 Bertrand Duvillié1,2,3,4,5*
Bertrand Duvillié1,2,3,4,5* Rayane Kourdoughli1,2,3,4,5Sabine Druillennec1,2,3,4,5Alain Eychène1,2,3,4,5Celio Pouponnot1,2,3,4,5
Rayane Kourdoughli1,2,3,4,5Sabine Druillennec1,2,3,4,5Alain Eychène1,2,3,4,5Celio Pouponnot1,2,3,4,5- 1Department of Signaling, Radiobiology and Cancer, Institut Curie, Orsay, France
- 2INSERM U1021, Centre Universitaire, Orsay, France
- 3CNRS UMR 3347, Centre Universitaire, Orsay, France
- 4Université Paris-Saclay, Orsay, France
- 5PSL Research University, Paris, France
Epidemiologic analyses have shed light on an association between type 2 diabetes (T2D) and pancreatic ductal adenocarcinoma (PDAC). Recent data also suggest a potential relationship between T2D and insulinoma. Under rare circumstances, type 1 diabetes (T1D) can also be implicated in tumorigenesis. The biological mechanisms underlying such relationships are extremely complex. Some genetic factors contributing to the development of T2D are shared with pancreatic exocrine and endocrine tumors. Obesity and overweight can also contribute to the initiation and severity of T2D, while aging may influence both endocrine and exocrine tumors. Finally, pharmacological treatments of T2D may have an impact on PDAC. On the other hand, some treatments for insulinoma can trigger diabetes. In the present minireview, we discuss the cellular and molecular mechanisms that could explain these interactions. This analysis may help to define new potential therapeutic strategies.
Introduction
Diabetes is a metabolic disorder characterized by chronic hyperglycemia. Type 1 diabetes (T1D) is less frequent (5.6%) than type 2 diabetes (T2D) and is caused by autoimmune destruction of pancreatic beta-cells. T2D represents 91.2% of diabetes cases and is generally associated with insulin resistance and compensatory hyperinsulinemia, an early indicator of metabolic dysfunction. In the longer term, T2D leads to progressive functional defects of beta-cells. The remaining cases are primarily gestational diabetes, which are represented by hyperglycemia that generally disappears after delivery. Pancreatic adenocarcinoma (PDAC), the most frequent (95%) exocrine pancreatic cancer, is also the most lethal, with a 5-year overall survival of less than 8% (1). Insulinomas are functional neuroendocrine tumors originating from beta-cells. They are generally benign but can metastasize in 5%–10% of cases (2). Interestingly, T2D and pancreatic cancers share several common risk factors, and long standing T2D represents a recognized risk for carcinogenesis. Inversely, PDAC may also be responsible for diabetes (3). This link between diabetes and cancer was first suggested by epidemiologic observations. In particular, PDAC is strongly associated with diabetes (4). Although T2D is a well-established risk factor for PDAC (5), this association is less clear for T1D. Indeed, a clinical prospective study on patients with T1D showed an increased risk of stomach, cervical and endometrium cancers, but only a very modest association with PDAC (6). These differences between T1D and T2D in the risk of developing PDAC may be attributed to differences in insulin levels, which is a risk factor (6). Finally, recent data has suggested a potential association between T2D and insulinoma (7). Despite this evidence, the causative link between T2D and PDAC, as well as insulinoma is not completely understood. The possible molecular mechanisms of this association will be discussed in this review.
Common Determinants of Diabetes and Pancreatic Cancer
T2D and PDAC have common determinants, including aging, obesity, and genetic factors (Figure 1), in addition to some environmental factors that include tobacco smocking, alcohol consumption and low level of physical activity (8). T2D and insulinoma also share causative signals.
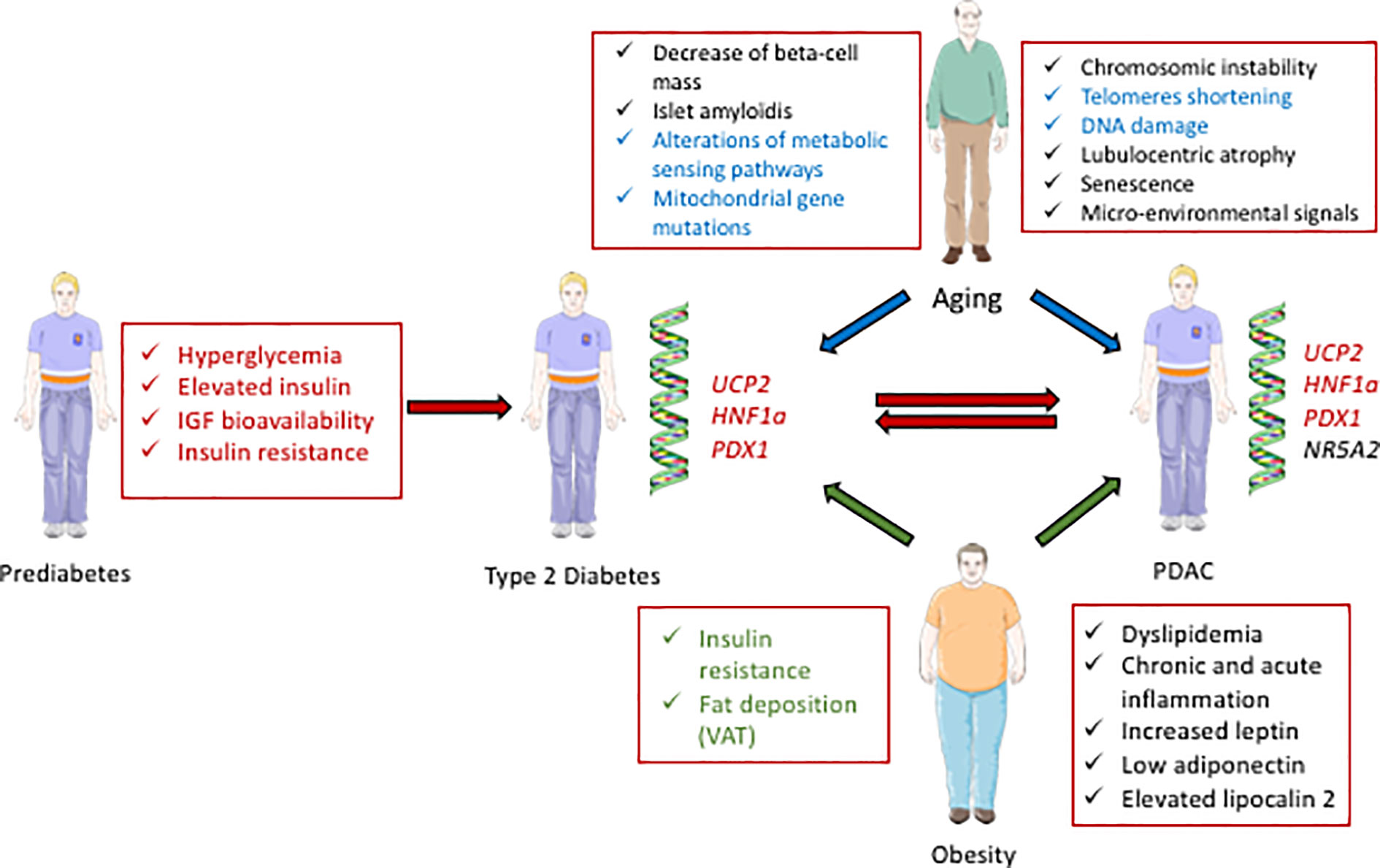
Figure 1 Schematic representation of the interactions between diabetes and PDAC. Some biological parameters occurring during prediabetes, including hyperglycemia, elevated insulin, and IGF bioavailability contribute to diabetes (T2D) and can further lead to PDAC (red arrows). Some genetic factors; UCP2, HNF1a, and PDX1, are also common determinants of diabetes and PDAC (written in red). Some parameters in relation with aging can cause T2D and/or PDAC (blue arrows, and the common determinants for T2D and PDAC development are written in blue). Some characteristics of obesity can contribute to T2D and/or PDAC (green arrows, and the common determinants for T2D and PDAC development are written in green).
