 Liza O’Donnell1
Liza O’Donnell1 Kate L. Loveland
Kate L. Loveland- 1Centre for Reproductive Health, Hudson Institute of Medical Research, Clayton, VIC, Australia
- 2Monash University, Clayton, VIC, Australia
The long-standing knowledge that Sertoli cells determine fetal testosterone production levels is not widespread, despite being first reported over a decade ago in studies of mice. Hence any ongoing use of testosterone as a marker of Leydig cell function in fetal testes is inappropriate. By interrogating new scRNAseq data from human fetal testes, we demonstrate this situation is also likely to be true in humans. This has implications for understanding how disruptions to either or both Leydig and Sertoli cells during the in utero masculinization programming window may contribute to the increasing incidence of hypospadias, cryptorchidism, testicular germ cell tumours and adult infertility. We recently discovered that activin A levels directly govern androgen production in mouse Sertoli cells, because the enzymes that drive the conversion of the precursor androgen androstenedione to generate testosterone are produced exclusively in Sertoli cells in response to activin A. This minireview addresses the implications of this growing understanding of how in utero exposures affect fetal masculinization for future research on reproductive health, including during programming windows that may ultimately be relevant for organ development in males and females.
Dogma Relating to Fetal Testis Biology: Moving and Stationary Targets
More than 30 years ago, the events that lead to gonad masculinization were shown to be triggered by the expression of SRY in pre-Sertoli cells of the fetal testis, in studies of humans (1) and mice (2). Since that time, we have learned just how complicated that process is, including the contribution of signals relating to SOX9 feedback (3), growth factors (4, 5) and retinoic acid (6). These cues direct primordial germ cells to differentiate into the earliest sperm precursors and instruct the somatic cells to multiply, differentiate, and form the testis cords that underpin adult spermatogenesis (7). Our established understanding was that many gonadal cell types were initially bipotential, but subsequently became set in their fate in fetal life. One type became either Sertoli cells in males or granulosa cells in females, providing direct support and instructions to germ cells. However, complementary break-through studies identified that somatic cell fate was actually pliable in both mice and humans. For example, in several studies published over a decade ago, the transcription factor Dmrt1 (Doublesex And Mab-3 Related Transcription Factor 1) was shown to be essential to stabilize the masculinized phenotype of Sertoli and Leydig cells (8), while Foxl2 serves a similar role in sustaining the ovarian phenotype in granulosa and thecal cell lineages (9). The reality of sex-reversal in somatic cell fate was thereby revealed.
Understanding how somatic cells instruct male or female differentiation has also been important for learning about human conditions which can be linked with sub- or infertility, due to impaired germline differentiation or reduced germ cell survival. The conceptualization of ‘Testicular Dysgenesis’ as a spectrum of phenotypes, including hypospadias, cryptorchidism, testicular germ cell tumours and infertility, has provided a basis for mechanistic studies of each of these conditions, as each could result from impaired somatic-germline communication in the fetal testis (10). The Testicular Dysgenesis Hypothesis has stood the test of time; the continuously increasing rates of these conditions worldwide highlights the vulnerability of the developing male reproductive tract to lifestyle and environmental exposures (11), in concert with a lesser, but relevant genetic determinant of disease risk. Mechanistically, this hypothesis has focussed on explaining the impact of disruptions to cell-cell communication in the testis in utero, particularly with regard to androgen production and signalling (12).
Crucial to understanding the origins of testicular dysgenesis outcomes has been delineation of a masculinization programming window (termed MPW), the period of development when the male fetus is sensitive to exposures that impact on reproductive health. During this window, multiple factors can influence the development of the male gonad and secondary sexual characteristics (Figure 1). Virilization of the fetal male is dependent on gonadal function but it is also vulnerable to alteration by maternal- and/or placental-derived factors (Figure 1). Best understood in rodent models, endocrine disruptor chemical exposures in the interval following sex determination and prior to birth have repeatedly been shown to increase the incidence of conditions including reduced anogenital distance, hypospadias, cryptorchidism and reduced germ cell numbers (13–15). These outcomes have been explored in data collected from boys and men, and from cultures of human fetal testes (16–18), with a growing understanding that not only exposure to endocrine disruptor chemicals, which can also include common pharmaceuticals, may negatively impact fetal and postnatal development. These agents can alter steroid composition and levels and may thereby influence local events in the testis. For example, ibuprofen alters human fetal Leydig cell function and testosterone production between weeks 7–17 of gestation, indicating that human male fetal steroidogenesis is vulnerable to alterations by a commonly used pharmacological compound (16). Nodal and activin signalling can also influence particular aspects of human fetal steroidogenesis in both the first and second trimesters (19), and as discussed below, these members of the TGFβ signalling pathway are amongst the key candidates for driving lifelong changes in male reproductive health.
