 Brian J. Iaffaldano
Brian J. Iaffaldano Michael P. Marino
Michael P. Marino Jakob Reiser
Jakob Reiser- Division of Cellular and Gene Therapies, Center for Biologics Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD, United States
Lentiviral (LV) vectors have emerged as powerful tools for treating genetic and acquired human diseases. As clinical studies and commercial demands have progressed, there has been a growing need for large amounts of purified LV vectors. To help meet this demand, we developed CRISPR library screening methods to identify genetic perturbations in human embryonic kidney 293 (HEK293) cells and their derivatives that may increase LV vector titers. Briefly, LV vector-based Human CRISPR Activation and Knockout libraries (Calabrese and Brunello) were used to modify HEK293 and HEK293T cells. These cell populations were then expanded, and integrated LV vector genomes were rescued by transfection. LV vectors were harvested, and the process of sequential transduction and rescue-transfection was iterated. Through this workflow, guide RNAs (gRNAs) that target genes that may suppress or enhance LV vector production were enriched and identified with Next-Generation Sequencing (NGS). Though more work is needed to test genes identified in this screen, we expect that perturbations of genes we identified here, such as TTLL12, which is an inhibitor of antiviral innate immunity may be introduced and multiplexed to yield cell lines with improved LV vector productivity.
1 Introduction
Lentiviral (LV) vectors have been utilized in numerous clinical trials to deliver transgenes into primary cells ex vivo, including T cells and hematopoietic stem cells (Milone and O’Doherty, 2018). Recently approved CAR T cell gene therapy products consisting of autologous patient T cells transduced with LV vectors include, Kymriah (tisagenlecleucel) to treat B-cell acute lymphoblastic leukemia (B-ALL) (Liu et al., 2017), Breyanzi (lisocabtagene maraleucel) to treat large B-cell lymphoma (LBCL) (Abramson et al., 2020), as well as Abecma (idecabtagene vicleucel) and Carvykti (ciltacabtagene autoleuce) to treat multiple myeloma (Munshi et al., 2021; U.S. Food and Drug Administration, 2022a). Zynteglo (betibeglogene autotemcel) consists of genetically modified autologous CD34+ cells containing haematopoietic stem cells (HSC) transduced with LV vectors bearing the βA−T87Q-globin transgene for the treatment of patients with transfusion-dependent beta-thalassemia (Schuessler-Lenz et al., 2020; U.S. Food and Drug Administration, 2022b). Skysona (elivaldogene autotemcel) is a LV vector-based gene therapy involving of genetically modified autologous CD34+ cells to treat boys with early, active cerebral adrenoleukodystrophy (CALD) (U.S. Food and Drug Administration, 2022c).
LV vector titers are often insufficient, and production is costly to scale (Perry and Rayat, 2021). The cells used to produce LV vectors are typically based on human embryonic kidney (HEK) 293 cells and its derivative cell lines such as HEK293T. Such cells have been rationally modified to further improve titers and suit specific vectors (Sosale et al., 2016, 47; Milani et al., 2017; Zhang et al., 2017; Hu et al., 2018; Ozog et al., 2018; Shirley et al., 2020). Notably, a multi-gene knockout clone of 293T cells resulted in a 7-fold increase in LV titers of complex vectors (Han et al., 2021). While thus far, rational approaches have been utilized to improve LV production, recently, an unbiased genome-wide CRISPR activation screen has been used to increase viral titer and infectious units of different adeno-associated virus (AAV) vector serotypes over 3-fold (Barnes et al., 2021); however, results of unbiased CRISPR library screens to increase LV vector titers have not been reported to date. However, OhAinle et al., have used a CRISPR library approach, where 15,348 gRNAs implicated in interferon response were screened by rescuing vectors containing Cas9 and gRNA with wild type HIV (OhAinle et al., 2018). Along similar lines, here, we have detailed a transfection-based workflow that can be used to identify genetic perturbations that can improve the ability of cells to produce lentiviral vectors. Using this approach, we identified candidate genes to be knocked out or overexpressed.
Using this modified knockout library, we first elected to conduct a pilot library screening experiment in HEK293 cells as a proof of concept, using a genome wide, LV vector-based CRISPR knockout library referred to as the Brunello library (Doench et al., 2016; Sanson et al., 2018). To do this, we developed an approach relevant to the production of LV vectors by introducing the entire Brunello gRNA library into HEK293 cells and rescuing integrated LV vector genomes encoding specific gRNAs by transient transfection. Using the smaller scale of the pilot screening we were able to generate sequence data for each cycle of rescue transfection and transduction, to observe changes in gRNA abundance over time. We then went on to broaden our methodology, by conducting full scale screenings using both the Brunello knockout library and Calabrese activation library in HEK293T cells (Lin et al., 2014). Through this approach, we observed the enrichment over time of gRNAs targeting defined genes, which introduce genetic perturbations that may increase LV titers.
2 Materials and methods
2.1 Construction of vectors and CRISPR libraries
LentiCRISPR v2 (Addgene Plasmid #52961, kindly provided by Feng Zhang) (Sanjana et al., 2014) was used as the backbone for a non-self-inactivating CRISPR library. This plasmid was digested with BamHI and PmeI and the large plasmid backbone fragment was gel extracted. A wild type LTR was added via a gBlock WPRE wt3′LTR sequence (Supplementary Figure S1) and a marker sequence, encoding either 2A Puro or 2 GFP, was added back via NEBuilder® HiFi DNA Assembly (New England Biolabs, Ipswich, MA). The 2A Puro fragment was amplified from LentiCRISPR v2 using the primers Hifi puro F (5′ gattacaaagacgatgacgataaggGATCCGGCGCAACAAACTTC 3′) and Hifi puro R (5′ ccagaggttgattGTCGACTTAACGCGTTCAG 3′). The 2a gfp fragment was amplified from TLCV2 (Addgene Plasmid #87360, kindly provided by Adam Karpf) (Barger et al., 2019) using the primers Hifi EGFP F (5′gattacaaagacgatgacgataaggGATCCGGAGAGGGCAGAG 3′) and Hifi EGFP R (5′cagaggttgattgtcgacttaacgcGTTTACTTGTACAGCTCGTCC 3′). PCR products were column purified prior to assembly. Assembly reactions were transformed into NEB® Stable Competent E. coli (New England Biolabs, Ipswich, MA) produced using a Zymo Mix & Go! E. coli Transformation Kit (Zymo Research, Irvine, CA). Colonies were verified by digestion with BamHI and PmeI, as well as Sanger sequencing. The assembled vector conferring puromycin resistance was designated as LentiCRISPR wt LTR Puro. Vectors were also constructed for a two-part CRISPR library system capable of gene knockout, activation and inhibition. This was achieved by modifying lentiGuide-Puro (Addgene Plasmid #52963, kindly provided by Feng Zhang) (Sanjana et al., 2014), pXPR_502 (Addgene Plasmid #96923, kindly provided by John Doench and David Root) (Sanson et al., 2018) and pXPR_050 (Addgene Plasmid #96925, kindly provided by John Doench and David Root) (Sanson et al., 2018), respectively. In this case, the wild type LTR sequence was introduced by PCR amplifying a fragment from pHR-CMV-nlsCRE (Addgene Plasmid #12265, kindly provided by Didier Trono) (Cudré-Mauroux et al., 2003), using the primers 3′LTR F (5′ taagaccaatgacttacaagGCAGCTGTAGATCTTAGC 3′) and 3′LTR R (5′ atgacatgaactactatacgTACTGCTAGAGATTTTCCAC 3′). Backbones were amplified from lentiGuide-Puro, pXPR_502 and pXPR_050 using the primers Library backbone F (5′ CGTATAGTAGTTCATGTCATC 3′) and Library backbone R (5′ CTTGTAAGTCATTGGTCTTAAAG 3′). Products were amplified using Q5® High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA). Fragments were gel purified and assembled using NEBuilder® HiFi DNA Assembly (New England Biolabs, Ipswich, MA).
