 Mirjana Kocova
Mirjana Kocova Paola Concolino
Paola Concolino Henrik Falhammar
Henrik Falhammar- 1Medical Faculty, University “Cyril & Methodius” Skopje, Skopje, North Macedonia
- 2Dipartimento di Scienze di Laboratorio e Infettivologiche, Unita' Operativa Complessa (UOC) Chimica, Biochimica e Biologia Molecolare Clinica, Fondazione Policlinico Universitario Agostino Gemelli Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
- 3Department of Endocrinology, Karolinska University Hospital, Stockholm, Sweden
- 4Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden
Substantial research has been performed during the last decades on the clinical and genetic variability of congenital adrenal hyperplasia (CAH) and its most common form, 21-hydroxylase deficiency (21OHD). CAH is one of the most prevalent autosomal recessive diseases in humans, and it can be divided into classic—further subdivided into salt wasting (SW) and simple virilizing (SV)—and non-classic (NC) forms. Pathogenic variants of CYP21A2 gene, encoding the 21-hydroxylase enzyme, have been reported with variable prevalence in different populations. NM_000500.9:c.293-13C/A>G (In2G) variant represents the most common CYP21A2 gene changes related to the classic 21OHD form. However, the phenotype of In2G carriers is variable depending on the variant homozygous/heterozygous status and combination with other CYP21A2 pathogenic variants. In addition, identical genotypes, harboring the homozygous In2G variant, can present with variable phenotypes including the SW and SV or rarely NC form of the disease. Here, we analyze and present the clinical aspects, genotype/phenotype correlations, and other characteristics related to the CYP21A2 In2G variant.
Introduction
Congenital adrenal hyperplasia (CAH) is one of the most common autosomal recessive diseases in humans with an incidence, evaluated on neonatal screening in different populations during the last decade or so, of 1:6,084 to 1:26,727 live births (1). It comprises several steroid enzyme deficiencies, among which 21-hydroxylase deficiency (21OHD) (OMIM # 201910) is by far the most common, affecting about 95%–99% of all CAH patients (1–5). In 21OHD, the hormonal disbalances consist of variable low cortisol and aldosterone levels, and compensatory high levels of 17-hydroxyprogesterone (17OHP), which converts to androgens. CAH appears in two clinical forms, classic and non-classic (NC) phenotypes (6). The classic form is further classified as salt-wasting (SW) and simple-virilizing (SV) forms. Patients affected by the SW form have SW with severe dehydration, hypoglycemia, failure to thrive, and hyperandrogenism. This can be clinically recognized early in girls due to atypical genitalia, while boys may first be identified, if not neonatally screened, when presenting with a life-threatening SW crisis within 2 weeks postnatally (2). The SV form has enough aldosterone production to avoid SW crisis and, prior to the introduction of neonatal screening, was identified due to atypical genitalia in girls and in male toddlers due to signs and symptoms of excessive androgen production, even though cases of diagnosis in adulthood occasionally happened (2, 7). The NC form is mild and can appear in numerous variants of the clinical picture, ranging from no signs in the newborn period, to mild virilization later in childhood, up to polycystic ovary syndrome or isolated hyperandrogenism and decreased fertility in adulthood (8). NC 21OHD is sometimes identified in neonatal screening (9), but most cases are identified due to symptoms and signs in adolescence or young adulthood, even though the majority probably never gets diagnosed. NC CAH shows an incidence of 1:200 to 1:1,000 (10, 11).
However, despite the traditional and generally accepted classification of CAH in different forms, due to a large variety of CYP21A2 mutations, the phenotype can have many variants, and it is clear that CAH phenotype represents a continuum between non-classic and classic forms (12). This is important for tailoring appropriate therapy in individual patients. The 21-hydroxylase enzyme is encoded by CYP21A2 gene, and the clinical 21OHD presentation depends upon the combination of pathogenic variants affecting this locus (13). Both CYP21A2 gene and CYP21A1P pseudogene are located on the short arm of chromosome 6 (6p21.3), in the human leukocyte antigen (HLA) class III region of the major histocompatibility (MHC) locus. These genes contain ten exons spaced over 3.4 kb with a sequence homology reaching 98% (12, 14, 15). Intergenic recombination events represent more than 95% of pathogenic variants causing 21OHD. Approximately 75% of the deleterious variants are transferred by small conversions from the pseudogene during meiosis (16, 17). Of the cases of 21OHD, 20%–25% are due to gene deletions, gene duplications, and deletions involving CYP21A2 and other contiguous genes (18). Finally, CYP21A2 pathogenic variants that are not apparently gene conversions account for 5%–10% of CAH alleles in most populations (16, 17).
To date, more than 230 CYP21A2 pathogenic variants have been identified (18). Most patients are compound heterozygous; and in this case, the phenotype correlates with the variant that predicts the higher residual enzyme activity (19). Based on the residual activity of the mutant enzyme, CYP21A2 variants are classified into specific groups (null, A, B, and C) (20, 21). While variants of the null group show 0% enzyme activity during in vitro assay, group A variants preserve a minimal (<1%) residual activity. These two groups are both associated with the SW form of 21OHD. Differently, group B (1%–5% enzyme activity) and group C (20%–50% enzyme activity) variants are related to the SV and NC forms, respectively (12, 22–25). Although there is good agreement between clinical phenotype and patient genotype, it is well-known that some exceptions exist (18–29).
The prevalence of 21OHD is highly variable among populations; some countries show higher (China and India) (19, 20) or lower prevalence (Japan and New Zealand) (3, 21–27). However, in most of the analyzed studies, the In2G variant is found to be, with rare exceptions (30–50), among the most common CYP21A2 pathogenic variants. It is usually related to the SW 21OHD; however, patients with a non-correspondent phenotype have been widely reported. In this review, we collected the most relevant evidence showing the phenotypic variability of the In2G variant.
CYP21A2 In2G Variant
To date, 18 intronic splicing variants, representing 7.7% of all disease-causing variants, have been reported in CYP21A2 gene (18, 51). In silico analysis or functional studies showed that all these variants are associated with the severe form of 21OHD due to the changed reading frame of the gene producing a non-functional enzyme (21, 52, 53). Generally, an intronic splicing variant causes the disruption of the acceptor/donor site, inducing activation of an intronic cryptic acceptor/donor site, retention of a whole intron or part of it, and exon skipping (51). Regarding intron 2 of CYP21A2 gene, five pathogenic splicing variants have been reported (51). Two of these, c.292+1G>A and c.293-2A>G, cause the SW phenotype to disrupt the donor and acceptor splicing sites, respectively (54, 55). Differently, the c.292+5G>A and c.293-7C>G variants were described, in SW patients, as reducing the consensus value for the intron 2 splice donor and acceptor sites, respectively (52, 56).
c.293-13C/A>G (In2G) is the most common splicing variant in CYP21A2 gene. It is usually transferred by microconversion from CYP21A2 pseudogene. At the −13 position, before the end of intron 2, the wild-type nucleotide is A or C. Substitution to G creates an additional splice acceptor site, causing aberrant splicing of intron 2 with retention of 19 intronic nucleotides. This results in a shift in the translational reading frame (21) (Figure 1). The In2G variant is typically related to the SW form of CAH. It is by far the most common CAH mutation in the majority of countries and ethnicities, predominantly Europeans followed by Middle Easterners and Hispanics (27), although there are exceptions (Table 1). In different studies, the prevalence of the In2G variant was between 20.6% and 30.3% (25, 27, 61, 63). In the homozygous form, it is more common in those with European and Middle Eastern ancestries than in Hispanic Americans, Asians, or East Indians (27). Some authors even refer up to 60.4% prevalence of this variant in certain ethnicities (32) (Table 1).