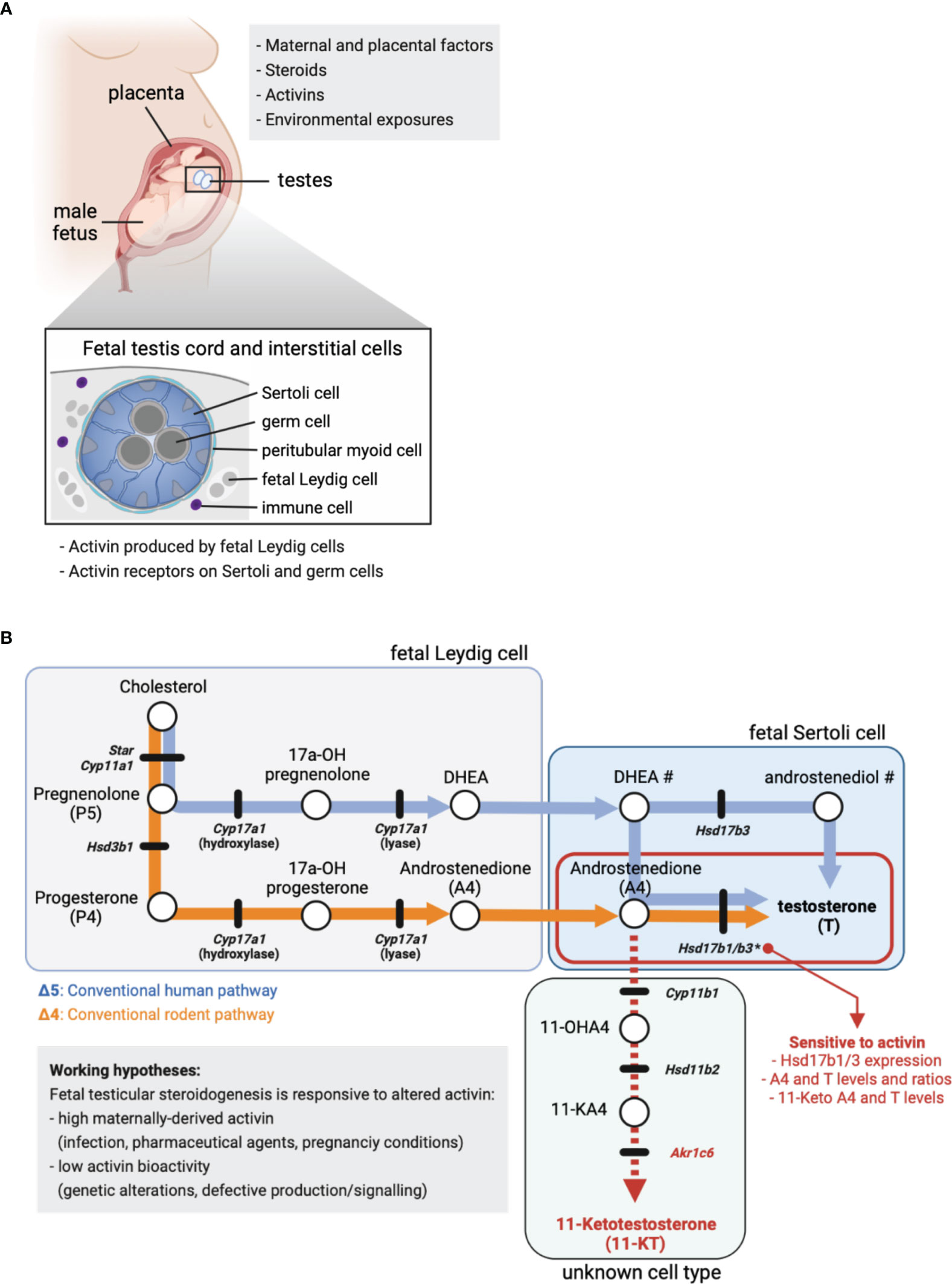
Figure 1 Fetal steroid production. (A) Maternal and placental factors, exogenous steroids, activins and environmental exposures each have the potential to alter fetal steroid production. Schematic of placenta, male fetus and testes in utero. Cross-section identifies key fetal testis cell types in the cord (germ and Sertoli cells) and in the surrounding interstitium (fetal Leydig, peritubular and immune cells). Activin is produced by fetal Leydig cells and signals via specific cell surface receptors present on Sertoli and germ cells. (B) Simplified summary of the conventional steroidogenic pathway in human (Δ5, blue line/arrows) and rodent (Δ4, orange line/arrows) fetal testes. The 11-Keto androgen pathway is indicated (red dashed line), as are 11-Hydroxyandrostenedione (11-OHA4), 11-Keto androstenedione (11-KA4) and 11-Ketotestosterone (11-KT). The cellular site of steroid production is indicated, where known. Circles denote cholesterol and steroids, while black lines indicate genes encoding enzymes involved in the conversion of one steroid to the next. Mouse nomenclature has been used throughout; Table 1 lists human nomenclature. # indicates the local site(s) of DHEA and androstenediol production in human is unclear. * indicates activin A-responsive genes; the ratio of A4 to T in mouse fetal Sertoli cells is dose-dependently reduced by low levels of Hsd17b1 and Hsd17b3 expression driven by altered activin A bioactivity. In these testes, excess A4 is associated with higher levels of 11-keto androgens.
