 Morten Krogh Herlin
Morten Krogh Herlin- Department of Clinical Genetics, Aarhus University Hospital, Aarhus N, Denmark
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a congenital anomaly characterized by agenesis/aplasia of the uterus and upper part of the vagina in females with normal external genitalia and a normal female karyotype (46,XX). Patients typically present during adolescence with complaints of primary amenorrhea where the diagnosis is established with significant implications including absolute infertility. Most often cases appear isolated with no family history of MRKH syndrome or related anomalies. However, cumulative reports of familial recurrence suggest genetic factors to be involved. Early candidate gene studies had limited success in their search for genetic causes of MRKH syndrome. More recently, genomic investigations using chromosomal microarray and genome-wide sequencing have been successful in detecting promising genetic variants associated with MRKH syndrome, including 17q12 (LHX1, HNF1B) and 16p11.2 (TBX6) deletions and sequence variations in GREB1L and PAX8, pointing towards a heterogeneous etiology with various genes involved. With uterus transplantation as an emerging fertility treatment in MRKH syndrome and increasing evidence for genetic etiologies, the need for genetic counseling concerning the recurrence risk in offspring will likely increase. This review presents the advancements in MRKH syndrome genetics from early familial occurrences and candidate gene searches to current genomic studies. Moreover, the review provides suggestions for future genetic investigations and discusses potential implications for clinical practice.
1 Introduction
In mammals, the Müllerian (paramesonephric) ducts give rise to the female reproductive tract, which consists of the Fallopian tubes (oviducts), uterus, cervix, and upper two-thirds of the vagina (1). Abnormalities in Müllerian duct (MD) development in women may result in congenital uterovaginal anomalies of various severity (2, 3).
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, also referred to as Müllerian aplasia, is a congenital disorder characterized by agenesis or aplasia of the uterus and upper part of the vagina. The patients are characterized by having a normal female karyotype (46,XX), normal external genitalia, and normal pubertal development of secondary sex characteristics (thelarche and pubarche) (4). MRKH syndrome is typically diagnosed during late adolescence when patients present with primary amenorrhea (5). The estimated birth prevalence of MRKH syndrome is 1 in 5,000 female live births (5, 6) and it is considered the second most common cause of primary amenorrhea (7). MRKH syndrome may present as an isolated anomaly (type I) or in association with extragenital malformations (type II), typically involving the kidneys, skeleton, and heart (4). Upon the diagnosis of MRKH syndrome, patients face life-impacting consequences related to their sexual identity, fear of coital difficulties, and grief of infertility (8–12), and it has been associated with a risk of depressive and anxiety symptoms (13–15). Psychological support and counseling are therefore crucial in patient care (16–18). Non-surgical and surgical treatments of vaginal hypoplasia are available to enable penetrative intercourse with non-surgical dilation considered the first-line choice (18–20). MRKH syndrome causes absolute uterine factor infertility (AUFI) but patients may achieve genetic motherhood through gestational surrogacy (21, 22). In 2014, the first baby was born following pregnancy after uterus transplantation (UTx) in Sweden, offering the first fertility treatment of AUFI achieving both gestational and genetic motherhood (23, 24).
The etiology of MRKH syndrome has long been an unanswered question in medical research, and both genetic and non-genetic factors have been considered. Despite substantial efforts to find explanations for the disorder, our current understanding of the underlying biology remains limited. However, discoveries during recent years do provide evidence for the importance of genetic factors and point towards a heterogeneous etiology with various genes involved in uterovaginal development in humans. This review presents the continuous advancements in our knowledge of genetics in MRKH syndrome, from early candidate gene studies to genome-wide gene discoveries, and provides recommendations for further progress and perspectives on potential clinical implications.
2 Embryology of the female reproductive tract
The human genitourinary system (including the kidneys, gonads, and reproductive tracts) develops from the intermediate mesoderm. At 3 weeks post gestation, the Wolffian (mesonephric) ducts, which develop into the male reproductive tracts, form and grow caudally from the primordial kidney (mesonephros) to reach the cloaca. At 5 weeks post gestation, bilateral invaginations of the coelomic epithelium of the urogenital ridges begin to form the MDs which extend caudally, guided by the Wolffian ducts, to reach the urogenital sinus in the midline (Müllerian tubercle). Here, the caudal parts of the two MDs start to fuse to form the uterus and upper vagina starting from week 8. The cranial openings of the invaginations remain and give rise to the fimbrial ends of the oviducts adjacent to the developing ovary (1, 25, 26).
The close relationship between kidney and uterovaginal development is also reflected by the high prevalence (~30%) of kidney malformations in MRKH syndrome (5, 27). Other common extragenital anomalies include the skeleton and heart, which do also develop from the mesoderm, with the paraxial mesoderm forming the axial skeleton (28) and the lateral plate mesoderm forming the heart and appendicular skeleton (29). Therefore, genes involved in the development of mesoderm and its derived structures are relevant candidates in the etiology of MRKH syndrome.
3 Evidence for genetic etiologies in MRKH syndrome
Most cases of MRKH syndrome appear isolated with no clear indications of a familial/genetic trait (5, 30). In addition, several reports of discordant monozygotic twin pairs (5, 31–35) and patient-reported outcomes of most surrogate pregnancies also support non-Mendelian causes (36). However, it is important to consider that the disease nature of MRKH syndrome implies absolute infertility, hindering mother-to-offspring inheritance of a genetic cause, which may cause an underestimation of the genetic component of MRKH syndrome from family histories. At this point of knowledge, a single mode of inheritance to cover all cases cannot be determined and monogenic, oligogenic, polygenic, multifactorial, and environmental factors should still be considered. Hypotheses on fetal exposures disturbing uterovaginal development have included thalidomide, diethylstilbestrol, organotins, and phthalates (37–40), but there is no firm evidence for a pathological role in MRKH syndrome.
In 1973, Buchta et al. (41) reported on familial occurrences of various renal malformations following autosomal dominant inheritance and coined the term “hereditary renal adysplasia”. One of the patients, who had eight children, was found to have left renal agenesis and a bicorn uterus during surgery for cervical carcinoma in situ. Interestingly, 14 years later, John M. Opitz (senior author of the first paper) reported that one daughter of this patient had been diagnosed with MRKH syndrome emphasizing the close link between uterovaginal malformations and renal malformations (42). This association was also described by Schimke & King, who instead proposed the term “hereditary urogenital adysplasia” (43). This pattern of both renal and MD anomalies within families, characterized by incomplete penetrance and variable sex-limited expressivity, has been reported several times in the literature (reviewed by Herlin et al. (44)). Taken together this emphasizes the importance of a detailed family history when investigating the genetics of MRKH syndrome.
Recently, the first case of mother-daughter inheritance of MRKH syndrome following gestational surrogacy was reported, when the daughter presented at 14 years old with primary amenorrhea and was diagnosed with type I MRKH syndrome as her mother. A 4 Mb deletion of 2q37.1q37.3 of unknown significance was identified in both patients, but not in the mother’s parents (22). This case exemplifies the potential risk of MRKH syndrome recurrence following assisted reproduction and with the expected increasing availability of UTx as this field moves towards clinical care (24), identifying causal variants and providing genetic counseling regarding recurrence risk and reproductive choices will become more relevant.
4 Genetic research in MRKH syndrome
One of the earliest reports describing genetic analysis in the diagnosis of MRKH syndrome is from Georges Andre Hauser in 1961. Hauser and colleagues described that sex-chromatin analysis could aid the differentiation of MRKH syndrome from Turner syndrome and defined MRKH syndrome (at the time termed ‘Mayer-Rokitansky-Küster syndrome’) to include normal female chromosomes (45, 46). Together with earlier anatomical descriptions of Mayer, Rokitansky, and Küster, his work led to the complete definition and its final name (4).
For over 25 years, researchers have searched for genetic alterations to cause uterovaginal agenesis in karyotypically normal women. Early studies investigated a wide range of candidate genes including GALT (47–49), WT1 (50), HNF1B (51, 52), AMH (49, 53), AMHR2 (49, 53), CFTR (54), WNT7A (55), HOXA7-13 genes (56–58), PBX1 (56, 59), RARG (60), RXRA (60), CTNNB1 (61), SHOX (62–64), PAX2 (65), LAMC1 (66), DLG1 (66), or ITIH5 (67). Most of these studies had negative results and provided limited evidence for genetic factors in MRKH syndrome. This included investigations of AMH and AMHR2, encoding anti-Müllerian hormone and its receptor, respectively, involved in physiological MD regression in males (49, 53). One gene, Wnt4, was reported to be involved in female development in mammals with mutant female mice displaying signs of masculinization and absent Müllerian ducts, hence a relevant candidate gene in MRKH syndrome. In 2004, Biason-Lauber et al. identified a WNT4 variant in an 18-year-old woman with Müllerian agenesis, renal agenesis, and clinical signs of hyperandrogenism. Functional analyses supported the variant pathogenicity and WNT4 was thereby the first identified gene in uterovaginal agenesis with firm evidence for monogenic causality (discussed further below).
The completion of the Human Genome Project in 2003 (68, 69) together with the concomitant development of genomic technologies including chromosomal microarray (CMA) and massively parallel sequencing (also referred to as Next-Generation Sequencing [NGS]) have allowed for genome-wide (“hypothesis-free”) searches of genetic variation in MRKH syndrome. CMA, including comparative genomic hybridization array and single nucleotide polymorphism (SNP) array, is used to detect copy number variations (CNVs) in the genome. These applications have identified various chromosomal imbalances (deletions/duplications) supporting the identification of candidate genes such as HNF1B, LHX1, and TBX6 (Table 1) (30, 71, 72, 81, 82). However, recurrent chromosomal imbalances in MRKH syndrome still only apply to a minor fraction of patients (around 10%). Optical genome mapping is a newer cytogenomic method with higher resolution for the detection of both imbalanced and balanced structural variation and has recently been applied in a study by Brakta et al. (73). In more recent years, whole-exome and whole-genome sequencing analyses have been applied for genome-wide detection of genetic variation at the nucleotide level (74, 75, 96–105). Investigations have included both extended familial cases and larger patient cohorts identifying variants of interesting genes, most notably GREB1L (99, 101, 102) and PAX8 (75).