Aging
The Effect of Aging in T2D
Aging is the time-dependent deterioration of physiological functions affecting both diabetes and PDAC. The incidence of diabetes increases with age: with 1.8 million patients aged 20–39, 11.7 millions at 40–59, to 19.3 millions at 60–79 in the European Union in 2017 (9). Moreover, the mass of islet cells increases during maturation, but slowly decreases after age 40 (10). Islet amyloidis, which is associated with insulin resistance and T2D, is more commonly found in older individuals (11). Finally, metabolic sensing pathways, such as the mTOR, AMP-activated protein kinase, and insulin/insulin-like growth factors (IGFs) pathways (12, 13) are age-dependent. Interestingly, mTOR, a kinase activated by metabolic signaling, also plays an important role in T2D (14) and PDAC (15).
Aging and PDAC
Aging dramatically increases the risk of pancreatic carcinogenesis (16). Indeed, pancreatic intraepithelial neoplasia (PanINs) is one precursor of PDAC. In an autopsy study, high grade PanIN lesions were found more frequently in T2D patients and old individuals, suggesting a role of both aging and diabetes (17). Moreover, PanINs are associated with chromosomal instability, telomere shortnening (18), and DNA damage, which all depend on aging (19). Several cellular mechanisms involved in aging also play an important role in PDAC. For example, lobulocentric atrophy, a combination of atrophy of acinar parenchyma, acinar to ductal metaplasia and fibrosis, promotes proliferation of small ductular structures and PanINs. This process is found in patients at high risk of PDAC and is age-dependent (20). Moreover, senescence is also important in cancer cells: after oncogenic transformation, cells can undergo senescence, with a reduction of their proliferation. However, some malignant cells often escape this process (21). In addition, age-related senescent-associated secretory phenotype (SASP) cells in the stromal micro-environment support cancer progression (21). In the KRASG12D model of PDAC, knock-out of the senescence-inducing factor SIN3B reduced the initiation and progression of pancreatic lesions, while decreasing secretion of the SASP factor IL-1α (22). Moreover, the conditional knock-out of IL-1α also reduces the number of neoplastic lesions. Finally, mitochondrial gene mutations, which accumulate with age, affect cell metabolism. Consequently, a selective growth advantage that promotes cancer is confered to the cells present in the aging environment (23, 24). Importantly, such mitochondrial events also enhance tumor progression in PDAC (25). Together, these data highlight the strong impact of aging in PDAC.
The Role of Aging in Insulinoma
Men1 knock-out mice provide a model of insulinoma, in which tumors develop late. Indeed, early inactivation of Men1 specific to beta-cells leads to multiple insulinoma only by 60 weeks (26), suggesting the requirement of additional somatic events. Notably, in Men1 knock-out tumors, an increase in the number of entire chromosome 11 was also found in insulinomas, and of chromosome 15 in pituitary prolactinomas. Several oncogenes, including c-MYC and ErbB2/Her2/Neu are present in these duplicated regions (26). The age-related penetrance of MEN1 in patients is 7%, 52%, 87%, 98%, 99%, and 100% at 10, 20, 30, 40, 50, and 60 years of age, respectively (27), suggesting that aging also influences tumorigenesis in human MEN1 tumors. Interestingly, different phenotypes in MEN1 monozygotic twins were observed: in (28), both twins developed parathyroidism, but only one had a pancreatic tumor. This observation suggests that one single mutation in MEN1 is insufficient to induce insulinoma. The variant V109G of p27 and inactivating mutations of CDKN1B were shown to influence the clinical phenotype of MEN1 patients (29). Moreover, some cell-cycle regulators, whose expression is age-dependent (30), are also differentially expressed in human beta-cells from insulinoma as compared to healthy tissue (31). For example, p16 is more heavily expressed at the adult stage than in prenatal beta-cells (32). Such expression restricts beta-cell proliferation with aging (30) and also promotes senescence. Interestingly, its expression is considerably reduced in insulinoma cells (31). Together, these data suggest that aging influences beta-cell proliferation, but that insulinoma cells develop a specific proliferation pattern evading this control.
Obesity
Obesity and Diabetes
Obesity, characterized by excessive accumulation of body fat, with a body mass index (BMI) of 30 kg/m2 or greater, is a well-known risk factor of diabetes (Figure 1) (33). Indeed, 87.5% of adults with T2D are also obese or overweight [BMI>25 (34, 35). The first causative link between obesity and T2D is insulin resistance. Indeed, both obesity and insulin resistance precede altered glycemia (36). Moreover, fat deposition has deleterious effects that depend on its anatomical location. The visceral adipose tissue (VAT), located in the abdominal cavity is linked to a higher risk of T2D as compared to subcutaneous adipose tissue (SCAT) (37).
Obesity and PDAC
Infiltration of adipose tissue favors pancreatic precancerous lesions (38). Indeed, in obese patients, fat has an effect on PanIN lesions and PDAC development. Obesity promotes inflammation that activates tumor associated neutrophils, and consequently pancreatic stellate cells, leading to increased desmoplasia and tumor growth (39). Moreover, chronic inflammation can promote EMT in PanIN cells, driving tumor progression and cell dissemination, leading to PDAC (40). Insulin resistance associated with obesity promotes dislypidemia (41), with elevated concentrations of triglycerides (42), and increased cholesterol synthesis (43). Hypertriglyceridemia is the third most common cause of acute pancreatitis (44), which also represents a risk for PDAC (45). Indeed, patients with acute pancreatitis had a 2-fold increased risk of pancreatic cancer when compared to the matched population. Moreover, adipokines, which include leptin, adiponectin, and lipocalin 2, also establish a connection between obesity and PDAC. In animals and humans, the leptin receptor Ob-Rb plays an important role in obesity (46). While leptin is produced by mature adipocytes, human PDAC cell lines and tissues both express the leptin receptor. Overexpression of leptin in an orthotopic model of human pancreatic cancer promotes tumor growth and lymph node metastasis (47), indicating that leptin is a key factor for PDAC. Recently, a possible interconnection between leptin and the Notch pathway, which is responsible for transformation, proliferation, tumor progression, EMT and chemoresistance, was described (48). Adiponectin is found in adipose tissue and its expression is very low in obese subjects. Prospective epidemiologic studies have shown that low concentration of adiponectin is linked to a higher risk of PDAC (49). Interestingly, adiponectin treatment inhibits the proliferation of human pancreatic cancer cells (50). Knocking-down adiponectin receptors abolished these effects, and enhanced the growth of human pancreatic cancer xenografts in nude mice. Moreover, this antiproliferative effect of adiponectin was shown to be mediated by the β-catenin signaling pathway. However, the roles of leptin and adiponectin are still debated, as a higher adiponectin/leptin ratio and lower leptin levels were found in patients with PDAC as compared to controls (51). Finally, lipocalin 2, a protein involved in innate immunity, also plays an important role in the cellular microenvironment that contributes to PDAC. Lipocalin 2 was found to be a regulator of VAT hypertrophy in animals treated with high fat diet (HFD). The deletion of Lipocalin2 decreases PDAC incidence in KRAS-G12D transgenic mice (52). More generally, diet has a strong impact on pancreatic cancer. Recently, Chang et al. showed that HFD dramatically increases the incidence of PDAC in KRAS-G12D mice. Indeed, the PanIN lesions express new genetic variants, suggesting that genetic alterations may participate to this process (53).