Activin A is a member of the TGFβ signalling superfamily that is emerging as an important regulator of male fetal development. Activin A was originally identified as a factor that was produced and acted locally to control release of follicle stimulating hormone from the pituitary gland (20, 21). It is synthesized as a disulfide-linked homodimer of two inhibin beta A subunits, encoded by the Inhba gene, and its structure and signalling pathway are characteristic of the more than 30 other members of the TGFβ superfamily of signalling molecules, many of which are simultaneously present in the fetal testis (5). It is now known to be produced throughout the body and serve functions in growth and development of many organs, while activin A overproduction is associated with severe pathologies such as cancer cachexia (22). Physiological regulation of activin signalling is complex, occurring through extracellular, transmembrane and intracellular means which are typically context-dependent. See (21, 23–27) for recent reviews on this topic.
Activin A performs multiple functions in testis development that include governance of Sertoli cell proliferation. This role was first revealed in the adult inhibin α knockout mouse strain with elevated activin A; males succumb to Sertoli cell tumours due to the absence of the potent activin inhibitor, inhibin, which forms as a heterodimer of one inhibin α subunit with an inhibin β A subunit (28). In addition to driving Sertoli cell proliferation in utero, activin A production by fetal Leydig cells also promotes the timely entry of fetal germ cells into a quiescent state following sex determination (29–31). Quiescence occurs in an important developmental interval, coincident with important aspects of normal male germ cell differentiation that include epigenetic reprograming and loss of pluripotency, each of which can be linked with reducing the risk of tumour formation (32). We have recently discovered that activin A is an important factor regulating androgen biosynthesis in the fetal testis (33); the mechanisms and significance of these findings will be discussed below.
Learning About Androgen Biosynthesis in the Fetal Testis
A Gold Mine of Data From Single Cell Transcriptome Analyses
The application of single cell RNA sequencing to study the mammalian fetal testis has revealed developmental relationships between somatic cells which highlight their common origins. Work in mouse (34) and human (35) have detailed the emergence of distinct Leydig and Sertoli cell populations from a single progenitor, extending findings from earlier lineage mapping studies (36). Important distinctions between fetal and adult Leydig cells have been progressively mapped, revealing that synthesis of the androgen precursor, androstenedione, is different in fetal Leydig cells compared to adults. Whereas luteinizing hormone (LH) stimulates steroid biosynthesis in adult Leydig cells, fetal cells can be stimulated by corticotrophin-releasing hormone (CRH) and adrenocorticotrophic hormone (ACTH), and their responsiveness to LH is probably not attained prior to E17 in mice (37, 38). Single cell RNASeq datasets have also shed new light on where, and which, steroids and androgens are produced in the fetal testis, as discussed below.
Sertoli Cells Are the Site of Testosterone Production in Fetal Mouse Testes
In the rodent testis, testosterone production occurs via the Δ4 steroidogenic pathway (Figure 1B, orange line) and concludes with conversion of androstenedione (A4) to testosterone (T) by the enzymes HSD17B3 (39) and HSD17B1 (40). The fetal and adult Leydig cell populations develop sequentially and are functionally distinct (41), so while T is produced in Leydig cells in adult mouse testes (42), fetal Leydig cells lack Hsd17b1 and Hsd17b3 and cannot convert A4 to T (39, 40). Instead, exclusive expression of Hsd17b1 and Hsd17b3 in fetal mouse Sertoli cells (43, 44) identifies Sertoli cells as the site of T synthesis in the fetal testis (33).
Since appropriate levels of steroids and steroidogenic enzymes during embryonic (E) development lay the foundation for the correct development of male sexual characteristics, factors that influence this process may illuminate important areas for research aimed at understanding the foundations of male reproductive disorders. Activin A (encoded by Inhba) is produced by fetal Leydig cells and is essential for normal embryonic mouse testis development. Inhba transcript levels in male testes increase directly after sex determination (E12.5) and the activin A dimer acts directly on receptors in Sertoli cells influencing their proliferation (30, 31, 45). Recently, our lab identified that activin A also positively regulates expression of Hsd17b1 and Hsd17b3 required to convert A4 to T; both are strongly reduced (E13.5 - E15.5) in the absence of activin A (Inhba KO) compared with wildtype controls (33). Analysis of intratesticular steroids demonstrated that a sustained decrease in Hsd17b1 and Hsd17b3 transcripts reduced conversion of A4 to T within E17.5 Inhba KO fetal testes; A4 levels were significantly increased and T tended to decrease. Additionally, the T/A4 ratio revealed a highly significant dose-dependent decrease in androgen production that correlated with reducing Inhba gene dosage (33). These data demonstrate that activin A fulfils a central role in determining local steroid levels and androgen production during fetal testis development. Lastly, Whiley et al. measured increased levels of the 11-oxygenated androgens, 11-KA4 (11-keto androstenedione, also named adrenosterone) and 11-KT (11-ketotestosterone) in Inhba KO testes (33). Since 11-KT can activate the androgen receptor (46), this finding could have important clinical significance.