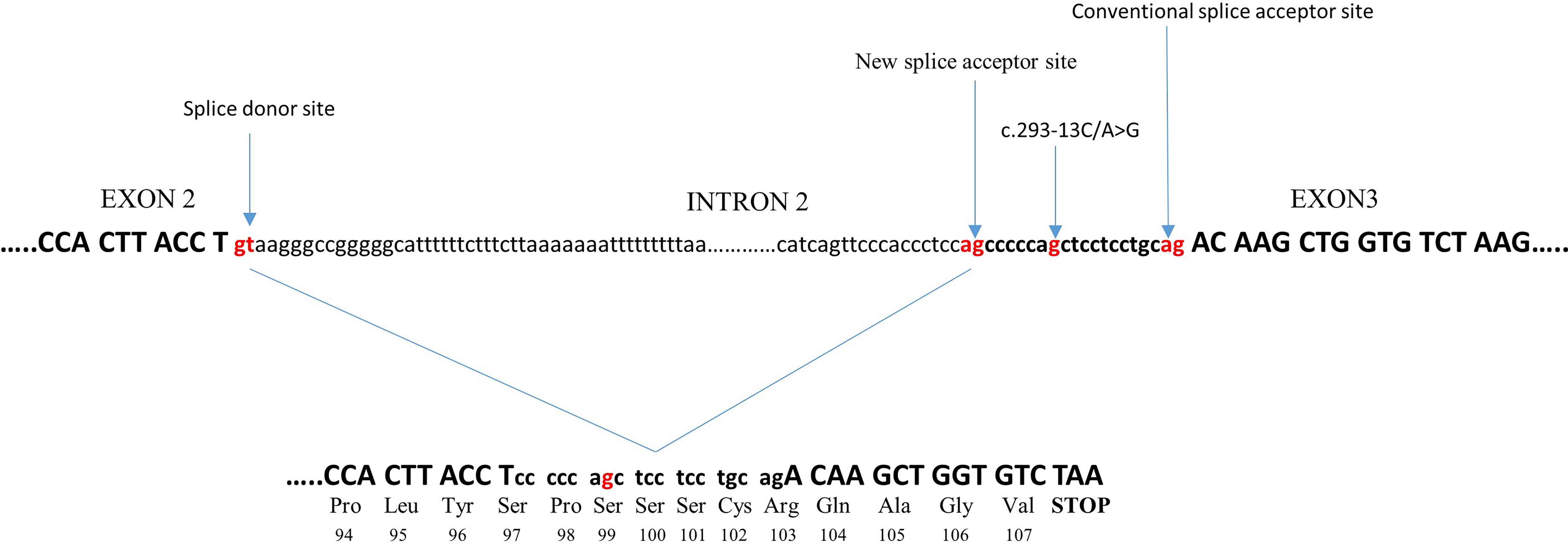
Figure 1 Effect of the c.293-13C/A>G variant on splicing. The c.293-13C/A>G variant, at a −13 position before the end of intron 2, creates an additional splice acceptor site, causing aberrant splicing of intron 2 with retention of 19 intronic nucleotides. This causes a frameshift in the following third exon to generate a stop codon after 107 amino acids, resulting in the production of a truncated protein.

Table 1 Prevalence of In2G variant in different countries.
In some populations, the In2G variant could be considered a founder variant. It is a unique finding in some enclosed populations such as Alaskan and one of the Roma populations from the Balkan region, although the number of explored individuals was rather small (62). Finally, in other populations, such as the Spanish population, the In2G variant appears to be related to recent conversion events (64).
Genotype/Phenotype Correlation in In2G Patients
There is a reasonable genotype/phenotype correlation in 21OHD despite disease-causing variant variability (3, 28–30). As mentioned previously, the phenotype is almost uniformly dependent on the milder variant in the genotype. However, some variants may occasionally confer an unexpected phenotype (27, 63, 65).
In the homozygous status or in trans with another null variant, the In2G variant usually causes severe SW CAH. When combined with a moderate or mild variant, it normally confers SV or NC CAH. However, a specific characteristic of the In2G variant is that the clinical picture might be less severe even in the homozygous form when it can present as an SV or NC phenotype (22, 24, 31, 32).