Obesity and Insulinoma
Obesity has thus far not been identified as a cause of insulinoma, but the reverse has been described. Insulinoma can be linked to hyperphagia in some cases by induction of hypoglycemia and hunger. This may lead to weight gain in 20%–40% of patients and even to overt obesity (54, 55). Interestingly, the orexigenic hormone ghrelin is associated with obesity (56). Co-expression of ghrelin and its receptor was detected in several pancreatic endocrine tumors, and specifically in insulinoma, but elevated circulating ghrelin is rare in these patients (57).
The Genetic Factors
The Genetics of Diabetes and PDAC
Genome wide association studies (GWAS) have been used to identify relationships between diabetes and pancreatic cancers. Several pancreatic developmental genes, NR5A2, PDX1, and HNF1A, were identified as susceptibility factors for PDAC (Figure 1) (58). Moreover, heterozygous mutations in some of these genes, PDX1 and HNF1α, are also responsible for different monogenic forms of maturity onset diabetes of the young (MODY 4 and MODY 5). Some variants of PDX1 and HNF1α are also associated with increased risks of T2D (59, 60), obesity (61), or hyperglycemia (62). The antioxidant mitochondrial uncoupling protein 2 (UCP2), which controls pancreatic development and insulin secretion (63), is overexpressed in PDAC tumors compared to normal adjacent tissues (64), suggesting that UCP2 overexpression is a biomarker of bad prognosis. However, other recent studies using the pancreatic cancer cell line Mia PACA2 showed that UCP2 inhibits cancer cell proliferation and tumorigenesis (65). This effect is mediated by retrograde mitochondrial signaling on the Warburg effect that reorients mitochondrial function toward oxidative phosphorylation rather than glycolysis. Additional analyses are needed to elucidate the discrepancy between these two studies involving UCP2. Taken together, these data indicate a link between genes controlling pancreas development, diabetes and PDAC.
The Genetic Links Between Diabetes and Insulinoma
Recently, a strong link between T2D and insulinoma has been established (Figure 2). A p.Ser64Phe mutation in MAFA that prevents GSK3-mediated MAFA phosphorylation (42, 44) was identified in 25 individuals from two independent families (7). These patients develop either insulinoma or diabetes with 90% penetrance. Interestingly, the MAF proteins are well established oncoproteins (66) and their tumorigenic activity is regulated by GSK3-mediated phosphorylation in different cancers (67, 68). Moreover, other studies have also revealed a role of MAFA in diabetes. Within the pancreas, MAFA is exclusively expressed in developing and mature beta-cells. MafA activates the insulin promoter in response to glucose, and regulates genes involved in beta-cell function such as glucose transporter 2, glucagon-like peptide 1 receptor and prohormone convertase 1/3 (69). Accordingly, glucose-stimulated insulin secretion (GSIS) is impaired in MafA knock-out mice, and the architecture of the islets is disorganized. Moreover, these mice develop T2D at 50 weeks after birth (70). In humans, data have also established a link between MAFA and different forms of diabetes. Indeed, expression of MAFA is decreased in islets from T2D patients and a polymorphism in MAFA is associated with T1D (71).
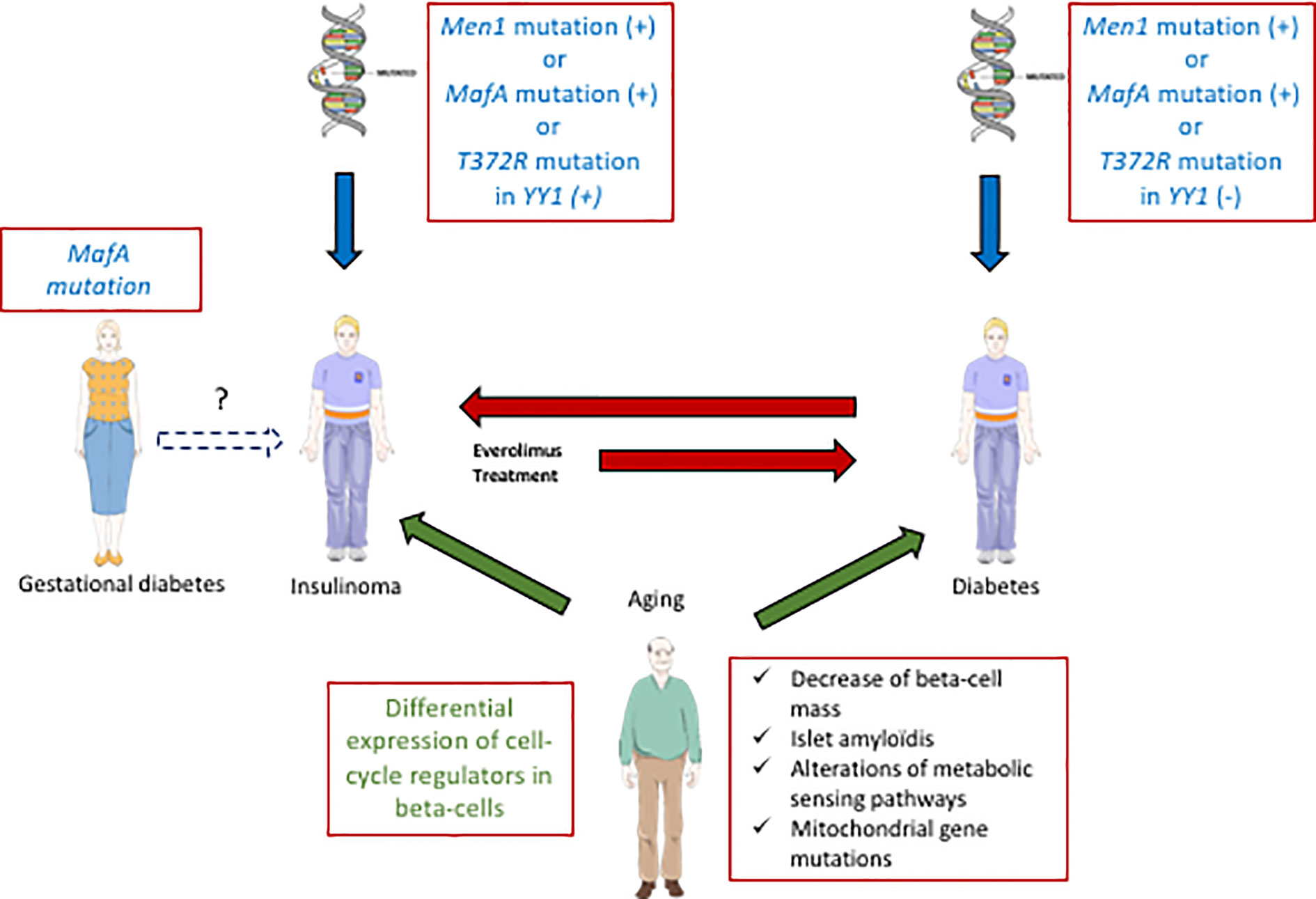
Figure 2 Interactions between diabetes and insulinoma. Some specific MafA mutations can predispose to diabetes (T2D) or insulinoma. In some rare cases with such mutations of MafA, gestational diabetes precedes insulinoma. Moreover, other genes (Men1, YY1) are also involved positively (+) or negatively (−) in the development of diabetes or insulinoma (blue arrows). Aging also contributes to both pathologies (green arrows, common aging determinant for T2D and insulinoma written in green). Some inulinoma treatments (everolimus) can enhance the risk of diabetes (red arrow). On the other hand, exceptional cases of T1D can induce insulinoma (red arrow).