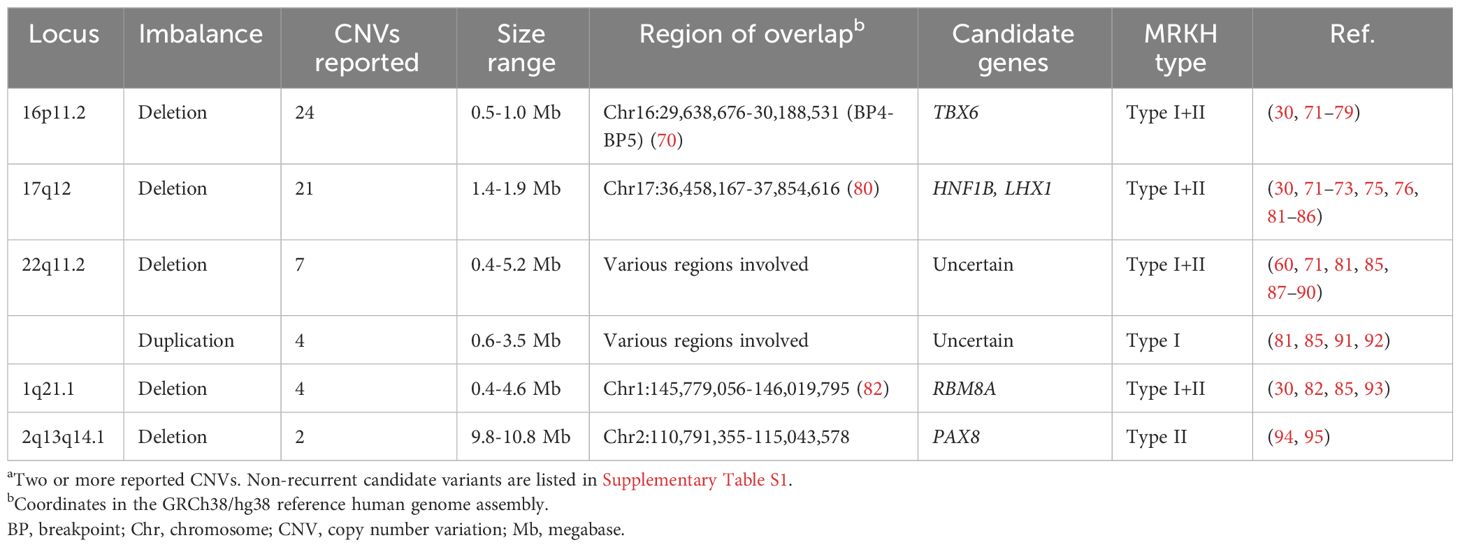
Table 1 Recurrenta copy number variations associated with MRKH syndrome.
The vast majority of studies to date have looked into germline genetic variation by analyzing DNA from blood samples. Due to the predominantly sporadic nature of MRKH syndrome and reports of discordant monozygotic twin pairs suggesting non-inherited genetic variation, researchers have also searched for somatic/mosaic gene variation and tissue-specific differential gene methylation/expression patterns in uterine remnants to explain the disorder (35, 98, 106–108).
5 Genetic findings in MRKH syndrome and evidence for causality
In this section, the most significant genetic findings to date and the current evidence for causality in MRKH syndrome are discussed. Table 1 summarizes the recurrent (reported in at least two cases) copy number variations in MRKH syndrome, whereas Table 2 lists the genes recurrently reported with sequence variants. Non-recurrent (only one case) candidate variants are listed in Supplementary Table S1.
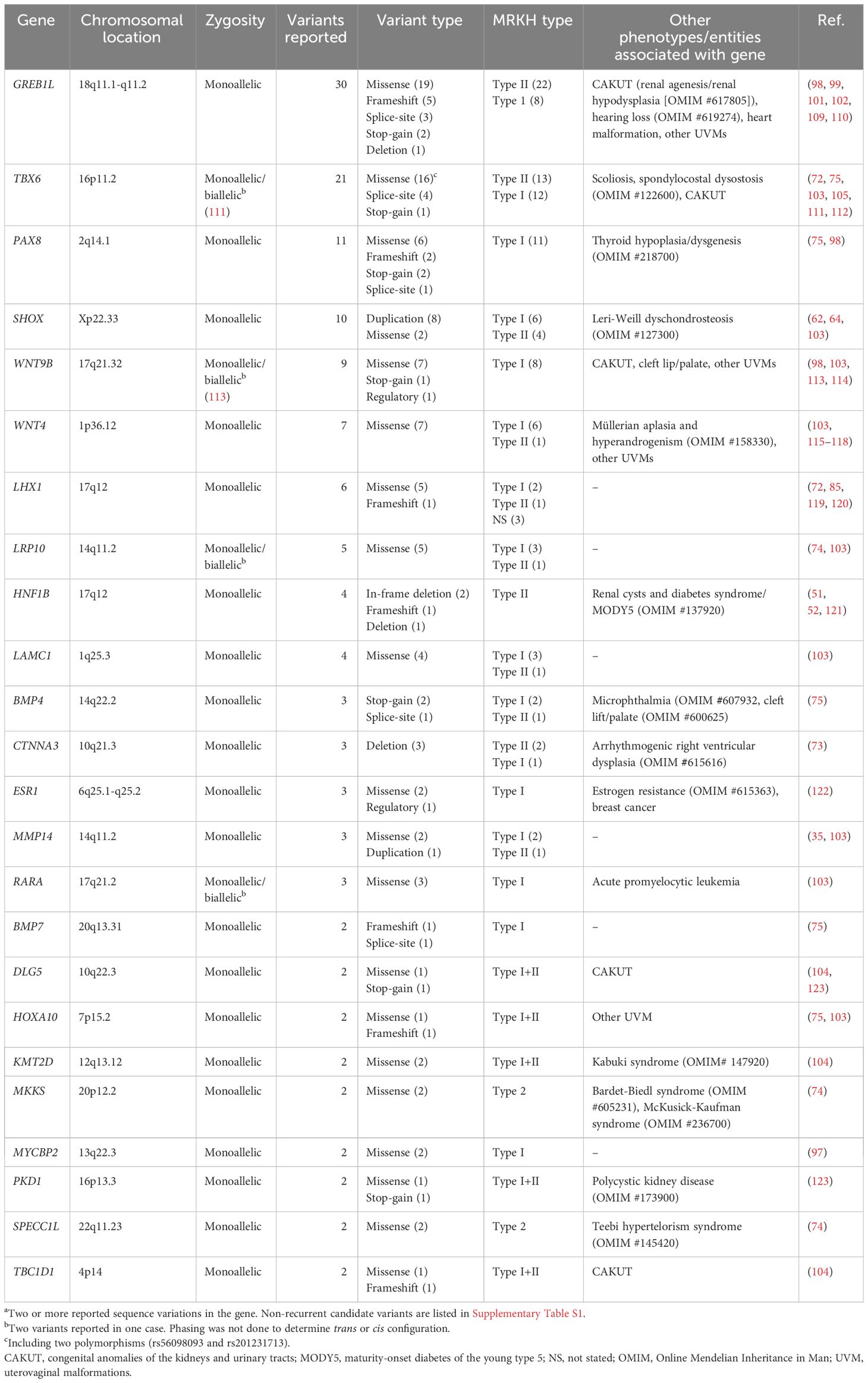
Table 2 Genes reported with recurrenta sequence variation associated with MRKH syndrome.
5.1 17q12 deletions, LHX1, and HNF1B
Deletion of 17q12 was first reported in MRKH syndrome by Cheroki et al. in 2008 (81) and to this date, 21 deletions have been reported (Table 1) (30, 71–73, 75, 76, 81–86). Ledig et al. reported a 17q12 deletion in one MRKH syndrome patient (85), whose mother and sister were later reported with the same deletion and other uterovaginal anomalies (124). Deletions of 17q12 are typically 1.4 Mb in size. They are highly penetrant with variable expressivity and cause a multisystem disorder which may include kidney disease, neurocognitive impairment, endocrinological disease including HNF1B-related maturity-onset diabetes of the young (MODY), genital malformations, liver disease, and other manifestations (80). Two candidate genes for MRKH syndrome, LHX1 and HNF1B, are located at this locus, both of them being involved in MD development.
Lhx1, formerly referred to as Lim1, encoding the transcription factor LIM homeobox 1, is involved in normal kidney and MD development (125, 126). Lhx1-null female mice have normal ovaries but lack their reproductive tract, which results from a disruption of MD elongation and epithelium formation (125–127). Besides deletions, six single nucleotide variants in LHX1 (Table 2) (72, 85, 119, 120) have been reported including one missense variant with functional evidence of decreased transcriptional activity (120). Still, LHX1 mutational analysis of larger cohorts did not report any variants, suggesting that sequence variants of LHX1 are no major cause of MRKH syndrome (30, 128).
HNF1B has long been a candidate gene for MRKH syndrome since Lindner et al. already in 1999 reported a Norwegian family with MODY type 5 and progressive parenchymal kidney disease following autosomal dominant inheritance caused by a 75 bp in-frame deletion in exon 2 of HNF1B. Notably, two of four female variant carriers also had uterovaginal agenesis, supporting MRKH syndrome as part of the HNF1B disease spectrum (51). Subsequent studies have reported HNF1B variants associated with various uterovaginal malformations (52, 121, 129, 130), including a ~59 kb whole-gene deletion of HNF1B found by WES analysis of a 9-year-old girl presenting with precocious puberty. After the genetic result, reverse phenotyping by imaging confirmed uterus agenesis (121). Recently, Thomson et al. (83) investigated the function of Hnf1b following conditional ablation in mice, which resulted in a hypoplastic uterus and renal anomalies (including renal agenesis) similar to an MRKH syndrome type II phenotype. They performed single-cell RNA sequencing of the Hnf1b-ablated embryonic uterine tissue and found dysregulated gene expression involved in cell proliferation, patterning, and differentiation. This supports HNF1B as a causative gene in MRKH syndrome independent of LHX1 involvement.