During the 1990s, the first evidences about the phenotypic heterogeneity related to the In2G variant were reported (66, 67). Witchel et al. compared the clinical and molecular findings in 38 individuals from 21 families. All patients carried two deleterious variants in trans, with the In2G present on at least 1 allele. A comparison of the phenotypic features with the molecular genotypes showed phenotypic heterogeneity extending from classic SW 21OHD to be asymptomatic (28). The authors hypothesized that other sequence variations influenced the competitive splicing signals at the intron 2/exon 3 junctions. However, experimental testing did not support this hypothesis, and the molecular basis of the phenotypic heterogeneity associated with the In2G variant remained to be elucidated (28). A few years later, Schulze et al. suggested that the putative asymptomatic In2G homozygous individuals were incorrectly typed due to the dropout of one allele during PCR amplification (68). Effectively, in the 1990s, it was too challenging to accurately genotype CYP21A2, which still presents as one of the most difficult and error-prone genes, even today. In fact, many old CYP21A2 genotyping results have been found to be incomplete or inaccurate by using up-to-date methodologies. For this reason, it could be necessary to re-evaluate the accuracy of some of the old literature. However, even with the use of more sophisticated techniques for genotyping, patients with the In2G variant and a non-correspondent phenotype have been reported. In a large study by New et al., out of 155 homozygous In2G patients, 143 (92.3%) had the SW form, 11 (7%) had the SV form, and 1 (0.6%) had the NC form (27). Even when the In2G variant was detected in trans with another severe mutation, such as p.(Gln319Ter), still 12% of patients (3/25) presented as SV (27). Finally, the genotype In2G/p.(Val282Leu) was related to the NC phenotype in 96.4% of patients, and only 4 (3.6%) subjects presented a severe phenotype (27). In a recent study by Riedl et al., of 62 In2G homozygous patients, 53 (85.5%) had the SW form, whereas 9 (14.5%) had the SV form (57). DumiK et al. described a comprehensive CYP21A2 mutation analysis in a large cohort of 93 Croatian patients with classic 21OHD (49). The most frequently detected mutation in this population was the In2G variant (34.9%) (Table 1). The concordance between observed and predicted clinical phenotype in Group A (In2G variant) patients was 85% (49). In particular, the authors described two families with genotype–phenotype discordance (49). In the first family, three sisters carried the In2G/In2G genotype. Two of them displayed an SW phenotype and were on hydrocortisone and 9-alpha-fludrohydrocortisone therapy. In contrast, the middle sisters had ambiguous genitalia, high levels of 17OHP and androgens, but repeat measurements of electrolytes, aldosterone, and plasma renin activity (PRA) were within the normal range, excluding the SW phenotype. In the second family, two siblings showed the In2G/p.(Arg358Trp) genotype. The brother was diagnosed with SV 21OHD at 3 years of age due to precocious pseudopuberty, high levels of 17OHP and androgens, and a normal level of aldosterone and PRA. His sister was diagnosed with SW CAH at birth, as she showed ambiguous genitalia, low levels of sodium and aldosterone, and high levels of potassium and PRA. In this case, hydrocortisone and 9-alpha-fludrohydrocortisone were introduced 10 days after birth (49). Also, in Argentinean CAH patients, the In2G variant was reported as the most prevalent mutation (35.2%) (Table 1), and while 83.8% of patients in group A (In2G variant) presented with the SW form, 16.2% showed the SV form of the disease (25). In this regard, also these authors described two siblings with the same genotype (In2G/In2G) but a different phenotype (25). Similar data from Brazilian, Hellenic, and Chinese CAH populations were provided by Carvalho et al., Dracopoulou-Vabouli et al., and Wang et al., respectively (Table 1) (20, 50, 58).
The In2G variant, in homozygous or in trans with a severe CYP21A2 mutation, was also related to the NC form of 21OHD. Bidet et al. analyzed the molecular spectrum of CYP21A2 gene in a large cohort of French NC CAH patients (37). The In2G variant was present on 10.9% of all chromosomes (Table 1), making it the second most frequent mutation in this study. Interestingly, the authors described a mild clinical and biological phenotype, related to the NC form of 21OHD, in a patient homozygous for the In2G variant.
Finally, a peculiar case was reported by Kohn et al. (55). These authors described two affected boys, both carrying the In2G/In2G genotype, who thrived in early infancy but suffered SW crises unusually late in infancy, at 3.5 and 5.5 months. At the onset of symptoms, the children showed hyponatremia, hyperkalemia, dehydration, and acidosis; serum aldosterone was low in spite of markedly elevated PRA. Baseline 17OHP levels were only moderately elevated; however, stimulated levels were consistent with the classic form of 21OHD. The authors speculated that the In2G variant could sometimes be associated with the delayed phenotypic expression of SW CAH and that the variable splicing may modify the clinical manifestations of the disease (55).
Outcomes of In2G Patients
Although all variants causing SW 21OHD induce similar clinical picture, require similar therapy, and produce similar outcomes, some of the specificities of the In2G variant are being confirmed in a number of larger studies. Here, we will mention some of them.
Fertility
Fertility is significantly decreased in all genetic forms of SW CAH in women due to high levels of androgens, problems after genital surgery, decreased sex drive, social adaptation issues such as not having a partner, or non-willingness to bear children (69). Only approximately 25% of women with CAH conceive a child compared with 45% of matched controls (70, 71). They give birth mostly by cesarean section (72%) and are prone to gestational diabetes. Elevated androgen concentrations impair the ability of progesterone to lower the activity of gonadotrophin-releasing hormone/luteinizing hormone (GnRH/LH) pulse generator, causing increased frequency of pulse amplitude of LH over follicle-stimulating hormone (FSH) production and also disrupting endometrial thickening, making the cervical mucus thicker, disrupting ovulation, and impairing embryo implantation, which all lead to impaired fertilization (69). Psychosexual factors also have a role. Women with CAH frequently present with masculine behavior, and approximately one-third do not have sexual interest and fantasies (72). Moreover, homosexuality is more common in women with CAH, and there is a direct relationship between the severity of the genotype and non-heterosexuality (73, 74). For example, in women with the null genotype, 50% had a non-heterosexual orientation; in the In2G genotype, 30%; and in matched controls, only 2% (74). Women with null and In2G genotypes were less often married and had fewer children than women with milder genotypes (75). However, fertility in women with the In2G genotype was better compared with the null genotype (71). Pregnancy in women with SW CAH was normal, and the offspring had a normal weight and development (76). The better fertility in females with In2G might have to do with the dose-dependent effects of prenatal androgens on the development of higher brain functions (74). Females with the null genotype scored lower on sexual function and satisfaction with their sexual life as well as had more genital surgical complications, compared with the In2G genotype (77).
Fertility is also compromised in males with CAH, mostly due to testicular adrenal rest tumor (TART) or sometimes hypogonadotropic hypogonadism (78). However, the remaining testicular tissue is larger, and the amount and quality of semen are better in patients with the In2G variant, although not reaching statistical difference (79), with male SW CAH having an increased number of adopted children (80). Nevertheless, the number of males with at least one biological child was equally low in both the null and In2G genotype groups (80).
Psychiatric Disorders
Research in animal models has demonstrated that sex differences in brain and behavior are induced by steroid hormones during specific, hormone-sensitive developmental periods (81). Steroid hormones permanently organize the brain for gender, including the pattern of sexuality, cognition, temperament, and specific interests according to sex, although these features can be modified by environmental and social factors (82). It has been demonstrated that typical male neural and behavioral characteristics develop under the influence of testosterone during perinatal development (81). The fetal hyperandrogenemia in females with CAH leads to male brain organization and subsequently to masculinized behavior and cognitive function (72, 83). Significant psychologic issues originate from these brain compositions in female patients with CAH, and they are dependent on the amount of prenatal and perinatal androgen levels. On the other hand, the disturbed hypothalamic–pituitary–adrenal (HPA) axis in patients with CAH may result in a hypersensitive stress system, making them vulnerable to addiction (84). Thus, three major psychiatric disturbances are present in female patients with CAH: high risk for psychiatric disorders, substance misuse disorders, and stress-adjustment disorders (85). Some authors find psychologic and psychiatric disturbances most expressed in patients with the null genotype (86). Having in mind the symbolic level of the 21-hydroxylase enzyme produced in some patients with the In2G genotype due to alternative splicing, it is expected that they will be less prone to psychologic or psychiatric disorders. In the study of Mueller et al. on a large sample of female patients with CAH, 44.4% met the criteria for at least one psychiatric diagnosis (87). Similar findings were reported in 221 adult females with CAH from six European countries (88). In the study of Engberg et al., females with CAH had high levels of psychiatric disorders as compared with matched controls, and interestingly, patients with the In2G mutation genotype were slightly more frequently affected than the null genotype (85). On the other hand, substance misuse, alcohol, drugs, and attention-deficit hyperactivity disorder were more frequent in patients with the null genotype. In contrast, in males with the In2G genotype, psychiatric disorders, personality disorders, and alcohol misuse were increased as compared with the null genotype (89). Similar findings were reported by Daae et al. (90). The reason for such discrepancy remains elusive. Interestingly, the parents of children with severe genotypes including null and In2G, i.e., obligate CYP21A2 variant carriers, are at much lower risk of being diagnosed with psychiatric disorders (91).
