 Ayman Eid
Ayman Eid Chakravarthi Mohan
Chakravarthi Mohan Sara Sanchez1,2
Sara Sanchez1,2 Fredy Altpeter
Fredy Altpeter- 1Agronomy Department, Institute of Food and Agricultural Sciences, University of Florida, Gainesville, FL, United States
- 2Department of Energy Center for Advanced Bioenergy and Bioproducts Innovation, Gainesville, FL, United States
- 3Genetics Institute, University of Florida, Gainesville, FL, United States
- 4Plant Molecular and Cellular Biology Program, Institute of Food and Agricultural Sciences, Gainesville, FL, United States
Genome editing with sequence-specific nucleases, such as clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9), is revolutionizing crop improvement. Developing efficient genome-editing protocols for highly polyploid crops, including sugarcane (x = 10–13), remains challenging due to the high level of genetic redundancy in these plants. Here, we report the efficient multiallelic editing of magnesium chelatase subunit I (MgCh) in sugarcane. Magnesium chelatase is a key enzyme for chlorophyll biosynthesis. CRISPR/Cas9-mediated targeted co-mutagenesis of 49 copies/alleles of magnesium chelatase was confirmed via Sanger sequencing of cloned PCR amplicons. This resulted in severely reduced chlorophyll contents, which was scorable at the time of plant regeneration in the tissue culture. Heat treatment following the delivery of genome editing reagents elevated the editing frequency 2-fold and drastically promoted co-editing of multiple alleles, which proved necessary to create a phenotype that was visibly distinguishable from the wild type. Despite their yellow leaf color, the edited plants were established well in the soil and did not show noticeable growth retardation. This approach will facilitate the establishment of genome editing protocols for recalcitrant crops and support further optimization, including the evaluation of alternative RNA-guided nucleases to overcome the limitations of the protospacer adjacent motif (PAM) site or to develop novel delivery strategies for genome editing reagents.
Introduction
The processing of sugarcane (Saccharum spp. hybrid) biomass provides 80% of the sugar and 26% of the ethanol produced globally. Sugarcane is one of the most productive crops under cultivation due to its superior light conversion and efficiencies of water and nitrogen use (Tew and Cobill, 2008; Byrt et al., 2011). It is also a prime candidate feedstock for the emerging bio-economy (Altpeter and Ratna, 2018). The highly polyploid (x = 10 − 13; 2n = 100 − 130), heterozygous, interspecific, and aneuploid sugarcane genome decelerates attempts at crop improvement (Le Cunff et al., 2008; de Setta et al., 2014). Most parental sugarcane clones lack pollen fertility and any synchrony of flowering, posing challenges to conventional breeding (Moore and Nuss, 1987; Horsley and Zhou, 2013). Elite cultivars display a high level of heterozygosity and polyploidy, requiring vegetative propagation to prevent the loss of favorable alleles and the accumulation of detrimental ones during the disruptive process of meiosis. Therefore, adding superior alleles to improve an elite cultivar with the use of conventional breeding is a demanding and time-consuming undertaking. Genome editing using sequence-specific nucleases (SSNs) is a powerful approach for the genetic improvement of crops (Zhang et al., 2018). It has great potential for sugarcane and other vegetatively propagated, heterozygous, and polyploid crops (Weeks, 2017) by enabling precision genome modifications in elite varieties while bypassing meiosis. Among the SSNs, RNA-guided nucleases, including CRISPR/Cas9, are the most widely used gene editing tools due to their target specificity, efficiency, simplicity of design, multiplexing capacity, and versatility (Chandrasegaran and Carroll, 2016). They have been repurposed to targeted mutagenesis, gene stacking, targeted nucleotide substitutions, chromosomal translocations, transcriptional or translational regulation, and viral interference (Jinek et al., 2012; Shan et al., 2013; Ali et al., 2015; Baltes et al., 2015; Svitashev et al., 2015; Zhang et al., 2018; Huang and Puchta, 2019; Beying et al., 2020; Gao et al., 2020).
Most approaches to genome editing require a DNA double-strand break (DSB) in or near the target sequence to be edited. Streptococcus pyogenes Cas9 (spCas9) possesses innate nuclease activity, which is targeted by an engineered, single 20 nt guide RNA molecule to the DNA cleavage site adjacent to a protospacer-associated motif (PAM) (Jinek et al., 2012). Then DNA cleavage triggers cellular repair mechanisms, including non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homology-directed repair (HDR), to rectify the DSB. The error-prone NHEJ and MMEJ repair pathways enable the construction of knockout alleles through frameshift mutations caused by indels. By contrast, HDR supports precision edits, including targeted codon replacements and gene stacking. HDR relies on recombination, using a template that displays homology to the break site (Puchta, 1998; Shrivastav et al., 2008; Chapman et al., 2012; Butt et al., 2019; Huang and Puchta, 2019).
Targeted mutagenesis is more challenging in highly polyploid crops such as sugarcane than in diploid crops. The large number of homeologs and homologs in sugarcane causes functional redundancy. However, this also offers an opportunity to generate a range of phenotypes, depending on the number of co-mutated copies/alleles, similar to RNAi. The creation of knockdown or knockout phenotypes requires an efficient multiallelic editing platform. We recently reported the TALEN-mediated targeted co-mutagenesis of more than 100 copies/alleles of the lignin biosynthetic gene caffeic acid O-methyltransferase (COMT) in sugarcane. This action resulted in drastically improved saccharification efficiency and greater bioethanol yields from the lignocellulosic biomass without compromising agronomic performance (Jung and Altpeter, 2016; Kannan et al., 2018; Ko et al., 2018).
The establishment of CRISPR/Cas9-mediated genome editing for sugarcane is desirable for improving multiplexing capacity, versatility, and ease of design relative to TALEN (Eid and Mahfouz, 2016). This will involve the optimization of genome editing reagents and their delivery to enable efficient co-editing of a large number of copies/alleles.
These optimizations are accelerated with the help of a rapidly scorable screening system that allows the visual identification and quantification of targeted mutations as soon as plants regenerate from tissue cultures. To establish genome editing protocols in other crops, the phytoene desaturase (PDS) gene in the carotenoid biosynthetic pathway was targeted for mutagenesis (Shan et al., 2013; Fan et al., 2015). Unlike dwarf and albino phenotypes of PDS knockouts, Mg-chelatase mutants display a light green to yellow leaf phenotype with similar growth rates to the wild type (WT) (Walker et al., 2018). Mg-chelatase catalyzes Mg2+ attachment to protoporphyrin IX, which is the major regulatory point for the chlorophyll biosynthesis pathway (Figure 1) (Willows et al., 1996).
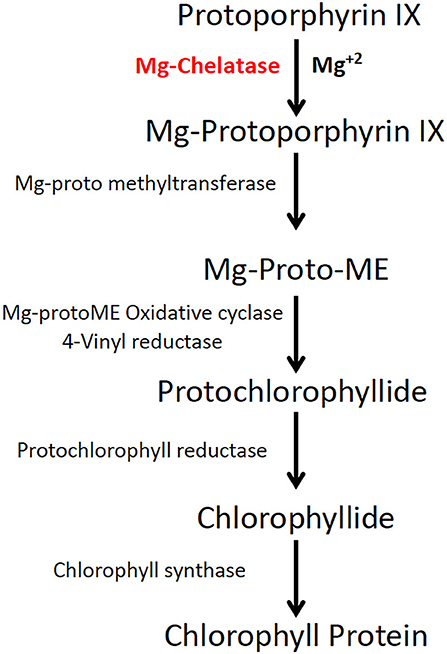
Figure 1. Magnesium chelatase is a key enzyme in the chlorophyll biosynthesis pathway. Magnesium chelatase catalyzes the conversion of protoporphyrin IX (PPIX) to magnesium protoporphyrin IX, a precursor of chlorophyll b and a in the presence of Mg2+ and ATP. The enzyme is a hexameric motor complex made up of three proteins, ChlI, ChlD, and ChlH.