Steroidogenesis in the Human Male Fetus: What We Know and What Are It’s Vulnerabilities?
Production of the androgen T by the human fetal testis is essential for masculinization. Its release from the testis drives virilization of the Wolffian ducts to form the seminal vesicles and ejaculatory ducts, whereas the 5α-reduction of T to the more potent androgen dihydrotestosterone (DHT) is necessary for virilization of the male external genitalia (47). The first trimester human testes can produce low levels of T via the Δ4 pathway prior to a switch to the Δ5 pathway at the beginning of the second trimester (48). Thereafter, the human fetal testis produces high levels of T during the second trimester via the classical Δ5 steroidogenic pathway (Figure 1B, blue lines) (48, 49). This switch coincides with the maturation of fetal Leydig cells as they acquire their capacity to produce androgen precursors (48). The precursor androstenedione (A4)) is converted to T mainly by the 17β-hydroxysteroid dehydrogenase activity of HSD17B3 (50) (Figure 1) and while HSD17B1 can perform this conversion, it is less efficient compared to the mouse enzyme (51). In the human fetal testis, HSD17B3 levels are comparable in first and second trimesters (48). However, the peak in T synthesis in the second trimester appears due to fetal Leydig cells becoming fully differentiated and acquiring the ability to synthesize androgen precursors (48).
Leydig cells are the major steroidogenic cells of the human fetal testis, expressing a wide range of steroid biosynthetic enzymes, and their differentiation coincides with elevated testicular T levels (49). Thus, these cells are often assumed to be responsible for androgen biosynthesis. However, as described in the section “Sertoli Cells Are the Site of Testosterone Production in Fetal Mouse Testes”, although the precursor steroids and A4 are produced by fetal Leydig cells in rodents, the conversion of A4 to T takes place in the Sertoli cells (Figure 1). Whether this also occurs in humans is not established (48). To address this possibility, we interrogated published single cell RNASeq datasets to determine in which cell types key steroidogenic enzymes are synthesized in mouse and human testes (35, 44). The expression patterns for HSD17B3 and HSD17B1 in human testicular cells provide strong evidence that, as in mice, human Sertoli cells are likely to be the primary site of androgen (T) synthesis in the second trimester fetal testis (Table 1; Supplementary Figure 1). Interestingly, human scRNASeq data suggest that only a small subset of human fetal Leydig cells produce androgen precursors between weeks 14–18 of gestation [Supplementary Figure 1 (35)]. This speculation is aligned with evidence that human fetal interstitial and Leydig-like cells are a heterogenous population (49).
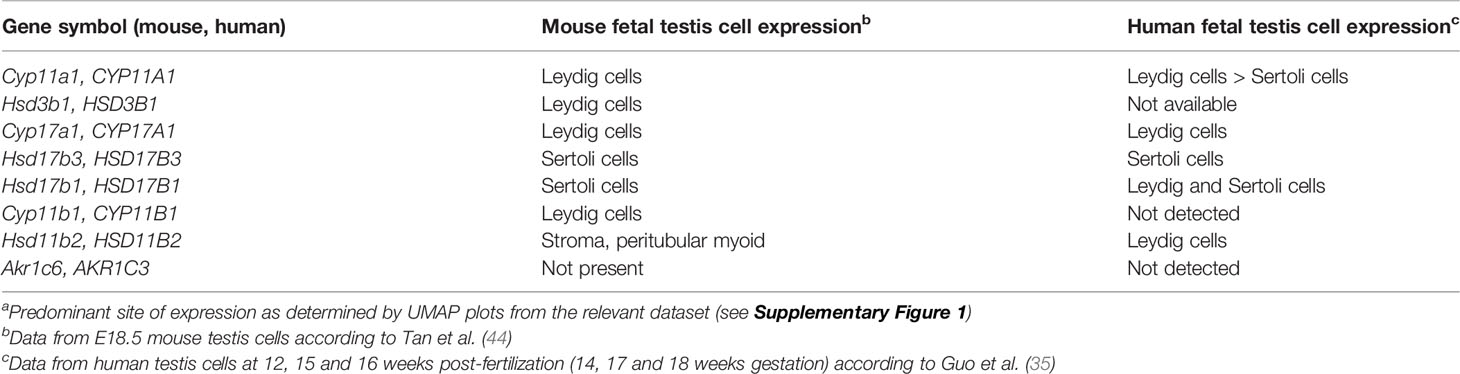
Table 1 mRNA expression patterns of key steroidogenic enzymes in fetal mouse and human testis cellsa.
Recent studies have revealed that alternative androgen production by the so-called “backdoor pathway” is also important for virilization of the human male fetus (52, 53). This pathway begins with progesterone, probably originating largely from the maternal side of the placenta (Figure 1A), followed by sequential biosynthetic steps in the adrenal glands and liver, that ultimately produce androsterone (53). This androgen is present in the human fetal male circulation at levels similar to testosterone (53). Thus, masculinization of the human male fetus depends on androgens produced via the classic and backdoor pathways, both within and outside of the testis. It involves a complex interplay of steroidogenic enzymes expressed in multiple tissues to produce testosterone, DHT and androsterone that act within different organs to control human fetal urogenital tract development (49, 52, 53).