Cardiovascular and Metabolic Disorders and Bone Health
As far as the metabolic outcomes and complications such as obesity, cardiovascular complications, and bone fragility, they are mostly associated with the therapy; therefore, delineating the influence of genotype is very complicated due to different treatment regimens and length of therapy. However, the risk is generally increased as compared with controls (92–95). In one large epidemiological study, only obesity and venous thromboembolism were significantly more common in patients with In2G than in controls, while patients with null variants had more cardiometabolic risk (96).
Discussion
The In2G variant is frequent in patients with 21OHD. It normally causes severe disease; however, the clinical presentation can vary from the SW form, through the SV form and rarely the NC form. Thus, there is a difference in the severity of 21OHD within group A. The mechanism underlying the variation in the clinical phenotype of the In2G variant was widely discussed. The most accredited hypothesis is that a small number of transcripts avoid aberrant splicing, providing a small amount of the 21-hydroxylase enzyme, which is sufficient for a milder clinical presentation of the disease. For this reason, in some patients, the phenotype appears as SV or even NC. In vitro experiments showed that the CYP21A2 intron 2 c.292+1G>A variant produces two different transcripts: the type I fragment lacked the entire introns 1 and 2 and exon 2, whereas the type II, representing approximately one-third of the mRNAs produced and generated by the use of a cryptic splice acceptor site downstream from exon 3, had a deletion of intron 1, entire exon 2, and part of intron 2 (97). These results supported the evidence that splicing is not a homogenous mechanism and that a single splicing variant can generate alternative transcripts, which might explain some unusual phenotypes (97, 98). In addition, the potential influence of extra-adrenal 21-hydroxylation on the CAH patient’s phenotype might be an additional cause to consider. In fact, it was demonstrated that hepatic CYP2C19 and CYP3A4 have the ability to 21-hydroxylate progesterone and thus may modulate mineralocorticoid deficiency (99).
In conclusion, although a good genotype–phenotype correlation exists in 21OHD, a disparity in phenotypic appearance is present in a portion of patients carrying the In2G/In2G or In2G/null genotypes. This evidence represents the most challenging issue in prenatal diagnosis and familiar counselling since the predictive value for different phenotypes can be uncertain.
Author Contributions
MK drafted the manuscript and participated in writing and editing. PC participated in writing and editing. HF participated in writing and editing. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Magnus Bergvall Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Speiser PW, Ng P, Sinaii N, Leschek EW, Green-Golan L, VanRyzin C, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2018) 103(11):4043–88. doi: 10.1210/jc.2018-01865
2. El-Maouche D, Arlt W, Merke DP. Congenital Adrenal Hyperplasia. Lancet (2017) 390(10108):2194–210. doi: 10.1016/S0140-6736(17)31431-9
3. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. One Hundred Years of Congenital Adrenal Hyperplasia in Sweden: A Retrospective, Population-Based Cohort Study. Lancet Diabetes Endocrinol (2013) 1(1):35–42. doi: 10.1016/S2213-8587(13)70007-X
4. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Health Status of Adults With Congenital Adrenal Hyperplasia: A Cohort Study of 203 Patients. J Clin Endocrinol Metab (2010) 95(11):5110–21. doi: 10.1210/jc.2010-0917
5. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, et al. Congenital Adrenal Hyperplasia - Current Insights in Pathophysiology, Diagnostics and Management. Endocr Rev (2021) 7:bnab016. doi: 10.1210/endrev/bnab016.
6. Falhammar H, Thorén M. Clinical Outcomes in the Management of Congenital Adrenal Hyperplasia. Endocrine (2012) 41(3):355–73. doi: 10.1007/s12020-011-9591-x
7. Falhammar H, Torpy DJ. Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Presenting as Adrenal Incidentaloma: A Systematic Review and Meta-Analysis. Endocr Pract (2016) 22(6):736–52. doi: 10.4158/EP151085.RA
8. Witchel SF, Azziz R. Nonclassic Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
9. Zetterström RH, Karlsson L, Falhammar H, Lajic S, Nordenström A. Update on the Swedish Newborn Screening for Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Int J Neonatal Screen (2020) 6(3):71. doi: 10.3390/ijns6030071
10. Nordenström A, Falhammar H. Management of Endocrine Disease Diagnosis and Management of the Patient With Non-Classic CAH Due to 21-Hydroxylase Deficiency. Eur J Endocrinol (2019) 180(3):R127–45. doi: 10.1530/EJE-18-0712
11. Hannah-Shmouni F, Morissette R, Sinaii N, Elman M, Prezant TR, Chen W, et al. Revisiting the Prevalence of Nonclassic Congenital Adrenal Hyperplasia in US Ashkenazi Jews and Caucasians. Genet Med (2017) 19(11):1276–9. doi: 10.1038/gim.2017.46
12. Falhammar H, Wedell A, Nordenstrom A. Biochemical and Genetic Diagnosis of 21-Hydroxylase Deficiency. Endocrine (2015) 50(2):306–14. doi: 10.1007/s12020-015-0731-6
13. White PC, New MI, Dupont B. Structure of Human Steroid 21-Hydroxylase Genes. Proc Natl Acad Sci USA (1986) 83(14):5111–5. doi: 10.1073/pnas.83.14.5111
14. Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete Nucleotide Sequence of Two Steroid 21-Hydroxylase Genes Tandemly Arranged in Human Chromosome: A Pseudogene and a Genuine Gene. Proc Natl Acad Sci U S A (1986) 83(9):2841–5. doi: 10.1073/pnas.83.9.2841
15. Speiser PW, White PC. Congenital Adrenal Hyperplasia. N Engl J Med (2003) 349(8):776–88. doi: 10.1056/NEJMra021561
16. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2 Mutation Update: Comprehensive Analysis of Databases and Published Genetic Variants. Hum Mutat (2018) 39(1):5–22. doi: 10.1002/humu.23351
17. Concolino P, Mello E, Zuppi C, Capoluongo E. Molecular Diagnosis of Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency: An Update of New CYP21A2 Mutations. Clin Chem Lab Med (2010) 48(8):1057–62. doi: 10.1515/CCLM.2010.239
18. Concolino P, Costella A. Congenital Adrenal Hyperplasia (CAH) Due to 21-Hydroxylase Deficiency: A Comprehensive Focus on 233 Pathogenic Variants of CYP21A2 Gene. Mol Diagn Ther (2018) 22(3):261–80. doi: 10.1007/s40291-018-0319-y
19. White PC, Speiser PW. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Endocr Rev (2000) 21(3):245–91. doi: 10.1210/edrv.21.3.0398
20. de Carvalho DF, Miranda MC, Gomes LG, Madureira G, Marcondes JA, Billerbeck AE, et al. Molecular CYP21A2 Diagnosis in 480 Brazilian Patients With Congenital Adrenal Hyperplasia Before Newborn Screening Introduction. Eur J Endocrinol (2016) 175(2):107–16. doi: 10.1530/EJE-16-0171
21. Higashi Y, Hiromasa T, Tanae A, Miki T, Nakura J, Kondo T, et al. Effects of Individual Mutations in the P-450(C21) Pseudogene on the P-450(C21) Activity and Their Distribution in the Patient Genomes of Congenital Steroid 21-Hydroxylase Deficiency. J Biochem (1991) 109(4):638–44. doi: 10.1093/oxfordjournals.jbchem.a123433
22. Krone N, Arlt W. Genetics of Congenital Adrenal Hyperplasia. Best Pract Res Clin Endocrinol Metab (2009) 23(2):181–92. doi: 10.1016/j.beem.2008.10.014
23. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou AM, et al. The Spectrum of Clinical, Hormonal and Molecular Findings in 280 Individuals With Nonclassical Congenital Adrenal Hyperplasia Caused by Mutations of the CYP21A2 Gene. Clin Endocrinol (Oxf) (2015) 82(4):543–9. doi: 10.1111/cen.12543
24. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease Expression and Molecular Genotype in Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. J Clin Invest (1992) 90(2):584–95. doi: 10.1172/JCI115897
25. Marino R, Ramirez P, Galeano J, Perez Garrido N, Rocco C, Ciaccio M, et al. Steroid 21-Hydroxylase Gene Mutational Spectrum in 454 Argentinean Patients: Genotype-Phenotype Correlation in a Large Cohort of Patients With Congenital Adrenal Hyperplasia. Clin Endocrinol (Oxf) (2011) 75(4):427–35. doi: 10.1111/j.1365-2265.2011.04123.x
26. Kocova M, Anastasovska V, Falhammar H. Clinical Outcomes and Characteristics of P30L Mutations in Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Endocrine (2020) 69(2):262–77. doi: 10.1007/s12020-020-02323-3
27. New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-Phenotype Correlation in 1,507 Families With Congenital Adrenal Hyperplasia Owing to 21-Hydroxylase Deficiency. Proc Natl Acad Sci USA (2013) 110(7):2611–6. doi: 10.1073/pnas.1300057110
28. Witchel SF, Bhamidipati DK, Hoffman EP, Cohen JB. Phenotypic Heterogeneity Associated With the Splicing Mutation in Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. J Clin Endocrinol Metab (1996) 81(11):4081–8. doi: 10.1210/jcem.81.11.8923864
29. Gong LF, Gao X, Yang N, Zhao JQ, Yang HH, Kong YY. A Pilot Study on Newborn Screening for Congenital Adrenal Hyperplasia in Beijing. J Pediatr Endocrinol Metab (2019) 32(3):253–8. doi: 10.1515/jpem-2018-0342
30. Maiti A, Chatterjee S. Congenital Adrenal Hyperplasia: An Indian Experience. J Paediatr Child Health (2011) 47(12):883–7. doi: 10.1111/j.1440-1754.2011.02104.x
31. Tsuji A, Konishi K, Hasegawa S, Anazawa A, Onishi T, Ono M, et al. Newborn Screening for Congenital Adrenal Hyperplasia in Tokyo, Japan From 1989 to 2013: A Retrospective Population-Based Study. BMC Pediatr (2015) 15:209. doi: 10.1186/s12887-015-0529-y
32. Anastasovska V, Kocova M. Genotype-Phenotype Correlation in CAH Patients With Severe CYP21A2 Point Mutations in the Republic of Macedonia. J Pediatr Endocrinol Metab (2010) 23(9):921–6. doi: 10.1515/jpem.2010.147
33. Milacic I, Barac M, Milenkovic T, Ugrin M, Klaassen K, Skakic A, et al. Molecular Genetic Study of Congenital Adrenal Hyperplasia in Serbia: Novel P.Leu129Pro and P.Ser165Pro CYP21A2 Gene Mutations. J Endocrinol Invest (2015) 38(11):1199–210. doi: 10.1007/s40618-015-0366-8
34. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Najmabadi H, et al. Ethnic-Specific Distribution of Mutations in 716 Patients With Congenital Adrenal Hyperplasia Owing to 21-Hydroxylase Deficiency. Mol Genet Metab (2007) 90(4):414–21. doi: 10.1016/j.ymgme.2006.12.005
35. Fardella CE, Poggi H, Soto J, Torrealba I, Cattani A, Ugarte F, et al. Mutations in the CYP21 B Gene in a Chilean Population With Simple Virilizing Congenital Adrenal Hyperplasia. J Endocrinol Invest (2000) 23(6):412–6. doi: 10.1007/BF03343746
36. Ohlsson G, Müller J, Skakkebaek NE, Schwartz M. Steroid 21-Hydroxylase Deficiency: Mutational Spectrum in Denmark, Three Novel Mutations, and In Vitro Expression Analysis. Hum Mutat (1999) 13(6):482–6. doi: 10.1002/(SICI)1098-1004(1999)13:6<482::AID-HUMU8>3.0.CO;2-0
37. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Tardy V, Billaud L, Laborde K, et al. Clinical and Molecular Characterization of a Cohort of 161 Unrelated Women With Nonclassical Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency and 330 Family Members. J Clin Endocrinol Metab (2009) 94(5):1570–8. doi: 10.1210/jc.2008-1582
38. Balsamo A, Cacciari E, Baldazzi L, Tartaglia L, Cassio A, Mantovani V, et al. CYP21 Analysis and Phenotype/Genotype Relationship in the Screened Population of the Italian Emilia-Romagna Region. Clin Endocrinol (Oxf) (2000) 53(1):117–25. doi: 10.1046/j.1365-2265.2000.01048.x
39. Ramazani A, Kahrizi K, Razaghiazar M, Mahdieh N, Koppens P. The Frequency of Eight Common Point Mutations in CYP21 Gene in Iranian Patients With Congenital Adrenal Hyperplasia. Iran BioMed J (2008) 12(1):49–53.