5.2 16p11.2 deletions and TBX6
In 2011, Nik-Zainal et al. (71) reported four cases with recurrent deletions of 16p11.2 and suggested TBX6, previously implicated in paraxial mesoderm development (131), as a candidate gene. To date, a large number of cases have been identified with both deletions (30, 71–79) and TBX6 sequence variants (72, 75, 103, 105, 111, 112). The 16p11.2/TBX6 locus is also associated with congenital scoliosis (132). The genetics hereof is complex and does not follow typical Mendelian inheritance, requiring one TBX6-null allele and a particular hypomorphic trans allele, as described in the compound inheritance gene dosage model (132, 133).
Ma et al. reported 16 rare TBX6 variants enriched in a large MRKH syndrome patient cohort compared to controls. They performed various functional analyses of 13 missense variants and found evidence for loss-of-function in 7 variants, which together do support the role of TBX6 variants. However, in contrast to null alleles associated with scoliosis, no second risk alleles were reported in MRKH syndrome (105). As of now, no clear biological mechanism for monoallelic TBX6 variants causing MRKH syndrome has been established, which challenges interpretation and warrants further studies.
5.3 22q11 deletions and duplications
Both deletions (60, 71, 81, 85, 87–90) and duplications (81, 85, 91, 92) of 22q11 have been reported in MRKH syndrome patients (Table 1). The CNVs vary in size and location with no single common overlapping region, and therefore no particular candidate genes at 22q11 have been identified. Of note, 22q11 duplication has also been associated with other uterovaginal malformations (124). 22q11 deletions are often associated with DiGeorge and velocardiofacial syndromes. Features hereof are partly thought to be caused by TBX1 haploinsufficiency, however, some of the deletions reported with MRKH syndrome do not include this particular gene. Overall, the evidence for 22q11 imbalances causing MRKH syndrome remains low.
5.4 1q21.1 and RBM8A
Variable-sized deletions at 1q21.1 have been associated with MRKH syndrome (30, 82, 85, 93). Duplications of 1q21.1 have also been identified in an MRKH syndrome patient (81) and one case with uterus didelphys (82). The overlapping region has been determined to be GRCh38: chr1:145,779,056-146,019,795 (82) with RBM8A as the proposed candidate gene in which sequence variants/polymorphisms also have been identified associated with MRKH syndrome (112).
Deletions of this region may also cause thrombocytopenia–absent radius (TAR) syndrome in compound heterozygosity with certain non-coding polymorphisms on the trans allele (134). Notably, TAR syndrome has previously been reported in a case with MRKH syndrome (135, 136). The possible causal role of 1q21.1 deletions/RBM8A gene variants in MRKH syndrome is, however, still unclear warranting further studies to establish causality.
5.5 2q13q14.1 deletions and PAX8
Pathogenic variants in PAX8 are an established monogenic cause of congenital hypothyroidism due to thyroid dysgenesis (CH, OMIM #218700), first described in 1998 by Macchia et al. (137). In an experimental study, Pax8-deficient female mice were reported with infertility independently of thyroid replacement therapy, and a possible role in MD development was suggested (138).
Years later, Ma et al. reported on the investigation of a 12-year-old girl with global developmental delay, MD agenesis, and hypothyroidism, found to carry a large 10.79 Mb deletion of 2q13q14.2 (94). A partially overlapping 2q12.1q14.1 deletion was reported in another case with CH, atrial septal defect, intellectual disability, and MD agenesis with anterior displacement of the anus (95). The two deletions share an overlapping region of 4.3 Mb spanning PAX8 as the proposed candidate gene for MRKH syndrome.
Most recently, Chen et al. (75) reported on a mutational burden analysis of 19 candidate genes based on WES data from 442 cases and 941 controls. Among cases, they found enrichment for predicted loss-of-function variants in PAX8. When including results from a replication cohort (n=150), a CH cohort (n=5), and missense variants, a total of 11 PAX8 variants were found associated with MRKH syndrome. In three cases with available parental DNA, paternal inheritance was confirmed, showing a sex-limited expressivity of infertility. Functional analysis of the five missense variants by luciferase reporter assay found evidence for loss-of-function in two variants. Finally, reverse-phenotyping of five female CH cases, revealed uterovaginal aplasia in one individual, emphasizing the pleiotropy of PAX8. This confirms MRKH syndrome as a part of the PAX8 disease spectrum in females, which the authors refer to as CH-MRKHS (75).
5.6 GREB1L
In 2017, three unrelated studies identified GREB1L variants as a new autosomal dominant cause of congenital anomalies of the kidney and urinary tract (CAKUT, OMIM #617805) (109, 110, 139). Some of the female cases, mainly fetuses affected by bilateral renal agenesis, were also described with uterovaginal malformations supporting a possible link to MD development (109, 110).
In 2019, Herlin et al. (101) reported on the investigation of a three-generational family with four cases of renal agenesis, including two adult female cousins with MRKH syndrome type II with unilateral renal agenesis. Whole-exome sequencing analysis in this family identified a segregating missense variant in GREB1L, supporting GREB1L variants as a novel monogenic cause of MRKH syndrome associated with incomplete penetrance and sex-limited expressivity and a phenotype mirroring the early descriptions of hereditary renal/urogenital adysplasia (41–43, 140, 141). Jacquinet et al. (102) identified GREB1L variants in 5 of 63 (7.9%) sporadic MRKH syndrome patients and segregating variants in four multiplex families presenting renal and uterovaginal malformations. Buchert et al. reported a stop-gain variant in a monozygotic twin-pair discordant for MRKH syndrome with the other twin having unilateral renal agenesis (98). Most recently, Jolly et al. performed an unbiased rare variant enrichment analysis based on WES data from a large American-European cohort (n=148), identifying GREB1L as the only gene approaching exome-wide significance based on seven detected variants. From a replication cohort of 442 Han Chinese cases, additional variants were found, including in six cases with type I MRKH syndrome. Of note, besides kidney and uterovaginal malformations, GREB1L variants have also been associated with inner ear malformations and deafness as well as complex congenital heart disease (142, 143).
GREB1L is considered to be involved in retinoic acid signaling, although its protein remains poorly characterized (109). Greb1l-knockdown in zebrafish causes abnormal pronephros morphogenesis (110, 139), and injection of wild-type human mRNA has been shown to rescue the phenotype (110). Knock-in mutagenesis of one missense variant in mice has also been shown to cause renal agenesis (139). Homozygous knock-out of Greb1l in mice has been shown to cause absence of the kidneys, Wolffian ducts, and Müllerian ducts (109). However, GREB1L variants reported in humans are monoallelic and predominantly missense variants (Table 2), and the pathogenic mechanism of how these missense variants cause MRKH syndrome is still unknown requiring further functional analysis.
Taken together, the current evidence with 30 reported variants (Table 2) including disease-segregating variants in extended pedigrees, epidemiological evidence of rare variant enrichment in larger cohorts, and functional evidence from knock-out mice, suggest GREB1L as a major causative gene in MRKH syndrome.
5.7 SHOX
Gervasini et al. (62) first reported on the association of partial SHOX duplications in MRKH syndrome in 2010. They investigated 30 cases and 53 controls and identified 5 cases with SHOX duplications including a sib-pair with MRKH syndrome. No duplications were identified among controls. Guerrier and Morcel (64) reported similar findings with three SHOX duplications and one duplication downstream of the gene. In contrast, Sandbacka et al. found no duplications among 101 Finnish cases questioning the role of SHOX in MD development (63). More recently, Mikhael et al. reported two missense variants from an investigation of 72 candidate genes in 111 cases (103). The functional role of these duplications (including their insertion sites) and missense variants is unknown and causality has not been established.
5.8 WNT9B
Wnt9b encodes a protein involved in MD development and Wnt9b knock-down in mice causes uterovaginal and renal agenesis. Wnt9b is expressed in the Wolffian duct epithelium providing signals guiding MD elongation (144). WNT9B has therefore been considered a candidate gene in MRKH syndrome and has been investigated in several studies identifying a total of 9 sequence variants in MRKH syndrome type I, of these 7 missense variants (Table 2) (98, 103, 113, 114). One patient has been reported with two variants (114). Other studies found no variants in MRKH syndrome patients (145, 146). WNT9B variants have also been reported in other uterovaginal malformations (114, 146). The functional role of these variants remains to be ascertained.
5.9 WNT4
As previously described, WNT4 was the first gene identified as a monogenic cause of MD agenesis in females (OMIM #158330) (115). Since its discovery, a total of seven missense variants have been reported (103, 115–118). A missense variant of WNT4 has also been reported with another uterovaginal anomaly with renal agenesis (Herlyn-Werner-Wunderlich syndrome) (147).
Importantly, MD agenesis caused by WNT4 variants is associated with clinical and biochemical hyperandrogenism, representing a phenotype distinct from MRKH syndrome in general as described by Biason-Lauber et al. (116), and is often considered a separate entity. This is also supported by several investigations reporting no variants in larger MRKH syndrome and MD anomaly cohorts (30, 148, 149).
5.10 LRP10
Lrp10 encodes low-density lipoprotein receptor-related protein 10, which has been proposed as a negative regulator of Wnt/β-catenin signaling (150), a pathway involved in MD development (151). In 2015, Rall et al. reported on a SNP-array analysis of CNVs in discordant twin pairs. In one affected twin, a 585 kb duplication at 14q11.2 spanning LPR10 was identified, not present in the other twin, and this gene was suggested as a candidate gene for MRKH syndrome. Two subsequent studies have reported a total of five missense variants in LRP10, hereof two variants in the same patient (74, 103). Functional evidence for these variants is lacking and causality has not been established.
6 Discussion
As described, knowledge of genetic variation in MRKH syndrome has increased considerably during recent years enabled by WES/WGS analysis. In the following, thoughts on how to continue the search for genetic causes are presented and potential implications for future clinical care are discussed.
6.1 Family history is key
The development of the field shows the importance of detailed family history in the genetic assessment, as it may include important information to suggest an underlying genetic trait or perhaps provide information on the genetic nature of the family’s trait. The early descriptions of hereditary urogenital adysplasia families, reported long before molecular genetic testing became available, is a good example hereof suggesting an autosomal dominant trait with sex-limited expressivity (41–43, 140, 141). In recent years, GREB1L variants have been identified as a major cause of MRKH syndrome, particularly in families with a hereditary urogenital adysplasia-like presentations (101, 102).
