 Chuxuan Li
Chuxuan Li Jian Li1*
Jian Li1* Quan Zou
Quan Zou- 1School of Mathematics and Computer Science, Zhejiang A&F University, Hangzhou, China
- 2Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, Quzhou, China
- 3Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China
Introduction: Accurately predicting the on-target activity of sgRNAs remains a challenge in CRISPR-Cas9 applications, due to the limited generalization of existing models across datasets, small-sample settings, and complex sequence contexts. Current methods often rely on shallow architectures or unimodal encodings, limiting their ability to capture the intricate dependencies underlying Cas9-mediated cleavage.
Methods: We present CRISPR-FMC, a dual-branch hybrid neural network that integrates One-hot encoding with contextual embeddings from a pre-trained RNA-FM model. Multi-scale convolution (MSC), BiGRU, and Transformer blocks are employed to extract hierarchical sequence features, while a bidirectional cross-attention mechanism with a residual feedforward network enhances multimodal fusion and generalization.
Results: Across nine public CRISPR-Cas9 datasets, CRISPR-FMC consistently outperforms existing baselines in both Spearman and Pearson correlation metrics, showing particularly strong performance under low-resource and cross-dataset conditions. Ablation experiments confirm the contribution of each module, and base substitution analysis reveals a pronounced sensitivity to the PAM-proximal region.
Discussion: The PAM-proximal sensitivity aligns with established biological evidence, indicating the model’s capacity to capture biologically relevant sequence determinants. These results demonstrate that CRISPR-FMC offers a robust and interpretable framework for sgRNA activity prediction across heterogeneous genomic contexts.
1 Introduction
The CRISPR-Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein 9) system is an adaptive immune mechanism originating from bacteria and archaea (Roberts and Barrangou, 2020). In recent years, it has emerged as a powerful tool for precise genome editing (Doudna and Charpentier, 2014). This system primarily relies on single-guide RNA (sgRNA) (Meier et al., 2017) to direct the Cas9 endonuclease to specific genomic loci, where it introduces double-strand breaks (DSBs) (Jinek et al., 2012). These breaks activate endogenous DNA repair pathways, resulting in insertions or deletions (indels) at the target site (Cong et al., 2013). Owing to its high editing efficiency, programmable specificity, and operational simplicity, CRISPR-Cas9 has been widely applied in gene function analysis, hereditary disease research, and genomic engineering (Shalem et al., 2014).
However, the activity of sgRNAs varies substantially across different target sequences and cell types, leading to inconsistencies in editing efficiency and experimental reproducibility (Doench et al., 2016). This variability underscores the necessity of developing accurate computational models to predict sgRNA on-target activity, thereby facilitating the rational design of highly effective sgRNAs and improving the reliability of genome editing experiments (Haeussler et al., 2016).
Computational methods for predicting on-target sgRNA activity are typically classified into two major categories: conventional approaches based on heuristic rules or classical machine learning algorithms, and more recent methodologies employing deep learning frameworks.
Traditional predictive models typically depend on manually engineered features—such as nucleotide frequency, GC content, and inferred secondary structures (Bochman et al., 2012)—and utilize algorithms like support vector machines (SVMs) (Vishwanathan and Murty, 2002) and logistic regression Hosmer (Hosmer Jr et al., 2013). Notable representatives of this category include the Rule Set family, WU-CRISPR (Wong et al., 2015), and CRISPRscan (Moreno-Mateos et al., 2015). Although these approaches provide a certain level of interpretability, their reliance on predefined features restricts their capacity to model intricate sequence characteristics and long-range contextual information.