Cases of insulinoma with pre-existing diabetes are very rare (72, 73). In (7), a 27 year-old female patient with a MAFA mutation preventing its phosphorylation by GSK3 first developed gestational diabetes, and subsequent insulinoma at 55 years old. This finding suggests that the MAFA mutation may have caused metabolic disorders in relation to diabetes, that would lead to insulinoma in the long term. Another genetic link between diabetes and insulinoma was suggested by the presence of a recurrent somatic T372R mutation in YY1 (Yin and Yang 1 protein) in 30% of tumors (74). YY1 is a transcription factor that belongs to the GLI-Kruppel class of zinc finger proteins and is a target of mTOR inhibitors. In beta-cells, YY1 regulates the transcription of CXCL12, which also has antidiabetogenic potential (75). In some cases, insulinoma develop in a context of hereditary predisposition to pancreatic endocrine tumors. Indeed, MEN1, type-1 multiple endocrine neoplasm, represents the most frequent predisposition gene for insulinoma (76). Moreover, a relationship between MEN 1 and MAFA genes was also established: altered MEN1 expression was shown to disrupt the MAFA differentiation pathway in human and mice insulinoma cells (77).
Is Diabetes a Risk Factor for Pancreatic Cancers?
Hyperglycemia
Hyperglycemia and PDAC
In T2D, hyperglycemia is caused by excessive hepatic gluconeogenesis, decreased incretin activity, and peripheral glucose uptake, as well as altered insulin signaling. T2D results from a long history of metabolic disorders before diagnosis. These events can cause carcinogenesis and particularly PDAC (Figure 1) (76). Indeed, patients can remain asymptomatic for many years, with undiscovered glucose intolerance and transient hyperglycemia (78). This prediabetic period considerably increases the probability of developing PDAC (79). One possible mechanism is the activation of the TGFβ pathway by glucose, leading to reduced E-cadherin levels in pancreatic ductal cells and to a pronounced mesenchymal phenotype promoting tumor growth and metastasis (5). Hyperglycemia may also increase genomic instability leading to KRAS mutations through activation of O-GlcNAcylation and nucleotides deficiency (80). Finally, the mTOR pathway controls protein synthesis and autophagy, and its deregulation is implicated in diabetes, cancer, and the aging process (81). Interestingly, mTOR inhibition in mouse models of KRAS-dependent PDAC subtypes reduces tumorigenesis (15).
Hyperglycemia and Insulinoma
Glucose controls beta-cell proliferation both in vitro and in vivo (82). In the context of insulin demand, beta-cells undergo hypertrophy or hyperplasia to normalize glycemia (83). One hypothesis is that pre-existing diabetes leads to insulinoma through hyperglycemia. However, such cases are extremely rare. Recently, two patients with pre-existing T1D developed insulinoma (84, 85). In (84), a 31-year-old man experienced T1D for 28 years. Surprisingly, frequent hypoglycemic episodes occurred, leading to the arrest of insulin therapy. After resection, histopathology revealed a grade-2 insulinoma. One unsolved issue is the absence of an autoimmune response against tumor cells in this patient (84). Further analyses will be necessary to investigate the mechanisms involved in tumor progression in such patients.
Insulin and Insulin-Like Growth Factors (IGFs) in Diabetes and Tumorigenesis
The insulin/IGF signalization plays an important role in diabetes. Epidemiological studies have associated serum level variations of IGF-1 (86, 87), IGF-2 (88), IGF binding proteins 1 (89, 90), 2 (91), 3 (86, 90), 4 (91) with T2D. Moreover, obese subjects also exhibit alterations of the IGF system (88, 92, 93) influenced by the presence or absence of T2D (93). Non-diabetic obese subjects have elevated free IGF-1 and IGF-2, total IGF-2, IGF-BP3, and reduced IGF-BP1 and 2 levels. In obese T2D patients, IGFBP-2 is further reduced (93).
PDAC
PDAC originates from both ductal and acinar cells of the pancreas (94), which are exposed to high levels of insulin. Such proxicrine signals promote growth of pancreatic cancer cells (Figure 1) (3). Indeed, the effects of insulin and IGFs 1 and 2 are mediated by the insulin receptor (IR) and IGF1 receptor (IGF1R) (95, 96). As previously discussed, obesity and T2D are associated with increased risk of PDAC. These metabolic disorders are characterized by insulin resistance, compensatory overproduction of insulin and increased bioavailability of IGF-1 (97). To examine the role of insulin in PDAC initiation, Ptf1aCreERLSL-KRASG12DIns1+/−Ins2−/− mice, which have a sustain reduction of insulin but no altered glycemia, were used (98). Mice with reduced insulin had a significant decrease in the number of PanINs and pancreatic tumors when compared to controls. Thus, these results demonstrate that insulin regulates PDAC development. Interestingly, altered expression of the tumor suppressor p53, observed in 50% to 75% of PDAC (99), was shown to stimulate the insulin/IGF1 pathway (100). Moreover, polymorphisms in the IGF genes have been associated with decreased survival of patients with PDAC (101). Taken together, these data strongly support a role of the insulin/IGF axis in pancreatic cancer.
Insulinoma
In animals and human, some associations between IGF2 and diabetes have been shown. In particular, IGF2 overexpression in transgenic mice leads to beta-cell dysfunction (102), by inducing beta-cell de-differentiation and reticulum stress. Moreover, in a mouse model of multistage carcinogenesis induced by the SV40 large T antigen in pancreatic beta-cells, IGF2 was increased and contributed to insulinoma development (103). Recently, studies have shown that the IGF pathway is activated in insulinoma (104). Glutamine can also stimulate biosynthesis and secretion of IGF2 in mouse insulinoma cells, which regulate beta-cell mass and function in an autocrine manner (105). Interestingly, hypermethylation of the differentially methylated region 2 of IGF2 was discovered in human insulinoma, leading to loss of imprinting and overexpression of IGF2 (106). Finally, IGF2 overexpression was also detected in Men1-mutated mouse insulinoma (107). IGF signaling thus appear to be an important hallmark of insulinoma.
Diabetes Treatment to Prevent Pancreatic Cancers?
PDAC
Metformin (108) is the most frequently prescribed, first-line treatment drug for T2D (109). Metformin decreases glycemia by lowering hepatic gluconeogenesis and improves insulin sensitivity by promoting glucose uptake in skeletal muscle and adipose tissue. Several epidemiological studies have demonstrated that metformin administration reduces incidence, recurrence and mortality of pancreatic cancer in diabetic patients (110, 111). Clinical trials using metformin in combination with other drugs used to treat PDAC are actually under investigation (https://clinicaltrials.gov/ct2/results?term=metformin&cond=Pancreatic+Cancer). To delineate the molecular mechanisms involved in this protective effect of metformin, animal models were used. Metformin significantly decreased the incidence of PDAC promoted by diet-induced obesity in the conditional KRAS-G12D knock-in mouse (112). Together, these findings demonstrate that metformin, a treatment for diabetes, represents an important pharmacological tool for PDAC prevention, strengthening the link between these two pathologies. Moreover, AdipoRon, which acts as an adiponectin receptor agonist (113), has antidiabetic properties and can inhibit tumor growth of pancreatic cancer cells MIAPACa-2 in xenografts. AdipoRon can also induce cell death in cells derived from PDAC patients (114). These data suggest that AdipoRon could be a therapeutic agent for both diabetes and PDAC. Finally, several studies analyzed the antidiabetic and protective effects of aspirin against PDAC. Indeed, inflammation is a hallmark of T2D, and aspirin reduces inflammation by regulating T-cell function (115). Interestingly, clinical analysis of a subgroup of patients with diabetes showed a protective role of aspirin against PDAC (116). However, the effects of aspirin against PDAC seem to be heterogenous and controversial (116–118). Further analysis is thus required to better understand these effects.