To alter the plasmid backbone of the Brunello library, the vector LentiCRISPR wt LTR Puro was digested with BsmBI and the large fragment was gel extracted. The Brunello library was acquired from Addgene as pooled plasmids (Addgene Pooled Library #73179, kindly provided by David Root and John Doench) (Doench et al., 2016; Sanson et al., 2018). The region containing the gRNA sequences was amplified using the primers Oligo-Fwd zhang 2017 (5′ GTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACC 3′) and Oligo-Knockout-Rev zhang 2017 (5′ ACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC 3′) (Joung et al., 2017). Eight 25 µL PCR reactions were carried out for 18 amplification cycles, using a total 50 ng of input plasmid DNA. The PCR products were pooled and gel purified. The modified backbone and gRNA fragments were assembled with an excess for gRNA inserts using NEBuilder® HiFi DNA Assembly (New England Biolabs, Ipswich, MA). The assemblies were electroporated into Endura™ Competent Cells (Lucigen Corporation, Middleton, WI), using 1.0 mm cuvettes with a Gene Pulser Xcell (Bio-Rad Laboratories, Hercules, CA) and parameters of 10 µf, 600 Ω and 1,800 V. Each electroporation consisted for 30 µL of competent cells and 40 ng of library plasmid DNA. Cells were allowed to recover in 2.5 mL of the provided recovery media in an incubator-shaker set to 37°C for 1 hour. After recovery, the electroporated cells were pooled and serial dilutions were plated. The remainder of cells were grown in 500 mL of liquid culture overnight at 30°C. Colony counts of the plated serial dilutions indicated that the liquid culture consisted of approximately three million colony forming units. Plasmid DNA was extracted from the 500 mL culture using a ZymoPURE™ II Plasmid Maxiprep Kit (Catalog #D4203, Zymo Research, Irvine, CA). To clone the Brunello library in a wild-type LTR lentiGuide-Puro (Addgene Plasmid #52963, kindly provide by Feng Zhang) (Sanjana et al., 2014) backbone, PCRs, digestions and HiFi reactions were conducted as described above. The gRNA from the Calabrese library were cloned into a wild type LTR backbone similarly; however, the reverse primer Calabrese Reverse (5′ GAGAGGAGCGACGCCATATCGTCTGCTCCCTCGTATTCGCAGCATAGCTCTTAAAC 3’) was used for PCR. The transformations for full scale two-part vector screening of Brunello and Calabrese part A and Calabrese part B were scaled as described above, with approximately 24 electroporations for each library and plated on 12,500 cm2 bioassay dishes for each library. Colonies were harvested by scraping with LB broth, pelleting and extracting via ZymoPURE™ II Plasmid Maxiprep Kit (Catalog #D4203, Zymo Research, Irvine, CA), as described in Joung et al., 2017 (Joung et al., 2017). At least 3 × 109 colony forming units were collected for each library. The new plasmids are available through Addgene; they include: lenticrispr-wt-ltr-puro (Addgene Plasmid #173428), lentiguide-puro-wt-ltr (Addgene Plasmid #182635), pxpr_050-wt-ltr (Addgene Plasmid #182646), and pxpr_502-wt-ltr (Addgene Plasmid #182647).
To construct doxycycline-inducible gene knockout vectors, the TLCV2 plasmid was digested with BsmBI. The linearized plasmid was gel extracted and ligated to gene specific oligo-duplexes formed using the primers listed in Supplementary Table S1. Plasmids were transformed as previously described and colonies were miniprepped and Sanger sequenced using the primer hU6-F (5′ GAGGGCCTATTTCCCATGATT 3’). All restriction enzymes used in this study were from New England Biolabs. All CRISPR nucleases used were S. pyogenes Cas9 (SpCas9).
2.2 Cell lines
Human embryonic kidney (HEK) 293T cells (CRL-11268; American Type Culture Collection [ATCC], Manassas, VA) and HEK293 cells (Catalog Number 103, NIH AIDS Reagent Program, Germantown, MD) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) containing high glucose (4.5 g/L), 2 mM L-glutamine, 10% heat inactivated fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin. Cell culture reagents were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA).
2.3 Preparation of CRISPR libraries using a lentiviral vector bearing an intact 3’ LTR
The cloned wtLTR Brunello and Calabrese plasmid libraries were first transfected into HEK293T cells. HEK293T cells were plated in four 225 cm2 flasks at 1 × 107 cells per flask 1 day prior to transfection. These flasks were transfected with library vector, third generation helper plasmids pCD/NL-BHΔ1 (Addgene Plasmid #41791) (Ou et al., 2012) and pCMV-Rev (NIH HIV Reagent Program ARP-1443) and VSV-G envelope plasmid pCEF-VSV-G (Addgene Plasmid #41792) (Ou et al., 2012), using polyethylenimine (PEI) transfection as described (Kuroda et al., 2009). Briefly, 12 µg of library vector, 9.6 µg of pCD/NL-BHΔ1, 12.48 µg of pCMV-Rev, and 4.08 µg of envelope was diluted with 2.4 mL of 150 mM NaCl and mixed with an equal volume of PEI (Catalog # 24765, Polysciences, Warrington, PA) per flask. This DNA: PEI mixture was incubated for 10min at RT and added to each flask. LV vector stocks were collected and concentrated as described (Kutner et al., 2009), and were titrated by puromycin selection. In order to titrate, 70,000 HEK 293 cells were seeded into 6 well plates 1 day prior to transduction. Cells were transduced with varying doses of LV vector samples as described above. The medium was changed the following day. After one additional day, cells were trypsinized and evenly split into media with or without 1 μg/mL puromycin. In parallel, non-transduced cells were added to selection media. After 3 days non-transduced cells were completely killed. Transduced cells were counted with or without puromycin selection to estimate LV vector titers. Approximately 100,000 cells were initially treated with LV vectors. After selection cells were trypsinized and counted by Cellometer® Auto T4 Bright Field Cell Counter or Celigo Imaging Cytometer (Nexcelom Bioscience LLC, Lawrence, MA) and LV titers were calculated. For two vector screening using the Brunello and Calabrese libraries, HEK293T cells were used for titration instead of HEK293 cells.
2.4 Carrying out rounds of pilot Brunello CRISPR library screening
To conduct the pilot Brunello screening, the concentrated LV library was then used to transduce HEK293 cells as described above. Approximately 50 million cells were transduced in four 150 cm2 flasks with a multiplicity of infection of approximately 0.4, based on the small-scale functional titration described above. After overnight transduction, the medium was replaced. The next day, cells were trypsinized and the cells from all four flasks and seeded into four new flasks containing 0.4 μg/mL puromycin (Thermo Fisher catalog # A1113803). After 2 days, the cells were trypsinized and pelleted. Half of these cells were frozen and reserved for genomic DNA extraction while the remaining cells were divided between four 225 cm2 flasks. After the cells reached approximately 80% confluency, they were PEI transfected with 32.64 µg of pCEF-VSV-G and 65.28 µg of second generation pCD/NL-BH*ΔΔΔ packaging plasmid (Addgene plasmid #17531) (Zhang et al., 2004). 32.64 µg of pCEF-VSV-G and 65.28 µg of pCD/NL-BH*ΔΔΔ was diluted with 9.6 mL of 150 mM NaCl and mixed with an equal volume of PEI. This DNA: PEI mixture was incubated for 10 min at RT and divided equally between each flask. The medium was replaced the morning after transfection, and LV vectors were harvested after an additional 2 days. Note that the amount of pCEF-VSV-G and pCD/NL-BH*ΔΔΔ are compared to a standard protocol, which maintains the DNA:PEI ratio as a vector plasmid is omitted. As before, LV vectors were concentrated by ultracentrifugation and titrated (Kutner et al., 2009). After titration these preparations could then be used to transduce fresh HEK293 cells that have not been exposed to library sequences. These cells could then in turn be selected and transfected. This process was repeated for 8 rescue cycles to allow differences in gRNA abundances to be compounded over time.
2.5 Carrying out rounds of large-scale Brunello and Calabrese CRISPR library screening
HEK293T cells carrying Cas9 vectors were first made by transducing with lentiCas9-Blast (Addgene Plasmid #52962, kindly provided by Feng Zhang) (Sanjana et al., 2014) or lenti dCAS-VP64_Blast (Addgene Plasmid #61425, kindly provided by Feng Zhang) (Konermann et al., 2015), for screening with the Brunello and Calabrese libraries, respectively. HEK293T cells were transduced with a multiplicity of infection of 0.4, based on the small-scale functional titration described above. Cells were then selected with 4 μg/mL blasticidin (Thermo Fisher catalog # A1113903). Cells were then seeded in 150 cm2 flasks, with each flask receiving approximately 12 million cells and selected with puromycin as described above. Each library, Brunello, Calabrese A, and Calabrese B was screened in triplicate, with each Brunello experimental unit consisting of four flasks and each Calabrese experimental unit consisting of three flasks, for a total of 30 concurrent 150 cm2 flasks. Iterative rounds of rescue transfection followed by transduction as described above were then conducted, with minimal gRNA population sizes of 40 million for Brunello experimental units and 30 million for Calabrese experimental units maintained. Notably, as HEK293T cells yield higher LV titers than HEK293 cells, concentration by ultracentrifugation was not required for this workflow. However, in order to prepare viral RNA following the eighth round of rescue, ultracentrifugation was used and RNA from at least 50 million transducing units was carried forward to PCR.