In contrast to traditional approaches, deep learning methods, empowered by end-to-end representation learning capabilities, can automatically extract high-order features from large-scale screening data and have demonstrated significant advantages in sgRNA on-target efficiency prediction tasks (Chuai et al., 2018). Existing approaches can be broadly classified into four main categories based on their model architectures and learning strategies. Firstly, models based on convolutional neural networks (CNNs) (Alzubaidi et al., 2021) are widely applied for local sequence pattern modeling. For instance, DeepCas9 (Kim et al., 2019) employs fixed-length convolutional kernels to extract localized nucleotide fragment features. Building upon this idea, CRISPR_HNN (Li et al., 2025) integrates a multi-scale convolutional module (MSC) (Jin et al., 2022), thereby enhancing the model’s capacity to capture local sequence patterns across diverse receptive fields. Secondly, to improve the identification of key functional bases, some methods integrate attention mechanisms. CRISPR-ONT (Zhang et al., 2021) and AttnToCrispr-CNN (Liu et al., 2019) apply attention modules to emphasize important base positions, thereby improving overall modeling and predictive performance. Thirdly, recurrent neural networks (RNNs) (Dhruv and Naskar, 2020) have been adopted to capture sequential and contextual dependencies. For instance, CrnnCrispr (Zhu et al., 2024) leverages Bidirectional Gated Recurrent Unit (BiGRU) modules (Yan et al., 2021) to enhance the modeling of contextual relationships along the sgRNA sequence. Lastly, to further strengthen structural modeling and cross-dataset generalization, several studies have explored more expressive architectures. TransCrispr (Wan and Jiang, 2022) employs a Transformer (Han et al., 2021) module to improve long-range dependency modeling, while C-RNNCrispr (Zhang et al., 2020) integrates external biological features to enhance performance across diverse data distributions.
Although recent deep learning methods have achieved promising results on various public CRISPR-Cas9 datasets, several key limitations remain. (1) Most existing encoding strategies are overly simplistic, failing to jointly capture shallow compositional and deep contextual features within sgRNA sequences. (2) Many feature extraction frameworks lack the architectural capacity to concurrently model both local sequence motifs and long-range dependencies. (3) Multimodal fusion strategies are typically shallow and semantically under-aligned, limiting model expressiveness and generalization, especially under complex sequence distributions or small-sample scenarios.
In response to these challenges, we propose CRISPR-FMC, an innovative dual-branch deep neural network with a novel structural design, systematically optimized along three key architectural dimensions: Pre-training, Multimodal Processing, and Cross-modal Interaction. (1) In the Pre-training stage, CRISPR-FMC integrates one-hot encoding and RNA-FM pre-trained embeddings (Chen et al., 2022) to construct two complementary input branches. The one-hot pathway captures low-level nucleotide composition, while the RNA-FM pathway encodes high-level contextual semantics, enabling the model to learn multi-level sequence representations from the outset. (2) During Multimodal Processing, each branch undergoes independent transformation through a sequence of modules specifically designed for its input characteristics. In particular, a multi-scale convolution (MSC) block extracts local motifs of varying lengths, and the Transformer and BiGRU layers are responsible for modeling long-range dependencies and sequential relationships. This parallel architecture enhances the model’s capacity to capture hierarchical and complementary features within each modality. (3) In the Cross-modal Interaction module, a bidirectional cross-attention mechanism (Chen et al., 2021) facilitates semantic alignment and feature-level integration between the two branches. The resulting fused features are subsequently refined by a residual feedforward network (FFN) (Xie S. et al., 2023), enabling high-order nonlinear transformations and improving the model’s generalization capability. Extensive evaluations across nine publicly available CRISPR-Cas9 benchmark datasets show that CRISPR-FMC consistently outperforms state-of-the-art baseline models in both predictive accuracy and robustness. To enhance practical utility, we developed an interactive web-based visualization platform that provides real-time interpretability and supports efficient sgRNA design.
The core contributions of this work can be summarized as follows:
1. We propose a dual-branch multimodal encoding scheme that integrates One-hot encoding and RNA-FM pre-trained embeddings, enabling the model to jointly capture low-level base composition and high-level contextual semantics. This design facilitates multi-level representation of sgRNA sequences and enhances the expressiveness of input features.
2. We construct a hybrid feature extraction framework that incorporates MSC modules for local motif detection, together with independent Transformer and BiGRU components for modeling long-range dependencies. This integration enables the model to simultaneously capture fine-grained local patterns and broader global sequence dependencies.
3. We design a Cross-modal Interaction mechanism that integrates a bidirectional cross-attention module and a FFN, which promotes deep semantic alignment and nonlinear feature fusion between modalities, improving representation stability and generalization capacity.