Family histories should not only include incidents of MRKH syndrome but also associated extragenital malformations and other uterovaginal malformations in family members. The subtle nature of many renal and uterovaginal malformations is, however, important to consider, as these may not yet have been revealed in asymptomatic relatives, requiring radiological imaging. The relevance of other uterovaginal malformations in the genetic assessment of MRKH syndrome patients is highlighted by the many genes reported in both phenotypes, including 17q12 deletion (85, 124), EMX2 (75, 152), GREB1L (102, 109, 110), HNF1B (52, 129, 130), HOXA10 (58, 75, 153), SPECC1L (74, 154, 155), WNT4 (115, 147), and WNT9B (114). This could suggest a spectrum of uterovaginal anomalies with shared causes.
6.2 Future genetic studies
Exome and genome sequencing have proven useful in the discovery of new genes during recent years, including GREB1L (99, 101, 102) and PAX8 (75). With the continuously developing field of genomic sequencing technologies and multi-omics analyses, there is reason to believe that additional genetic causes remain to be identified. Recently, the first complete telomere-to-telomere genome reference was published, which will support even better detection of genetic variation in future genetic studies (156). Also, our understanding of the role of non-coding parts in the genome such as topologically associated domains and long non-coding RNAs is increasing (157, 158). Genome sequencing today is typically based on short-read sequencing due to its cost-effectiveness and accuracy. Long-read sequencing has increased in use and provides some advantages in terms of de novo assembly, read-mapping, detection of structural variants, phasing of variants, and transcript isoform identification (159). Another technology for improved analysis of structural variation is optical genome mapping, which has been applied by Brakta et al. (73). All along the technical developments, it will still be relevant to revisit sequencing data of unsolved cases, as new knowledge emerges supporting variant interpretation. Sequenced MRKH syndrome cohorts will increase in size, as will available genomic data resources, which will improve rare variant discovery from association analyses.
It is also relevant to look for somatic variation or differential gene expression in affected tissues as previously performed on surgically-removed uterine remnants (35, 98, 106–108). This may also be helpful in functional analyses of candidate variants of unknown significance to provide evidence for pathogenicity. However, it should be considered that organogenesis in the embryo is an accurately orchestrated process with precise spatiotemporal control of gene expression (160). Therefore, gene expression in adult uterine remnants may not be representative of the gene expression occurring during embryonic development, which in that case would require mouse modeling. As the causal effect of many gene variants remains unknown, functional analyses will be important for further progress.
Finally, it will be important to investigate candidate variants in extended pedigrees to understand the mode of inheritance and disease segregation, also considering more complex mechanisms in developmental genomics as proposed in the compound inheritance gene dosage model (161).
6.3 Importance of a genetic diagnosis – possible implications for clinical practice
Identifying genetic causes of MRKH syndrome is of immense importance including both academic pursuits towards improving our understanding of genetic factors underlying human uterovaginal development as well as establishing the necessary evidence to provide informed care and counseling for the individual patient and their families in clinical settings. Rare disease patients often undergo lengthy diagnostic odysseys from the first suspicion until the time of (genetic) diagnosis. Although the diagnostics in MRKH syndrome primarily involve finding the gynecological cause for primary amenorrhea, questions of “why did this happen?” remain and feelings of guilt and responsibility are prevalent among patients and their parents (11, 12). Identifying the underlying cause of malformation syndromes may reduce the burden for patients and families, even if it does not lead to changes in treatment (162).
Genetic counseling and management following the identification of a gene variant depends on the ‘clinical actionability’ of the variant, which requires both a valid gene-condition association and sufficient evidence for pathogenicity (163). As evidence for genetic causes in MRKH syndrome continues to increase, it may contribute to better disease classification in the future based on molecular causes instead of the current clinical distinction of type I and II. It can also be expected to guide future patient management such as examinations for associated diseases and anomalies specific to the particular gene. Clinically actionable variants may lead to genetic testing of family members at risk, and variant carriers may then be referred to relevant specialists for further examination. Finally, genetic counseling may address the recurrence risk if patients/couples with an identified genetic cause wish to pursue genetic parenthood either through gestational surrogacy or UTx. The possibility of recurrence in offspring was demonstrated in a recent report, describing the first case of mother-to-daughter inheritance following surrogacy (22). As UTx approaches clinical implementation (24) and the evidence for genetic causes increases, questions about offspring recurrence risk and reproductive choices will likely become more prevalent and so will requests for preimplantation genetic testing as part of the in vitro fertilization preceding the UTx procedure.
However, in the current state of knowledge, many reported variants associated with MRKH syndrome are still to be considered as variants of uncertain significance, which warrant cautious interpretations and counseling in clinical care.
7 Conclusion
In conclusion, recent advancements provide evidence for genetic causes of MRKH syndrome. The combined evidence points towards a heterogeneous etiology with various genes implicated. With the identification of monogenic causes in MRKH syndrome and increasing fertility options allowing couples to pursue genetic parenthood, the need for genetic counseling will likely increase.
Author contributions
MKH: Conceptualization, Data curation, Formal analysis, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Lundbeck Foundation (grant # R403-2022-1385) and the Health Research Foundation of Central Denmark Region (grant # A4204).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1368990/full#supplementary-material.
References
1. Habiba M, Heyn R, Bianchi P, Brosens I, Benagiano G. The development of the human uterus: morphogenesis to menarche. Hum Reprod Update. (2021) 27:1–26. doi: 10.1093/humupd/dmaa036
2. Grimbizis GF, Gordts S, Di Spiezio Sardo A, Brucker S, De Angelis C, Gergolet M, et al. The ESHRE/ESGE consensus on the classification of female genital tract congenital anomalies. Hum Reprod. (2013) 28:2032–44. doi: 10.1093/humrep/det098
3. Pfeifer SM, Attaran M, Goldstein J, Lindheim SR, Petrozza JC, Rackow BW, et al. ASRM müllerian anomalies classification 2021. Fertil Steril. (2021) 116:1238–52. doi: 10.1016/j.fertnstert.2021.09.025
4. Herlin MK, Petersen MB, Brännström M. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis. (2020) 15:214. doi: 10.1186/s13023-020-01491-9
5. Herlin M, Bjorn AM, Rasmussen M, Trolle B, Petersen MB. Prevalence and patient characteristics of Mayer-Rokitansky-Kuster-Hauser syndrome: a nationwide registry-based study. Hum Reprod. (2016) 31:2384–90. doi: 10.1093/humrep/dew220
6. Aittomaki K, Eroila H, Kajanoja P. A population-based study of the incidence of Mullerian aplasia in Finland. Fertil Steril. (2001) 76:624–5. doi: 10.1016/s0015-0282(01)01963-x
7. Timmreck LS, Reindollar RH. Contemporary issues in primary amenorrhea. Obstet Gynecol Clin North Am. (2003) 30:287–302. doi: 10.1016/S0889-8545(03)00027-5
8. Ernst ME, Sandberg DE, Keegan C, Quint EH, Lossie AC, Yashar BM. The lived experience of MRKH: sharing health information with peers. J Pediatr Adolesc Gynecol. (2016) 29:154–8. doi: 10.1016/j.jpag.2015.09.009