40. Grigorescu Sido A, Weber MM, Grigorescu Sido P, Clausmeyer S, Heinrich U, Schulze E. 21-Hydroxylase and 11beta-Hydroxylase Mutations in Romanian Patients With Classic Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2005) 90(10):5769–73. doi: 10.1210/jc.2005-0379
41. Ezquieta B, Oliver A, Gracia R, Gancedo PG. Analysis of Steroid 21-Hydroxylase Gene Mutations in the Spanish Population. Hum Genet (1995) 96(2):198–204. doi: 10.1007/BF00207379
42. Dolzan V, Sólyom J, Fekete G, Kovács J, Rakosnikova V, Votava F, et al. Mutational Spectrum of Steroid 21-Hydroxylase and the Genotype-Phenotype Association in Middle European Patients With Congenital Adrenal Hyperplasia. Eur J Endocrinol (2005) 153(1):99–106. doi: 10.1530/eje.1.01944
43. Asanuma A, Ohura T, Ogawa E, Sato S, Igarashi Y, Matsubara Y, et al. Molecular Analysis of Japanese Patients With Steroid 21-Hydroxylase Deficiency. J Hum Genet (1999) 44(5):312–7. doi: 10.1007/s100380050167
44. Jääskeläinen J, Levo A, Voutilainen R, Partanen J. Population-Wide Evaluation of Disease Manifestation in Relation to Molecular Genotype in Steroid 21-Hydroxylase (CYP21) Deficiency: Good Correlation in a Well Defined Population. J Clin Endocrinol Metab (1997) 82(10):3293–7. doi: 10.1210/jcem.82.10.4271
45. Baş F, Kayserili H, Darendeliler F, Uyguner O, Günöz H, Yüksel Apak M, et al. CYP21A2 Gene Mutations in Congenital Adrenal Hyperplasia: Genotype-Phenotype Correlation in Turkish Children. J Clin Res Pediatr Endocrinol (2009) 1(3):116–28. doi: 10.4008/jcrpe.v1i3.49
46. Lako M, Ramsden S, Campbell RD, Strachan T. Mutation Screening in British 21-Hydroxylase Deficiency Families and Development of Novel Microsatellite Based Approaches to Prenatal Diagnosis. J Med Genet (1999) 36(2):119–24.
47. Marumudi E, Sharma A, Kulshreshtha B, Khadgawat R, Khurana ML, Ammini AC. Molecular Genetic Analysis of CYP21A2 Gene in Patients With Congenital Adrenal Hyperplasia. Indian J Endocrinol Metab (2012) 16(3):384–8. doi: 10.4103/2230-8210.95679
48. Kotaska K, Lisá L, Průsa R. Common CYP21 Gene Mutations in Czech Patients and Statistical Analysis of Worldwide Mutation Distribution. Cent Eur J Public Health (2003) 11(3):124–8.
49. Dumic KK, Grubic Z, Yuen T, Wilson RC, Kusec V, Barisic I, et al. Molecular Genetic Analysis in 93 Patients and 193 Family Members With Classical Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency in Croatia. J Steroid Biochem Mol Biol (2017) 165(Pt A):51–6. doi: 10.1016/j.jsbmb.2016.03.035
50. Dracopoulou-Vabouli M, Maniati-Christidi M, Dacou-Voutetakis C. The Spectrum of Molecular Defects of the CYP21 Gene in the Hellenic Population: Variable Concordance Between Genotype and Phenotype in the Different Forms of Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2001) 86(6):2845–8. doi: 10.1210/jcem.86.6.7574
51. Concolino P, Rizza R, Costella A, Carrozza C, Zuppi C, Capoluongo E. CYP21A2 Intronic Variants Causing 21-Hydroxylase Deficiency. Metabolism (2017) 71:46–51. doi: 10.1016/j.metabol.2017.03.003
52. Rubtsov PM, Igudin EL, Pichugina M, Spirin PV, Prasolov VS, Tul'pakov AN. [Characterization of New Splicing Mutation in Steroid 21-Hydroxylase Gene]. Bioorg Khim (2011) 37(6):815–20. doi: 10.1134/S1068162011060124
53. Katsumata N, Shinagawa T, Horikawa R, Fujikura K. Novel Intronic CYP21A2 Mutation in a Japanese Patient With Classic Salt-Wasting Steroid 21-Hydroxylase Deficiency. Metabolism (2010) 59(11):1628–32. doi: 10.1016/j.metabol.2010.03.012
54. Lee HH, Chao HT, Lee YJ, Shu SG, Chao MC, Kuo JM, et al. Identification of Four Novel Mutations in the CYP21 Gene in Congenital Adrenal Hyperplasia in the Chinese. Hum Genet (1998) 103(3):304–10. doi: 10.1007/s004390050821
55. Kohn B, Day D, Alemzadeh R, Enerio D, Patel SV, Pelczar JV, et al. Splicing Mutation in CYP21 Associated With Delayed Presentation of Salt-Wasting Congenital Adrenal Hyperplasia. Am J Med Genet (1995) 57(3):450–4. doi: 10.1002/ajmg.1320570318
56. Friães A, Rêgo AT, Aragüés JM, Moura LF, Mirante A, Mascarenhas MR, et al. CYP21A2 Mutations in Portuguese Patients With Congenital Adrenal Hyperplasia: Identification of Two Novel Mutations and Characterization of Four Different Partial Gene Conversions. Mol Genet Metab (2006) 88(1):58–65. doi: 10.1016/j.ymgme.2005.11.015
57. Riedl S, Röhl FW, Bonfig W, Brämswig J, Richter-Unruh A, Fricke-Otto S, et al. Genotype/phenotype Correlations in 538 Congenital Adrenal Hyperplasia Patients From Germany and Austria: Discordances in Milder Genotypes and in Screened Versus Prescreening Patients. Endocr Connect (2019) 8(2):86–94. doi: 10.1530/EC-18-0281
58. Wang R, Yu Y, Ye J, Han L, Qiu W, Zhang H, et al. 21-Hydroxylase Deficiency-Induced Congenital Adrenal Hyperplasia in 230 Chinese Patients: Genotype-Phenotype Correlation and Identification of Nine Novel Mutations. Steroids (2016) 108:47–55. doi: 10.1016/j.steroids.2016.01.007