4. We conducted a comprehensive evaluation of the proposed method across nine publicly available CRISPR-Cas9 benchmark datasets. The results demonstrate that CRISPR-FMC consistently outperforms state-of-the-art approaches in both predictive accuracy and cross-dataset generalization, exhibiting particularly strong performance under limited-sample conditions.
2 Materials and methods
2.1 Datasets
To comprehensively evaluate the predictive performance of the CRISPR-FMC model across diverse experimental settings and sample sizes, nine publicly available CRISPR-Cas9 on-target efficiency datasets were employed. These datasets span multiple Cas9 variants, various cell types, and high-throughput screening experiments of different scales. All datasets are widely adopted in prior studies and serve as benchmark resources for modeling CRISPR-Cas9 on-target activity.
The datasets are categorized into three groups based on sample size (see Table 1):
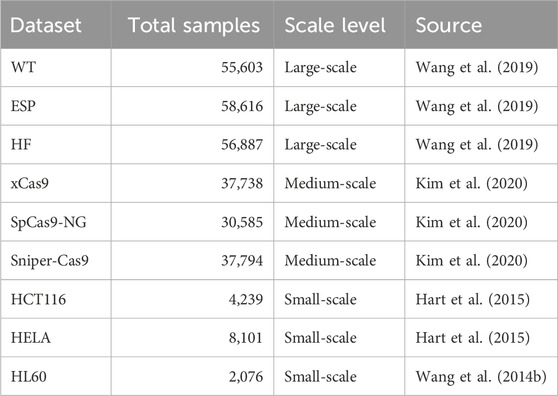
Table 1. A comprehensive summary of the CRISPR-Cas9 on-target efficiency datasets employed in this study. Detailed information, including Cas9 variants, cell lines, and experimental methods, are provided in Supplementary Table 4.
Large-scale datasets. The WT (Wang et al., 2019), ESP (Wang et al., 2019), and HF (Wang et al., 2019) datasets comprise 55,603, 58,616, and 56,887 sgRNA sequences, respectively. These datasets originate from genome-wide screening experiments conducted by Wang et al. (2019) using SpCas9 and its high-fidelity variants (e.g., eSpCas9(1.1), SpCas9-HF1). Due to their broad genomic coverage and substantial sequence diversity, they serve as ideal benchmarks for evaluating model performance and stability under large-sample conditions.
Medium-scale datasets. The xCas9 Kim et al. (2020), SpCas9-NG (Kim et al., 2020), and Sniper-Cas9 Kim et al. (2020) datasets contain 37,738, 30,585, and 37,794 sgRNA sequences, respectively. These datasets were derived from experiments summarized in Kim et al. (2020), where predictive models were built for a series of PAM-variant SpCas9 systems.
Small-scale datasets. The HCT116 (Hart et al., 2015), HELA (Hart et al., 2015), and HL60 (Wang T. et al., 2014) datasets contain 4,239, 8,101, and 2,076 sgRNA sequences, respectively. These datasets were originally compiled by Chuai et al. (2018), who integrated four independently validated CRISPR screening datasets involving 1,071 genes across multiple human cell lines. We selected three of these cell line-specific datasets—HCT116, HELA, and HL60—for our experiments. Redundant entries were removed to ensure non-overlapping, high-confidence training samples.
Each sample consists of a 23-nucleotide sequence, comprising a 20-nt sgRNA protospacer and a 3-nt PAM (e.g., NGG) located at the 3′ end. The sgRNA guides the Cas9 protein to the target site via complementary base pairing, while the PAM—though not directly involved in hybridization—is essential for Cas9 binding and cleavage. To preserve the integrity of both sequence context and cleavage specificity, the full 23-nt sgRNA-PAM sequence was used as input to our model, enabling richer representation learning.
To maintain consistency across datasets originating from heterogeneous experimental conditions, all indel frequencies were scaled to the
2.2 Sequence encoding
To comprehensively characterize sgRNA sequences and enhance the model’s capacity to capture both fine-grained local patterns and broader contextual dependencies, we propose a dual-branch sequence encoding strategy that integrates conventional one-hot encoding with semantic embeddings derived from a pre-trained RNA-FM module, as illustrated in Figure 1. This hybrid representation framework is designed to provide the model with both interpretable low-level compositional signals and enriched high-level contextual semantics, thereby enhancing predictive performance and feature expressiveness (Song X. et al., 2024).