2.6 Characterization of pilot CRISPR libraries
The primers Brunello Seq F (5′ TTGTGGAAAGGACGAAACACCG 3′) and Brunello Seq R (5′ CCAATTCCCACTCCTTTCAAGACCT 3’) were used to amplify the region containing the gRNA sequences. For plasmid templates, Q5® High-Fidelity DNA Polymerase (Catalog #M0491, New England Biolabs, Ipswich, MA) was used for 20 cycles of amplification in four 50 µL reactions. To sequence vector RNAs, RNA was extracted from concentrated LV vector stocks, each representing approximately 40 million transducing units (TUs), using a Quick-RNA Viral Kit (Catalog #D4033, Zymo Research, Irvine, CA). Half of the extracted RNA was used in four 50 µL OneTaq® One-Step RT-PCR reactions (Catalog #E5315, New England Biolabs, Ipswich, MA) with 20 cycles of amplification. In all cases, products were visualized on a 2% agarose gel and then column purified (Catalog #D4033, Zymo Research, Irvine, CA). Purified products were sent for CRISPR sequencing at the Massachusetts General Hospital DNA Core. FastQ files were then analyzed to determine the abundance of library members using a modified python script, with KEY_REGION_START = 10 and KEY_REGION_END = 35 (Joung et al., 2017).
2.7 Characterization of large-scale CRISPR libraries
Sequencing libraries were generated as described by (Joung et al., 2017). The ultramers identified as NGS-Fwd #1-10 and NGS-KO-Rev #one to eight were used for both the Brunello and Calabrese libraries. When viral RNA was used as a template, OneTaq® One-Step RT-PCR reactions (Catalog #E5315, New England Biolabs, Ipswich, MA) was used for 12 cycles, with each experimental unit amplified in four separate 50 µL reactions. PCR products were then concentrated and gel extracted as described and sequenced using the NovaSeq platform (Illumina, San Diego, CA) (Joung et al., 2017). Approximately 200 million reads were generated for each experimental unit, as well as wild type LTR Brunello and Calabrese libraries. The reads were counted and gRNA, gene-level analysis and Gene Set Enrichment Analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005) was conducted with default parameters using the MAGeCK(Li et al., 2014) toolkit hosted on usegalaxy. eu (The Galaxy Community, 2022).
2.8 Development of bulk knockout cell populations using doxycycline inducible lentiviral vectors
Vectors harboring gRNA targeting control safe harbor sites or specific exons identified in the library screening were transduced into HEK293 cells in a 6-well format at a MOI of less than 0.1 using transduction methods described above. Cells were passaged using media containing 0.4 μg/mL puromycin. After 3 days, cells were transferred into media containing puromycin and 1 μg/mL doxycycline. They were maintained in this medium for 1 week. The presence of gene editing was confirmed for the TRIP12 locus, as described below. Cells were then grown in 75 cm2 flasks with medium containing 0.2 μg/mL puromycin for an additional week.
2.9 Quantification of editing frequency
The targeted TRIP12 locus was PCR amplified using the primers TRIP12 Long F (5′ TGAGGTTGGGAGTTGGAGAC 3′) and TRIP12 Long R (5′ ACCGATCCTGAACTGGAGTG 3′). The amplicons were cleaned using an Exo-CIP™ Rapid PCR Cleanup Kit (New England Biolabs, Ipswich, MA) and sequenced using an internal primer, TRIP12 Seq (5′ AGCTAGTAATACTGCTGGAGAGGT 3′). The frequency of indel formation was evaluated by using ICE Analysis (https://ice.synthego.com/#/). Alternatively, editing frequency was quantified by generating a 198 bp PCR product using the primers, TRIP12 NGS F (5′ TTGATGAAAGTTGAAATTGAGCA 3′) and TRIP12 NGS R (5′ CACTTTTCCATTTTCTGTGGA3’). Such products were column purified and sent for CRISPR sequencing at the Massachusetts General Hospital DNA Core. Editing frequency was then quantified by using CRISPResso2 (Clement et al., 2019). An example of an agarose gel-based assay to measure indel formation in clonal populations is also provided in Supplementary Figure S2.
2.10 Quantification of cell line LV titers
Prior to characterization of LV vector productivity, HEK 293 cells were cultured in 75 cm2 flasks. These cells were then plated in a 6-well or 24-well format and tested for LV productivity using an EGFP vector, as described above for third generation transfection (Kuroda et al., 2009). EGFP positive cells were counted by flow cytometry or Celigo Imaging Cytometer (Nexcelom Bioscience LLC, Lawrence, MA).
2.11 Statistical analysis
Pairwise comparison was conducted using a two-tailed Student’s t-test. Significant differences (p < 0.05) between two treatments are indicated by asterisks.
3 Results
3.1 Establishment of CRISPR library screening workflow
CRISPR libraries are commonly distributed in a LV vector context; however, they are typically established using third generation, self-inactivating LV vector backbones with the U3 regions of the vector long terminal repeats (LTRs) deleted. As the workflow we implemented requires iterative rounds of vector production and transduction, we first modified an existing LV vector-based CRISPR knockout plasmid library (Doench et al., 2016; Sanson et al., 2018), by sub-cloning it into a modified LV vector backbone that harbors an intact 3’ LTR to facilitate rescue of infectious LV vector particles.
An outline of the LV vector production CRISPR knockout LV vector library screening workflow approach using knockout libraries is provided in Figure 1. After one initial round of LV vector production, iterative rounds of LV production were carried out by rescue transfection using LV packaging and VSV-G-encoding plasmids to rescue integrated vector genomes. Fresh HEK293 cells, which were not yet exposed to the CRISPR library, were then transduced with a multiplicity of infection (MOI) of less than 0.4, such that on average, transduced cells only contained a single gRNA, targeting a single gene. During this pilot screening experiment using HEK293 cells, titers for the rescued LV vectors typically ranged from 5 to 8 × 105 transducing units (TU)/ml and the total yields of rescued vectors were typically greater than 108 TU.
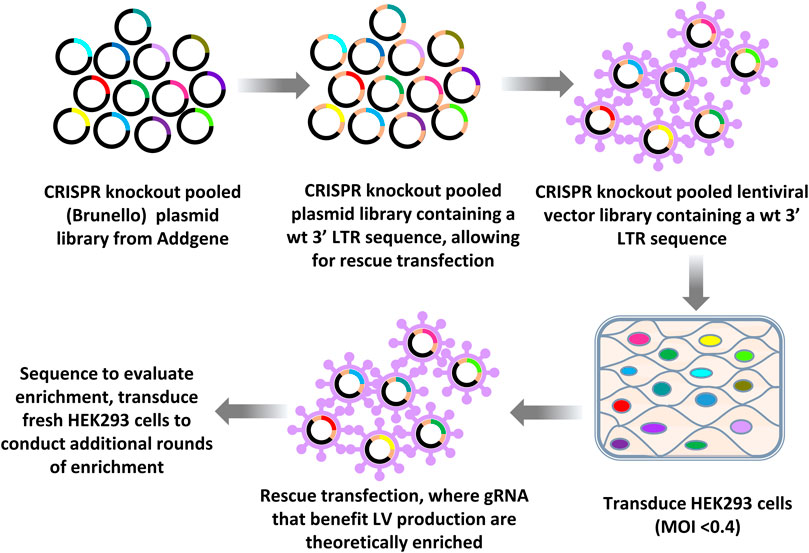
FIGURE 1. Workflow of CRISPR knockout library screening to identify genomic targets affecting LV vector production. The CRISPR knockout (Brunello) plasmid library (top panel left) was first subcloned into a LV vector plasmid backbone containing an intact (wild type) 3′ LTR (top panel middle). LV vectors were produced by transfecting this vector plasmid library plus plasmids encoding LV packaging functions and VSV-G into HEK293T cells (top right panel) to generate LV vector particles. The resulting LV vector library was then used to transduce HEK293 cells at an MOI of less than 0.4. LV vector particles were rescued from transduced cells by rescue transfection using LV packaging and VSV-G-encoding plasmids. This process of transduction followed by rescue transfection was iterated. The gRNAs encoded by the LV vectors were sequenced by performing RT-PCR using vector RNA from concentrated vector stocks or by using plasmid DNA as a template. PCR products were then sequenced.