9. Patterson CJ, Crawford R, Jahoda A. Exploring the psychological impact of Mayer-Rokitansky-Küster-Hauser syndrome on young women: An interpretative phenomenological analysis. J Health Psychol. (2016) 21:1228–40. doi: 10.1177/1359105314551077
10. Tsitoura A, Michala L. The sexuality of adolescents and young women with MRKH syndrome: A qualitative study. J Sex Med. (2021) 18:2012–9. doi: 10.1016/j.jsxm.2021.09.006
11. Hatim H, Zainuddin AA, Anizah A, Kalok A, Daud TIM, Ismail A, et al. The missing uterus, the missed diagnosis, and the missing care. Mayer-rokitansky-küster-hauser syndrome in the lives of women in Malaysia. J Pediatr Adolesc Gynecol. (2021) 34:161–7. doi: 10.1016/j.jpag.2020.11.009
12. Jensen AH, Herlin MK, Vogel I, Lou S. A life course perspective on Mayer-Rokitansky-Küster-Hauser syndrome: women’s experiences and negotiations of living with an underdeveloped uterus and vagina. Disabil Rehabil. (2023) 46(6):1130–40. doi: 10.1080/09638288.2023.2191014
13. Laggari V, Diareme S, Christogiorgos S, Deligeoroglou E, Christopoulos P, Tsiantis J, et al. Anxiety and depression in adolescents with polycystic ovary syndrome and Mayer-Rokitansky-Küster-Hauser syndrome. J Psychosom Obstet Gynaecol. (2009) 30:83–8. doi: 10.1080/01674820802546204
14. Chen N, Song S, Duan Y, Kang J, Deng S, Pan H, et al. Study on depressive symptoms in patients with Mayer-Rokitansky-Küster-Hauser syndrome: an analysis of 141 cases. Orphanet J Rare Dis. (2020) 15:121. doi: 10.1186/s13023-020-01405-9
15. Song S, Chen N, Duan Y-P, Kang J, Deng S, Pan H-X, et al. Anxiety symptoms in patients with Mayer-Rokitansky-Küster-Hauser syndrome: a cross-sectional study. Chin Med J (Engl). (2020) 133:388–94. doi: 10.1097/CM9.0000000000000648
16. Weijenborg PT, ter Kuile MM. The effect of a group programme on women with the Mayer-Rokitansky-Küster-Hauser syndrome. BJOG. (2000) 107:365–8. doi: 10.1111/j.1471-0528.2000.tb13232.x
17. Heller-Boersma JG, Schmidt UH, Edmonds DK. A randomized controlled trial of a cognitive-behavioural group intervention versus waiting-list control for women with uterovaginal agenesis (Mayer-Rokitansky-Küster-Hauser syndrome: MRKH). Hum Reprod. (2007) 22:2296–301. doi: 10.1093/humrep/dem167
18. ACOG. ACOG committee opinion no. 728: müllerian agenesis: diagnosis, management, and treatment. Obstet Gynecol. (2018) 131:e35–42. doi: 10.1097/AOG.0000000000002458
19. Herlin M, Bay Bjørn A-M, Jørgensen LK, Trolle B, Petersen MB. Treatment of vaginal agenesis in Mayer-Rokitansky-Küster-Hauser syndrome in Denmark: a nationwide comparative study of anatomical outcome and complications. Fertil Steril. (2018) 110(4):746–53. doi: 10.1016/j.fertnstert.2018.05.015
20. Cheikhelard A, Bidet M, Baptiste A, Viaud M, Fagot C, Khen-Dunlop N, et al. Surgery is not superior to dilation for the management of vaginal agenesis in Mayer-Rokitansky-Küster-Hauser syndrome: a multicenter comparative observational study in 131 patients. Am J Obstet Gynecol. (2018) 219:281.e1–9. doi: 10.1016/j.ajog.2018.07.015
21. Friedler S, Grin L, Liberti G, Saar-Ryss B, Rabinson Y, Meltzer S. The reproductive potential of patients with Mayer-Rokitansky-Küster-Hauser syndrome using gestational surrogacy: a systematic review. Reprod BioMed Online. (2016) 32:54–61. doi: 10.1016/j.rbmo.2015.09.006
22. Daum H, Kremer E, Frumkin A, Meiner V, Diamant H, Harel I, et al. A case report of familial mayer-rokitansky-küster-hauser syndrome as part of the phenotypic spectrum of the 2q37 deletion. J Pediatr Adolesc Gynecology. (2023) 37(1):95–7. doi: 10.1016/j.jpag.2023.09.006
23. Brännström M, Johannesson L, Bokström H, Kvarnström N, Mölne J, Dahm-Kähler P, et al. Livebirth after uterus transplantation. Lancet (London England). (2015) 385:607–16. doi: 10.1016/S0140-6736(14)61728-1
24. Brännström M, Racowsky C, Carbonnel M, Wu J, Gargiulo A, Adashi EY, et al. Uterus transplantation: from research, through human trials and into the future. Hum Reprod Update. (2023) 29:521–44. doi: 10.1093/humupd/dmad012
25. Robboy SJ, Kurita T, Baskin L, Cunha GR. New insights into human female reproductive tract development. Differentiation. (2017) 97:9–22. doi: 10.1016/j.diff.2017.08.002
26. Kobayashi A, Behringer RR. Developmental genetics of the female reproductive tract in mammals. Nat Rev Genet. (2003) 4:969–80. doi: 10.1038/nrg1225
27. Preibsch H, Rall K, Wietek BM, Brucker SY, Staebler A, Claussen CD, et al. Clinical value of magnetic resonance imaging in patients with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: diagnosis of associated malformations, uterine rudiments and intrauterine endometrium. Eur Radiol. (2014) 24:1621–7. doi: 10.1007/s00330-014-3156-3
28. Tani S, Chung U, Ohba S, Hojo H. Understanding paraxial mesoderm development and sclerotome specification for skeletal repair. Exp Mol Med. (2020) 52:1166–77. doi: 10.1038/s12276-020-0482-1
29. Prummel KD, Nieuwenhuize S, Mosimann C. The lateral plate mesoderm. Development. (2020) 147(12):dev175059. doi: 10.1242/dev.175059
30. Williams LS, Demir Eksi D, Shen Y, Lossie AC, Chorich LP, Sullivan ME, et al. Genetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families. Fertil Steril. (2017) 108:145–151.e2. doi: 10.1016/j.fertnstert.2017.05.017
31. Lischke JH, Curtis CH, Lamb EJ. Discordance of vaginal agenesis in monozygotic twins. Obstet Gynecol. (1973) 41:920–4.
32. Regenstein AC, Berkeley AS. Discordance of müllerian agenesis in monozygotic twins. A case report. J Reprod Med. (1991) 36:396–7.
33. Duru UA, Laufer MR. Discordance in Mayer-von Rokitansky-Küster-Hauser Syndrome noted in monozygotic twins. J Pediatr Adolesc Gynecol. (2009) 22:e73-5. doi: 10.1016/j.jpag.2008.07.012
34. Milsom SR, Ogilvie CM, Jefferies C, Cree L. Discordant Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome in identical twins - a case report and implications for reproduction in MRKH women. Gynecol Endocrinol Off J Int Soc Gynecol Endocrinol. (2015) 31:684–7. doi: 10.3109/09513590.2015.1032928
35. Rall K, Eisenbeis S, Barresi G, Rückner D, Walter M, Poths S, et al. Mayer-Rokitansky-Küster-Hauser syndrome discordance in monozygotic twins: matrix metalloproteinase 14, low-density lipoprotein receptor-related protein 10, extracellular matrix, and neoangiogenesis genes identified as candidate genes in a tissue-specific. Fertil Steril. (2015) 103:494–502.e3. doi: 10.1016/j.fertnstert.2014.10.053
36. Petrozza JC, Gray MR, Davis AJ, Reindollar RH. Congenital absence of the uterus and vagina is not commonly transmitted as a dominant genetic trait: outcomes of surrogate pregnancies. Fertil Steril. (1997) 67:387–9. doi: 10.1016/S0015-0282(97)81927-9
37. Hoffmann W, Grospietsch G, Kuhn W. Thalidomide and female genital malformations. Lancet (London England). (1976) 2:794. England. doi: 10.1016/S0140-6736(76)90618-8
38. Robboy SJ, Taguchi O, Cunha GR. Normal development of the human female reproductive tract and alterations resulting from experimental exposure to diethylstilbestrol. Hum Pathol. (1982) 13:190–8. doi: 10.1016/S0046-8177(82)80177-9
39. Ema M, Miyawaki E, Kawashima K. Suppression of uterine decidualization as a cause of implantation failure induced by triphenyltin chloride in rats. Arch Toxicol. (1999) 73:175–9. doi: 10.1007/s002040050603
40. Hannas BR, Howdeshell KL, Furr J, Gray LEJ. In utero phthalate effects in the female rat: a model for MRKH syndrome. Toxicol Lett. (2013) 223:315–21. doi: 10.1016/j.toxlet.2013.03.021
41. Buchta RM, Viseskul C, Gilbert EF, Sarto GE, Opitz JM. Familial bilateral renal agenesis and hereditary renal adysplasia. Z Kinderheilkd. (1973) 115:111–29. doi: 10.1007/BF00440537
42. Opitz JM. Vaginal atresia (von Mayer-Rokitansky-Küster or MRK anomaly) in hereditary renal adysplasia (HRA). Am J Med Genet. (1987) 26:873–6. doi: 10.1002/ajmg.1320260414
43. Schimke RN, King CR. Hereditary urogenital adysplasia. Clin Genet. (1980) 18:417–20. doi: 10.1111/j.1399-0004.1980.tb01786.x
44. Herlin M, Hojland AT, Petersen MB. Familial occurrence of Mayer-Rokitansky-Kuster-Hauser syndrome: a case report and review of the literature. Am J Med Genet A. (2014) 164a:2276–86. doi: 10.1002/ajmg.a.36652
45. Hauser GA, Schreiner WE. [Mayer-Rokitansky-Kuester syndrome. Rudimentary solid bipartite uterus with solid vagina]. Schweiz Med Wochenschr. (1961) 91:381–4.
46. Hauser GA, Keller M, Koller T, Wenner R. [The Rokitansky-Kuester-syndrome. Uterus bipartitus solidus rudimentarius cum vagina solida]. Gynaecologia. (1961) 151:111–2.