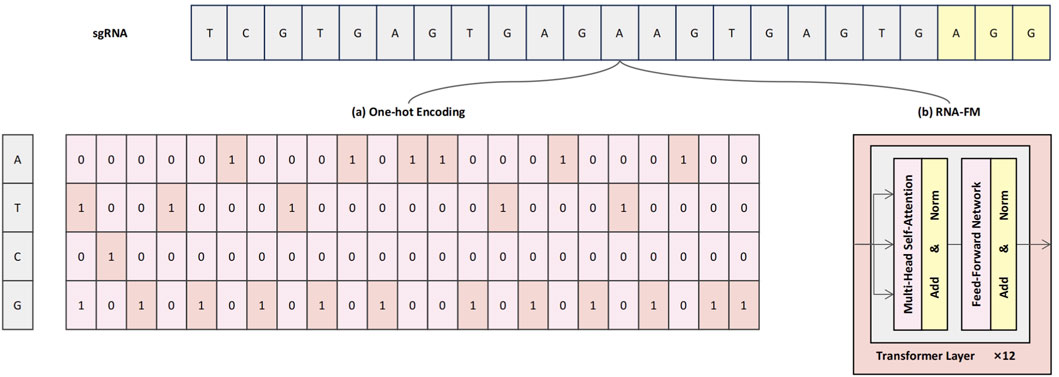
Figure 1. Schematic illustration of the dual-branch sequence encoding framework. (a) One-hot encoding represents each nucleotide as a sparse four-dimensional binary vector, preserving base identity and positional specificity. (b) RNA-FM embeddings provide high-level contextual representations, extracted from a pre-trained Transformer-based language model.
Specifically, the One-hot encoding branch (Figure 1a) preserves the exact nucleotide composition of each base in the sgRNA sequence. In this scheme, each nucleotide—A, T, G, and C—is represented by a four-dimensional One-hot vector: A = [1, 0, 0, 0], T = [0, 1, 0, 0], C = [0, 0, 1, 0], and G = [0, 0, 0, 1]. As a result, the full 23-nucleotide sgRNA sequence—comprising a 20-base protospacer and a 3-base PAM—is encoded as a
However, One-hot encoding alone lacks the capacity to capture semantic relationships or contextual dependencies among nucleotides. To address this limitation, RNA-FM embeddings are incorporated as a complementary representation (Figure 1b). RNA-FM is a Transformer-based RNA language model that was pre-trained on a large-scale corpus of over 240 million non-coding RNA sequences, capable of modeling rich contextual semantics through its core self-attention mechanisms (Huang et al., 2023). This allows the model to learn complex relationships between nucleotides that are distant in the primary sequence, thereby implicitly capturing information regarding potential secondary structures and other conserved functional motifs. During encoding, the mean vector from the final Transformer module is extracted to produce a 640-dimensional dense embedding that encapsulates these learned long-range dependencies, structural preferences, and contextual sequence features.
By integrating the explicit base identity information from One-hot encoding with the context-aware semantic representations provided by RNA-FM, the proposed dual-branch encoding scheme enables complementary feature fusion at both compositional and semantic levels. This multimodal sequence representation (Xie J. et al., 2023) allows the subsequent network modules to jointly learn localized structural features and global contextual dependencies, thereby improving the model’s ability to accurately predict sgRNA on-target editing efficiency (Xiang et al., 2021).
2.3 Model
Recent advances in deep learning have catalyzed the development of a diverse array of models designed to improve the accuracy and robustness of CRISPR-Cas9 on-target activity prediction (Elkayam and Orenstein, 2022; Bao and Liu, 2024; Zhu et al., 2024). However, existing methods still face significant challenges in modeling the high-dimensional structure of biological sequences and effectively integrating multimodal information. Specifically, the sgRNA sequence encodes hierarchical semantic features (Sun et al., 2024) that are difficult to fully represent through a single encoding scheme. In addition, local pattern recognition and global dependency modeling are often treated independently, which hinders joint optimization. Furthermore, the absence of deep semantic alignment (Ates et al., 2023) during heterogeneous modality fusion can lead to feature redundancy and semantic inconsistency, ultimately limiting predictive performance.