3.2 Characterization of a modified knockout library
The modified LV vector library was characterized by calculating the abundance of gRNAs targeting essential genes for a pool of 1,734 genes considered essential for cell survival (Figure 2) (Blomen et al., 2015). Our expectation was that gRNAs targeting genes considered essential for cell survival would decrease in abundance if the library was functional. In a plasmid context of the library, the proportion of gRNAs targeting essential genes did not change, and thus met expectations, as CRISPR reagents were not yet expressed or acting on mammalian cells. The number of gRNA reads mapping to essential genes was proportional to their representation of the total gRNA population. The gRNAs targeting essential genes were modestly reduced when vectors were produced using the LV vector-based knockout library containing wild type (wt) LTR sequences and transient transfection of third-generation LV packaging constructs in HEK293T cells. In LV vector populations generated by rescue transfection using LV packaging and VSV-G-encoding plasmids, gRNAs targeting essential genes were reduced by roughly 50%. The regular enrichment of select gRNAs was observed over 8 rounds of LV vector rescue (Figure 3). Normalized read counts for pilot Brunello screening are available in Supplementary Table S2. Having observed that our modified library was active in HEK293 cells, as the abundance of gRNA targeting essential genes was reduced and that select gRNAs regularly increased in abundance over time, we went on to pursue additional validation of identified gRNAs, as well as conduct additional library screening experiments on a larger scale.
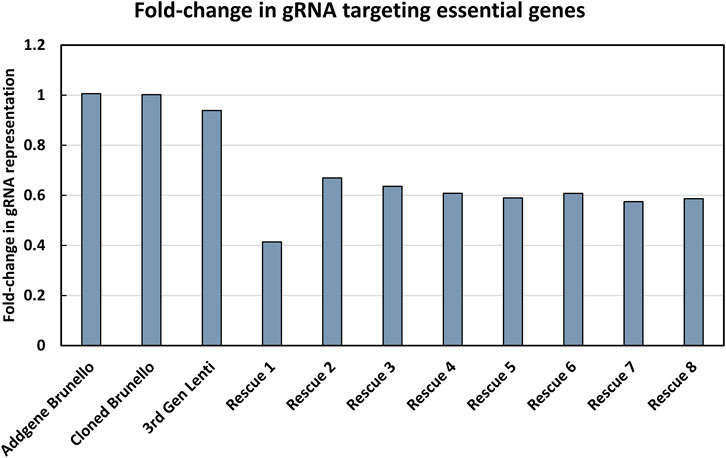
FIGURE 2. Characterization of the modified LV vector CRISPR knockout library. The fold-change in gRNAs targeting essential genes was determined. The y-axis shows the fold-change in gRNA representation of essential genes, where a value of 1 indicates that the number of reads in the library mapping to essential genes is proportional to their abundance in the library. The x-axis shows the progress of the workflow, where each name represents a pool of PCR products that was sequenced, starting from the initial knockout plasmid library from Addgene (referred to as Addgene Brunello), the plasmid library involving a LV vector plasmid with a wild type LTR (referred to as “Cloned Brunello”), lentiviral particles produced by using the LV vector plasmid library (referred to as “third Gen Lenti”) and ending with iterative rounds of viral rescue (referred to as “Rescue 1-8”).
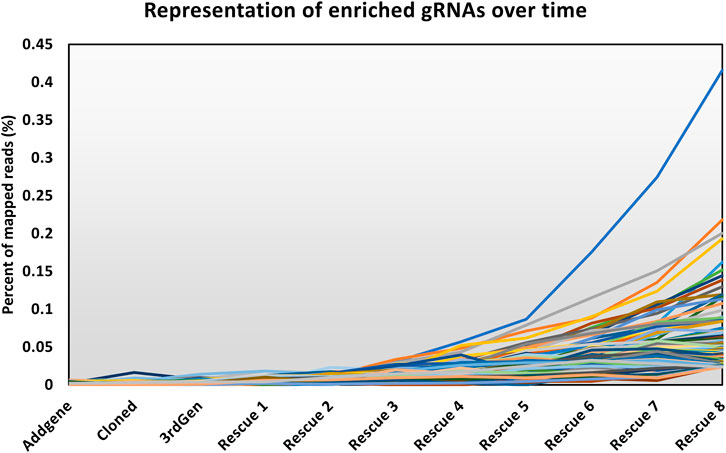
FIGURE 3. Abundance of enriched gRNAs over time. The line chart shows the representation of the top 255 gRNAs throughout the screen, where the y-axis shows the percentage of the mapped reads attributed to an individual gRNA among all the mapped reads. The x-axis shows the progress of the workflow as described in Figure 1.
3.3 Characterization of library hits in HEK293 cells for impact on lentiviral vector titers
Enriched gRNAs were further characterized by generating knockout cell populations and observing the capacity of such cells to produce LV vectors (Figure 4). gRNAs were selected for this characterization based on their degree of enrichment, previously described interactions with HIV-1 that suggested a loss of gene function benefitted HIV productivity (Ptak et al., 2008; Fu et al., 2009; Pinney et al., 2009) and if multiple gRNAs targeting the same gene were enriched (Figure 5A). We elected to generate bulk knockout populations using the doxycycline inducible CRISPR/Cas9 LV vector, TLCV2-LoxP. This allowed for Cas9 nuclease expression and knockouts to be generated in response to doxycycline addition, and the testing of LV production in the absence of nuclease activity. This emulates the heterogeneity of knockouts that would be present during library screening, as in both cases a population of unedited cells and cells edited to varying degrees are established. Accordingly, we expected that increases in vector titers from gene knockouts using this method may be similar to those bioinformatically inferred from the initial library screening. A sequence on chromosome 12 was selected as a control target, such that control cells still experience Cas9 nuclease activity, but known genes were avoided (Chaikind et al., 2016).
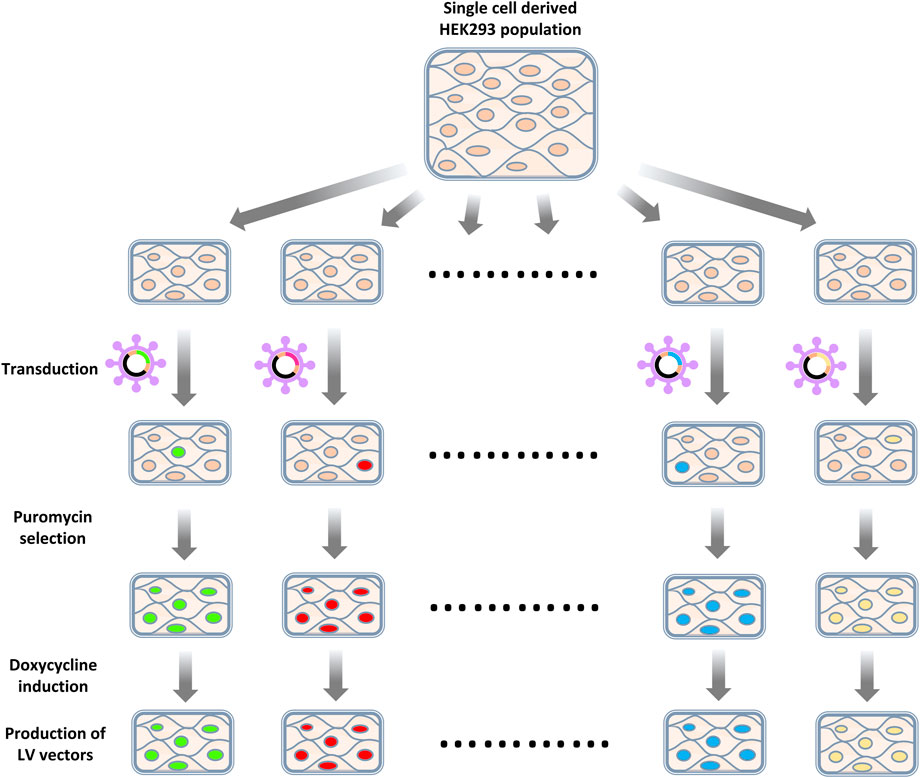
FIGURE 4. Workflow of knockout cell population generation. HEK293 cells were first transduced with LV vectors targeting selected exons or a control locus. Transduced cells were then selected by puromycin selection. Cas9 expression was induced by introducing doxycycline. After a recovery period, cells were co-transfected with an LV packaging plasmid and a VSV-G-encoding plasmid to assess the titers of LV vectors produced.