47. Cramer DW, Goldstein DP, Fraer C, Reichardt JK. Vaginal agenesis (Mayer-Rokitansky-Kuster-Hauser syndrome) associated with the N314D mutation of galactose-1-phosphate uridyl transferase (GALT). Mol Hum Reprod. (1996) 2:145–8. doi: 10.1093/molehr/2.3.145
48. Klipstein S, Bhagavath B, Topipat C, Sasur L, Reindollar RH, Gray MR. The N314D polymorphism of the GALT gene is not associated with congenital absence of the uterus and vagina. Mol Hum Reprod. (2003) 9:171–4. doi: 10.1093/molehr/gag018
49. Zenteno JC, Carranza-Lira S, Kofman-Alfaro S. Molecular analysis of the anti-Müllerian hormone, the anti-Müllerian hormone receptor, and galactose-1-phosphate uridyl transferase genes in patients with the Mayer-Rokitansky-Küster-Hauser syndrome. Arch Gynecol Obstet. (2004) 269:270–3. doi: 10.1007/s00404-002-0456-7
50. van Lingen BL, Reindollar RH, Davis AJ, Gray MR. Further evidence that the WT1 gene does not have a role in the development of the derivatives of the müllerian duct. Am J Obstet Gynecol. (1998) 179(3 Pt 1):597–603. doi: 10.1016/s0002-9378(98)70051-1
51. Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. (1999) 8:2001–8. doi: 10.1093/hmg/8.11.2001
52. Oram RA, Edghill EL, Blackman J, Taylor MJO, Kay T, Flanagan SE, et al. Mutations in the hepatocyte nuclear factor-1β (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am J Obstet Gynecol. (2010) 203:364.e1–5. doi: 10.1016/j.ajog.2010.05.022
53. Resendes BL, Sohn SH, Stelling JR, Tineo R, Davis AJ, Gray MR, et al. Role for anti-Müllerian hormone in congenital absence of the uterus and vagina. Am J Med Genet. (2001) 98:129–36. doi: 10.1002/(ISSN)1096-8628
54. Timmreck LS, Gray MR, Handelin B, Allito B, Rohlfs E, Davis AJ, et al. Analysis of cystic fibrosis transmembrane conductance regulator gene mutations in patients with congenital absence of the uterus and vagina. Am J Med Genet A. (2003) 120A:72–6. doi: 10.1002/ajmg.a.20197
55. Timmreck LS, Pan HA, Reindollar RH, Gray MR. WNT7A mutations in patients with Müllerian duct abnormalities. J Pediatr Adolesc Gynecol. (2003) 16:217–21. doi: 10.1016/S1083-3188(03)00124-4
56. Burel A, Mouchel T, Odent S, Tiker F, Knebelmann B, Pellerin I, et al. Role of HOXA7 to HOXA13 and PBX1 genes in various forms of MRKH syndrome (congenital absence of uterus and vagina). J Negat Results Biomed. (2006) 5:4. doi: 10.1186/1477-5751-5-4
57. Lalwani S, Wu H, Reindollar RH, Gray MR. HOXA10 mutations in congenital absence of uterus and vagina. Fertil Steril. (2008) 89:325–30. doi: 10.1016/j.fertnstert.2007.03.033
58. Ekici AB, Strissel PL, Oppelt PG, Renner SP, Brucker S, Beckmann MW, et al. HOXA10 and HOXA13 sequence variations in human female genital malformations including congenital absence of the uterus and vagina. Gene. (2013) 518:267–72. doi: 10.1016/j.gene.2013.01.030
59. Ma J, Qin Y, Liu W, Duan H, Xia M, Chen Z-J. Analysis of PBX1 mutations in 192 Chinese women with Müllerian duct abnormalities. Fertil Steril. (2011) 95:2615–7. doi: 10.1016/j.fertnstert.2011.04.074
60. Cheroki C, Krepischi-Santos AC, Rosenberg C, Jehee FS, Mingroni-Netto RC, Pavanello Filho I, et al. Report of a del22q11 in a patient with Mayer-Rokitansky-Küster-Hauser (MRKH) anomaly and exclusion of WNT-4, RAR-gamma, and RXR-alpha as major genes determining MRKH anomaly in a study of 25 affected women. Am J Med Genet A. (2006) 140:1339–42. doi: 10.1002/ajmg.a.31254
61. Drummond JB, Rezende CF, Peixoto FC, Carvalho JS, Reis FM, De Marco L. Molecular analysis of the beta-catenin gene in patients with the Mayer-Rokitansky-Küster-Hauser syndrome. J Assist Reprod Genet. (2008) 25:511–4. doi: 10.1007/s10815-008-9261-y
62. Gervasini C, Grati FR, Lalatta F, Tabano S, Gentilin B, Colapietro P, et al. SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet Med Off J Am Coll Med Genet. (2010) 12:634–40. doi: 10.1097/GIM.0b013e3181ed6185
63. Sandbacka M, Halttunen M, Jokimaa V, Aittomäki K, Laivuori H. Evaluation of SHOX copy number variations in patients with Müllerian aplasia. Orphanet J Rare Dis. (2011) 6:53. doi: 10.1186/1750-1172-6-53
64. Guerrier D, Morcel K. Partial SHOX duplications associated with various cases of congenital uterovaginal aplasia (MRKH syndrome): A tangible evidence but a puzzling mechanism. J Genet Med Gene Ther. (2021) 4:1–8. doi: 10.29328/journal.jgmgt
65. Wang P, Zhao H, Sun M, Li Y, Chen Z-J. PAX2 in 192 Chinese women with Müllerian duct abnormalities: mutation analysis. Reprod BioMed Online. (2012) 25:219–22. doi: 10.1016/j.rbmo.2012.04.010
66. Ravel C, Bashamboo A, Bignon-Topalovic J, Siffroi J-P, McElreavey K, Darai E. Polymorphisms in DLGH1 and LAMC1 in mayer-rokitansky-kuster-hauser syndrome. Reprod BioMed Online. (2012) 24:462–5. doi: 10.1016/j.rbmo.2011.12.008
67. Morcel K, Watrin T, Jaffre F, Deschamps S, Omilli F, Pellerin I, et al. Involvement of ITIH5, a candidate gene for congenital uterovaginal aplasia (Mayer-Rokitansky-Küster-Hauser syndrome), in female genital tract development. Gene Expr. (2012) 15:207–14. doi: 10.3727/105221613x13571653093169
68. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. (2001) 409:860–921. doi: 10.1038/35057062
69. Consortium IHGS. Finishing the euchromatic sequence of the human genome. Nature. (2004) 431:931–45. doi: 10.1038/nature03001
70. Taylor CM, Smith R, Lehman C, Mitchel MW, Singer K, Weaver WC, et al. “16p11.2 recurrent deletion”. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle (1993). Available at: https://www.ncbi.nlm.nih.gov/books/NBK11167/.
71. Nik-Zainal S, Strick R, Storer M, Huang N, Rad R, Willatt L, et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Mullerian aplasia. J Med Genet. (2011) 48:197–204. doi: 10.1136/jmg.2010.082412
72. Sandbacka M, Laivuori H, Freitas E, Halttunen M, Jokimaa V, Morin-Papunen L, et al. TBX6, LHX1 and copy number variations in the complex genetics of Mullerian aplasia. Orphanet J Rare Dis. (2013) 8:125. doi: 10.1186/1750-1172-8-125
73. Brakta S, Hawkins ZA, Sahajpal N, Seman N, Kira D, Chorich LP, et al. Rare structural variants, aneuploidies, and mosaicism in individuals with Mullerian aplasia detected by optical genome mapping. Hum Genet. (2023) 142:483–94. doi: 10.1007/s00439-023-02522-8
74. Backhouse B, Hanna C, Robevska G, van den Bergen J, Pelosi E, Simons C, et al. Identification of candidate genes for mayer-rokitansky-küster-hauser syndrome using genomic approaches. Sex Dev Genet Mol Biol Evol Endocrinol Embryol Pathol sex Determ Differ. (2019) 13:26–34. doi: 10.1159/000494896
75. Chen N, Zhao S, Jolly A, Wang L, Pan H, Yuan J, et al. Perturbations of genes essential for Müllerian duct and Wölffian duct development in Mayer-Rokitansky-Küster-Hauser syndrome. Am J Hum Genet. (2021) 108:337–45. doi: 10.1016/j.ajhg.2020.12.014
76. Pontecorvi P, Bernardini L, Capalbo A, Ceccarelli S, Megiorni F, Vescarelli E, et al. Protein-protein interaction network analysis applied to DNA copy number profiling suggests new perspectives on the aetiology of Mayer-Rokitansky-Küster-Hauser syndrome. Sci Rep. (2021) 11:448. doi: 10.1038/s41598-020-79827-5
77. Demir Eksi D, Shen Y, Erman M, Chorich LP, Sullivan ME, Bilekdemir M, et al. Copy number variation and regions of homozygosity analysis in patients with MÜLLERIAN aplasia. Mol Cytogenet. (2018) 11:13. doi: 10.1186/s13039-018-0359-3
78. Gatti M, Tolva G, Bergamaschi S, Giavoli C, Esposito S, Marchisio P, et al. Mayer-rokitansky-küster-hauser syndrome and 16p11.2 recurrent microdeletion: A case report and review of the literature. J Pediatr Adolesc Gynecol. (2018) 31:533–5. doi: 10.1016/j.jpag.2018.04.003
79. Su K, Liu H, Ye X, Jin H, Xie Z, Yang C, et al. Recurrent human 16p11.2 microdeletions in type I Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome patients in Chinese Han population. Mol Genet Genomic Med. (2023) 12(1):e2280. doi: 10.1002/mgg3.2280
80. Mitchel MW, Moreno-De-Luca D, Myers SM, Levy RV, Turner S, Ledbetter DH, et al. “17q12 recurrent deletion syndrome”. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle (1993). Available at: https://www.ncbi.nlm.nih.gov/books/NBK401562/.