In this study, we explored whether targeted mutagenesis of magnesium chelatase subunit I with CRISPR/Cas9 provides a rapidly scorable phenotype for predicting the extent of multiallelic editing in highly polyploid sugarcane.
Materials and Methods
Isolation of Allelic Variants of Mg Chelatase and Design of sgRNAs
The MgCh sequence in sugarcane was compared to sorghum and maize MgCh sequences via tBLASTn. This allowed the conserved domains to be identified, informing the primer design for the PCR amplification of multiple allelic MgCh variants from sugarcane target cultivar CP88-1762 (WT) (Supplementary Table 1). The amplicons were cloned into the p-GEMT® easy vector (A1360) (Promega, WI, USA), followed by the Sanger sequencing of multiple colonies. The sgRNAs were selected in silico using CRISPOR (http://crispor.tefor.net/). sgRNA1 was designed to cleave to a highly conserved region, while the sgRNA2 target was less conserved and included three allelic variants that differed in the number of mismatches (0, 1, or 2) in the genomic target sequence of the sgRNA (Figure 2A).
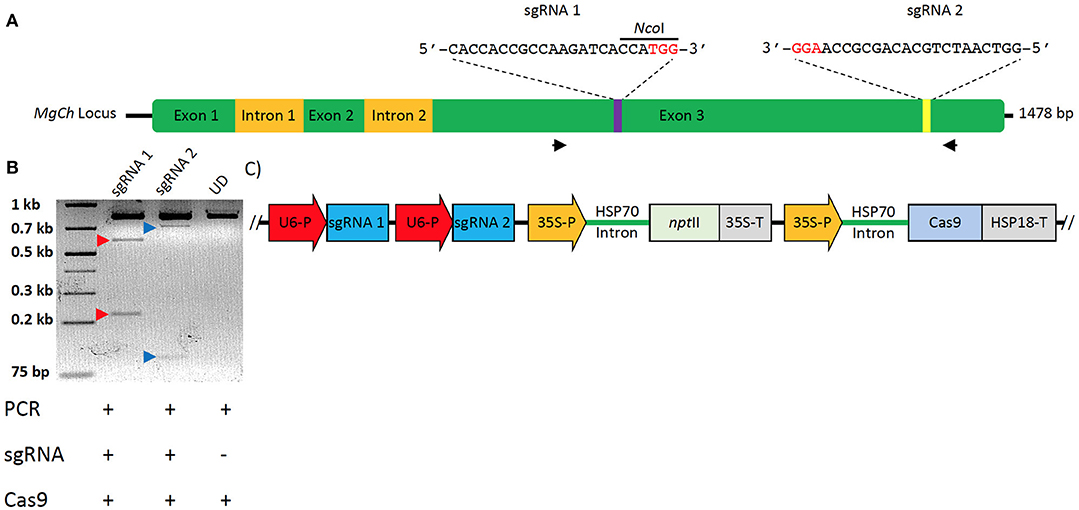
Figure 2. Strategy for targeted mutagenesis of sugarcane MgCh and confirmation of In-vitro cleavage activity of sgRNAs. (A) Schematic representation of sugarcane MgCh locus and sgRNAs' target sites, sgRNA1 targeting nts 731–750 and sgRNA2 targeting nts 1223–1242. Mutations at sgRNA target site 1 would disrupt the NcoI restriction recognition site. Exons are indicated with green boxes, and introns with yellow boxes. (B) In vitro cleavage assay to validate sgRNA activity, the (810 nts) MgCh PCR amplicon is digested by ribonucleoprotein complex (RNP) of Cas9 and either sgRNA1 or sgRNA2 into ~581 and 229 nts or 701 and 109 nts, respectively. (C) Map of sugarcane gene editing plasmid (pMGE); Two sgRNAs are monoscistronically expressed under Oryza sativa U6 promoter, nptII is under transcriptional control of cauliflower mosaic virus (CaMV) 35S promoter and CaMV terminator, Cas9 is under transcriptional control of CaMV 35S promoter and Sorghum bicolor HSP18 terminator. Protospacer adjacent motifs (PAMs) are indicated in red font.
sgRNA Synthesis and in vitro Cleavage Assay
sgRNA templates were generated via PCR using Q5® High-Fidelity DNA Polymerase (NEB, MA, USA) using a DNA template encoding T7 promoter sequence corresponding to the target sequence. The optimized sgRNA scaffold (Chen et al., 2013) was assembled from oligonucleotides (Eurofins Genomics, KY, USA) through overlapping PCR, as previously described (Lin et al., 2014). Primer T7MgCh1F was combined with T7F, ScaffoldR1, and ScaffoldR2 to generate sgRNA1 DNA. The primer T7MgCh2F was combined with T7F, ScaffoldR1, and ScaffoldR2 to generate sgRNA2 DNA using the following PCR conditions: 30 cycles of 95°C for 10 s, 57°C for 10 s, and 72°C for 10 s (Lin et al., 2014). The reactions were purified with the GeneJET PCR Purification Kit (K0701) (Thermo Fisher Scientific, MA, USA) and were electrophoresed with 1% agarose gel. In vitro transcription was done using the HiScribe™ T7 Quick High Yield RNA Synthesis Kit (E2050S) (NEB) with 75 ng sgRNA DNA template. DNase I treatment and RNA cleanup were performed using Monarch® Total RNA Miniprep Kit (T2010) (NEB). The Mg-chelatase template PCR was amplified using Q5® High-Fidelity DNA Polymerase (M0491) (NEB) with primers C27 and C28 and the following cycle: initial denaturation at 98°C for 30 s, then 35 cycles of 98°C for 5 s, 68°C for 10 s, and 72°C for 20 s, with a final extension of 72°C for 2 min. sgRNAs validation was done by incubating 200 ng Mg-Chelatase template DNA, 250 ng sgRNA, 250 ng Cas9 protein (PNA Bio), and 2 μL NEB buffer 3 in a 20 μL reaction for 3 h at 37° C. The reaction was stopped with the addition of 1 μL PureLink™ RNase A (20 mg/mL) (12091021) (Thermo Fisher Scientific) and was incubated for 10 min at 65°C prior to electrophoresis with 2% agarose gel (Figure 2B).
Vector Construction
sgRNA vectors containing Oryza sativa U6 promoter were designed and custom synthesized in the pUC57 backbone (Genscript, NJ, USA) to generate pUCMg12. The Mg-chelatase target guide sequences were simultaneously cloned into the vector using annealed primer-dimers (Supplementary Table 1) holding 5′ overhangs to ligate into BbsI and BsaI restriction sites of pUC57. pUCMg12 vector was subcloned into the CRISPR backbone vector through digestion with Srf I and NotI enzymes, and the resultant colonies were confirmed via Sanger sequencing. The resulting vector MGE harbors a sugarcane codon-optimized Streptococcus pyogenes Cas9 driven by CaMV35S promoter with ZmHSP70 (heat shock protein) intron and AtHSP terminator. pMGE expresses the nptII (neomycin phosphotransferase II) gene as a plant selectable marker under transcriptional control of CaMV35S promoter with ZmHSP70 intron and CaMV35S terminator (Figure 2C).