To address these limitations, we propose a structurally innovative dual-branch deep neural network (Luo et al., 2024), CRISPR-FMC, which integrates MSC, Transformer encoders, BiGRU, and a bidirectional cross-attention mechanism. The architecture is designed to enhance model expressiveness along three key dimensions: diversity of input representations, coordination of feature extraction, and depth of semantic fusion (Qin and Song, 2022). As illustrated in Figure 2, CRISPR-FMC consists of four primary modules: (a) Pre-training (Figure 2a, b) Multimodal Processing (Figure 2b, c) Cross-modal Interaction (Figure 2c,d) Prediction Output (Figure 2d).
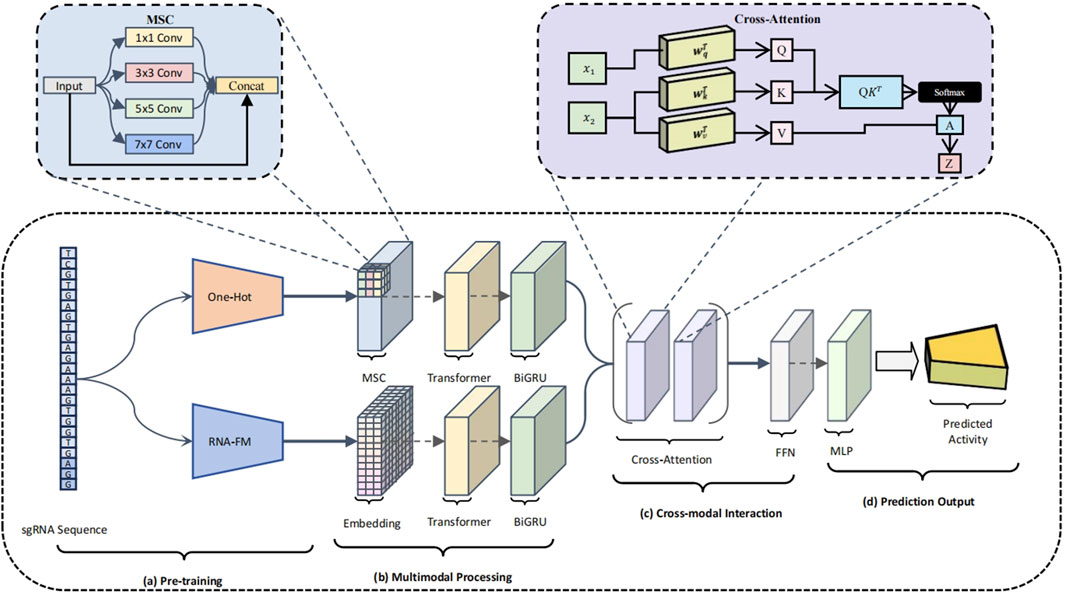
Figure 2. Schematic overview of the CRISPR-FMC model architecture. The model consists of four key modules: (a) Pre-training, where the sgRNA sequence is encoded through both one-hot vectors and RNA-FM embeddings, forming a dual-channel input that captures structural and semantic information; (b) Multimodal Processing, which applies MSC modules, Transformer encoders, and BiGRU units within each branch to extract local structural patterns and global contextual dependencies; (c) Cross-modal Interaction, which enables deep semantic fusion between modalities via a bidirectional cross-attention mechanism and further refines the joint representation through a residual FFN; (d) Prediction Output, where a MLP performs regression to estimate the on-target activity score of the input sgRNA.