81. Cheroki C, Krepischi-Santos AC, Szuhai K, Brenner V, Kim CA, Otto PA, et al. Genomic imbalances associated with mullerian aplasia. J Med Genet. (2008) 45:228–32. doi: 10.1136/jmg.2007.051839
82. McGowan R, Tydeman G, Shapiro D, Craig T, Morrison N, Logan S, et al. DNA copy number variations are important in the complex genetic architecture of müllerian disorders. Fertil Steril. (2015) 103:1021–30. doi: 10.1016/j.fertnstert.2015.01.008
83. Thomson E, Tran M, Robevska G, Ayers K, van der Bergen J, Gopalakrishnan Bhaskaran P, et al. Functional genomics analysis identifies loss of HNF1B function as a cause of Mayer-Rokitansky-Küster-Hauser syndrome. Hum Mol Genet. (2023) 32:1032–47. doi: 10.1093/hmg/ddac262
84. Bernardini L, Gimelli S, Gervasini C, Carella M, Baban A, Frontino G, et al. Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports. Orphanet J Rare Dis. (2009) 4:25. doi: 10.1186/1750-1172-4-25
85. Ledig S, Schippert C, Strick R, Beckmann MW, Oppelt PG, Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. (2011) 95:1589–94. doi: 10.1016/j.fertnstert.2010.07.1062
86. Hinkes B, Hilgers KF, Bolz HJ, Goppelt-Struebe M, Amann K, Nagl S, et al. A complex microdeletion 17q12 phenotype in a patient with recurrent de novo membranous nephropathy. BMC Nephrol. (2012) 13:27. doi: 10.1186/1471-2369-13-27
87. Devriendt K, Moerman P, Van Schoubroeck D, Vandenberghe K, Fryns JP. Chromosome 22q11 deletion presenting as the Potter sequence. J Med Genet. (1997) 34:423–5. doi: 10.1136/jmg.34.5.423
88. Le Caignec C, Boceno M, Saugier-Veber P, Jacquemont S, Joubert M, David A, et al. Detection of genomic imbalances by array based comparative genomic hybridisation in fetuses with multiple malformations. J Med Genet. (2005) 42:121–8. doi: 10.1136/jmg.2004.025478
89. Morcel K, Watrin T, Pasquier L, Rochard L, Le Caignec C, Dubourg C, et al. Utero-vaginal aplasia (Mayer-Rokitansky-Kuster-Hauser syndrome) associated with deletions in known DiGeorge or DiGeorge-like loci. Orphanet J Rare Dis. (2011) 6:9. doi: 10.1186/1750-1172-6-9
90. Sundaram UT, McDonald-McGinn DM, Huff D, Emanuel BS, Zackai EH, Driscoll DA, et al. Primary amenorrhea and absent uterus in the 22q11.2 deletion syndrome. Am J Med Genet A. (2007) 143A:2016–8. doi: 10.1002/ajmg.a.31736
91. AlSubaihin A, VanderMeulen J, Harris K, Duck J, McCready E. Müllerian agenesis in cat eye syndrome and 22q11 chromosome abnormalities: A case report and literature review. J Pediatr Adolesc Gynecol. (2018) 31:158–61. doi: 10.1016/j.jpag.2017.09.004
92. Dell’Edera D, Allegretti A, Ventura M, Mercuri L, Mitidieri A, Cuscianna G, et al. Mayer-Rokitansky-Küster-Hauser syndrome with 22q11.21 microduplication: a case report. J Med Case Rep. (2021) 15:208. doi: 10.1186/s13256-021-02716-6
93. Miclea D, Alkhzouz C, Bucerzan S, Grigorescu-Sido P, Popp RA, Pascanu IM, et al. Molecular and cytogenetic analysis of Romanian patients with differences in sex development. Diagnostics (Basel Switzerland). (2021) 11(11):2107. doi: 10.3390/diagnostics11112107
94. Ma D, Marion R, Punjabi NP, Pereira E, Samanich J, Agarwal C, et al. A de novo 10.79 Mb interstitial deletion at 2q13q14.2 involving PAX8 causing hypothyroidism and mullerian agenesis: A novel case report and literature review. Mol Cytogenet. (2014) 7:4–9. doi: 10.1186/s13039-014-0085-4
95. Smol T, Ribero-Karrouz W, Edery P, Gorduza DB, Catteau-Jonard S, Manouvrier-Hanu S, et al. Mayer-Rokitansky-Künster-Hauser syndrome due to 2q12.1q14.1 deletion: PAX8 the causing gene? Eur J Med Genet. (2020) 63:103812. doi: 10.1016/j.ejmg.2019.103812
96. Chen M-J, Wei S-Y, Yang W-S, Wu T-T, Li H-Y, Ho H-N, et al. Concurrent exome-targeted next-generation sequencing and single nucleotide polymorphism array to identify the causative genetic aberrations of isolated Mayer-Rokitansky-Küster-Hauser syndrome. Hum Reprod. (2015) 30:1732–42. doi: 10.1093/humrep/dev095
97. Takahashi K, Hayano T, Sugimoto R, Kashiwagi H, Shinoda M, Nishijima Y, et al. Exome and copy number variation analyses of Mayer-Rokitansky-Küster- Hauser syndrome. Hum Genome variation. (2018) 5:27. England. doi: 10.1038/s41439-018-0028-4
98. Buchert R, Schenk E, Hentrich T, Weber N, Rall K, Sturm M, et al. Genome sequencing and transcriptome profiling in twins discordant for mayer-rokitansky-küster-hauser syndrome. J Clin Med. (2022) 11(19):5598. doi: 10.1101/2022.06.01.22275812
99. Jolly A, Du H, Borel C, Chen N, Zhao S, Grochowski CM, et al. Rare variant enrichment analysis supports GREB1L as a contributory driver gene in the etiology of Mayer-Rokitansky-Küster-Hauser syndrome. HGG Adv. (2023) 4:100188. doi: 10.1016/j.xhgg.2023.100188
100. Pan H-X, Luo G-N, Wan S-Q, Qin C-L, Tang J, Zhang M, et al. Detection of de novo genetic variants in Mayer-Rokitansky-Küster-Hauser syndrome by whole genome sequencing. Eur J Obstet Gynecol Reprod Biol X. (2019) 4:100089. doi: 10.1016/j.eurox.2019.100089
101. Herlin MK, Le VQ, Højland AT, Ernst A, Okkels H, Petersen AC, et al. Whole-exome sequencing identifies a GREB1L variant in a three-generation family with Müllerian and renal agenesis: A novel candidate gene in Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. A case report. Hum Reprod. (2019) 34:1838–46. doi: 10.1093/humrep/dez126
102. Jacquinet A, Boujemla B, Fasquelle C, Thiry J, Josse C, Lumaka A, et al. GREB1L variants in familial and sporadic hereditary urogenital adysplasia and Mayer-Rokitansky-Kuster-Hauser syndrome. Clin Genet. (2020) 98:126–37. doi: 10.1111/cge.13769
103. Mikhael S, Dugar S, Morton M, Chorich LP, Tam KB, Lossie AC, et al. Genetics of agenesis/hypoplasia of the uterus and vagina: narrowing down the number of candidate genes for Mayer-Rokitansky-Küster-Hauser Syndrome. Hum Genet. (2021) 140:667–80. doi: 10.1007/s00439-020-02239-y
104. Chu C, Li L, Li S, Zhou Q, Zheng P, Zhang Y-D, et al. Variants in genes related to development of the urinary system are associated with Mayer-Rokitansky-Küster-Hauser syndrome. Hum Genomics. (2022) 16:10. doi: 10.1186/s40246-022-00385-0
105. Ma C, Chen N, Jolly A, Zhao S, Coban-Akdemir Z, Tian W, et al. Functional characteristics of a broad spectrum of TBX6 variants in Mayer-Rokitansky-Küster-Hauser syndrome. Genet Med Off J Am Coll Med Genet. (2022) 24:2262–73. doi: 10.1016/j.gim.2022.08.012
106. Rall K, Barresi G, Walter M, Poths S, Haebig K, Schaeferhoff K, et al. A combination of transcriptome and methylation analyses reveals embryologically-relevant candidate genes in MRKH patients. Orphanet J Rare Dis. (2011) 6:32. doi: 10.1186/1750-1172-6-32
107. Hentrich T, Koch A, Weber N, Kilzheimer A, Maia A, Burkhardt S, et al. The endometrial transcription landscape of MRKH syndrome. Front Cell Dev Biol. (2020) 8:572281. doi: 10.3389/fcell.2020.572281
108. Brucker SY, Hentrich T, Schulze-Hentrich JM, Pietzsch M, Wajngarten N, Singh AR, et al. Endometrial organoids derived from Mayer-Rokitansky-Küster-Hauser syndrome patients provide insights into disease-causing pathways. Dis Model Mech. (2022) 15(5):dmm049379. doi: 10.1242/dmm.049379
109. De Tomasi L, David P, Humbert C, Silbermann F, Arrondel C, Tores F, et al. Mutations in GREB1L cause bilateral kidney agenesis in humans and mice. Am J Hum Genet. (2017) 101:803–14. doi: 10.1016/j.ajhg.2017.09.026
110. Sanna-Cherchi S, Khan K, Westland R, Krithivasan P, Fievet L, Rasouly HM, et al. Exome-wide association study identifies GREB1L mutations in congenital kidney malformations. Am J Hum Genet. (2017) 101:789–802. doi: 10.1016/j.ajhg.2017.09.018
111. Tewes A-C, Hucke J, Römer T, Kapczuk K, Schippert C, Hillemanns P, et al. Sequence variants in TBX6 are associated with disorders of the müllerian ducts: an update. Sex Dev Genet Mol Biol Evol Endocrinol Embryol Pathol sex Determ Differ. (2019) 13:35–40. doi: 10.1159/000496819
112. Tewes AC, Rall KK, Romer T, Hucke J, Kapczuk K, Brucker S, et al. Variations in RBM8A and TBX6 are associated with disorders of the mullerian ducts. Fertil Steril. (2015) 103:1313–8. doi: 10.1016/j.fertnstert.2015.02.014
113. Wang M, Li Y, Ma W, Li H, He F, Pu D, et al. Analysis of WNT9B mutations in Chinese women with Mayer-Rokitansky-Küster-Hauser syndrome. Reprod BioMed Online. (2014) 28:80–5. doi: 10.1016/j.rbmo.2013.09.022
114. Waschk DE, Tewes AC, Romer T, Hucke J, Kapczuk K, Schippert C, et al. Mutations in WNT9B are associated with Mayer-Rokitansky-Kuster-Hauser syndrome. Clin Genet. (2016) 89(5):590–6. doi: 10.1111/cge.12701
115. Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N Engl J Med. (2004) 351:792–8. doi: 10.1056/NEJMoa040533
116. Biason-Lauber A, De Filippo G, Konrad D, Scarano G, Nazzaro A, Schoenle EJ. WNT4 deficiency–a clinical phenotype distinct from the classic Mayer-Rokitansky-Kuster-Hauser syndrome: a case report. Hum Reprod. (2007) 22:224–9. doi: 10.1093/humrep/del360
117. Philibert P, Biason-Lauber A, Rouzier R, Pienkowski C, Paris F, Konrad D, et al. Identification and functional analysis of a new WNT4 gene mutation among 28 adolescent girls with primary amenorrhea and müllerian duct abnormalities: a French collaborative study. J Clin Endocrinol Metab. (2008) 93:895–900. doi: 10.1210/jc.2007-2023
118. Philibert P, Biason-Lauber A, Gueorguieva I, Stuckens C, Pienkowski C, Lebon-Labich B, et al. Molecular analysis of WNT4 gene in four adolescent girls with mullerian duct abnormality and hyperandrogenism (atypical Mayer-Rokitansky-Küster-Hauser syndrome). Fertil Steril. (2011) 95:2683–6. doi: 10.1016/j.fertnstert.2011.01.152