Biolistic Transformation of Sugarcane
The MGE plasmid was linearized using the AscI enzyme, and the minimal cassette was introduced into the embryogenic callus of sugarcane cultivar CP88-1762 through biolistic gene transfer, as described previously (Taparia et al., 2012). To evaluate the impact of heat treatment on mutation frequency, 50% of the calli were heat-treated at 37°C for 48 h, 4 days after bombardment, and they were compared to bombarded calli from the same experiment that were kept at the usual incubation temperature (28°C). The calli were subsequently incubated at 28°C and selected with geneticin (20 mg/L), as described by Taparia et al. (2012). Plantlets 5–10 cm in height were sampled for molecular analyses.
DNA Isolation, PCR, and Sanger and Next-Generation Sequencing
Genomic DNA was isolated from leaf tissues using the CTAB method (Murray and Thompson, 1980). The C5 and C9 primers (Supplementary Table 1) used for PCR amplified a region of MgCh, spanning exon 3 (~1,100 bp) from the genomic DNA template, including targets for sgRNAs 1 and 2 (Figure 2A). Q5® High-Fidelity DNA Polymerase (NEB, MA, USA) was used for PCR under the following conditions: 98°C for 30 s, 35 cycles of amplification at 98°C for 15 s, 55°C for 10 s, 72°C for 20 s, and final extension at 72°C for 2 min. The PCR amplicons used in the restriction enzyme assays to detect targeted mutations were amplified using C27 and C28 primers (Supplementary Table 1) and the Phire Plant Direct PCR Kit, under the following conditions: 98°C for 5 min, 35 cycles of amplification at 98°C for 5 s, 65.1°C for 5 s, 72°C for 20 s, and final extension at 72°C for 2 min, using PCR amplicons for Sanger sequencing ligated to pJET 1.2 blunt vector (Thermo Fisher Scientific, MA, USA). The plasmid DNA was prepared from cloned amplicons using the GeneJET miniprep kit (Thermo Fisher Scientific). Sanger sequencing of the cloned PCR amplicons was performed at Eurofins Genomics. The sequence chromatograms were visually checked for quality.
For next-generation sequencing, amplicons of 574 bp were generated using primers C31 and C32 (Supplementary Table 1) with Phire polymerase (Thermo Fisher Scientific) and the following amplification conditions: 98°C for 5 min, 35 cycles of amplification at 98°C for 5 s, 64.2°C for 5 s, 72°C for 20 s, and a final extension at 72°C for 2 min. The reactions were purified using a GeneJET PCR Purification Kit (K0701) (Thermo Fisher Scientific). Complete amplicon sequencing was performed using the CCIB DNA Core Facility at Massachusetts General Hospital (Cambridge, MA). Adapters with unique barcodes were ligated onto each sample during the library construction. The libraries were pooled into equimolar concentrations for multiplexed sequencing on the Illumina MiSeq platform (Illumina, San Diego, CA) with 2×150 run parameters.
To detect edited sgRNA target sites, all reads were examined in the corresponding fastq file to identify either the 5′ primer (C31) or the 3′ primer (C32) close to the beginning or the end of the read, respectively (exact match required). If primer C31 was found, a local alignment algorithm was run with the parameters match score = 1, mismatch penalty = −0.5, gap opening penalty = −0.5, and gap extension penalty = −0.2 to search for the sgRNA1 sequence in the 65 bp region downstream of C31. A minimum score of 15 was required to accept the alignment, in addition to a perfect match on the first two bases. If C32 was found, a search for the sgRNA2 sequence in the 50 bp region downstream of C32 was initiated, with the same parameters. If a match for the sgRNA sequence was found, the alignment was examined to determine the number of base substitutions, insertions, and deletions, and the number of reads that contained every possible combination of events (e.g., perfect match, substitutions only, substitutions and insertions, substitutions and deletions, etc.) was reported. In addition, matrices showing the frequencies of all observed substitutions in each sample were generated.
For the sgRNA2 target, the results were computed separately for each of the three known sgRNA2 variants, based on single-nucleotide changes at positions 4 (C → T) and 15 (T → C). Only the three haplotypes CT, TT, and TC were observed at a significant frequency. To determine the indel sizes, the number of reads in which the sgRNA target sequence contained insertions (or deletions) totaling 1, 2, 3, 4, 5, or more than 5 base pairs (based on the local alignment) were also reported, as well as the average size of all insertions or deletions. The results for each of the three sgRNA2 variants were reported separately. To record the reads with severely modified sgRNA targets while preventing alignment with the sgRNA sequence, all reads that contained a valid primer (C31 or C32) but did not contain the sgRNA sequence were examined. This included searching for an 11 bp conserved sequence that could be located downstream of the sgRNA (at positions 56 and 61 downstream of C31 or C32, respectively). This search was performed using a local alignment algorithm with the following parameters: match score = 1, mismatch penalty = −1, gap opening penalty = −2, gap extension penalty = −2, and minimum score required for hit = 9. All analyses were performed with custom Python scripts using the Biopython package (specifically, the Bio.pairwise2 library).
Detecting Targeted Mutations Using Restriction Enzyme Digestion of PCR Amplicons
PCR products from C27–C28 were purified using GeneJET PCR Purification Kit (K0701) (Thermo Fisher Scientific). Following this, 200 ng of each purified product was incubated with 0.2 μL NcoI-HF (NEB) for 3 h at 37°C. The reactions were deactivated by incubation at 80°C for 20 min prior to loading on 2% agarose gel for visualization.
Phenotypic Evaluations
The plants were transferred from tissue culture media to Sunshine mix #8 (Sungrow Horticulture, Agawam, MA, USA) potting mix and grown in a walk-in growth chamber when the shoots were ~10 cm long. During the first week after the transfer, a level of relative humidity near 100% was maintained, and then it was adjusted to 75% humidity. Plant growth occurred on a 16/8 h light/dark photoperiod and a light intensity of 400 μmol m−2 s−1, at 28/22°C day/night temperature. The plants were fertilized every 2 weeks after their transfer to the soil by irrigation with Miracle Grow All Purpose Plant Food (ScottsMiracle-Gro, Maryville, OH, USA). Leaf greenness was measured on the fully expanded top leaf from three tillers per plant using a SPAD chlorophyll meter (Minolta SPAD-502, Konica-Minolta), and this was repeated twice at 3-week intervals.
Statistical Analyses
The means were compared using Fisher's least significant difference test. A minimum of three independent biological replicates were used for the statistical analyses.
Results
Identification of Allelic Variants of MgCh in Sugarcane and Confirming sgRNA Activity in vitro
Sanger sequencing of cloned MgCh amplicons from WT DNA led to the identification of allelic variants in WT and informed the design of two sgRNAs that targeted exon 3. sgRNA1 targeted MgCh at nts 731–750, and sgRNA2 targeted MgCh at nts 1223–1242 (Figure 2A). Both sgRNAs were validated through in vitro cleavage assay. sgRNA1 guided the cleavage of the 810 nt long partial MgCh amplicons into 581 and 229 nt fragments. Targeting via sgRNA2 generates 701 and 109 nts fragments upon cleavage (Figure 2B).
Visual Detection of Mutant Phenotypes
Events that feature the depletion of chlorophyll are visually distinguishable as soon as the plantlets regenerate from tissue culture (Figure 3A). This included light green or yellow leaves, by contrast to the dark green shoots from the non-bombarded control plates.

Figure 3. Detection of chlorophyll depletion phenotype during in vitro propagation. (A) Development of chlorophyll depletion phenotype compared to the WT. Some of the genome-edited events (e.g., line HY1 [heat-treated, yellow 1]) displayed a distinguishable phenotype with yellow leaf color in plantlets that regenerated from tissue culture. (B) Comparison of line HY1 (right) to WT (left) after establishment in soil under greenhouse conditions. (C) Close-up of leaves from NG1, NG2, HG1, HG2, HY1, and HY2 compared to WT. HY = heat-treated yellow, NG = non-heat treated green, HG = heat treated green, WT = wild type.