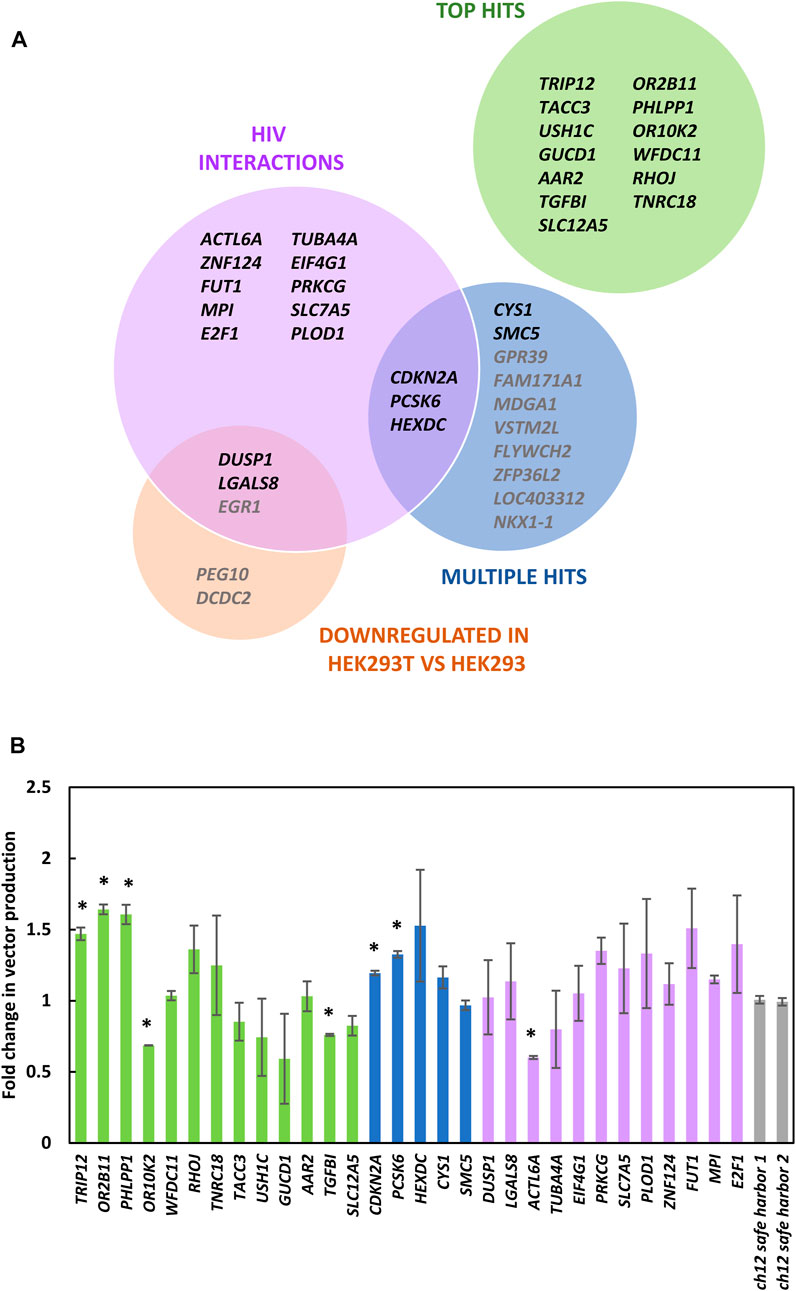
FIGURE 5. Characterization of bulk knockout populations. (A) 30 gRNAs enriched in the library screen that were selected for further characterization. The Top Hits category are the genes with individual gRNA that were most enriched. Multiple hits show genes that had multiple gRNAs within the top 500 of library hits. The HIV interactions category shows genes that had enriched gRNA within the top 1,000 of library members and may increase HIV titers when perturbed based on previously described HIV interactions (Ptak et al., 2008; Fu et al., 2009; Pinney et al., 2009). Genes downregulated in HEK293T vs. HEK293 lists genes downregulated by a factor of four or more within the top 1,000 library members (Lin et al., 2014). (B) LV vector titers of bulk knockout populations from panel (A) relative to cells transduced with gRNAs targeting a safe harbor locus (Chaikind et al., 2016). Genes from panel (A) listed in gray were not tested in panel (B). Bars represent standard error of the mean of three biological replicates.
Using this method knockout populations were generated for 30 of the gRNAs identified. As the most enriched gRNA targeted TRIP12 (Gudjonsson et al., 2012), we characterized the editing efficiency achieved in the bulk TRIP12 knockout population, revealing a knockout efficiency of 50% both by Next-Generation and Sanger Sequencing. The ability of these bulk populations to produce LV vectors was compared (Figure 5B). Among the most enriched gRNAs, the top three hits, targeting TRIP12, OR2B11 and PHLPP1 yielded significantly higher vector titers than cells transduced with control gRNAs. We observed that genes within the multiple gRNA hit category seemed to perform better than control cells most consistently, suggesting that this methodology would benefit from additional replicates with greater sequencing depth, to accommodate formal gene-level analysis.
3.4 Replicated knockout and activation screens in HEK293T cells
We went on to conduct larger scale, replicated screens using the Brunello and Calabrese libraries in HEK293T cells. We elected to use HEK293T cells as they are highly relevant to LV vector production and have much greater LV yields (Bae et al., 2020), allowing for the large number of transducing units required to be generated. The Brunello rescues typically yielded titers near 1 × 107 TU/mL, while the Calabrese rescues yielded two to three x 106 TU/mL. After sequencing, the enrichment of gRNAs targeting select genes was observed (Figure 6). The most enriched knockout target was PLAC4, while the most enriched activation target was TTLL12. In comparing the results of the pilot Brunello screen to the replicated screen in HEK293T cells, we observed that the top 10 gRNA from the “top hits” and “multiple hits” categories of the pilot averaged within the top 15% and 7.5% of enriched genes, respectively. Raw counts and gene-level analysis for replicated Brunello and Calabrese library screenings is available in Supplementary Table S3. Similarly, Gene Set Enrichment Analysis (GSEA) was carried out and is available in Supplementary Table S4.