To effectively capture both the structural and semantic characteristics of sgRNA sequences, CRISPR-FMC adopts a dual-branch input framework (Figure 2A). This architecture enables the model to simultaneously learn from low-level compositional features and high-level contextual representations. The first branch applies conventional one-hot encoding, converting each sgRNA into a
To effectively capture multi-scale sequence dependencies, CRISPR-FMC constructs separate feature extraction pipelines for the One-hot and RNA-FM branches (Figure 2B), each comprising an MSC module, a Transformer encoder, and a BiGRU module arranged sequentially. In the One-hot encoding branch, input sequences are first processed by a MSC module composed of parallel convolutional layers with kernel sizes of
To enable deep semantic alignment and effective integration of heterogeneous feature representations, CRISPR-FMC incorporates a bidirectional cross-attention mechanism within its cross-modal interaction module (Figure 2C). Feature embeddings from both branches are linearly projected into a shared space of dimension
The attention-refined features are further processed via residual connections and layer normalization to stabilize training and preserve gradient flow. Bidirectional outputs are concatenated along the feature dimension to form a unified representation of shape
2.4 Experimental settings
Model training and evaluation were conducted on a local high-performance computing platform with an NVIDIA GeForce RTX 2080Ti GPU (22 GB VRAM), an Intel Core i5-12400F CPU, and 32 GB RAM, running Windows 11. The model was implemented in PyTorch 1.8.1 with Python 3.9. All dependencies, including numpy, pandas, scikit-learn, matplotlib, scipy, and fair-esm, were managed via Conda and PyPI to ensure experimental reproducibility.
Based on the datasets described in Section 2.1 and summarized in Table 1, model training and evaluation were performed independently on each of the nine publicly available CRISPR-Cas9 datasets. These datasets encompass a wide range of cell types and sgRNA activity measurements across varying experimental scales. A dual-branch architecture was employed to capture multi-level sequence representations, with the corresponding encoding strategies detailed in Section 2.2.
All target values were scaled to the
The regression objective was defined based on the Log-Cosh loss (Equation 1), which combines the smooth optimization characteristics of the mean squared error (MSE) Marmolin (1986) with the robustness of the mean absolute error (MAE) Chai and Draxler (2014) against outliers. The Log-Cosh loss is defined as:
where
Compared to MSE and MAE, the Log-Cosh (Xu et al., 2020) loss behaves similarly to MSE for small residuals, providing smooth gradients, and transitions to MAE-like behavior for large residuals, thereby enhancing robustness to outliers. This property makes it well suited for biological prediction tasks that involve experimental noise or measurement variability.
Throughout training, the loss at each epoch was recorded to monitor convergence and assess training dynamics. For each cross-validation fold, the model with the lowest validation loss was selected and retained for subsequent inference.
2.5 Model effect evaluation
To comprehensively assess the predictive performance of CRISPR-FMC for sgRNA on-target activity, two regression-based evaluation metrics were employed: Spearman’s rank correlation coefficient (SCC) (Equation 3) (Zar, 2005) and Pearson correlation coefficient (PCC) (Equation 4) (Benesty et al., 2008). SCC quantifies the strength of a monotonic relationship between predicted and actual values, reflecting the model’s ability to preserve the relative ranking of sgRNA activity. In contrast, PCC measures the linear correlation between predicted scores and ground truth, indicating the model’s capacity to capture numerical agreement in regression.
Let
where
where
All evaluations were conducted on the test folds of five-fold cross-validation. Metric computation was performed with the scipy.stats module in Python to ensure standardized and reproducible evaluation.
3 Results and analysis
3.1 Model comparison
To rigorously evaluate the effectiveness of the proposed CRISPR-FMC model in predicting CRISPR-Cas9 sgRNA activity, we conducted comparative experiments against several state-of-the-art baseline methods across nine publicly available datasets (see Table 1 for details). The competing methods include CrnnCrispr (Zhu et al., 2024), CRISPR-ONT (Zhang et al., 2021), TransCrispr (Wan and Jiang, 2022), and C-RNNCrispr (Zhang et al., 2020), each representing a distinct network design paradigm and feature modeling strategy. To ensure a fair and reproducible comparison, all baseline results presented in this study were independently reproduced by our team. We utilized publicly available source code for each model and trained and evaluated them under the same five-fold cross-validation protocol used for CRISPR-FMC. Specifically, CrnnCrispr integrates CNN and BiGRU modules to capture local sequence motifs and contextual dependencies. CRISPR-ONT incorporates an attention mechanism to enhance the identification of key functional sites. TransCrispr employs a hybrid CNN–Transformer architecture to strengthen the modeling of long-range dependencies. C-RNNCrispr, on the other hand, adopts a recursive convolutional structure to facilitate hierarchical feature extraction across multiple semantic levels.