Finally, as noted above, certain 11-keto steroids have the capacity to contribute to androgen action in the human fetal testis and other androgen-responsive tissues. Originally identified as androgens in fish, the 11-keto androgens 11-keto testosterone (11-KT) and 11-keto DHT (11-KDHT) are potent agonists of the human androgen receptor (46, 54, 55). They are likely to be synthesized in human peripheral tissues from the abundant adrenal steroid 11b-hydroxyandrostenedione (11OHA4) (Figure 1B) (46, 54). CYP11B1 is a key enzyme in 11-keto steroid biosynthesis (Figure 1B) and is expressed in mouse and human testis (Table 1; Supplementary Figure 1). This enzyme can be stimulated by hCG in immature mouse Leydig cells leading to an increase in testicular levels of 11-KT (55). In mice, 11-KT is shows similar androgen activity to T, suggesting it is an important bioactive androgen (55). The enzyme responsible for the biosynthesis of 11-keto androstenedione (11-KA4), HSD11B2 (Figure 1B), is expressed in both mouse and human testis cells (Table 1; Supplementary Figure 1) whereas the enzyme that converts 11-KA4 to 11-KT (AKR1C6 in mice and AKR1C3 in humans (Figure 1B), is not expressed in the testis (Table 1, Supplementary Figure 1). These data suggest that fetal mouse and human testes express the enzymes that produce 11-KA4 and that this precursor can be converted to the androgen 11-KT in peripheral tissues. We have detected both 11-KA4 and 11-KT in fetal mouse testes, indicating that 11-KT could act as bioactive androgen during fetal testis development (33). Importantly, we showed the levels of these keto steroids are responsive to activin A deficiency; reduced conversion of A4 to T led to increased levels of A4 that were accompanied by increased testicular levels of 11-KA4 and 11-KT (33). Thus, in activin A-deficient mice, the reduced capacity to produce the androgen T, as a consequence of reduced Hsd17b1 and Hsd17b3 expression in Sertoli cells, is accompanied by an increased capacity to produce the androgen 11-KT (33). Further studies are needed to understand the contribution of 11-KT to androgen action in the human fetal testis and in the masculinization of the male fetus.
Conclusions: Why Does This Matter?
The complexity of human fetal steroidogenesis means that there are multiple cell types and thus sites vulnerable to disruption that could alter androgen levels and activities in different fetal tissues. In the mouse testis, activin A regulation of the T biosynthetic enzymes Hsd17b1 and Hsd17b3 in Sertoli cells is now known to alter the ratio of A4 to T and the production of keto-steroids with androgenic bioactivity (Figure 1B), and this is likely to be important to some extent in humans (Table 1). Changes to activin A bioactivity at the start of gonad development, as in the condition of pre-eclampsia, would be ultimately expected to influence adult fertility and development of other organs; this may also affect the developing ovary. The fact that fetal T is produced by the Sertoli cells and thus T levels are not an accurate measure of fetal Leydig cell function needs to be more widely appreciated. Recent data showing that androgens in the human male fetus testis can originate via different biosynthetic pathways adds complexity to the mechanisms via which external and intrinsic factors could modulate androgen action during male fetal development. This knowledge will help to identify a roadmap of developmental vulnerability to environmental exposures, focussed appropriately on the key affected cell types. This should ultimately be used to develop strategies that can protect reproductive health.