59. Espinosa Reyes TM, Collazo Mesa T, Lantigua Cruz PA, Agramonte Machado A, Domínguez Alonso E, Falhammar H. Genotype-Phenotype Correlation in Patients With Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency in Cuba. Int J Endocrinol (2021) 2021:9316284. doi: 10.1155/2021/9316284
60. Ordoñez-Sánchez ML, Ramírez-Jiménez S, López-Gutierrez AU, Riba L, Gamboa-Cardiel S, Cerrillo-Hinojosa M, et al. Molecular Genetic Analysis of Patients Carrying Steroid 21-Hydroxylase Deficiency in the Mexican Population: Identification of Possible New Mutations and High Prevalence of Apparent Germ-Line Mutations. Hum Genet (1998) 102(2):170–7. doi: 10.1007/s004390050672
61. Stikkelbroeck NM, Hoefsloot LH, de Wijs IJ, Otten BJ, Hermus AR, Sistermans EA. CYP21 Gene Mutation Analysis in 198 Patients With 21-Hydroxylase Deficiency in The Netherlands: Six Novel Mutations and a Specific Cluster of Four Mutations. J Clin Endocrinol Metab (2003) 88(8):3852–9. doi: 10.1210/jc.2002-021681
62. Kocova M, Anastasovska V, Petlichkovski A, Falhammar H. First Insights Into the Genetics of 21-Hydroxylase Deficiency in the Roma Population. Clin Endocrinol (2021) 95(1):41–6. doi: 10.1111/cen.14447
63. Wedell A, Thilén A, Ritzén EM, Stengler B, Luthman H. Mutational Spectrum of the Steroid 21-Hydroxylase Gene in Sweden: Implications for Genetic Diagnosis and Association With Disease Manifestation. J Clin Endocrinol Metab (1994) 78(5):1145–52. doi: 10.1097/00006254-199410000-00020
64. Ezquieta B, Cueva E, Oyarzábal M, Oliver A, Varela JM, Jariego C. Gene Conversion (655G Splicing Mutation) and the Founder Effect (Gln318Stop) Contribute to the Most Frequent Severe Point Mutations in Congenital Adrenal Hyperplasia (21-Hydroxylase Deficiency) in the Spanish Population. Clin Genet (2002) 62(2):181–8. doi: 10.1034/j.1399-0004.2002.620213.x
65. Kocova M, Anastasovska V, Bitovska I. The Impact of CYP21A2 (P30L/I172N) Genotype on Female Fertility in One Family. Eur J Med Res (2019) 24(1):21. doi: 10.1186/s40001-019-0379-4
66. Schulze E, Scharer G, Rogatzki A, Priebe L, Lewicka S, Bettendorf M, et al. Divergence Between Genotype and Phenotype in Relatives of Patients With the Intron 2 Mutation of Steroid-21-Hydroxylase. Endocr Res (1995) 21(1-2):359–64. doi: 10.3109/07435809509030452
67. Witchel SS, Lee PA, Trucco M. Who Is a Carrier? Detection of Unsuspected Mutations in 21-Hydroxylase Deficiency. Am J Med Genet (1996) 61(1):2–9. doi: 10.1002/(SICI)1096-8628(19960102)61:1<2::AID-AJMG1>3.0.CO;2-1
68. Schulze E, Bettendorf M, Maser-Gluth C, Decker M, Schwabe U. Allele-Dropout Using PCR-Based Diagnosis for the Splicing Mutation in Intron-2 of the CYP21B-Gene: Successful Amplification With a Taq/Pwo-Polymerase Mixture. Endocr Res (1998) 24(3-4):637–41. doi: 10.3109/07435809809032662
69. Gomes LG, Bachega TASS, Mendonca BB. Classic Congenital Adrenal Hyperplasia and Its Impact on Reproduction. Fertil Steril (2019) 111(1):7–12. doi: 10.1016/j.fertnstert.2018.11.037
70. Hirschberg AL, Gidlöf S, Falhammar H, Frisén L, Almqvist C, Nordenskjöld A, et al. Reproductive and Perinatal Outcomes in Women With Congenital Adrenal Hyperplasia: A Population-Based Cohort Study. J Clin Endocrinol Metab (2021) 106(2):e957–65. doi: 10.1210/clinem/dgaa801
71. Hagenfeldt K, Janson PO, Holmdahl G, Falhammar H, Filipsson H, Frisén L, et al. Fertility and Pregnancy Outcome in Women With Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Hum Reprod (2008) 23(7):1607–13. doi: 10.1093/humrep/den118
72. Meyer-Bahlburg HF, Dolezal C, Baker SW, New MI. Sexual Orientation in Women With Classical or Non-Classical Congenital Adrenal Hyperplasia as a Function of Degree of Prenatal Androgen Excess. Arch Sex Behav (2008) 37(1):85–99. doi: 10.1007/s10508-007-9265-1
73. Daae E, Feragen KB, Waehre A, Nermoen I. Falhammar H Sexual Orientation in Individuals With Congenital Adrenal Hyperplasia: A Systematic Review. Front Behav Neurosci (2020) 14:38. doi: 10.3389/fnbeh.2020.00038
74. Frisén L, Nordenström A, Falhammar H, Filipsson H, Holmdahl G, Janson PO, et al. Gender Role Behavior, Sexuality, and Psychosocial Adaptation in Women With Congenital Adrenal Hyperplasia Due to CYP21A2 Deficiency. J Clin Endocrinol Metab (2009) 94(9):3432–9. doi: 10.1210/jc.2009-0636
75. Strandqvist A, Falhammar H, Lichtenstein P, Hirschberg AL, Wedell A, Norrby C, et al. Suboptimal Psychosocial Outcomes in Patients With Congenital Adrenal Hyperplasia: Epidemiological Studies in a Nonbiased National Cohort in Sweden. J Clin Endocrinol Metab (2014) 99(4):1425–32. doi: 10.1210/jc.2013-3326
76. Bothou C, Anand G, Li D, Kienitz T, Seejore K, Simeoli C, et al. Current Management and Outcome of Pregnancies in Women With Adrenal Insufficiency: Experience From a Multicenter Survey. J Clin Endocrinol Metab (2020) 105(8):e2853–63. doi: 10.1210/clinem/dgaa266.