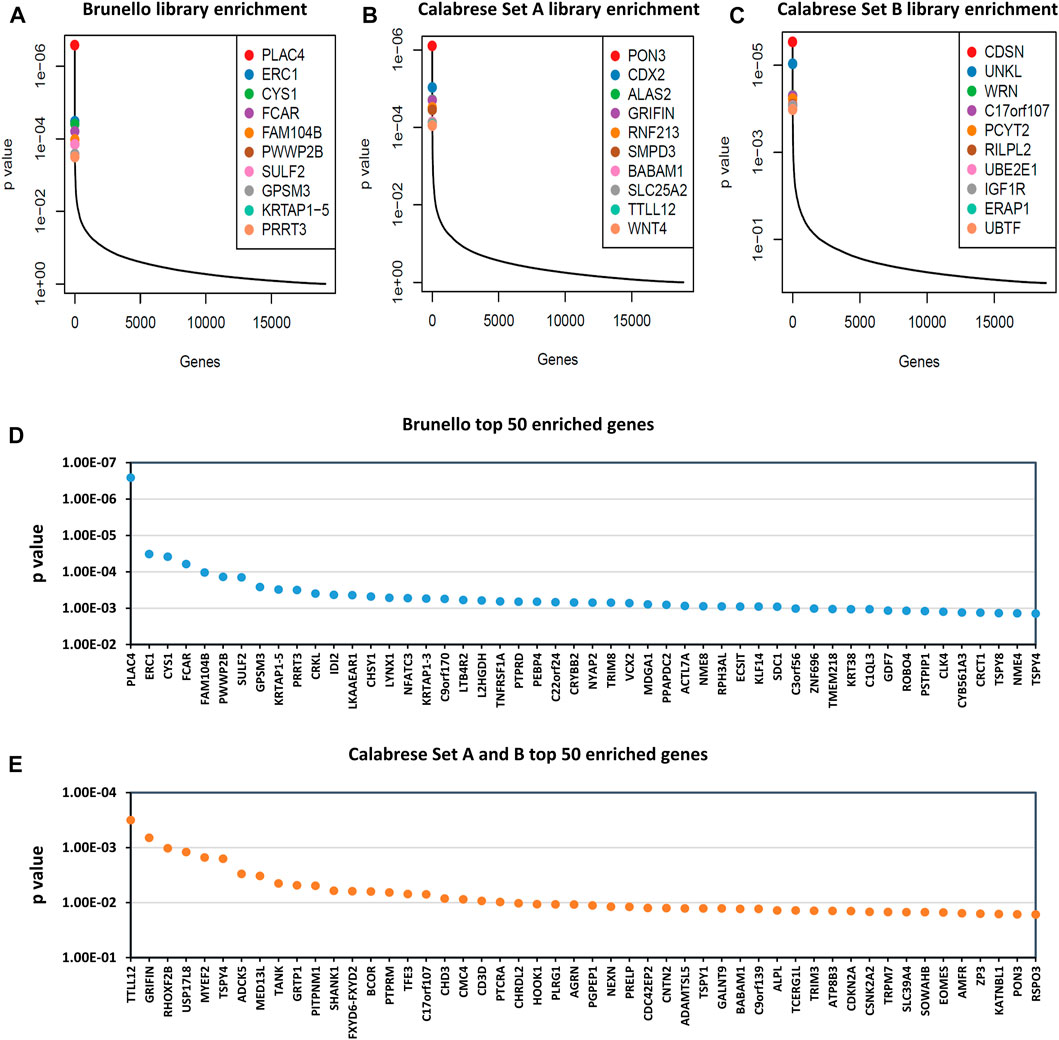
FIGURE 6. Gene enrichment of Brunello and Calabrese gRNA after 8 rounds of rescue transfection. Read counts were analyzed by MAGeCK (Li et al., 2014), where reads generated from cloned library plasmids were classed as controls and reads generated from viral supernatant were classed as treatment. Outputs of the most enriched genes are shown in plots: (A) Brunello library enrichment, (B) Calabrese Set A library enrichment and (C) Calabrese Set B library enrichment, where the y-axis indicates p-value and the x-axis represents genes ordered from most to least enriched. (D) Brunello library enrichment, showing top 50 most enriched genes on x-axis. (E) Combined Calabrese library enrichment, showing top 50 most enriched genes on x-axis and average p-value on the y-axis.
4 Discussion
HEK293 derived cells were shown to yield higher vector titers compared to other cell lines such as African Green monkey-derived COS-7 cells (Reiser et al., 1996) and human TE571 cells (Chang et al., 1999). Notably, HEK293T cells, which contain SV40 large T-antigen sequences yield substantially higher LV vector titers than the parental cell line, HEK293 (Gama-Norton et al., 2011; Bae et al., 2020). The presence of T-antigen-encoding sequences has been shown to increase LV vector titers in HEK293 cells (Ferreira et al., 2020); The knockdown or removal of T-antigen-encoding sequences in HEK293Ts by short hairpin RNA (shRNA) or CRISPR/Cas9 respectively, were found to not appreciably decrease LV vector titers (Bae et al., 2020; Ferreira et al., 2020). This suggests that genetic perturbations created by SV40 large T-antigen may positively impact LV vector production. This phenomenon was repeated by the addition of a SV40 T-antigen sequence to HEK293 cells (Gama-Norton et al., 2011), confirming that SV40 T-antigen sequences underly the increase in LV vector titers. In addition, there are large-scale differences in chromosome copy number between HEK293 and HEK293T cells, such as a notable high degree of duplication of a region of chromosome 13 in HEK293T cells (Lin et al., 2014). This duplicated region contains the miRNA-17-92 cluster, which plays a pivotal role in many cellular processes (Mogilyansky and Rigoutsos, 2013). In addition, there are 155 upregulated and 334 downregulated genes in HEK293T vs. HEK293 (Lin et al., 2014), some of which may impact vector titers. Such genes represent potential rational targets to improve LV vector titers in HEK293 cells. Moreover, the large increase in LV vector titers resulting from genetic differences between HEK293 and HEK293T suggests that genetic perturbations that increase titer exist and may be identified by an unbiased approach.
While HEK293 and HEK293 cell derivatives are commonly used to produce LV vectors, there are many genetic improvements that increase LV vector production likely yet to be realized. Such cells contain cellular bottlenecks and viral restriction factors that may impact LV vector titers. In the case of HIV infection, many host restriction factors limit virus production and transduction (Zotova et al., 2019). These restriction factors are mitigated by viral accessory proteins, which are purposefully omitted in LV vectors due to safety concerns. As lentiviral vectors are HIV derived, it may be expected that restriction factors are present in human cell lines such as HEK293 (Uhlén et al., 2015). Expression data from the Human Protein Atlas (https://www.proteinatlas.org/about/licence) (Uhlén et al., 2015) suggests that most are expressed to varying degrees in HEK293 cells, with the notable exceptions of BST-2, Trim22, and Mx2. It was also recently reported that HEK293T cells lack an intact innate immune response, which may contribute to their amenability to LV vector production. However, while differences in titer when an innate immune response was or was not activated were observed, they did not reach statistical significance (Ferreira et al., 2020).
In our pilot gene knockout screen, we observed the regular enrichment of select gRNA throughout multiple rounds of rescue transfections. This affirmed that the changes observed in gRNA frequency had biological underpinnings and were not simply stochastic effects. When using such gRNA to generate gene knockouts in bulk populations, we were able to observe modest increases in LV vector titers for some gRNAs. To explore this approach more fully, we conducted additional screening in HEK293T cells using both knockout and activation libraries.
Gene-level analysis revealed that the most enriched knockout target was PLAC4. Interestingly, PLAC4 is an RNA gene that has recently been implicated in interferon response and reduced covid-19 loads (Rouchka et al., 2021). In addition, gRNA targeting genes FCAR, PRRT3 and NFATC3, which are downregulated by HIV-1 infection were enriched (Dürrbaum-Landmann et al., 1994). Lastly, genes that were identified in the pilot screen, CYS1, CDKN2A, MDGA1, and VSTM2L, were again enriched in the larger scale screening, suggesting they may have merit as potential targets to improve LV titers.
When combining the enrichment data for Calabrese Set A and Set B, we identified the most enriched activation target was TTLL12, which was ranked as the 9th and 30th most enriched gene within the two experiments. TTLL12 has been shown to inhibit antiviral innate immunity (Ju et al., 2017). Along these lines, Gene Set Enrichment Analysis found significance enrichment of a gene set implicated in nucleic acid metabolism and innate immune sensing for both Calabrese Set A & B (p ≤ 0.1) and for the Brunello data (p ≤ 0.05). Moreover, TTLL12 is also upregulated by HIV-1 Tat (Jarboui et al., 2012). In addition to TTLL12, gRNAs targeting the three genes MYEF2, GRTP1, and TFE3, were highly enriched. The knockdown of these genes has been demonstrated to diminish HIV-1 replication (Brass et al., 2008; König et al., 2008).
One potential source of false positives in this workflow is caused by using a combined (one-vector) system, expressing both Cas9 and a gRNA (Sanjana et al., 2014). This may result in vectors lacking expression of Cas9 due to inactivation or deletion of Cas9 sequence. Both events would likely be interpreted as an increase in vector titers, as cells without Cas9 would be expected to proliferate faster and since smaller vector genomes may result in higher vector titers. To mitigate this potential shortcoming, when conducting our large-scale screening, we proceeded with a two-vector system where cell lines are made to stably express Cas9 prior to initiating screening. Similarly, another important consideration in this workflow when conducting gene knockouts, is the limited exposure of cells to Cas nuclease reagents. This limited exposure may be insufficient to yield a phenotypic effect, depending on factors such as the efficiency of each gRNA, the length of the indels introduced, the copy number of the targeted gene and mRNA and protein turnover rates of a given gene. Using a two-vector system, where cells are already producing Cas9 nuclease prior to transduction may increase the extent of editing. Similarly, this approach may benefit from screening in cell lines with reduced ploidy levels. However, this is less of a concern for activation screening. Given the potential for truncated vectors to emerge that still contain gRNA sequences that could be amplified, biological replicates were used to control for such rare events where, for example, a vector harboring a given gRNA may gain a mutation that increases productivity. By including replicates, it would become unlikely that a given gRNA would independently acquire such mutations multiple times. While beyond the scope of this paper, it would be worthwhile to generate full length reads of library vectors and observe the abundance of such mutations over time. Such analysis could potentially be used after screening to remove false positives.