Author Contributions
KL designed content, co-wrote text, and edited text and figure. PW co-wrote and edited text, designed and produced figure. LO’D co-wrote text, and edited text and figure. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the NHMRC (Ideas 1181516).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful for excellent bioinformatics support from Ben Nathaniel. We apologise to colleagues whose work we were unable to cite for reasons of brevity.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.898876/full#supplementary-material
References
1. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, et al. A Gene From the Human Sex-Determining Region Encodes a Protein With Homology to a Conserved DNA-Binding Motif. Nature (1990) 346(6281):240–4. doi: 10.1038/346240a0
2. Koopman P, Münsterberg A, Capel B, Vivian N, Lovell-Badge R. Expression of a Candidate Sex-Determining Gene During Mouse Testis Differentiation. Nature (1990) 348(6300):450–2. doi: 10.1038/348450a0
3. Gonen N, Lovell-Badge R. The Regulation of Sox9 Expression in the Gonad. Curr Top Dev Biol (2019) 134:223–52. doi: 10.1016/bs.ctdb.2019.01.004
4. Wilhelm D, Palmer S, Koopman P. Sex Determination and Gonadal Development in Mammals. Physiol Rev (2007) 87(1):1–28. doi: 10.1152/physrev.00009.2006
5. Young JC, Wakitani S, Loveland KL. TGF-Beta Superfamily Signaling in Testis Formation and Early Male Germline Development. Semin Cell Dev Biol (2015) 45:94–103. doi: 10.1016/j.semcdb.2015.10.029
6. Bowles J, Knight D, Smith C, Wilhelm D, Richman J, Mamiya S, et al. Retinoid Signaling Determines Germ Cell Fate in Mice. Science (2006) 312(5773):596–600. doi: 10.1126/science.1125691
7. Spiller C, Bowles J. Sexually Dimorphic Germ Cell Identity in Mammals. Curr Top Dev Biol (2019) 134:253–88. doi: 10.1016/bs.ctdb.2019.01.011
8. Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 Prevents Female Reprogramming in the Postnatal Mammalian Testis. Nature (2011) 476(7358):101–4. doi: 10.1038/nature10239
9. Uhlenhaut NH, Jakob S, Anlag K, Eisenberger T, Sekido R, Kress J, et al. Somatic Sex Reprogramming of Adult Ovaries to Testes by FOXL2 Ablation. Cell (2009) 139(6):1130–42. doi: 10.1016/j.cell.2009.11.021
10. Sharpe RM, Skakkebaek NE. Testicular Dysgenesis Syndrome: Mechanistic Insights and Potential New Downstream Effects. Fertil Steril (2008) 89(2 Suppl):e33–8. doi: 10.1016/j.fertnstert.2007.12.026
11. Skakkebaek NE, Lindahl-Jacobsen R, Levine H, Andersson AM, Jorgensen N, Main KM, et al. Environmental Factors in Declining Human Fertility. Nat Rev Endocrinol (2022) 18(3):139–57. doi: 10.1038/s41574-021-00598-8
12. Skakkebaek NE, Rajpert-De Meyts E, Buck Louis GM, Toppari J, Andersson AM, Eisenberg ML, et al. Male Reproductive Disorders and Fertility Trends: Influences of Environment and Genetic Susceptibility. Physiol Rev (2016) 96(1):55–97. doi: 10.1152/physrev.00017.2015
13. van den Driesche S, Kilcoyne KR, Wagner I, Rebourcet D, Boyle A, Mitchell R, et al. Experimentally Induced Testicular Dysgenesis Syndrome Originates in the Masculinization Programming Window. JCI Insight (2017) 2(6):e91204. doi: 10.1172/jci.insight.91204
14. Walker C, Garza S, Papadopoulos V, Culty M. Impact of Endocrine-Disrupting Chemicals on Steroidogenesis and Consequences on Testicular Function. Mol Cell Endocrinol (2021) 527:111215. doi: 10.1016/j.mce.2021.111215
15. Welsh M, Saunders PT, Fisken M, Scott HM, Hutchison GR, Smith LB, et al. Identification in Rats of a Programming Window for Reproductive Tract Masculinization, Disruption of Which Leads to Hypospadias and Cryptorchidism. J Clin Invest (2008) 118(4):1479–90. doi: 10.1172/JCI34241
16. Ben Maamar M, Lesné L, Hennig K, Desdoits-Lethimonier C, Kilcoyne KR, Coiffec I, et al. Ibuprofen Results in Alterations of Human Fetal Testis Development. Sci Rep (2017) 7:44184. doi: 10.1038/srep44184
17. Eladak S, Moison D, Guerquin MJ, Matilionyte G, Kilcoyne K, N'Tumba-Byn T, et al. Effects of Environmental Bisphenol A Exposures on Germ Cell Development and Leydig Cell Function in the Human Fetal Testis. PloS One (2018) 13(1):e0191934. doi: 10.1371/journal.pone.0191934
18. Rodprasert W, Toppari J, Virtanen HE. Endocrine Disrupting Chemicals and Reproductive Health in Boys and Men. Front Endocrinol (Lausanne) (2021) 12:706532. doi: 10.3389/fendo.2021.706532
