 10.1515/jomb-2016-0008
99. Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Eyre S, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheumatic Dis (2014) 73:1202–10. doi: 10.1136/annrheumdis-2013-203276
100. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature (2014) 512:69–73. doi: 10.1038/nature13322
101. Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10:210–5. doi: 10.1038/nri2725
102. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem (2007) 282:2871–9. doi: 10.1074/jbc.M608083200
103. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (New York NY) (2008) 320:674–7. doi: 10.1126/science.1156995
104. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxidants Redox Signaling (2015) 22:1111–29. doi: 10.1089/ars.2014.5994
105. Abais JM, Zhang C, Xia M, Liu Q, Gehr TW, Boini KM, et al. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxidants Redox Signaling (2013) 18:1537–48. doi: 10.1089/ars.2012.4666
106. Callaway DA, Jiang JX. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J Bone Min Metab (2015) 33:359–70. doi: 10.1007/s00774-015-0656-4
107. Schröder K. NADPH oxidases in bone homeostasis and osteoporosis. Cell Mol Life Sci (2015) 72:25–38. doi: 10.1007/s00018-014-1712-2
108. Yoshida K, Okamura H, Hiroshima Y, Abe K, Kido J-I, Shinohara Y, et al. PKR induces the expression of NLRP3 by regulating the NF-κB pathway in Porphyromonas gingivalis-infected osteoblasts. Exp Cell Res (2017) 354:57–64. doi: 10.1016/j.yexcr.2017.03.028
109. Ran S, Chu M, Gu S, Wang J, Liang J. Enterococcus faecalis induces apoptosis and pyroptosis of human osteoblastic MG63 cells via the NLRP3 inflammasome. Int Endod J (2019) 52:44–53. doi: 10.1111/iej.12965
110. Zhu X, Zhang K, Lu K, Shi T, Shen S, Chen X, et al. Inhibition of pyroptosis attenuates Staphylococcus aureus-induced bone injury in traumatic osteomyelitis. Ann Transl Med (2019) 7:170–0. doi: 10.21037/atm.2019.03.40
111. Greene E, Flees J, Dhamad A, Alrubaye A, Hennigan S, Pleimann J, et al. Double-Stranded RNA Is a Novel Molecular Target in Osteomyelitis Pathogenesis: A Translational Avian Model for Human Bacterial Chondronecrosis with Osteomyelitis. Am J Pathol (2019) 189:2077–89. doi: 10.1016/j.ajpath.2019.06.013
112. Xu L, Shen L, Yu X, Li P, Wang Q, Chengqian L. Effects of irisin on osteoblast apoptosis and osteoporosis in postmenopausal osteoporosis rats through upregulating Nrf2 and inhibiting NLRP3 inflammasome. Exp Ther Med (2020) 19(2):1084–90. doi: 10.3892/etm.2019.8313
113. Xu L, Zhang L, Wang Z, Li C, Li S, Li L, et al. Melatonin Suppresses Estrogen Deficiency-Induced Osteoporosis and Promotes Osteoblastogenesis by Inactivating the NLRP3 Inflammasome. Calcif Tissue Int (2018) 103:400–10. doi: 10.1007/s00223-018-0428-y
114. Faienza MF, Chiarito M, D’Amato G, Colaianni G, Colucci S, Grano M, et al. Monoclonal antibodies for treating osteoporosis. Expert Opin Biol Ther (2018) 18:149–57. doi: 10.1080/14712598.2018.1401607
115. Elshaier AM, Hakimiyan AA, Rappoport L, Rueger DC, Chubinskaya S. Effect of interleukin-1beta on osteogenic protein 1-induced signaling in adult human articular chondrocytes. Arthritis Rheumatol (2009) 60:143–54. doi: 10.1002/art.24151
116. Liu D, Yao S, Wise GE. MyD88 expression in the rat dental follicle: implications for osteoclastogenesis and tooth eruption. Eur J Oral Sci (2010) 118:333–41. doi: 10.1111/j.1600-0722.2010.00751.x
117. An Y, Zhang H, Wang C, Jiao F, Xu H, Wang X, et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J (2019) 33:12515–27. doi: 10.1096/fj.201802805RR
118. Pei J, Fan L, Nan K, Li J, Zhibin S, Dang X, et al. Excessive Activation of TLR4/NF-κB Interactively Suppresses the Canonical Wnt/β-catenin Pathway and Induces SANFH in SD Rats. Sci Rep (2017) 7:11928–8. doi: 10.1038/s41598-017-12196-8
119. Queiroz-Junior CM, Silva MJ, Corrêa JD, Madeira MF, Garlet TP, Garlet GP, et al. A controversial role for IL-12 in immune response and bone resorption at apical periodontal sites. Clin Dev Immunol (2010) 2010:327417. doi: 10.1155/2010/327417
120. Miossec P. Interleukin-17 in rheumatoid arthritis: if T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheumatol (2003) 48:594–601. doi: 10.1002/art.10816
Keywords: pyroptosis, osteoporosis, IL-1β, IL-18, NLRP3
Citation: Tao Z, Wang J, Wen K, Yao R, Da W, Zhou S, Meng Y, Qiu S, Yang K, Zhu Y and Tao L (2021) Pyroptosis in Osteoblasts: A Novel Hypothesis Underlying the Pathogenesis of Osteoporosis. Front. Endocrinol. 11:548812. doi: 10.3389/fendo.2020.548812
Received: 04 May 2020; Accepted: 24 November 2020;
Published: 08 January 2021.
Edited by:
Jonathan H. Tobias, University of Bristol, United KingdomReviewed by:
Maria Felicia Faienza, University of Bari Aldo Moro, ItalySudip Sen, All India Institute of Medical Sciences, India
Copyright © 2021 Tao, Wang, Wen, Yao, Da, Zhou, Meng, Qiu, Yang, Zhu and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Tao, taolindr@163.com
†These authors have contributed equally to this work