Characterization of Mutant Lines With Restriction Enzyme Assay
A total of 52 transgenic lines were regenerated, including 22 lines from non-heat-treated tissue and 30 lines from heat-treated tissue from 10 bombardments with the pMGE construct for each treatment. The PCR amplicons of MgCh from these lines were analyzed for the loss of restriction sites in the target region of sgRNA1. Loss of the NcoI restriction site was expected if indels or nucleotide substitutions were generated in the target site of the sgRNA1 (Figure 2A). A total of nine lines were identified, including a partially undigested MgCh amplicon following NcoI treatment, by contrast to WT, which displayed a completely digested amplicon (Figure 4). Among the nine lines with altered NcoI restriction digest pattern, three lines originated from no-heat treatment (NG1, NG2, and NG3), and six lines were from heat-treated calli (HY1, HY2, and HY3 and HG1, HG2, and HG4; Figure 4).

Figure 4. Identification of mutants by detection of restriction site loss in sgRNA1 target site of MgCh PCR amplicons. Cleavage in sgRNA1 target site is likely to disrupt NcoI restriction site due to the creation of indels through an error-prone NHEJ-mediated repair process. A functional NcoI site would allow the cleavage of 810 bp PCR amplicon of MgCh into ~578 bp and 232 bp products. Blue lines indicate transgenic lines generated without heat treatment, and red lines indicate transgenic lines that regenerate following heat treatment of bombarded callus at 37°C for 48 h. N = non-heat treated, G = green, H = heat-treated, Y = yellow. The NcoI digested MgCh PCR amplicon was electrophoresed on 2% agarose gel. Lines NG1, NG2, NG3, HY1, HG1 HG2, HG3, HY3, and HY2 all display partially undigested MgCh PCR amplicons following NcoI restriction digestions indicated by yellow arrows. HY = heat-treated, yellow; NG = non-heat treated, green; HG = heat treated, green; WT = wild type.
Sanger Sequencing of Cloned PCR Amplicons of MgCh
Sanger sequencing of cloned PCR amplicons, including the 2 sgRNA target regions in exon 3 of MgCh (Figure 2A), confirmed the targeted mutations from the non-heat-treated set. Three mutant lines were confirmed by Sanger sequencing and restriction enzyme assay (14% of the regenerated lines; Table 1 and Supplementary Figure 1). The heat-treated tissues regenerated six mutant lines (20% of the regenerated lines, Table 1 and Supplementary Figure 1). All three of the lines with the most severe chlorophyll depletion HY1, HY2, and HY3 (heat-treated yellow 1, 2, and 3; Table 1) were derived from the heat treatment. In addition, three mutants with mild to moderate chlorophyll reduction were also regenerated from the heat treatment (HG1, HG2, and HG3; Table 1).
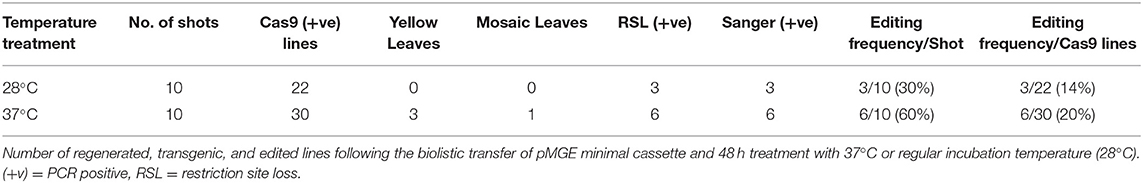
Table 1. Summary of phenotyping and genotyping of regenerated plants following biolistic transfer of pMGE and treatment with 37°C or regular incubation temperature (28°C).
Single-nucleotide polymorphism outside of the target regions for sgRNA1 and sgRNA2 allowed the identification of unique reads to represent individual MgCh copies/alleles. This allowed differentiation between single-edited and co-edited events within MgCh variants/alleles in the analyzed lines.
In mutant line HY1 (Figure 3B), 59 unique reads were identified from 175 cloned MgCh PCR amplicons, representing individual MgCh copies/alleles. In total, 49 of the 59 copies/alleles from HY1 were edited (83.1%), and 10 were not edited (16.9%). Among the 49 edited copies/alleles, 27 copies/alleles (45.8%) only displayed edits at the target site of sgRNA1 (PAM1), 3 (5.1%) only displayed edits at the target site of sgRNA2 (PAM2), and 19 (32.2%) displayed edits at both target sites (Figure 5, Supplementary Tables 2, 3). Among the 49 edited copies/alleles, 40 (67.7% of all unique reads) carried frame-shift mutations, and 9 (15.3% of all unique reads) displayed a single amino acid deletion or in-frame isoform (Supplementary Tables 2, 4). Indel analyses revealed that the most dominant deletions were 2–3 nt long, the longest deletion was 14 nt, and the detected insertions were all 1 nt long (Figure 5, Supplementary Tables 2–4).
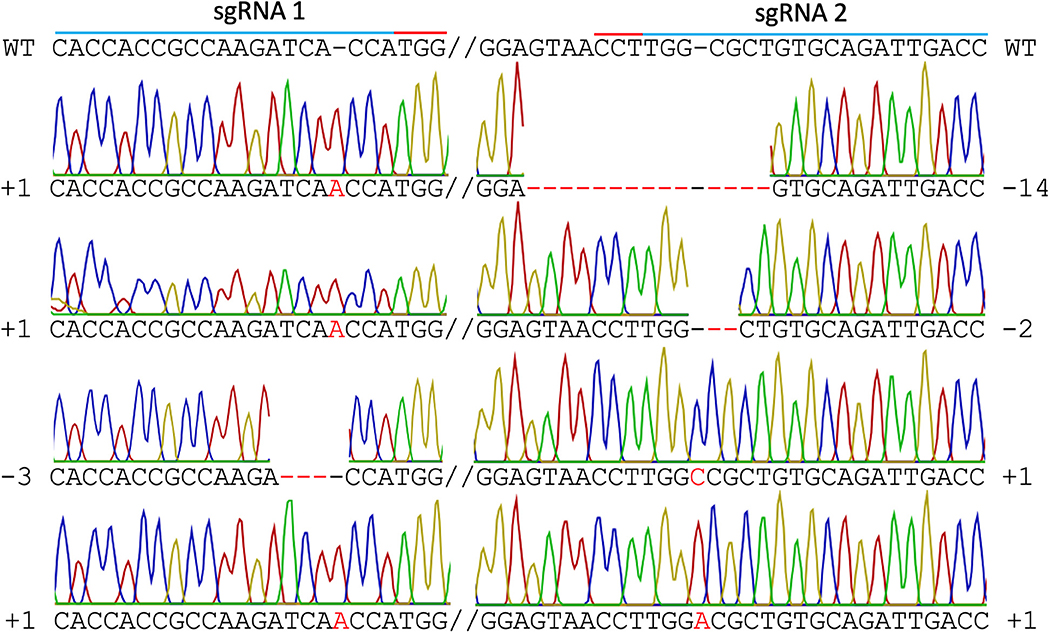
Figure 5. Sanger sequencing of line HY1. Different types of edits are displayed. The blue line indicates both sgRNA target sites, the red line indicates PAM sites, the red font indicates insertions, and the red dashed line indicates deletions. WT = wild type.