119. Ledig S, Brucker S, Barresi G, Schomburg J, Rall K, Wieacker P. Frame shift mutation of LHX1 is associated with Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Hum Reprod. (2012) 27:2872–5. doi: 10.1093/humrep/des206
120. Zhang W, Zhou X, Liu L, Zhu Y, Liu C, Pan H, et al. Identification and functional analysis of a novel LHX1 mutation associated with congenital absence of the uterus and vagina. Oncotarget. (2017) 8:8785–90. doi: 10.18632/oncotarget.v8i5
121. Ai Z, Zhu X, Chen H, Chen R. Precocious puberty or growth hormone deficiency as initial presentation in Mayer-Rokitansky-kuster-Hauser syndrome: a clinical report of 5 cases. BMC Pediatr. (2022) 22:418. doi: 10.1186/s12887-022-03474-0
122. Brucker SY, Frank L, Eisenbeis S, Henes M, Wallwiener D, Riess O, et al. Sequence variants in ESR1 and OXTR are associated with Mayer-Rokitansky-Küster-Hauser syndrome. Acta Obstet Gynecol Scand. (2017) 96:1338–46. doi: 10.1111/aogs.13202
123. Tian W, Chen N, Ye Y, Ma C, Qin C, Niu Y, et al. A genotype-first analysis in a cohort of Mullerian anomaly. J Hum Genet. (2022) 67:347–52. doi: 10.1038/s10038-021-00996-w
124. Ledig S, Tewes AC, Hucke J, Römer T, Kapczuk K, Schippert C, et al. Array-comparative genomic hybridization analysis in patients with Müllerian fusion anomalies. Clin Genet. (2018) 93:640–6. doi: 10.1111/cge.13160
125. Kobayashi A, Shawlot W, Kania A, Behringer RR. Requirement of Lim1 for female reproductive tract development. Development. (2004) 131:539–49. doi: 10.1242/dev.00951
126. Huang C-C, Orvis GD, Kwan KM, Behringer RR. Lhx1 is required in Müllerian duct epithelium for uterine development. Dev Biol. (2014) 389:124–36. doi: 10.1016/j.ydbio.2014.01.025
127. Kobayashi A, Kwan K-M, Carroll TJ, McMahon AP, Mendelsohn CL, Behringer RR. Distinct and sequential tissue-specific activities of the LIM-class homeobox gene Lim1 for tubular morphogenesis during kidney development. Development. (2005) 132:2809–23. doi: 10.1242/dev.01858
128. Xia M, Zhao H, Qin Y, Mu Y, Wang J, Bian Y, et al. LHX1 mutation screening in 96 patients with müllerian duct abnormalities. Fertil Steril. (2012) 97:682–5. doi: 10.1016/j.fertnstert.2011.12.005
129. Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet. (2006) 43:84–90. doi: 10.1136/jmg.2005.032854
130. Haumaitre C, Fabre M, Cormier S, Baumann C, Delezoide A-L, Cereghini S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum Mol Genet. (2006) 15:2363–75. doi: 10.1093/hmg/ddl161
131. White PH, Farkas DR, McFadden EE, Chapman DL. Defective somite patterning in mouse embryos with reduced levels of Tbx6. Development. (2003) 130:1681–90. doi: 10.1242/dev.00367
132. Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. (2015) 372:341–50. doi: 10.1056/NEJMoa1406829
133. Liu J, Wu N, Yang N, Takeda K, Chen W, Li W, et al. TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet Med. (2019) 21:1548–58. doi: 10.1038/s41436-018-0377-x
134. Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. (2012) 44:435–9. doi: 10.1038/ng.1083
135. Griesinger G, Dafopoulos K, Schultze-Mosgau A, Schroder A, Felberbaum R, Diedrich K. Mayer-Rokitansky-Küster-Hauser syndrome associated with thrombocytopenia-absent radius syndrome. Fertil Steril. (2005) 83:452–4. doi: 10.1016/j.fertnstert.2004.06.077
136. Ahmad R, Pope S. Association of Mayer-Rokitansky-Küster-Hauser syndrome with Thrombocytopenia Absent Radii syndrome: a rare presentation. Eur J obstetrics gynecology Reprod Biol. (2008) 139:257–8. Ireland. doi: 10.1016/j.ejogrb.2007.01.018
137. Macchia PE, Lapi P, Krude H, Pirro MT, Missero C, Chiovato L, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet. (1998) 19:83–6. doi: 10.1038/ng0598-83
138. Mittag J, Winterhager E, Bauer K, Grümmer R. Congenital hypothyroid female pax8-deficient mice are infertile despite thyroid hormone replacement therapy. Endocrinology. (2007) 148:719–25. doi: 10.1210/en.2006-1054
139. Brophy PD, Rasmussen M, Parida M, Bonde G, Darbro BW, Hong X, et al. A gene implicated in activation of retinoic acid receptor targets is a novel renal agenesis gene in humans. Genetics. (2017) 207:215–28. doi: 10.1534/genetics.117.1125
140. Pavanello R de C, Eigier A, Otto PA. Relationship between Mayer-Rokitansky-Küster (MRK) anomaly and hereditary renal adysplasia (HRA). Am J Med Genet. (1988) 29:845–9. doi: 10.1002/ajmg.1320290414
141. Battin J, Lacombe D, Leng JJ. Familial occurrence of hereditary renal adysplasia with müllerian anomalies. Clin Genet. (1993) 43:23–4. doi: 10.1111/j.1399-0004.1993.tb04420.x
142. Schrauwen I, Kari E, Mattox J, Llaci L, Smeeton J, Naymik M, et al. De novo variants in GREB1L are associated with non-syndromic inner ear malformations and deafness. Hum Genet. (2018) 137:459–70. doi: 10.1007/s00439-018-1898-8
143. Zhao E, Bomback M, Khan A, Krishna Murthy S, Solowiejczyk D, Vora NL, et al. The expanded spectrum of human disease associated with GREB1L likely includes complex congenital heart disease. Prenat Diagn. (2024) 44:343–51. doi: 10.1002/pd.6527
144. Carroll TJ, Park J-S, Hayashi S, Majumdar A, McMahon AP. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell. (2005) 9:283–92. doi: 10.1016/j.devcel.2005.05.016
145. Ravel C, Lorenço D, Dessolle L, Mandelbaum J, McElreavey K, Darai E, et al. Mutational analysis of the WNT gene family in women with Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. (2009) 91:1604–7. doi: 10.1016/j.fertnstert.2008.12.006
146. Tang R, Dang Y, Qin Y, Zou S, Li G, Wang Y, et al. WNT9B in 542 Chinese women with Müllerian duct abnormalities: mutation analysis. Reprod BioMed Online. (2014) 28:503–7. doi: 10.1016/j.rbmo.2013.11.011
147. Li L, Chu C, Li S, Lu D, Zheng P, Sheng J, et al. Renal agenesis-related genes are associated with Herlyn-Werner-Wunderlich syndrome. Fertil Steril. (2021) 116:1360–9. doi: 10.1016/j.fertnstert.2021.06.033
148. Clément-Ziza M, Khen N, Gonzales J, Crétolle-Vastel C, Picard J-Y, Tullio-Pelet A, et al. Exclusion of WNT4 as a major gene in Rokitansky-Küster-Hauser anomaly. Am J Med Genet Part A. (2005) 137:98–9. doi: 10.1002/ajmg.a.30833
149. Chang X, Qin Y, Xu C, Li G, Zhao X, Chen Z-J. Mutations in WNT4 are not responsible for Müllerian duct abnormalities in Chinese women. Reprod BioMed Online. (2012) 24:630–3. doi: 10.1016/j.rbmo.2012.03.008
150. Jeong Y-H, Sekiya M, Hirata M, Ye M, Yamagishi A, Lee S-M, et al. The low-density lipoprotein receptor-related protein 10 is a negative regulator of the canonical Wnt/β-catenin signaling pathway. Biochem Biophys Res Commun. (2010) 392:495–9. doi: 10.1016/j.bbrc.2010.01.049
151. Deutscher E, Hung-Chang Yao H. Essential roles of mesenchyme-derived beta-catenin in mouse Müllerian duct morphogenesis. Dev Biol. (2007) 307:227–36. doi: 10.1016/j.ydbio.2007.04.036
152. Liu S, Gao X, Qin Y, Liu W, Huang T, Ma J, et al. Nonsense mutation of EMX2 is potential causative for uterus didelphysis: first molecular explanation for isolated incomplete müllerian fusion. Fertil Steril. (2015) 103:769–74.e2. doi: 10.1016/j.fertnstert.2014.11.030
153. Cheng Z, Zhu Y, Su D, Wang J, Cheng L, Chen B, et al. A novel mutation of HOXA10 in a Chinese woman with a Mullerian duct anomaly. Hum Reprod. (2011) 26:3197–201. doi: 10.1093/humrep/der290
154. Kruszka P, Li D, Harr MH, Wilson NR, Swarr D, McCormick EM, et al. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J Med Genet. (2015) 52:104–10. doi: 10.1136/jmedgenet-2014-102677
155. Bhoj EJ, Li D, Harr MH, Tian L, Wang T, Zhao Y, et al. Expanding the SPECC1L mutation phenotypic spectrum to include Teebi hypertelorism syndrome. Am J Med Genet A. (2015) 167A:2497–502. doi: 10.1002/ajmg.a.37217
156. Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science. (2022) 376:44–53. doi: 10.1126/science.abj6987
157. Beagan JA, Phillips-Cremins JE. On the existence and functionality of topologically associating domains. Nat Genet. (2020) 52:8–16. doi: 10.1038/s41588-019-0561-1
158. Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen L-L, et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. (2023) 24:430–47. doi: 10.1038/s41580-022-00566-8
159. Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. (2020) 21:30. doi: 10.1186/s13059-020-1935-5
160. Sampath Kumar A, Tian L, Bolondi A, Hernández AA, Stickels R, Kretzmer H, et al. Spatiotemporal transcriptomic maps of whole mouse embryos at the onset of organogenesis. Nat Genet. (2023) 55:1176–85. doi: 10.1038/s41588-023-01435-6
161. Lupski JR. Clan genomics: From OMIM phenotypic traits to genes and biology. Am J Med Genet A. (2021) 185:3294–313. doi: 10.1002/ajmg.a.62434
162. Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med Off J Am Coll Med Genet. (2021) 23:2029–37. doi: 10.1038/s41436-021-01242-6
Keywords: DNA copy number variations, genetics, genitourinary development, infertility, Mayer-Rokitansky-Küster-Hauser syndrome, MRKH syndrome, MRKHS, Müllerian aplasia
Citation: Herlin MK (2024) Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: advancements and implications. Front. Endocrinol. 15:1368990. doi: 10.3389/fendo.2024.1368990
Received: 11 January 2024; Accepted: 04 April 2024;
Published: 18 April 2024.
Edited by:
Anna Lauber-Biason, University of Fribourg, SwitzerlandReviewed by:
Paola Pontecorvi, Sapienza University of Rome, ItalyKarina Kapczuk, Poznan University of Medical Sciences, Poland
Copyright © 2024 Herlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Morten Krogh Herlin, mortherl@rm.dk