Insulinoma
Anti-cancer drugs are used to treat insulinoma. Recently, a combination of mTOR inhibitors and streptozotocin was shown to have synergistic antitumor effects in insulinoma cells, both in vitro and in vivo (119). Everolimus, an mTOR inhibitor, was successfully used to treat advanced pancreatic neuroendocrine tumors in a phase 3 clinical trial (120) (RADIANT-3 ClinicalTrials.gov number, NCT00510068). However, other data indicate that such anti-cancer therapy also has endocrine side effects, such as increased plasma triglycerides, LDL cholesterol, and high incidence of hyperglycemia (121). Thus, despite its benefits in cancer, this treatment may enhance the risk of diabetes.
Conclusion
Pre-clinical and clinical data provide clear evidence of common characteristics shared by T2D and PDAC, as well as T2D and insulinoma. The association between diabetes and PDAC is frequent, while it is more unusual between diabetes and insulinoma. Some specific gene mutations contribute to both T2D and insulinoma, strengthening the link between these diseases, while others mutations have opposite effects on T2D and insulinoma. Diabetes and PDAC share several metabolical disorders, that are also found during obesity. Accordingly, obesity often contributes to PDAC initiation, whereas obesity is a consequence of insulinoma. Understanding the relation between T2D and PDAC and between T2D and insulinoma may have important consequences. Indeed, treatments of T2D can limit PDAC progression, while treatment for insulinoma may induce T2D. These important findings should be taken into consideration to develop new pharmacological strategies to limit tumor progression.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
BD is supported by Société Francophone du Diabète (SFD) (Grant number 26866).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
For the figures, SERVIER MEDICAL ART was used. We thank Eleanor Hawkins for proofreading our manuscript.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018: Cancer Statistics, 2018. CA: A Cancer J Clinicians (2018) 68:7–30. doi: 10.3322/caac.21442
2. Halfdanarson TR, Rubin J, Farnell MB, Grant CS, Petersen GM. Pancreatic endocrine neoplasms: epidemiology and prognosis of pancreatic endocrine tumors. Endoc Relat Cancer (2008) 15:409–27. doi: 10.1677/ERC-07-0221
3. Andersen DK, Korc M, Petersen GM, Eibl G, Li D, Rickels MR, et al. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes (2017) 66:1103–10. doi: 10.2337/db16-1477
4. Aggarwal G, Kamada P, Chari ST. Prevalence of Diabetes Mellitus in Pancreatic Cancer Compared to Common Cancers. Pancreas (2013) 42:198–201. doi: 10.1097/MPA.0b013e3182592c96
5. Rahn S, Zimmermann V, Viol F, Knaack H, Stemmer K, Peters L, et al. Diabetes as risk factor for pancreatic cancer: Hyperglycemia promotes epithelial-mesenchymal-transition and stem cell properties in pancreatic ductal epithelial cells. Cancer Lett (2018) 415:129–50. doi: 10.1016/j.canlet.2017.12.004
6. Zendehdel K. Cancer Incidence in Patients With Type 1 Diabetes Mellitus: A Population-Based Cohort Study in Sweden. Cancer Spectrum Knowledge Environ (2003) 95:1797–800. doi: 10.1093/jnci/djg105
7. Iacovazzo D, Flanagan SE, Walker E, Quezado R, de Sousa Barros FA, Caswell R, et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc Natl Acad Sci USA (2018) 115:1027–32. doi: 10.1073/pnas.1712262115
8. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and Cancer: A Consensus Report. CA: A Cancer J Clinicians (2010) 60:207–21. doi: 10.3322/caac.20078
9. OECD/EU. Health at a Glance: Europe 2018: State of Health in the EU Cycle. Paris: OECD Publishing (2018). doi: 10.1787/health_glance_eur-2018-en
10. Mizukami H, Takahashi K, Inaba W, Osonoi S, Kamata K, Tsuboi K, et al. Age-associated changes of islet endocrine cells and the effects of body mass index in Japanese. J Diabetes Invest (2014) 5:38–47. doi: 10.1111/jdi.12118
11. Campbell F, Verbeke CS. Pathology of the Pancreas. London: Springer London (2013). doi: 10.1007/978-1-4471-2449-8
12. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature (2013) 493:338–45. doi: 10.1038/nature11861
13. Kennedy BK, Lamming DW. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab (2016) 23:990–1003. doi: 10.1016/j.cmet.2016.05.009
14. Guillén C, Benito M. mTORC1 Overactivation as a Key Aging Factor in the Progression to Type 2 Diabetes Mellitus. Front Endocrinol (2018) 9:621. doi: 10.3389/fendo.2018.00621
15. Iriana S, Ahmed S, Gong J, Annamalai AA, Tuli R, Hendifar AE. Targeting mTOR in Pancreatic Ductal Adenocarcinoma. Front Oncol (2016) 6:99. doi: 10.3389/fonc.2016.00099
16. Matsuda Y. Age-related morphological changes in the pancreas and their association with pancreatic carcinogenesis. Pathol Int (2019) 69:450–62. doi: 10.1111/pin.12837
17. Matsuda Y, Furukawa T, Yachida S, Nishimura M, Seki A, Nonaka K, et al. The Prevalence and Clinicopathological Characteristics of High-Grade Pancreatic Intraepithelial Neoplasia: Autopsy Study Evaluating the Entire Pancreatic Parenchyma. Pancreas (2017) 46:658–64. doi: 10.1097/MPA.0000000000000786
18. van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol (2002) 161:1541–7. doi: 10.1016/S0002-9440(10)64432-X
19. Aunan JR, Cho WC, Søreide K. The Biology of Aging and Cancer: A Brief Overview of Shared and Divergent Molecular Hallmarks. A&D (2017) 8:628. doi: 10.14336/AD.2017.0103
20. Shi C, Hruban RH, Klein AP. Familial pancreatic cancer. Arch Pathol Lab Med (2009) 133:365–74. doi: 10.1043/1543-2165-133.3.365
21. Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer (2020) 20:89–106. doi: 10.1038/s41568-019-0222-9
22. Rielland M, Cantor DJ, Graveline R, Hajdu C, Mara L, Diaz B de D, et al. Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression. J Clin Invest (2014) 124:2125–35. doi: 10.1172/JCI72619
23. Rozhok AI, Salstrom JL, DeGregori J. Stochastic modeling indicates that aging and somatic evolution in the hematopoietic system are driven by non-cell-autonomous processes. Aging (2014) 6:1033–48. doi: 10.18632/aging.100707
24. Tidwell TR, Søreide K, Hagland HR. Aging, Metabolism, and Cancer Development: from Peto’s Paradox to the Warburg Effect. A&D (2017) 8:662. doi: 10.14336/AD.2017.0713
25. Hopkins JF, Denroche RE, Aguiar JA, Notta F, Connor AA, Wilson JM, et al. Mutations in Mitochondrial DNA From Pancreatic Ductal Adenocarcinomas Associate With Survival Times of Patients and Accumulate as Tumors Progress. Gastroenterology (2018) 154:1620–4. doi: 10.1053/j.gastro.2018.01.029
26. Crabtree JS, Scacheri PC, Ward JM, McNally SR, Swain GP, Montagna C, et al. Of Mice and MEN1: Insulinomas in a Conditional Mouse Knockout. Mol Cell Biol (2003) 23:6075–85. doi: 10.1128/MCB.23.17.6075-6085.2003
27. Bassett JH, Forbes SA, Pannett AA, Lloyd SE, Christie PT, Wooding C, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet (1998) 62:232–44. doi: 10.1086/301729
28. Namihira H, Sato M, Miyauchi A, Ohye H, Matsubara S, Bhuiyan MM, et al. Different phenotypes of multiple endocrine neoplasia type 1 (MEN1) in monozygotic twins found in a Japanese MEN1 family with MEN1 gene mutation. Endocr J (2000) 47:37–43. doi: 10.1507/endocrj.47.37
29. Longuini VC, Lourenço DM, Sekiya T, Meirelles O, Goncalves TD, Coutinho FL, et al. Association between the p27 rs2066827 variant and tumor multiplicity in patients harboring MEN1 germline mutations. Eur J Endocrinol (2014) 171:335–42. doi: 10.1530/EJE-14-0130
30. Köhler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of β-cell replication in the prenatal and postnatal human pancreas. Am J Physiol-Endocrinol Metab (2011) 300:E221–30. doi: 10.1152/ajpendo.00496.2010
31. Ueberberg S, Tannapfel A, Schenker P, Viebahn R, Uhl W, Schneider S, et al. Differential expression of cell-cycle regulators in human beta-cells derived from insulinoma tissue. Metab Clin Exp (2016) 65:736–46. doi: 10.1016/j.metabol.2016.02.007
32. Helman A, Avrahami D, Klochendler A, Glaser B, Kaestner KH, Ben-Porath I, et al. Effects of ageing and senescence on pancreatic β-cell function. Diabetes Obes Metab (2016) 18 Suppl 1:58–62. doi: 10.1111/dom.12719
33. Ozcan U. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science (2004) 306:457–61. doi: 10.1126/science.1103160
34. World Health Organization. Obesity: preventing and managing the global epidemic: report of a WHO consultation. Geneva: World Health Organization (2000).