77. Nordenström A, Frisén L, Falhammar H, Filipsson H, Holmdahl G, Janson PO, et al. Sexual Function and Surgical Outcome in Women With Congenital Adrenal Hyperplasia Due to CYP21A2 Deficiency: Clinical Perspective and the Patients’ Perception. J Clin Endocrinol Metab (2010) 95(8):3633–40. doi: 10.1210/jc.2009-2639
78. Claahsen-van der Grinten HL, Stikkelbroeck N, Falhammar H, Reisch N. MANAGEMENT OF ENDOCRINE DISEASE: Gonadal Dysfunction in Congenital Adrenal Hyperplasia. Eur J Endocrinol (2021) 184(3):R85–97. doi: 10.1530/EJE-20-1093
79. Falhammar H, Nyström HF, Ekström U, Granberg S, Wedell A, Thorén M. Fertility, Sexuality and Testicular Adrenal Rest Tumors in Adult Males With Congenital Adrenal Hyperplasia. Eur J Endocrinol (2012) 166(3):441–9. doi: 10.1530/EJE-11-0828
80. Falhammar H, Frisén L, Norrby C, Almqvist C, Hirschberg AL, Nordenskjöld A, et al. Reduced Frequency of Biological and Increased Frequency of Adopted Children in Males With 21-Hydroxylase Deficiency: A Swedish Population-Based National Cohort Study. J Clin Endocrinol Metab (2017) 102(11):4191–9. doi: 10.1210/jc.2017-01139
81. Bakker J. The Role of Steroid Hormones in the Sexual Differentiation of the Human Brain. J Neuroendocrinol (2021) p:e13050. doi: 10.1111/jne.13050
82. Jordan-Young RM. Hormones, Context, and “Brain Gender”: A Review of Evidence From Congenital Adrenal Hyperplasia. Soc Sci Med (2012) 74(11):1738–44. doi: 10.1016/j.socscimed.2011.08.026
83. Meyer-Bahlburg HFL. Brain Development and Cognitive, Psychosocial, and Psychiatric Functioning in Classical 21-Hydroxylase Deficiency. Endocr Dev (2011) 20:88–95. doi: 10.1159/000321225
84. Charmandari E, Merke DP, Negro PJ, Keil MF, Martinez PE, Haim A, et al. Endocrinologic and Psychologic Evaluation of 21-Hydroxylase Deficiency Carriers and Matched Normal Subjects: Evidence for Physical and/or Psychologic Vulnerability to Stress. J Clin Endocrinol Metab (2004) 89(5):2228–36. doi: 10.1210/jc.2003-031322
85. Engberg H, Butwicka A, Nordenström A, Hirschberg AL, Falhammar H, Lichtenstein P, et al. Congenital Adrenal Hyperplasia and Risk for Psychiatric Disorders in Girls and Women Born Between 1915 and 2010: A Total Population Study. Psychoneuroendocrinology (2015) 60:195–205. doi: 10.1016/j.psyneuen.2015.06.017
86. Mueller SC, Grissom EM, Dohanich GP. Assessing Gonadal Hormone Contributions to Affective Psychopathologies Across Humans and Animal Models. Psychoneuroendocrinology (2014) 46:114–28. doi: 10.1016/j.psyneuen.2014.04.015
87. Mueller SC, Ng P, Sinaii N, Leschek EW, Green-Golan L, VanRyzin C, et al. Psychiatric Characterization of Children With Genetic Causes of Hyperandrogenism. Eur J Endocrinol (2010) 163(5):801–10. doi: 10.1530/EJE-10-0693
88. de Vries ALC, Roehle R, Marshall L, Frisén L, van de Grift TC, Kreukels BPC, et al. Mental Health of a Large Group of Adults With Disorders of Sex Development in Six European Countries. Psychosom Med (2019) 81(7):629–40. doi: 10.1097/PSY.0000000000000718
89. Falhammar H, Butwicka A, Landén M, Lichtenstein P, Nordenskjöld A, Nordenström A, et al. Increased Psychiatric Morbidity in Men With Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. J Clin Endocrinol Metab (2014) 99(3):E554–60. doi: 10.1210/jc.2013-3707
90. Daae E, Feragen KB, Nermoen I, Falhammar H. Psychological Adjustment, Quality of Life, and Self-Perceptions of Reproductive Health in Males With Congenital Adrenal Hyperplasia: A Systematic Review. Endocrine (2018) 62(1):3–13. doi: 10.1007/s12020-018-1723-0
91. Nordenström A, Butwicka A, Lindén Hirschberg A, Almqvist C, Nordenskjöld A, Falhammar H, et al. Are Carriers of CYP21A2 Mutations Less Vulnerable to Psychological Stress? A Population-Based National Cohort Study. Clin Endocrinol (Oxf) (2017) 86(3):317–24. doi: 10.1111/cen.13242
92. Tamhane S, Rodriguez-Gutierrez R, Iqbal AM, Prokop LJ, Bancos I, Speiser PW, et al. Cardiovascular and Metabolic Outcomes in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab (2018) 103(11):4097–103. doi: 10.1210/jc.2018-01862
93. Li L, Bensing S, Falhammar H. Rate of Fracture in Patients With Glucocorticoid Replacement Therapy: A Systematic Review and Meta-Analysis. Endocrine (2021) 74(1):29–37. doi: 10.1007/s12020-021-02723-z
94. Rangaswamaiah S, Gangathimmaiah V, Nordenstrom A, Falhammar H. Bone Mineral Density in Adults With Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. Front Endocrinol (Lausanne) (2020) 11:493. doi: 10.3389/fendo.2020.00493
95. Falhammar H, Frisén L, Hirschberg AL, Nordenskjöld A, Almqvist C, Nordenström A. Increased Prevalence of Fractures in Congenital Adrenal Hyperplasia: A Swedish Population-Based National Cohort Study. J Clin Endocrinol Metab (2021) 3:dgab712. doi: 10.1210/clinem/dgab712
96. Falhammar H, Frisén L, Hirschberg AL, Norrby C, Almqvist C, Nordenskjöld A, et al. Increased Cardiovascular and Metabolic Morbidity in Patients With 21-Hydroxylase Deficiency: A Swedish Population-Based National Cohort Study. J Clin Endocrinol Metab (2015) 100(9):3520–8. doi: 10.1210/JC.2015-2093
97. Lee HH, Chang SF. Multiple Transcripts of the CYP21 Gene Are Generated by the Mutation of the Splicing Donor Site in Intron 2 From GT to AT in 21-Hydroxylase Deficiency. J Endocrinol (2001) 171(3):397–402. doi: 10.1677/joe.0.1710397
98. Annalora AJ, Marcus CB, Iversen PL. Alternative Splicing in the Cytochrome P450 Superfamily Expands Protein Diversity to Augment Gene Function and Redirect Human Drug Metabolism. Drug Metab Dispos (2017) 45(4):375–89. doi: 10.1124/dmd.116.073254
Keywords: CYP21A2, c.293-13C/A>G, splicing variant, CAH, genetic counselling
Citation: Kocova M, Concolino P and Falhammar H (2022) Characteristics of In2G Variant in Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Front. Endocrinol. 12:788812. doi: 10.3389/fendo.2021.788812
Received: 03 October 2021; Accepted: 29 November 2021;
Published: 24 January 2022.
Edited by:
Maria Fragoso, Institute of Cancer of Sao Paulo, BrazilReviewed by:
Rodolfo A. Rey, Hospital de Niños Ricardo Gutiérrez, ArgentinaTania Bachega, University of São Paulo, Brazil
Gabriela Paula Finkielstain, Takeda Pharmaceutical Company Limited, Argentina
Copyright © 2022 Kocova, Concolino and Falhammar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjana Kocova, mirjanakocova@yahoo.com
†ORCID: Mirjana Kocova, orcid.org/0000-0001-7097-3439
Paola Concolino, orcid.org/0000-0002-0523-5744
Henrik Falhammar, orcid.org/0000-0002-5622-6987