We expect that some hits identified using approaches similar to those described here may be specific to a given experimental setup. Library screening using different vector pseudotypes, cell lines or culture conditions could potentially yield different results. It is also notable that this approach involved the use of a second-generation packaging plasmid encoding Tat during transfection, while applications of screening would most likely be utilized in a third-generation context. Additionally, library screening could be conducted where vectors harboring gRNA also carry a transgene of interest to probe for transgene specific responses that reduce titers. As there are currently gRNA libraries for gene knockout, inhibition and activation, as well as libraries targeting long non-coding RNA (Zhu et al., 2016), there is much potential yet to be realized for improved cell lines for LV vector production.
In the future, we imagine that libraries that delete large regions of genomic DNA using CRISPR Cascade will become available (Dolan et al., 2019). Events from several CRISPR library variants could potentially be stacked to identify improved cell lines for LV vector production. Here we have listed several knockout and overexpression candidate genes that may be used to rationally develop improved LV production cell lines in the future. Furthermore, our results are a first step that demonstrates a feasible, unbiased workflow to identify genetic modifications that improve LV vector titers.
Data availability statement
The plasmids presented in the study are deposited in addgene.org, Addgene Plasmid #173428, #182635, #182646 and #182647.
Author contributions
BI and JR conceived of and designed the experiments. BI and MM conducted the experiments. BI, MM and JR wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The following was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HEK 293 cells from Dr. Andrew Rice. This work was supported by the Intramural Research Program of the Center for Biologics Evaluation and Research (CBER), US Food and Drug Administration. This project was supported in part by the appointment of BI to the Research Participation Program at CBER administered by the Oak Ridge Institute for Science and Education through the US Department of Education and the US Food and Drug Administration. The Galaxy server that was used for some calculations is in part funded by Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and German Federal Ministry of Education and Research [BMBF grants 031 A538A/A538C RBC, 031L0101B/031L0101C de. NBI-epi, 031L0106 de. STAIR (de.NBI)].
Acknowledgments
We thank Andrew Byrnes and Pankaj Mandal for reviewing the manuscript. The following reagent was obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: pCMV-rev (pRev-1), ARP-1443, contributed by Dr. Marie-Louise Hammarskjöld and Dr. David Rekosh.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgeed.2023.1218328/full#supplementary-material
References
Abramson, J. S., Palomba, M. L., Gordon, L. I., Lunning, M. A., Wang, M., Arnason, J., et al. (2020). Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet lond. Engl. 396, 839–852. doi:10.1016/S0140-6736(20)31366-0
Bae, D. H., Marino, M., Iaffaldano, B., Fenstermaker, S., Afione, S., Argaw, T., et al. (2020). Design and testing of vector-producing HEK293T cells bearing a genomic deletion of the SV40 T antigen coding region. Mol. Ther. - Methods Clin. Dev. 18, 631–638. doi:10.1016/j.omtm.2020.07.006
Barger, C. J., Branick, C., Chee, L., and Karpf, A. R. (2019). Pan-cancer analyses reveal genomic features of FOXM1 overexpression in cancer. Cancers 11, 251. doi:10.3390/cancers11020251
Barnes, C. R., Lee, H., Ojala, D. S., Lewis, K. K., Limsirichai, P., and Schaffer, D. V. (2021). Genome-wide activation screens to increase adeno-associated virus production. Mol. Ther. - Nucleic Acids 26, 94–103. doi:10.1016/j.omtn.2021.06.026
Blomen, V. A., Májek, P., Jae, L. T., Bigenzahn, J. W., Nieuwenhuis, J., Staring, J., et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. doi:10.1126/science.aac7557
Brass, A. L., Dykxhoorn, D. M., Benita, Y., Yan, N., Engelman, A., Xavier, R. J., et al. (2008). Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926. doi:10.1126/science.1152725
Chaikind, B., Bessen, J. L., Thompson, D. B., Hu, J. H., and Liu, D. R. (2016). A programmable Cas9-serine recombinase fusion protein that operates on DNA sequences in mammalian cells. Nucleic Acids Res. 44, 9758–9770. doi:10.1093/nar/gkw707
Chang, L. J., Urlacher, V., Iwakuma, T., Cui, Y., and Zucali, J. (1999). Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 6, 715–728. doi:10.1038/sj.gt.3300895
Clement, K., Rees, H., Canver, M. C., Gehrke, J. M., Farouni, R., Hsu, J. Y., et al. (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226. doi:10.1038/s41587-019-0032-3
Cudré-Mauroux, C., Occhiodoro, T., König, S., Salmon, P., Bernheim, L., and Trono, D. (2003). Lentivector-mediated transfer of Bmi-1 and telomerase in muscle satellite cells yields a Duchenne myoblast cell line with long-term genotypic and phenotypic stability. Hum. Gene Ther. 14, 1525–1533. doi:10.1089/104303403322495034
Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191. doi:10.1038/nbt.3437
Dolan, A. E., Hou, Z., Xiao, Y., Gramelspacher, M. J., Heo, J., Howden, S. E., et al. (2019). Introducing a spectrum of long-range genomic deletions in human embryonic stem cells using Type I CRISPR-Cas. Mol. Cell. 74, 936–950.e5. doi:10.1016/j.molcel.2019.03.014
Dürrbaum-Landmann, I., Kaltenhäuser, E., Flad, H. D., and Ernst, M. (1994). HIV-1 envelope protein gp120 affects phenotype and function of monocytes in vitro. J. Leukoc. Biol. 55, 545–551. doi:10.1002/jlb.55.4.545
Ferreira, C. B., Sumner, R. P., Rodriguez-Plata, M. T., Rasaiyaah, J., Milne, R. S., Thrasher, A. J., et al. (2020). Lentiviral vector production titer is not limited in HEK293T by induced intracellular innate immunity. Mol. Ther. - Methods Clin. Dev. 17, 209–219. doi:10.1016/j.omtm.2019.11.021
Fu, W., Sanders-Beer, B. E., Katz, K. S., Maglott, D. R., Pruitt, K. D., and Ptak, R. G. (2009). Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 37, D417–D422. doi:10.1093/nar/gkn708
Gama-Norton, L., Botezatu, L., Herrmann, S., Schweizer, M., Alves, P. M., Hauser, H., et al. (2011). Lentivirus production is influenced by SV40 large T-antigen and chromosomal integration of the vector in HEK293 cells. Hum. Gene Ther. 22, 1269–1279. doi:10.1089/hum.2010.143
Gudjonsson, T., Altmeyer, M., Savic, V., Toledo, L., Dinant, C., Grøfte, M., et al. (2012). TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 150, 697–709. doi:10.1016/j.cell.2012.06.039
Han, J., Tam, K., Tam, C., Hollis, R. P., and Kohn, D. B. (2021). Improved lentiviral vector titers from a multi-gene knockout packaging line. Mol. Ther. - Oncolytics 23, 582–592. doi:10.1016/j.omto.2021.11.012
Hu, P., Bi, Y., Ma, H., Suwanmanee, T., Zeithaml, B., Fry, N. J., et al. (2018). Superior lentiviral vectors designed for BSL-0 environment abolish vector mobilization. Gene Ther. 25, 454–472. doi:10.1038/s41434-018-0039-2
Jarboui, M. A., Bidoia, C., Woods, E., Roe, B., Wynne, K., Elia, G., et al. (2012). Nucleolar protein trafficking in response to HIV-1 Tat: Rewiring the nucleolus. PLOS ONE 7, 48702. doi:10.1371/journal.pone.0048702
Joung, J., Konermann, S., Gootenberg, J. S., Abudayyeh, O. O., Platt, R. J., Brigham, M. D., et al. (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863. doi:10.1038/nprot.2017.016