SPAD Chlorophyll Meter Analyses to Quantify Leaf Greenness in Mutant MgCh Lines
Leaf greenness from mutant and WT plants was determined using the SPAD chlorophyll meter (Minolta SPAD-502, Konica-Minolta). Two lines were derived from incubation at 28°C following gene transfer (non-heat-treated green lines NG1 and NG3) that displayed SPAD values comparable to the WT leaves which was detected as 43.39 SPAD units (Table 2). Line NG2 displayed a 9% lower SPAD value than WT, but its greenness was not visibly distinguishable from WT (Figure 3C). Therefore, mutant plants derived from incubation at 28°C were considered to be lacking a chlorophyll-depletion phenotype. The green lines originating from the heat-treated callus (37°C, HG1, HG2, and HG3) displayed SPAD values of 30.63, 34.7, and 31.07, respectively. This indicated a 20 to 29% reduction of chlorophyll compared to WT, although this was not visibly distinguishable from WT (Table 2; Figure 3C). The yellow lines (HY1, HY2, and HY3) originating from the heat-treated callus displayed SPAD values of 5.5, 6.63, and 12.53, respectively. This indicates a 71 to 87% reduction in chlorophyll compared to WT and was visibly distinguishable from the WT, NG, and HG lines (Table 2, Figure 3C).
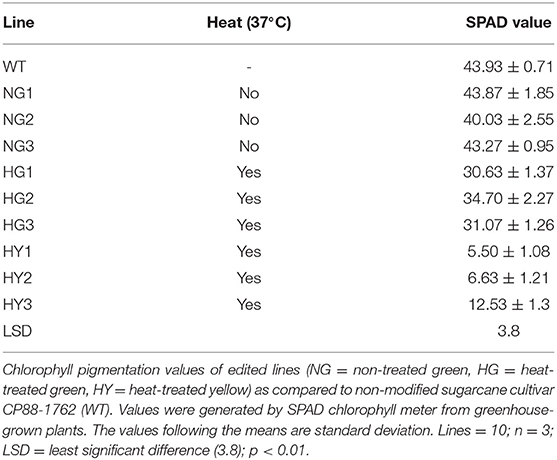
Table 2. Evaluation of leaf pigmentation of edited lines using a SPAD meter.
Quantifying Multiallelic Co-editing Efficiency of MgCh With Next-Generation Sequencing
Next-generation sequencing data showed that the observed level of chlorophyll depletion largely corresponded to the proportion of edited reads. The exception to this was line HG3, which had only 4.4–4.8% of the reads edited at sgRNA target site 2 or 1, respectively, and displayed a 28% reduction of chlorophyll (Tables 2, 3). MgCh reads aligning to the sgRNA1 target site displayed editing efficiencies ranging from 6.9 to 20.8% for NG lines, 4.8 to 28.8% for HG lines, and 42 to 82.3% for HY lines (Table 3). The editing efficiencies at the sgRNA2 target ranged from 5.7 to 8.7% for the NG lines, 4.4 to 29% for the HG lines, and 18.7 to 19.7% for the HY lines (Table 3). Taking the highest editing efficiency for each line at either the sgRNA1 or sgRNA2 target sites, NG lines displayed a range from 6.9 to 20.8%, HG lines from 4.8 to 29%, and HY lines from 42 to 82.3% of the MgCh reads as edited NGS reads (Table 3). The most common edit detected at the sgRNA1 target site was insertion, with an average of 24.3% of the total events across all of the lines. The most frequent editing event at sgRNA2 target site was a combination of substitution and insertion, with 8.6% of the total events across all of the lines (Table 3).
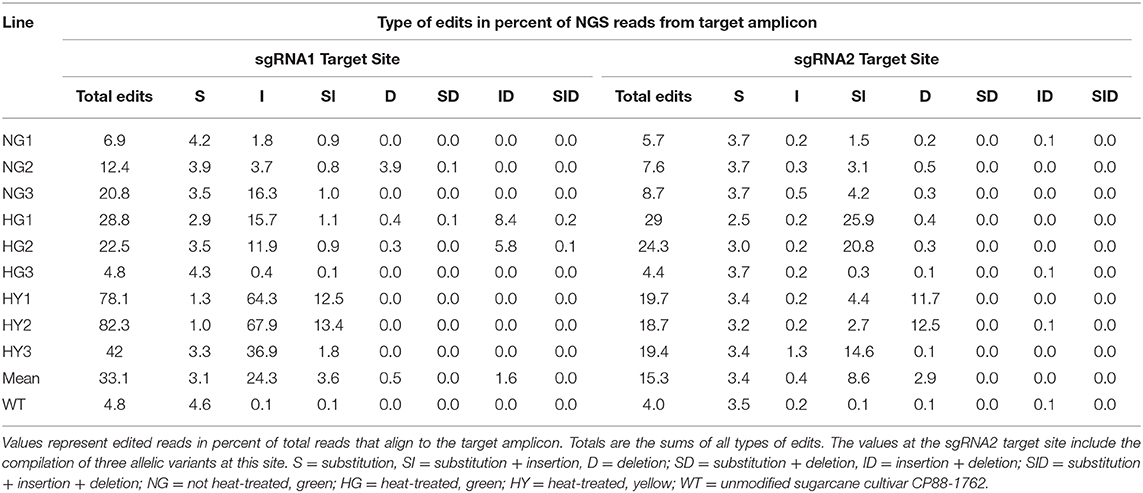
Table 3. Summary of editing events detected by next-generation sequencing in the 20-nt sgRNA target sites.
On average of all the edited lines, 33.1 or 15.3% of all NGS reads, that aligned to sgRNA1 or sgRNA2 displayed edits, respectively (Table 3). The sgRNA1 target site was located in a highly conserved region of MgCh with no sequence variants in WT. sgRNA2 was designed to target a site where three sequence variants were present in WT, with 0, 1, or 2 nt mismatches against the corresponding sgRNA. The editing efficiency was highest at the variant, with no mismatches to the sgRNA, reaching up to 28% of the MgCh reads in line HG1. Both variants, displayed low but detectable editing efficiencies with 1 or 2 mismatches (Supplementary Table 5). The NGS reads were also analyzed to detect long insertions and long deletions at both sgRNA1 and sgRNA2 target sites. Long insertions were detected only at sgRNA site 1, in up to 6.5% of the MgCh reads as edited NGS reads for line HG1 (Supplementary Table 6). In lines with significant more long insertions than the wild type, the average length of long insertions ranged from 6.8 bp in NG1 to 9.4 bp in HG1 (Supplementary Table 6). Long deletions at sgRNA1 target site were detected in <0.1% of the NGS reads. At sgRNA2 target site long deletions were detected in 0.5% (HG3) to 19.4% (HY2) of the MgCh reads. In lines with significant more long deletions than the wild type, the average length of long deletions ranged from 5.9 bp in HY3 to 14.3 in HY2 (Supplementary Table 6).
Discussion
Polyploidy is a common challenge in functional genomics and genetic improvement for many important crops. Sugarcane is an interspecific hybrid with a highly polyploid genome (x = 10 − 13; 2n = 100 − 130) typically containing 10 or more homo(eo)logs at each locus (Le Cunff et al., 2008). This high level of genetic redundancy requires very efficient co-editing of multiple copies/alleles for the generation of knockout or knockdown mutant phenotypes. Here, we describe CRISPR/Cas9-mediated targeted mutagenesis of the magnesium chelatase gene (MgCh), which is a high-copy gene in sugarcane. Co-editing of up to 49 of the 59 detected copies/alleles was confirmed by Sanger sequencing. Of the 30 transgenic lines harboring the MgCh gene editing (MGE) construct, six were confirmed to have targeted mutations for an editing efficiency of 20%. Three events with the co-editing of the majority of the MgCh copies/alleles displayed severe chlorophyll depletion, which was visibly scorable as the plantlets regenerated from tissue culture.