35. Malone JI, Hansen BC. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatr Diabetes (2019) 20:5–9. doi: 10.1111/pedi.12787
36. Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev (1995) 75:473–86. doi: 10.1152/physrev.1995.75.3.473
37. Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev (2010) 11:11–8. doi: 10.1111/j.1467-789X.2009.00623.x
38. Rebours V, Gaujoux S, d’Assignies G, Sauvanet A, Ruszniewski P, Levy P, et al. Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN). Clin Cancer Res (2015) 21:3522–8. doi: 10.1158/1078-0432.CCR-14-2385
39. Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discovery (2016) 6:852–69. doi: 10.1158/2159-8290.CD-15-1177
40. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell (2012) 148:349–61. doi: 10.1016/j.cell.2011.11.025
41. Klop B, Elte J, Cabezas M. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients (2013) 5:1218–40. doi: 10.3390/nu5041218
42. Otvos JD, Mora S, Shalaurova I, Greenland P, Mackey RH, Goff DC. Clinical implications of discordance between low-density lipoprotein cholesterol and particle number. J Clin Lipidol (2011) 5:105–13. doi: 10.1016/j.jacl.2011.02.001
43. Pihlajamäki J, Gylling H, Miettinen TA, Laakso M. Insulin resistance is associated with increased cholesterol synthesis and decreased cholesterol absorption in normoglycemic men. J Lipid Res (2004) 45:507–12. doi: 10.1194/jlr.M300368-JLR200
44. Mosztbacher D, Hanák L, Farkas N, Szentesi A, Mikó A, Bajor J, et al. Hypertriglyceridemia-induced acute pancreatitis: A prospective, multicenter, international cohort analysis of 716 acute pancreatitis cases. Pancreatology (2020) 20:608–16. doi: 10.1016/j.pan.2020.03.018
45. Kirkegård J, Cronin-Fenton D, Heide-Jørgensen U, Mortensen FV. Acute Pancreatitis and Pancreatic Cancer Risk: A Nationwide Matched-Cohort Study in Denmark. Gastroenterology (2018) 154:1729–36. doi: 10.1053/j.gastro.2018.02.011
46. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi: 10.1038/372425a0
47. Fan Y, Gan Y, Shen Y, Cai X, Song Y, Zhao F, et al. Leptin signaling enhances cell invasion and promotes the metastasis of human pancreatic cancer via increasing MMP-13 production. Oncotarget (2015) 6:16120–34. doi: 10.18632/oncotarget.3878
48. Cascetta P, Cavaliere A, Piro G, Torroni L, Santoro R, Tortora G, et al. Pancreatic Cancer and Obesity: Molecular Mechanisms of Cell Transformation and Chemoresistance. IJMS (2018) 19:3331. doi: 10.3390/ijms19113331
49. Grote VA, Rohrmann S, Dossus L, Nieters A, Halkjaer J, Tjønneland A, et al. The association of circulating adiponectin levels with pancreatic cancer risk: A study within the prospective EPIC cohort. Int J Cancer (2012) 130:2428–37. doi: 10.1002/ijc.26244
50. Jiang J, Fan Y, Zhang W, Shen Y, Liu T, Yao M, et al. Adiponectin Suppresses Human Pancreatic Cancer Growth through Attenuating the β-Catenin Signaling Pathway. Int J Biol Sci (2019) 15:253–64. doi: 10.7150/ijbs.27420
51. Krechler T, Zeman M, Vecka M, Macasek J, Jachymova M, Zima T, et al. Leptin and adiponectin in pancreatic cancer: connection with diabetes mellitus. neo (2011) 58:58–64. doi: 10.4149/neo_2011_01_58
52. Gomez-Chou SB, Swidnicka-Siergiejko AK, Badi N, Chavez-Tomar M, Lesinski GB, Bekaii-Saab T, et al. Lipocalin-2 Promotes Pancreatic Ductal Adenocarcinoma by Regulating Inflammation in the Tumor Microenvironment. Cancer Res (2017) 77:2647–60. doi: 10.1158/0008-5472.CAN-16-1986
53. Chang H-H, Moro A, Takakura K, Su H-Y, Mo A, Nakanishi M, et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PloS One (2017) 12:e0184455. doi: 10.1371/journal.pone.0184455
54. Bonfig W, Kann P, Rothmund M, Schwarz HP. Recurrent hypoglycemic seizures and obesity: delayed diagnosis of an insulinoma in a 15 year-old boy–final diagnostic localization with endosonography. J Pediatr Endocrinol Metab (2007) 20:1035–8. doi: 10.1515/jpem.2007.20.9.1035
55. Mathur A, Gorden P, Libutti SK. Insulinoma. Surg Clin North Am (2009) 89:1105–21. doi: 10.1016/j.suc.2009.06.009
56. Cummings DE. Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol Behav (2006) 89:71–84. doi: 10.1016/j.physbeh.2006.05.022
57. Ekeblad S, Lejonklou MH, Grimfjärd P, Johansson T, Eriksson B, Grimelius L, et al. Co-expression of ghrelin and its receptor in pancreatic endocrine tumours. Clin Endocrinol (2006) 66(1):115–22 doi: 10.1111/j.1365-2265.2006.02695.x
58. Li D, Duell EJ, Yu K, Risch HA, Olson SH, Kooperberg C, et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis (2012) 33:1384–90. doi: 10.1093/carcin/bgs151
59. Holmkvist J, Cervin C, Lyssenko V, Winckler W, Anevski D, Cilio C, et al. Common variants in HNF-1 α and risk of type 2 diabetes. Diabetologia (2006) 49:2882–91. doi: 10.1007/s00125-006-0450-x
60. Tallapragada DSP, Bhaskar S, Chandak GR. New insights from monogenic diabetes for “common” type 2 diabetes. Front Genet (2015) 6:251. doi: 10.3389/fgene.2015.00251
61. MAGIC, on behalf of Procardis Consortium, Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet (2010) 42:937–48. doi: 10.1038/ng.686
62. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, The Multiple Tissue Human Expression Resource (MUTHER) Consortium, Manning AK, Hivert M-F, Scott RA, Grimsby JL, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet (2012) 44:659–69. doi: 10.1038/ng.2274
63. Broche B, Ben Fradj S, Aguilar E, Sancerni T, Bénard M, Makaci F, et al. Mitochondrial Protein UCP2 Controls Pancreas Development. Diabetes (2018) 67:78–84. doi: 10.2337/db17-0118
64. Donadelli M, Dando I, Pozza ED, Palmieri M. Mitochondrial uncoupling protein 2 and pancreatic cancer: A new potential target therapy. WJG (2015) 21:3232–8. doi: 10.3748/wjg.v21.i11.3232
65. Esteves P, Pecqueur C, Ransy C, Esnous C, Lenoir V, Bouillaud F, et al. Mitochondrial Retrograde Signaling Mediated by UCP2 Inhibits Cancer Cell Proliferation and Tumorigenesis. Cancer Res (2014) 74:3971–82. doi: 10.1158/0008-5472.CAN-13-3383
66. Eychène A, Rocques N, Pouponnot C. A new MAFia in cancer. Nat Rev Cancer (2008) 8:683–93. doi: 10.1038/nrc2460
67. Herath NI, Rocques N, Garancher A, Eychène A, Pouponnot C. GSK3-mediated MAF phosphorylation in multiple myeloma as a potential therapeutic target. Blood Cancer J (2014) 4:e175–5. doi: 10.1038/bcj.2013.67