Ju, L. G., Zhu, Y., Lei, P. J., Yan, D., Zhu, K., Wang, X., et al. (2017). TTLL12 inhibits the activation of cellular antiviral signaling through interaction with VISA/MAVS. J. Immunol. 198, 1274–1284. doi:10.4049/jimmunol.1601194
Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. doi:10.1038/nature14136
König, R., Zhou, Y., Elleder, D., Diamond, T. L., Bonamy, G. M. C., Irelan, J. T., et al. (2008). Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 135, 49–60. doi:10.1016/j.cell.2008.07.032
Kuroda, H., Kutner, R. H., Bazan, N. G., and Reiser, J. (2009). Simplified lentivirus vector production in protein-free media using polyethylenimine-mediated transfection. J. Virol. Methods 157, 113–121. doi:10.1016/j.jviromet.2008.11.021
Kutner, R. H., Zhang, X. Y., and Reiser, J. (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 4, 495–505. doi:10.1038/nprot.2009.22
Li, W., Xu, H., Xiao, T., Cong, L., Love, M. I., Zhang, F., et al. (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. doi:10.1186/s13059-014-0554-4
Lin, Y. C., Boone, M., Meuris, L., Lemmens, I., Van Roy, N., Soete, A., et al. (2014). Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 5, 4767–4812. doi:10.1038/ncomms5767
Liu, Y., Chen, X., Han, W., and Zhang, Y. (2017). Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today Barc. Spain 53, 597–608. doi:10.1358/dot.2017.53.11.2725754
Milani, M., Annoni, A., Bartolaccini, S., Biffi, M., Russo, F., Di Tomaso, T., et al. (2017). Genome editing for scalable production of alloantigen-free lentiviral vectors for in vivo gene therapy. EMBO Mol. Med. 9, 1558–1573. doi:10.15252/emmm.201708148
Milone, M. C., and O’Doherty, U. (2018). Clinical use of lentiviral vectors. Leukemia 32, 1529–1541. doi:10.1038/s41375-018-0106-0
Mogilyansky, E., and Rigoutsos, I. (2013). The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell. Death Differ. 20, 1603–1614. doi:10.1038/cdd.2013.125
Mootha, V. K., Lindgren, C. M., Eriksson, K. F., Subramanian, A., Sihag, S., Lehar, J., et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. doi:10.1038/ng1180
Munshi, N. C., Anderson, L. D., Shah, N., Madduri, D., Berdeja, J., Lonial, S., et al. (2021). Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 384, 705–716. doi:10.1056/NEJMoa2024850
OhAinle, M., Helms, L., Vermeire, J., Roesch, F., Humes, D., Basom, R., et al. (2018). A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. eLife 7, e39823. doi:10.7554/eLife.39823
Ou, W., Marino, M. P., Suzuki, A., Joshi, B., Husain, S. R., Maisner, A., et al. (2012). Specific targeting of human interleukin (IL)-13 receptor α2-positive cells with lentiviral vectors displaying IL-13. Hum. Gene Ther. Methods 23, 137–147. doi:10.1089/hgtb.2012.054
Ozog, S., Chen, C. X., Simpson, E., Garijo, O., Timberlake, N. D., Minder, P., et al. (2018). CD46 null packaging cell line improves measles lentiviral vector production and gene delivery to hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev. 13, 27–39. doi:10.1016/j.omtm.2018.11.006
Perry, C., and Rayat, A. C. M. E. (2021). Lentiviral vector bioprocessing. Viruses 13, 268. doi:10.3390/v13020268
Pinney, J. W., Dickerson, J. E., Fu, W., Sanders-Beer, B. E., Ptak, R. G., and Robertson, D. L. (2009). HIV-Host interactions: A map of viral perturbation of the host system. AIDS Lond. Engl. 23, 549–554. doi:10.1097/QAD.0b013e328325a495
Ptak, R. G., Fu, W., Sanders-Beer, B. E., Dickerson, J. E., Pinney, J. W., Robertson, D. L., et al. (2008). Cataloguing the HIV type 1 human protein interaction network. AIDS Res. Hum. Retroviruses 24, 1497–1502. doi:10.1089/aid.2008.0113
Reiser, J., Harmison, G., Kluepfel-Stahl, S., Brady, R. O., Karlsson, S., and Schubert, M. (1996). Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc. Natl. Acad. Sci. 93, 15266–15271. doi:10.1073/pnas.93.26.15266
Rouchka, E. C., Chariker, J. H., Alejandro, B., Adcock, R. S., Singhal, R., Ramirez, J., et al. (2021). Induction of interferon response by high viral loads at early stage infection may protect against severe outcomes in COVID-19 patients. Sci. Rep. 11, 15715. doi:10.1038/s41598-021-95197-y
Sanjana, N. E., Shalem, O., and Zhang, F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. doi:10.1038/nmeth.3047
Sanson, K. R., Hanna, R. E., Hegde, M., Donovan, K. F., Strand, C., Sullender, M. E., et al. (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416–5515. doi:10.1038/s41467-018-07901-8
Schuessler-Lenz, M., Enzmann, H., and Vamvakas, S. (2020). Regulators’ advice can make a difference: European medicines agency approval of zynteglo for beta thalassemia. Clin. Pharmacol. Ther. 107, 492–494. doi:10.1002/cpt.1639
Shirley, J. L., de Jong, Y. P., Terhorst, C., and Herzog, R. W. (2020). Immune responses to viral gene therapy vectors. Mol. Ther. 28, 709–722. doi:10.1016/j.ymthe.2020.01.001
Sosale, N. G., Ivanovska, I. I., Tsai, R. K., Swift, J., Hsu, J. W., Alvey, C. M., et al. (2016). Marker of Self CD47 on lentiviral vectors decreases macrophage-mediated clearance and increases delivery to SIRPA-expressing lung carcinoma tumors. Mol. Ther. Methods Clin. Dev. 3, 16080. doi:10.1038/mtm.2016.80
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550. doi:10.1073/pnas.0506580102
The Galaxy Community (2022). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 50, W345–W351. doi:10.1093/nar/gkac247
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. doi:10.1126/science.1260419
U.S. Food and Drug Administration (2022a). Carvykti fda. Available: at: https://www.fda.gov/vaccines-blood-biologics/carvykti (Accessed September 13, 2022).
U.S. Food and Drug Administration (2022b). FDA approves first cell-based gene therapy to treat adult and pediatric patients with beta-thalassemia who require regular blood transfusions FDA. Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-treat-adult-and-pediatric-patients-beta-thalassemia-who (Accessed September 13, 2022).
U.S. Food and Drug Administration (2022c). Skysona fda. Available at: https://www.fda.gov/vaccines-blood-biologics/skysona (Accessed December 20, 2022).
Zhang, W., Xiao, D., Shan, L., Zhao, J., Mao, Q., and Xia, H. (2017). Generation of apoptosis-resistant HEK293 cells with CRISPR/Cas mediated quadruple gene knockout for improved protein and virus production. Biotechnol. Bioeng. 114, 2539–2549. doi:10.1002/bit.26382
Zhang, X. Y., Russa, V. F. L., and Reiser, J. (2004). Transduction of bone-marrow-derived mesenchymal stem cells by using lentivirus vectors pseudotyped with modified RD114 envelope glycoproteins. J. Virol. 78, 1219–1229. doi:10.1128/JVI.78.3.1219-1229.2004
Zhu, S., Li, W., Liu, J., Chen, C. H., Liao, Q., Xu, P., et al. (2016). Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat. Biotechnol. 34, 1279–1286. doi:10.1038/nbt.3715
Keywords: lentiviral vectors, CRISPR library screening, vector titers, gene knockout, gene activation
Citation: Iaffaldano BJ, Marino MP and Reiser J (2023) CRISPR library screening to develop HEK293-derived cell lines with improved lentiviral vector titers. Front. Genome Ed. 5:1218328. doi: 10.3389/fgeed.2023.1218328
Received: 07 May 2023; Accepted: 03 July 2023;
Published: 13 July 2023.
Edited by:
Yizong Hu, Massachusetts Institute of Technology, United StatesReviewed by:
Rui Li, Johns Hopkins University, United StatesRekha Garg, University of Florida, United States
Prasad Trivedi, University of Florida, Gainesville, United States, in collaboration with reviewer RG
Copyright © 2023 Iaffaldano, Marino and Reiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian J. Iaffaldano, YnJpYW4uaWFmZmFsZGFub0BnbWFpbC5jb20=