In hexaploidy wheat, a tri-genome targeted sgRNA to the PDS gene was co-introduced with Cas9 in 38 independent transgenic lines, but no photobleaching phenotype was identified. Only 2 of the 38 transgenic wheat lines displayed targeted mutagenesis (editing efficiency of 5%), and none of them displayed co-editing of multiple copies/alleles (co-editing efficiency of 0%) (Howells et al., 2018). By contrast, the diploid barley displayed an editing efficiency that was three times higher than wheat, with the same construct. However, in barley, only chimeric events were identified, displaying the photo-bleaching phenotype in sections of the leaves that were associated with progressive somatic edits (Howells et al., 2018). Generally, short indels that include insertions, substitutions, and deletions are highly reported events in gene editing in the polyploid plant genomes (Naim et al., 2018; Shan et al., 2018; Wang et al., 2020; Wolabu et al., 2020).
To the best of our knowledge, this report is the first to describe the targeted mutagenesis of MgCh in plants. Naturally occurring mutations in the MgCh gene have previously been reported to cause impaired chlorophyll biosynthesis and thus to result in phenotypes with light green or yellow foliage (Campbell et al., 2014). RNA interference of MgCh in tobacco and peach results in light green phenotypes with significantly reduced chlorophyll contents (Papenbrock et al., 2000). By contrast to MgCh mutants, PDS mutants display impaired chlorophyll, carotenoid, and gibberellin biosynthesis resulting in dwarf or albino plantlets in both biallelic, homozygous, and biallelic heterozygous events (Qin et al., 2007). Albino plants may also be caused by somaclonal variation, and the dwarfing resulting from PDS suppression makes tissue collection to confirm molecular analyses more challenging. By contrast, Mg-chelatase-impaired natural mutants have been described with yields similar to (Slattery et al., 2017) or higher than WT plants (Pettigrew et al., 1989) despite 50% reduced chlorophyll content. The latter is associated with increased light penetration into the canopy, causing an increase in the CO2 exchange rate there. The former results in a reduced nitrogen requirement. Therefore, manipulating chlorophyll content has been proposed as a strategy for improving canopy-level photosynthesis or nitrogen use efficiency under the dense canopies of tall biomass plants such as sugarcane (Kirst et al., 2017; Walker et al., 2018).
In this study, we demonstrated that the targeted mutagenesis of magnesium chelatase with CRISPR/Cas9 provides a rapidly scorable phenotype without leading to obvious growth retardation. Notably, the level of chlorophyll depletion was predictive of the extent of multiallelic co-editing of MgCh, which enables a rapid readout of the editing outcome.
The efficiencies of Cas9- and Cas12a-mediated mutagenesis can be elevated by the heat treatment of the callus following the delivery of editing reagents, as previously reported for Arabidopsis, rice, maize, and wheat (LeBlanc et al., 2018; Malzahn et al., 2019; Milner et al., 2020). Therefore, we deployed an MGE-based rapid readout system to compare co-editing efficiencies in response to heat treatment (37°C) or standard incubation temperature (28°C) for sugarcane callus. Three transgenic lines emerging from the heat treatment and none of the transgenic lines that regenerate under standard incubation temperatures displayed a phenotype with visible chlorophyll depletion. Genotyping with assaying restriction enzyme loss in the sgRNA target region, Sanger sequencing, and next-generation sequencing revealed a total of three mutant lines per 10 shots from callus regeneration under standard incubation temperatures and six mutant lines per 10 shots from heat-treated tissue. The events which were visibly distinguishable from WT due to severe chlorophyll depletion following heat treatment displayed editing of more than 40% of the MgCh NGS target amplicon reads. The events from standard temperature treatment displayed an editing rate of 6.9–20.8% of the MgCh NGS target amplicon reads without displaying a phenotype that could be visibly distinguished from WT. This suggested that heat treatment elevated the editing frequency 2-fold and drastically promoted the co-editing of multiple alleles to create a phenotype that was visibly distinguishable from WT. Efficient co-editing of multiple copies/alleles is of major importance for the generation of a distinct mutant phenotype in vegetatively propagated polyploid crops such as sugarcane and potato, where the combination of mutant alleles via sexual hybridization would disrupt the highly heterozygous, elite cultivar.
Multiallelic gene editing via CRISPR/Cas9 has been reported in several polyploid crops. In studies of both tetraploid potato and tetraploid switchgrass, only 2.0% of the T0 transgenic plants displayed co-mutation of all four targeted alleles of the granule-bound starch synthase gene or phosphoglycerate mutase gene (Andersson et al., 2017; Liu et al., 2018). Using polycistronic delivery for four sgRNAs instead of a single one dramatically elevated the editing efficiency in the tetraploid alfalfa. None of the 339 transgenic lines harboring a single sgRNA or Cas9 displayed tetra-allelic editing or the desired stay-green phenotype. By contrast, seven lines were confirmed with co-editing of all four alleles of the stay-green gene (MsSGR) in T0 from 492 transgenic lines (Wolabu et al., 2020).
Co-delivery of two sgRNA instead of a single sgRNA also elevated the co-editing efficiency in this study. Analyses of unique reads following the Sanger sequencing of cloned PCR amplicons of line HY1 revealed that 45.8% of the reads were edited only at sgRNA site 1, 5.1% were edited only at sgRNA site 2, and 32.2% were edited at both sites. Both Sanger and next-generation sequencing analyses suggested that functional knockouts are mostly composed of short insertions and short deletions resulting in out of frame mutations. For example, in mutant line HY1, with severe depletion of chlorophyll, 67.8% of the alleles were out of frame.
To exploit the reduction in chlorophyll content for elevating the canopy level photosynthesis, it may be desirable to target a limited number of MgCh copies/alleles for mutagenesis. The choice of sgRNAs allows the targeting of specific alleles. Unlike sgRNA1, which was targeted to a highly conserved region with no allelic variants, sgRNA2 was targeted to a region that had three allelic variants with zero, one, or two mismatches to the sgRNA (Supplementary Table 5). Co-delivering both sgRNAs allowed a comparison of the impact of the individual sgRNA on the editing efficiency and the editing of the different allelic variants. Higher editing efficiency was found for all mutants for the target of sgRNA1 (33.1% edited NGS reads) than for sgRNA2 (15.3% edited NGS reads). The allelic variants with SNPs in sgRNA2 target region displayed very few edits and contributed to the overall lower editing efficiency at this site. This suggests that the choice of gRNA and the combination of sgRNAs offers opportunities to tailor the desired co-editing efficiency in sugarcane, similar to what is reported for other polyploid crops (Andersson et al., 2017; Liu et al., 2018; Wolabu et al., 2020).
The described approach will facilitate the establishment of genome-editing protocols for recalcitrant crops and will support important optimizations for the elevation of gene-editing efficiencies, including the evaluation of alternative tissue culture protocols, genome editing reagents, and their delivery.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, Bioproject ID PRJNA704370.
Author Contributions
FA conceived, designed, and managed the research project. DW and FA designed the sgRNAs. DW and AE validated sgRNAs with in vitro assays. CM generated the recombinant DNA constructs. CM and SS carried out the tissue cultures and generated the transgenic sugarcane plants. AE carried out the molecular and phenotypic analyses of the regenerated sugarcane lines. FA and AE wrote the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by the DOE Center for Advanced Bioenergy and Bioproducts Innovation (U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research) under award number DE-SC0018420. Any opinions, findings and conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the U.S. Department of Energy. This work was also supported by the USDA National Institute of Food and Agriculture, Hatch project 1020425. We acknowledge the São Paulo Research Foundation for the visiting research fellowship (FAPESP Proc No. 2018/11544–5) to CM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Hardev Sandhu at The Everglades Research and Education Center, UF-IFAS, for providing sugarcane tops, and Sun Gro Horticulture, Apopka, FL, for the donation of potting mix. We would like to thank Alberto Riva from University of Florida Interdisciplinary Center for Biotechnology Research (ICBR) Bioinformatics Core for bioinformatics support with NGS data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgeed.2021.654996/full#supplementary-material
Supplementary Figure 1. Sanger sequencing reads exemplifying targeted mutations in different lines. The blue line indicates both sgRNA target sites, the orange line indicates PAM sites, the red font indicates insertions, and the red dashed line indicates deletions. Lines HY2, HY3, NG1, NG2, NG3, HG1, and HG3. HY = heat-treated, yellow; NG = non-heat treated, green; HG = heat treated, green; WT = wild type.