19. Jorgensen A, Macdonald J, Nielsen JE, Kilcoyne KR, Perlman S, Lundvall L, et al. Nodal Signaling Regulates Germ Cell Development and Establishment of Seminiferous Cords in the Human Fetal Testis. Cell Rep (2018) 25(7):1924–37 e4. doi: 10.1016/j.celrep.2018.10.064
20. Ling N, Ying SY, Ueno N, Shimasaki S, Esch F, Hotta M, et al. Pituitary FSH is Released by a Heterodimer of the Beta-Subunits From the Two Forms of Inhibin. Nature (1986) 321(6072):779–82. doi: 10.1038/321779a0
21. Robertson DM, Klein R, de Vos FL, McLachlan RI, Wettenhall RE, Hearn MT, et al. The Isolation of Polypeptides With FSH Suppressing Activity From Bovine Follicular Fluid Which are Structurally Different to Inhibin. Biochem Biophys Res Commun (1987) 149(2):744–9. doi: 10.1016/0006-291X(87)90430-X
22. Ries A, Schelch K, Falch D, Pany L, Hoda MA, Grusch M. Activin A: An Emerging Target for Improving Cancer Treatment? Expert Opin Ther Targets (2020) 24(10):985–96. doi: 10.1080/14728222.2020.1799350
23. Chen W, Ten Dijke P. Immunoregulation by Members of the TGFbeta Superfamily. Nat Rev Immunol (2016) 16(12):723–40. doi: 10.1038/nri.2016.112
24. Lodberg A. Principles of the Activin Receptor Signaling Pathway and its Inhibition. Cytokine Growth Factor Rev (2021) 60:1–17. doi: 10.1016/j.cytogfr.2021.04.001
25. Namwanje M, Brown CW. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb Perspect Biol (2016) 8(7). doi: 10.1101/cshperspect.a021881
26. Vale W, Rivier J, Vaughan J, McClintock R, Corrigan A, Woo W, et al. Purification and Characterization of an FSH Releasing Protein From Porcine Ovarian Follicular Fluid. Nature (1986) 321(6072):776–9. doi: 10.1038/321776a0
27. Wijayarathna R, Hedger MP. Activins, Follistatin and Immunoregulation in the Epididymis. Andrology (2019) 7(5):703–11. doi: 10.1111/andr.12682
28. Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. Alpha-Inhibin is a Tumour-Suppressor Gene With Gonadal Specificity in Mice. Nature (1992) 360(6402):313–9. doi: 10.1038/360313a0
29. Archambeault DR, Tomaszewski J, Childs AJ, Anderson RA, Yao HH. Testicular Somatic Cells, Not Gonocytes, are the Major Source of Functional Activin A During Testis Morphogenesis. Endocrinology (2011) 152(11):4358–67. doi: 10.1210/en.2011-1288
30. Archambeault DR, Yao HH. Activin A, a Product of Fetal Leydig Cells, is a Unique Paracrine Regulator of Sertoli Cell Proliferation and Fetal Testis Cord Expansion. Proc Natl Acad Sci U S A (2010) 107(23):10526–31. doi: 10.1073/pnas.1000318107
31. Mendis SH, Meachem SJ, Sarraj MA, Loveland KL. Activin A Balances Sertoli and Germ Cell Proliferation in the Fetal Mouse Testis. Biol Reprod (2011) 84(2):379–91. doi: 10.1095/biolreprod.110.086231
32. Webster NJ, Maywald RL, Benton SM, Dawson EP, Murillo OD, LaPlante EL, et al. Testicular Germ Cell Tumors Arise in the Absence of Sex-Specific Differentiation. Development (2021) 148(9). doi: 10.1242/dev.197111
33. Whiley PAF, O'Donnell L, Moody SC, Handelsman DJ, Young JC, Richards EA, et al. Activin A Determines Steroid Levels and Composition in the Fetal Testis. Endocrinology (2020) 161(7):1–16. doi: 10.1210/endocr/bqaa058
34. Stévant I, Kühne F, Greenfield A, Chaboissier MC, Dermitzakis ET, Nef S. Dissecting Cell Lineage Specification and Sex Fate Determination in Gonadal Somatic Cells Using Single-Cell Transcriptomics. Cell Rep (2019) 26(12):3272–83.e3. doi: 10.1016/j.celrep.2019.02.069
35. Guo J, Sosa E, Chitiashvili T, Nie X, Rojas EJ, Oliver E, et al. Single-cell analysis of the developing human testis reveals somatic niche cell specification and fetal germline stem cell establishment. Cell Stem Cell (2021) 28(4):764–78 e4. doi: 10.1016/j.stem.2020.12.004
36. Liu C, Rodriguez K, Yao HH. Mapping Lineage Progression of Somatic Progenitor Cells in the Mouse Fetal Testis. Development (2016) 143(20):3700–10. doi: 10.1242/dev.135756
37. O'Shaughnessy PJ, Baker P, Sohnius U, Haavisto AM, Charlton HM, Huhtaniemi I. Fetal Development of Leydig Cell Activity in the Mouse is Independent of Pituitary Gonadotroph Function. Endocrinology (1998) 139(3):1141–6. doi: 10.1210/endo.139.3.5788
38. Sararols P, Stévant I, Neirijnck Y, Rebourcet D, Darbey A, Curley MK, et al. Specific Transcriptomic Signatures and Dual Regulation of Steroidogenesis Between Fetal and Adult Mouse Leydig Cells. Front Cell Dev Biol (2021) 9:695546. doi: 10.3389/fcell.2021.695546