68. Rocques N, Abou Zeid N, Sii-Felice K, Lecoin L, Felder-Schmittbuhl M-P, Eychène A, et al. GSK-3-Mediated Phosphorylation Enhances Maf-Transforming Activity. Mol Cell (2007) 28:584–97. doi: 10.1016/j.molcel.2007.11.009
69. Aramata S, Han S-I, Kataoka K. Roles and regulation of transcription factor MafA in islet beta-cells. Endocr J (2007) 54:659–66. doi: 10.1507/endocrj.kr-101
70. Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, et al. MafA Is a Key Regulator of Glucose-Stimulated Insulin Secretion. Mol Cell Biol (2005) 25:4969–76. doi: 10.1128/MCB.25.12.4969-4976.2005
71. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest (2013) 123:3305–16. doi: 10.1172/JCI65390
72. Kamocki ZK, Wodyńska NA, Pryczynicz A. Co-existence of insulinoma and diabetes: A case report. Oncol Lett (2014) 8:1697–700. doi: 10.3892/ol.2014.2338
73. Sakurai A, Aizawa T, Katakura M, Sato Y, Kaneko G, Yoshizawa K, et al. Insulinoma in a Patient with Non-Insulin-Dependent Diabetes Mellitus. Endocr J (1997) 44:473–7. doi: 10.1507/endocrj.44.473
74. Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai J, et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun (2013) 4:2810. doi: 10.1038/ncomms3810
75. Marković J, Grdović N, Dinić S, Karan-Djurašević T, Uskoković A, Arambašić J, et al. PARP-1 and YY1 Are Important Novel Regulators of CXCL12 Gene Transcription in Rat Pancreatic Beta Cells. PloS One (2013) 8:e59679. doi: 10.1371/journal.pone.0059679
76. Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer (2008) 113:1807–43. doi: 10.1002/cncr.23648
77. Hamze Z, Vercherat C, Bernigaud-Lacheretz A, Bazzi W, Bonnavion R, Lu J, et al. Altered MENIN expression disrupts the MAFA differentiation pathway in insulinoma. Endocr-Relat Cancer (2013) 20:833–48. doi: 10.1530/ERC-13-0164
78. Paternoster S, Falasca M. The intricate relationship between diabetes, obesity and pancreatic cancer. Biochim Biophys Acta (BBA) - Rev Cancer (2020) 1873:188326. doi: 10.1016/j.bbcan.2019.188326
79. Pang Y, Kartsonaki C, Guo Y, Bragg F, Yang L, Bian Z, et al. Diabetes, plasma glucose and incidence of pancreatic cancer: A prospective study of 0.5 million C hinese adults and a meta-analysis of 22 cohort studies. Int J Cancer (2017) 140:1781–8. doi: 10.1002/ijc.30599
80. Hu C-M, Tien S-C, Hsieh P-K, Jeng Y-M, Chang M-C, Chang Y-T, et al. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metab (2019) 29:1334–49. doi: 10.1016/j.cmet.2019.02.005
81. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
82. Alonso LC, Yokoe T, Zhang P, Scott DK, Kim SK, O’Donnell CP, et al. Glucose Infusion in Mice: A New Model to Induce -Cell Replication. Diabetes (2007) 56:1792–801. doi: 10.2337/db06-1513
83. Cerf ME. High Fat Programming of β-Cell Failure. Adv Exp Med Biol (2010) 654:77–89. doi: 10.1007/978-90-481-3271-3_5
84. Lablanche S, Chobert-Bakouline M, Risse O, Laverrière M-H, Chabre O, Benhamou P-Y. Malignant insulinoma may arise during the course of type 1 diabetes mellitus: A case report. Diabetes Metab (2015) 41:258–61. doi: 10.1016/j.diabet.2014.08.004
85. Oikawa Y, Katsuki T, Kawasaki M, Hashiguchi A, Mukai K, Handa K, et al. Insulinoma may mask the existence of Type 1 diabetes: Insulinoma may mask Type 1 diabetes. Diabetic Med (2012) 29:e138–41. doi: 10.1111/j.1464-5491.2012.03615.x
86. Drogan D, Schulze MB, Boeing H, Pischon T. Insulin-Like Growth Factor 1 and Insulin-Like Growth Factor–Binding Protein 3 in Relation to the Risk of Type 2 Diabetes Mellitus: Results From the EPIC–Potsdam Study. Am J Epidemiol (2016) 183:553–60. doi: 10.1093/aje/kwv188
87. Sandhu MS, Heald AH, Gibson JM, Cruickshank JK, Dunger DB, Wareham NJ. Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet (2002) 359:1740–5. doi: 10.1016/S0140-6736(02)08655-5
88. Sandhu MS, Gibson JM, Heald AH, Dunger DB, Wareham NJ. Low Circulating IGF-II Concentrations Predict Weight Gain and Obesity in Humans. Diabetes (2003) 52:1403–8. doi: 10.2337/diabetes.52.6.1403
89. Heald AH, Cruickshank JK, Riste LK, Cade JE, Anderson S, Greenhalgh A, et al. Close relation of fasting insulin-like growth factor binding protein-1 (IGFBP-1) with glucose tolerance and cardiovascular risk in two populations. Diabetologia (2001) 44:333–9. doi: 10.1007/s001250051623
90. Rajpathak SN, He M, Sun Q, Kaplan RC, Muzumdar R, Rohan TE, et al. Insulin-Like Growth Factor Axis and Risk of Type 2 Diabetes in Women. Diabetes (2012) 61:2248–54. doi: 10.2337/db11-1488
91. Hjortebjerg R, Laugesen E, Høyem P, Oxvig C, Stausbøl-Grøn B, Knudsen ST, et al. The IGF system in patients with type 2 diabetes: associations with markers of cardiovascular target organ damage. Eur J Endocrinol (2017) 176:521–31. doi: 10.1530/EJE-16-0940
92. Heald AH, Kärvestedt L, Anderson SG, McLaughlin J, Knowles A, Wong L, et al. Low Insulin-like Growth Factor-II Levels Predict Weight Gain in Normal Weight Subjects with Type 2 Diabetes. Am J Med (2006) 119:167.e9–167.e15. doi: 10.1016/j.amjmed.2005.08.001
93. Frystyk J, Skjaerbaek C, Vestbo E, Fisker S, Orskov H. Circulating levels of free insulin-like growth factors in obese subjects: the impact of type 2 diabetes. Diabetes Metab Res Rev (1999) 15:314–22. doi: 10.1002/(sici)1520-7560(199909/10)15:5<314::aid-dmrr56>3.0.co;2-e
94. Xu Y, Liu J, Nipper M, Wang P. Ductal vs. acinar? Recent insights into identifying cell lineage of pancreatic ductal adenocarcinoma. Ann Pancreat Cancer (2019) 2:11–1. doi: 10.21037/apc.2019.06.03
95. Duvillié B, Cordonnier N, Deltour L, Dandoy-Dron F, Itier JM, Monthioux E, et al. Phenotypic alterations in insulin-deficient mutant mice. Proc Natl Acad Sci USA (1997) 94:5137–40. doi: 10.1073/pnas.94.10.5137
96. Leroux L, Desbois P, Lamotte L, Duvillié B, Cordonnier N, Jackerott M, et al. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes (2001) 50 Suppl 1:S150–153. doi: 10.2337/diabetes.50.2007.s150
97. Alemán JO, Eusebi LH, Ricciardiello L, Patidar K, Sanyal AJ, Holt PR. Mechanisms of Obesity-Induced Gastrointestinal Neoplasia. Gastroenterology (2014) 146:357–73. doi: 10.1053/j.gastro.2013.11.051
98. Zhang AMY, Magrill J, de Winter TJJ, Hu X, Skovsø S, Schaeffer DF, et al. Endogenous Hyperinsulinemia Contributes to Pancreatic Cancer Development. Cell Metab (2019) 30:403–4. doi: 10.1016/j.cmet.2019.07.003
99. Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res (1997) 57:1731–4.
100. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol (2010) 20:427–34. doi: 10.1016/j.tcb.2010.03.004
101. Dong X, Javle M, Hess KR, Shroff R, Abbruzzese JL, Li D. Insulin-Like Growth Factor Axis Gene Polymorphisms and Clinical Outcomes in Pancreatic Cancer. Gastroenterology (2010) 139:464–73. doi: 10.1053/j.gastro.2010.04.042
102. Casellas A, Mallol C, Salavert A, Jimenez V, Garcia M, Agudo J, et al. Insulin-like Growth Factor 2 Overexpression Induces β-Cell Dysfunction and Increases Beta-cell Susceptibility to Damage. J Biol Chem (2015) 290:16772–85. doi: 10.1074/jbc.M115.642041
103. Christofori G, Naik P, Hanahan D. Deregulation of both imprinted and expressed alleles of the insulin–like growth factor 2 gene during β–cell tumorigenesis. Nat Genet (1995) 10:196–201. doi: 10.1038/ng0695-196
104. Henfling MER, Perren AA, Schmitt AM, Saddig CM, Starke AA, Riedl RG, et al. The IGF pathway is activated in insulinomas but downregulated in metastatic disease. Endocr-Relat Cancer (2018) 25:1005–18. doi: 10.1530/ERC-18-0222
105. Modi H, Cornu M, Thorens B. Glutamine Stimulates Biosynthesis and Secretion of Insulin-like Growth Factor 2 (IGF2), an Autocrine Regulator of Beta Cell Mass and Function. J Biol Chem (2014) 289:31972–82. doi: 10.1074/jbc.M114.587733
106. Dejeux E, Olaso R, Dousset B, Audebourg A, Gut IG, Terris B, et al. Hypermethylation of the IGF2 differentially methylated region 2 is a specific event in insulinomas leading to loss-of-imprinting and overexpression. Endocr-Relat Cancer (2009) 16:939–52. doi: 10.1677/ERC-08-0331
107. Fontanière S, Tost J, Wierinckx A, Lachuer J, Lu J, Hussein N, et al. Gene expression profiling in insulinomas of Men1 β-cell mutant mice reveals early genetic and epigenetic events involved in pancreatic β-cell tumorigenesis. Endocr-Relat Cancer (2006) 13:1223–36. doi: 10.1677/erc.1.01294
108. Palmer SC, Strippoli GFM. Metformin as first-line treatment for type 2 diabetes. Lancet (2018) 392:120. doi: 10.1016/S0140-6736(18)31541-1
109. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab (2005) 1:15–25. doi: 10.1016/j.cmet.2004.12.003
110. Li D, Yeung SJ, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic Therapies Affect Risk of Pancreatic Cancer. Gastroenterology (2009) 137:482–8. doi: 10.1053/j.gastro.2009.04.013
111. Li X, Li T, Liu Z, Gou S, Wang C. The effect of metformin on survival of patients with pancreatic cancer: a meta-analysis. Sci Rep (2017) 7:5825. doi: 10.1038/s41598-017-06207-x
112. Chang H-H, Moro A, Chou CEN, Dawson DW, French S, Schmidt AI, et al. Metformin Decreases the Incidence of Pancreatic Ductal Adenocarcinoma Promoted by Diet-induced Obesity in the Conditional KrasG12D Mouse Model. Sci Rep (2018) 8:5899. doi: 10.1038/s41598-018-24337-8
113. Sapio L, Nigro E, Ragone A, Salzillo A, Illiano M, Spina A, et al. AdipoRon Affects Cell Cycle Progression and Inhibits Proliferation in Human Osteosarcoma Cells. J Oncol (2020) 2020:1–12. doi: 10.1155/2020/7262479
114. Akimoto M, Maruyama R, Kawabata Y, Tajima Y, Takenaga K. Antidiabetic adiponectin receptor agonist AdipoRon suppresses tumour growth of pancreatic cancer by inducing RIPK1/ERK-dependent necroptosis. Cell Death Dis (2018) 9:804. doi: 10.1038/s41419-018-0851-z
115. Nyambuya TM, Dludla PV, Mxinwa V, Mokgalaboni K, Ngcobo SR, Tiano L, et al. The impact of metformin and aspirin on T-cell mediated inflammation: A systematic review of in vitro and in vivo findings. Life Sci (2020) 255:117854. doi: 10.1016/j.lfs.2020.117854
116. Khalaf N, Yuan C, Hamada T, Cao Y, Babic A, Morales-Oyarvide V, et al. Regular Use of Aspirin or Non-Aspirin Nonsteroidal Anti-Inflammatory Drugs Is Not Associated With Risk of Incident Pancreatic Cancer in Two Large Cohort Studies. Gastroenterology (2018) 154:1380–1390.e5. doi: 10.1053/j.gastro.2017.12.001
117. Sun J, Li Y, Liu L, Jiang Z, Liu G. Aspirin use and pancreatic cancer risk: A systematic review of observational studies. Medicine (2019) 98:e18033. doi: 10.1097/MD.0000000000018033
118. Zhang Y-P, Wan Y-D, Sun Y-L, Li J, Zhu R-T. Aspirin might reduce the incidence of pancreatic cancer: A meta-analysis of observational studies. Sci Rep (2015) 5:15460. doi: 10.1038/srep15460
119. Bollard J, Patte C, Massoma P, Goddard I, Gadot N, Benslama N, et al. Combinatorial Treatment with mTOR Inhibitors and Streptozotocin Leads to Synergistic In Vitro and In Vivo Antitumor Effects in Insulinoma Cells. Mol Cancer Ther (2018) 17:60–72. doi: 10.1158/1535-7163.MCT-17-0325
120. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N Engl J Med (2011) 364:514–23. doi: 10.1056/NEJMoa1009290
Keywords: diabetes, pancreas, cancer, aging, insulinoma, obesity
Citation: Duvillié B, Kourdoughli R, Druillennec S, Eychène A and Pouponnot C (2020) Interplay Between Diabetes and Pancreatic Ductal Adenocarcinoma and Insulinoma: The Role of Aging, Genetic Factors, and Obesity. Front. Endocrinol. 11:563267. doi: 10.3389/fendo.2020.563267
Received: 18 May 2020; Accepted: 14 September 2020;
Published: 30 September 2020.
Edited by:
Jianfeng Liu, Huazhong University of Science and Technology, ChinaReviewed by:
Eva Surmacz, Allysta Pharmaceuticals, Inc., United StatesSilvio Naviglio, University of Campania Luigi Vanvitelli, Italy
Copyright © 2020 Duvillié, Kourdoughli, Druillennec, Eychène and Pouponnot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bertrand Duvillié, bertrand.duvillie@curie.fr