Supplementary Table 1. Primers used in the experiment.
Supplementary Table 2. Summary of edits of mutant line HY1 as detected by Sanger sequencing of cloned PCR amplicons of MgCh.
Supplementary Table 3A. Genotyping of 59 alleles/variants as detected by Sanger sequencing of cloned PCR amplicons of MgCh in mutant line HY1.
Supplementary Table 3B. Genotyping of 59 alleles/variants as detected by Sanger sequencing of cloned PCR amplicons of MgCh in mutant line HY1.
Supplementary Table 4. Types of edits in MgCh gene in line HY1.
Supplementary Table 5. Summary of Next Generation Sequencing revealing the editing frequency at target site for sgRNA2 for 3 different allelic variants of the MgCh gene.
Supplementary Table 6. Analysis of NGS reads that did not align to sgRNA target sequence due to long insertions or long deletions.
References
Ali, Z., Abul-faraj, A., Li, L., Ghosh, N., Piatek, M., Mahjoub, A., et al. (2015). Efficient Virus-Mediated Genome Editing in Plants Using the CRISPR/Cas9 System. Mol. Plant 8, 1288–1291. doi: 10.1016/j.molp.2015.02.011
Altpeter, F., and Ratna, K. (2018). “Genetic improvement of sugarcane by transgenic, intragenic and genome editing technologies,” in Achieving Sustainable Cultivation of Sugarcane, Vol. 2, ed P. Rott (Cambridge: Burleigh Dodds Science Publishing), 133–154. doi: 10.19103/AS.2017.0035.07
Andersson, M., Turesson, H., Nicolia, A., Falt, A. S., Samuelsson, M., and Hofvander, P. (2017). Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient CRISPR-Cas9 expression in protoplasts. Plant Cell Rep. 36, 117–128. doi: 10.1007/s00299-016-2062-3
Baltes, N. J., Hummel, A. W., Konecna, E., Cegan, R., Bruns, A. N., Bisaro, D. M., et al. (2015). Conferring resistance to geminiviruses with the CRISPR-Cas prokaryotic immune system. Nat. Plants 1:15145. doi: 10.1038/nplants.2015.145
Beying, N., Schmidt, C., Pacher, M., Houben, A., and Puchta, H. (2020). CRISPR-Cas9-mediated induction of heritable chromosomal translocations in Arabidopsis. Nat. Plants 6, 638–645. doi: 10.1038/s41477-020-0663-x
Butt, H., Eid, A., Momin, A. A., Bazin, J., Crespi, M., Arold, S. T., et al. (2019). CRISPR directed evolution of the spliceosome for resistance to splicing inhibitors. Genome Biol. 20, 73. doi: 10.1186/s13059-019-1680-9
Byrt, C. S., Grof, C. P., and Furbank, R. T. (2011). C4 plants as biofuel feedstocks: optimising biomass production and feedstock quality from a lignocellulosic perspective. J. Integr. Plant Biol. 53, 120–135. doi: 10.1111/j.1744-7909.2010.01023.x
Campbell, B. W., Mani, D., Curtin, S. J., Slattery, R. A., Michno, J. M., Ort, D. R., et al. (2014). Identical substitutions in magnesium chelatase paralogs result in chlorophyll-deficient soybean mutants. G3 5, 123–131. doi: 10.1534/g3.114.015255
Chandrasegaran, S., and Carroll, D. (2016). Origins of Programmable Nucleases for Genome Engineering. J Mol Biol. 428(5 Pt B), 963–989. doi: 10.1016/j.jmb.2015.10.014
Chapman, J. R., Taylor, M. R., and Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 47, 497–510. doi: 10.1016/j.molcel.2012.07.029
Chen, B., Gilbert, L. A., Cimini, B. A., Schnitzbauer, J., Zhang, W., Li, G. W., et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. doi: 10.1016/j.cell.2013.12.001
de Setta, N., Monteiro-Vitorello, C. B., Metcalfe, C. J., Cruz, G. M., Del Bem, L. E., Vicentini, R., et al. (2014). Building the sugarcane genome for biotechnology and identifying evolutionary trends. BMC Genomics 15:540. doi: 10.1186/1471-2164-15-540
Eid, A., and Mahfouz, M. M. (2016). Genome editing: the road of CRISPR/Cas9 from bench to clinic. Exp. Mol. Med. 48:e265. doi: 10.1038/emm.2016.111
Fan, D., Liu, T., Li, C., Jiao, B., Li, S., Hou, Y., et al. (2015). Efficient CRISPR/Cas9-mediated targeted mutagenesis in populus in the first generation. Sci. Rep. 5:12217. doi: 10.1038/srep12217
Gao, H., Mutti, J., Young, J. K., Yang, M., Schroder, M., Lenderts, B., et al. (2020). Complex Trait Loci in Maize Enabled by CRISPR-Cas9 Mediated Gene Insertion. Front. Plant Sci. 11:535. doi: 10.3389/fpls.2020.00535
Horsley, T., and Zhou, M. (2013). Effect of photoperiod treatments onpollen viability and flowering at the South African sugarcane research institute. Proc. South Afr. Sugar Technol. Assoc. 86, 286–290.