39. Shima Y, Miyabayashi K, Haraguchi S, Arakawa T, Otake H, Baba T, et al. Contribution of Leydig and Sertoli Cells to Testosterone Production in Mouse Fetal Testes. Mol Endocrinol (2013) 27(1):63–73. doi: 10.1210/me.2012-1256
40. Hakkarainen J, Zhang FP, Jokela H, Mayerhofer A, Behr R, Cisneros-Montalvo S, et al. Hydroxysteroid (17beta) Dehydrogenase 1 Expressed by Sertoli Cells Contributes to Steroid Synthesis and is Required for Male Fertility. FASEB J (2018) 32(6):3229–41. doi: 10.1096/fj.201700921R
41. O'Shaughnessy PJ, Willerton L, Baker PJ. Changes in Leydig Cell Gene Expression During Development in the Mouse. Biol Reprod (2002) 66(4):966–75. doi: 10.1095/biolreprod66.4.966
42. Hall PF, Irby DC, De Kretser DM. Conversion of Cholesterol to Androgens by Rat Testes: Comparison of Interstitial Cells and Seminiferous Tubules. Endocrinology (1969) 84(3):488–96. doi: 10.1210/endo-84-3-488
43. Jameson SA, Natarajan A, Cool J, DeFalco T, Maatouk DM, Mork L, et al. Temporal Transcriptional Profiling of Somatic and Germ Cells Reveals Biased Lineage Priming of Sexual Fate in the Fetal Mouse Gonad. PloS Genet (2012) 8(3):e1002575. doi: 10.1371/journal.pgen.1002575
44. Tan K, Song HW, Wilkinson MF. Single-Cell RNAseq Analysis of Testicular Germ and Somatic Cell Development During the Perinatal Period. Development (2020) 147(3). doi: 10.1242/dev.183251
45. Itman C, Small C, Griswold M, Nagaraja AK, Matzuk MM, Brown CW, et al. Developmentally Regulated SMAD2 and SMAD3 Utilization Directs Activin Signaling Outcomes. Dev Dyn (2009) 238(7):1688–700. doi: 10.1002/dvdy.21995
46. Pretorius E, Arlt W, Storbeck KH. A New Dawn for Androgens: Novel Lessons From 11-Oxygenated C19 Steroids. Mol Cell Endocrinol (2017) 441:76–85. doi: 10.1016/j.mce.2016.08.014
47. Wilson JD. Sexual Differentiation. Annu Rev Physiol (1978) 40:279–306. doi: 10.1146/annurev.ph.40.030178.001431
48. Savchuk I, Morvan ML, Antignac JP, Kurek M, Le Bizec B, Söder O, et al. Ontogenesis of Human Fetal Testicular Steroidogenesis at Early Gestational Age. Steroids (2019) 141:96–103. doi: 10.1016/j.steroids.2018.12.001
49. Connan-Perrot S, Léger T, Lelandais P, Desdoits-Lethimonier C, David A, Fowler PA, et al. Six Decades of Research on Human Fetal Gonadal Steroids. Int J Mol Sci (2021) 22(13). doi: 10.3390/ijms22136681
50. Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, et al. Male Pseudohermaphroditism Caused by Mutations of Testicular 17 Beta-Hydroxysteroid Dehydrogenase 3. Nat Genet (1994) 7(1):34–9. doi: 10.1038/ng0594-34
51. Nokelainen P, Puranen T, Peltoketo H, Orava M, Vihko P, Vihko R. Molecular Cloning of Mouse 17 Beta-Hydroxysteroid Dehydrogenase Type 1 and Characterization of Enzyme Activity. Eur J Biochem (1996) 236(2):482–90. doi: 10.1111/j.1432-1033.1996.00482.x
52. Miller WL, Auchus RJ. The "Backdoor Pathway" of Androgen Synthesis in Human Male Sexual Development. PloS Biol (2019) 17(4):e3000198. doi: 10.1371/journal.pbio.3000198
53. O'Shaughnessy PJ, Antignac JP, Le Bizec B, Morvan ML, Svechnikov K, Söder O, et al. Alternative (Backdoor) Androgen Production and Masculinization in the Human Fetus. PloS Biol (2019) 17(2):e3000002. doi: 10.1371/journal.pbio.3000002
54. Pretorius E, Africander DJ, Vlok M, Perkins MS, Quanson J, Storbeck KH. 11-Ketotestosterone and 11-Ketodihydrotestosterone in Castration Resistant Prostate Cancer: Potent Androgens Which Can No Longer Be Ignored. PloS One (2016) 11(7):e0159867. doi: 10.1371/journal.pone.0159867
55. Yazawa T, Uesaka M, Inaoka Y, Mizutani T, Sekiguchi T, Kajitani T, et al. Cyp11b1 is Induced in the Murine Gonad by Luteinizing Hormone/Human Chorionic Gonadotropin and Involved in the Production of 11-Ketotestosterone, a Major Fish Androgen: Conservation and Evolution of the Androgen Metabolic Pathway. Endocrinology (2008) 149(4):1786–92. doi: 10.1210/en.2007-1015
Keywords: androgens, activin A, Sertoli cell, Leydig cell, fetal steroidogenesis
Citation: O’Donnell L, Whiley PAF and Loveland KL (2022) Activin A and Sertoli Cells: Key to Fetal Testis Steroidogenesis. Front. Endocrinol. 13:898876. doi: 10.3389/fendo.2022.898876
Received: 18 March 2022; Accepted: 13 April 2022;
Published: 24 May 2022.
Edited by:
Andrew Pask, The University of Melbourne, AustraliaReviewed by:
Terje Svingen, Technical University of Denmark, DenmarkCopyright © 2022 O’Donnell, Whiley and Loveland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kate L. Loveland, kate.loveland@monash.edu