Howells, R. M., Craze, M., Bowden, S., and Wallington, E. J. (2018). Efficient generation of stable, heritable gene edits in wheat using CRISPR/Cas9. BMC Plant Biol. 18:215. doi: 10.1186/s12870-018-1433-z
Huang, T. K., and Puchta, H. (2019). CRISPR/Cas-mediated gene targeting in plants: finally a turn for the better for homologous recombination. Plant Cell Rep. 38, 443–453. doi: 10.1007/s00299-019-02379-0
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Jung, J. H., and Altpeter, F. (2016). TALEN mediated targeted mutagenesis of the caffeic acid O-methyltransferase in highly polyploid sugarcane improves cell wall composition for production of bioethanol. Plant Mol. Biol. 92, 131–142. doi: 10.1007/s11103-016-0499-y
Kannan, B., Jung, J. H., Moxley, G. W., Lee, S. M., and Altpeter, F. (2018). TALEN-mediated targeted mutagenesis of more than 100 COMT copies/alleles in highly polyploid sugarcane improves saccharification efficiency without compromising biomass yield. Plant Biotechnol. J. 16, 856–866. doi: 10.1111/pbi.12833
Kirst, H., Gabilly, S. T., Niyogi, K. K., Lemaux, P. G., and Melis, A. (2017). Photosynthetic antenna engineering to improve crop yields. Planta 245, 1009–1020. doi: 10.1007/s00425-017-2659-y
Ko, J. K., Jung, J. H., Altpeter, F., Kannan, B., Kim, H. E., Kim, K. H., et al. (2018). Largely enhanced bioethanol production through the combined use of lignin-modified sugarcane and xylose fermenting yeast strain. Bioresour. Technol. 256, 312–320. doi: 10.1016/j.biortech.2018.01.123
Le Cunff, L., Garsmeur, O., Raboin, L. M., Pauquet, J., Telismart, H., Selvi, A., et al. (2008). Diploid/polyploid syntenic shuttle mapping and haplotype-specific chromosome walking toward a rust resistance gene (Bru1) in highly polyploid sugarcane (2n approximately 12x approximately 115). Genetics 180, 649–660. doi: 10.1534/genetics.108.091355
LeBlanc, C., Zhang, F., Mendez, J., Lozano, Y., Chatpar, K., Irish, V. F., et al. (2018). Increased efficiency of targeted mutagenesis by CRISPR/Cas9 in plants using heat stress. Plant J. 93, 377–386. doi: 10.1111/tpj.13782
Lin, S., Staahl, B. T., Alla, R. K., and Doudna, J. A. (2014). Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3:e04766. doi: 10.7554/eLife.04766.010
Liu, Y., Merrick, P., Zhang, Z., Ji, C., Yang, B., and Fei, S. Z. (2018). Targeted mutagenesis in tetraploid switchgrass (Panicum virgatum L.) using CRISPR/Cas9. Plant Biotechnol. J. 16, 381–393. doi: 10.1111/pbi.12778
Malzahn, A. A., Tang, X., Lee, K., Ren, Q., Sretenovic, S., Zhang, Y., et al. (2019). Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol. 17:9. doi: 10.1186/s12915-019-0629-5
Milner, M. J., Craze, M., Hope, M. S., and Wallington, E. J. (2020). Turning up the temperature on CRISPR: increased temperature can improve the editing efficiency of wheat using CRISPR/Cas9. Front. Plant Sci. 11:583374. doi: 10.3389/fpls.2020.583374
Moore, P. H., and Nuss, K. J. (1987). “Flowering and flower synchronization,” in Sugarcane Improve-ment Through Breeding, ed D. J. Heinz (Amsterdam: Elsevier), 273–311. doi: 10.1016/B978-0-444-42769-4.50012-6
Murray, M. G., and Thompson, W. F. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325. doi: 10.1093/nar/8.19.4321
Naim, F., Dugdale, B., Kleidon, J., Brinin, A., Shand, K., Waterhouse, P., et al. (2018). Gene editing the phytoene desaturase alleles of Cavendish banana using CRISPR/Cas9. Transgenic Res. 27, 451–460. doi: 10.1007/s11248-018-0083-0
Papenbrock, J., Pfundel, E., Mock, H. P., and Grimm, B. (2000). Decreased and increased expression of the subunit CHL I diminishes Mg chelatase activity and reduces chlorophyll synthesis in transgenic tobacco plants. Plant J. 22, 155–164. doi: 10.1046/j.1365-313x.2000.00724.x
Pettigrew, W. T., Hesketh, J. D., Peters, D. B., and Woolley, J. T. (1989). Characterization of canopy photosynthesis of chlorophyll-deficient soybean isolines. Crop Sci. 29, 1025–1029. doi: 10.2135/cropsci1989.0011183X002900040040x
Puchta, H. (1998). Repair of genomic double-strand breaks in somatic plant cells by one-sided invasion of homologous sequences. Plant J. 13, 331–339. doi: 10.1046/j.1365-313X.1998.00035.x
Qin, G., Gu, H., Ma, L., Peng, Y., Deng, X. W., Chen, Z., et al. (2007). Disruption of phytoene desaturase gene results in albino and dwarf phenotypes in Arabidopsis by impairing chlorophyll, carotenoid, and gibberellin biosynthesis. Cell Res. 17, 471–482. doi: 10.1038/cr.2007.40
Shan, Q., Wang, Y., Li, J., Zhang, Y., Chen, K., Liang, Z., et al. (2013). Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 31, 686–688. doi: 10.1038/nbt.2650
Shan, S., Mavrodiev, E. V., Li, R., Zhang, Z., Hauser, B. A., Soltis, P. S., et al. (2018). Application of CRISPR/Cas9 to Tragopogon (Asteraceae), an evolutionary model for the study of polyploidy. Mol. Ecol. Resour. 18, 1427–1443. doi: 10.1111/1755-0998.12935
Shrivastav, M., De Haro, L. P., and Nickoloff, J. A. (2008). Regulation of DNA double-strand break repair pathway choice. Cell Res. 18, 134–147. doi: 10.1038/cr.2007.111
Slattery, R. A., VanLoocke, A., Bernacchi, C. J., Zhu, X. G., and Ort, D. R. (2017). Photosynthesis, Light Use Efficiency, and Yield of Reduced-Chlorophyll Soybean Mutants in Field Conditions. Front. Plant Sci. 8:549. doi: 10.3389/fpls.2017.00549
Svitashev, S., Young, J. K., Schwartz, C., Gao, H., Falco, S. C., and Cigan, A. M. (2015). Targeted mutagenesis, precise gene editing, and site-specific gene insertion in maize using Cas9 and guide RNA. Plant Physiol. 169, 931–945. doi: 10.1104/pp.15.00793
Taparia, Y., Gallo, M., and Altpeter, F. (2012). Comparison of direct and indirect embryogenesis protocols, biolistic gene transfer and selection parameters for efficient genetic transformation of sugarcane. Plant Cell Tissue Organ. Cult. 111, 131–141. doi: 10.1007/s11240-012-0177-y
Tew, T. L., and Cobill, R. M. (2008). “Genetic improvement of sugarcane (Saccharum spp.) as an energy crop,” in Genetic Improvement of Bioenergy Crops, ed W. Vermerris (New York, NY: Springer New York), 273–294. doi: 10.1007/978-0-387-70805-8_9
Walker, B. J., Drewry, D. T., Slattery, R. A., VanLoocke, A., Cho, Y. B., and Ort, D. R. (2018). Chlorophyll can be reduced in crop canopies with little penalty to photosynthesis. Plant Physiol. 176, 1215–1232. doi: 10.1104/pp.17.01401
Wang, J., Wu, H., Chen, Y., and Yin, T. (2020). Efficient CRISPR/Cas9-mediated gene editing in an interspecific hybrid poplar with a highly heterozygous genome. Front. Plant Sci. 11:996. doi: 10.3389/fpls.2020.00996
Weeks, D. P. (2017). “Chapter Four - Gene editing in polyploid crops: wheat, camelina, canola, potato, cotton, peanut, sugar cane, and citrus,” in Progress in Molecular Biology and Translational Science, Vol. 149, eds D. P. Weeks, and B. Yang (San Diego, CA: Academic Press), 65–80. doi: 10.1016/bs.pmbts.2017.05.002
Willows, R. D., Gibson, L. C., Kanangara, C. G., Hunter, C. N., and von Wettstein, D. (1996). Three separate proteins constitute the magnesium chelatase of Rhodobacter sphaeroides. Eur. J. Biochem. 235, 438–443. doi: 10.1111/j.1432-1033.1996.00438.x
Wolabu, T. W., Cong, L., Park, J. J., Bao, Q., Chen, M., Sun, J., et al. (2020). Development of a highly efficient multiplex genome editing system in outcrossing tetraploid alfalfa (Medicago sativa). Front. Plant Sci. 11:1063. doi: 10.3389/fpls.2020.01063
Keywords: CRISPR/Cas9, genome editing, polyploid, magnesium chelatase, sugarcane, biolistic gene transfer, heat treatment
Citation: Eid A, Mohan C, Sanchez S, Wang D and Altpeter F (2021) Multiallelic, Targeted Mutagenesis of Magnesium Chelatase With CRISPR/Cas9 Provides a Rapidly Scorable Phenotype in Highly Polyploid Sugarcane. Front. Genome Ed. 3:654996. doi: 10.3389/fgeed.2021.654996
Received: 18 January 2021; Accepted: 15 March 2021;
Published: 29 April 2021.
Edited by:
Wendy Harwood, John Innes Centre, United KingdomReviewed by:
Anshu Alok, University of Minnesota Twin Cities, United StatesHongliang Zhu, China Agricultural University, China
Copyright © 2021 Eid, Mohan, Sanchez, Wang and Altpeter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fredy Altpeter, YWx0cGV0ZXJAdWZsLmVkdQ==