The experimental results are summarized in Figure 3. CRISPR-FMC consistently achieved the highest performance across all nine datasets. On large-scale datasets such as WT, ESP, and HF, the model attained SCC values of 0.861, 0.851, and 0.851, along with PCC scores of 0.889, 0.845, and 0.866, respectively—showcasing robust predictive capability in data-rich settings. The margins over the best-performing baseline (e.g., CrnnCrispr or TransCrispr) were moderate, typically ranging from 1.5% to 2.5%.
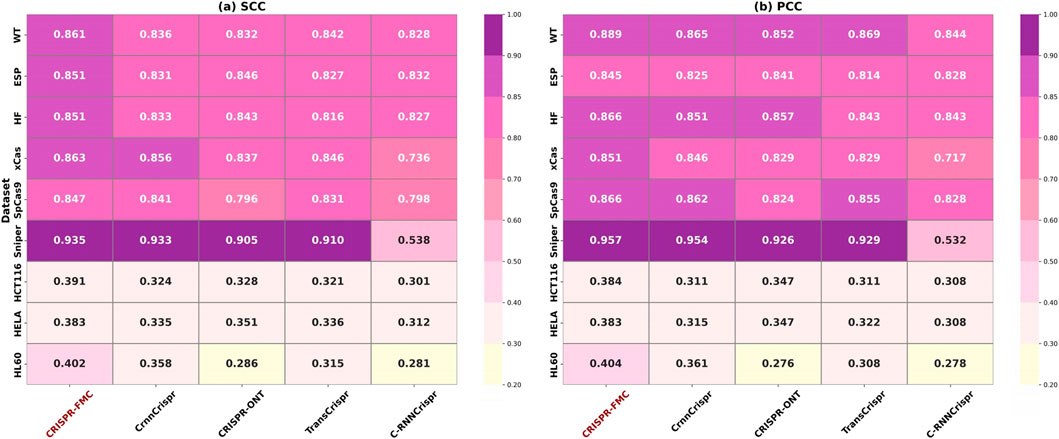
Figure 3. The color corresponding to the highest numerical values in both panels (a) SCC and (b) PCC has been adjusted to a darker shade to accurately reflect the color scale. Darker colors indicate stronger predictive performance. CRISPR-FMC consistently achieves superior results across most datasets in both metrics.
Notably, on the Sniper dataset, CRISPR-FMC achieved the highest performance with SCC = 0.935 and PCC = 0.957, outperforming the second-best model (CrnnCrispr) which scored 0.933 and 0.954, respectively. This result demonstrates the model’s effectiveness on variant-specific Cas9 datasets.
The model also performed well on small-scale datasets such as HCT116, HELA, and HL60. On the HL60 dataset, CRISPR-FMC reached SCC = 0.402 and PCC = 0.404, surpassing CrnnCrispr, which achieved SCC = 0.358 and PCC = 0.361. These results confirm that CRISPR-FMC maintains strong generalization ability even under limited data availability.
The overall performance advantage can be attributed to the model’s dual-branch architecture and multimodal feature fusion, combining One-hot encoded features and RNA-FM embeddings. The design enables the capture of both low-level sequence motifs and high-level contextual dependencies through MSC, BiGRU, and cross-attention modules, thereby enhancing prediction robustness across different datasets.
3.2 Ablation experiments
To systematically assess the contribution of each architectural component within the CRISPR-FMC model to sgRNA activity prediction, we performed a series of comprehensive ablation experiments across nine publicly available CRISPR-Cas9 datasets. In each experiment, a single submodule was removed or replaced, while the remaining model architecture and training protocol were kept unchanged. This controlled design ensures that any observed performance variation can be directly attributed to the specific structural modification. The key components examined are summarized in Table 2, and the selected datasets (Table 1) encompass a wide range of sequence complexities and sample sizes.
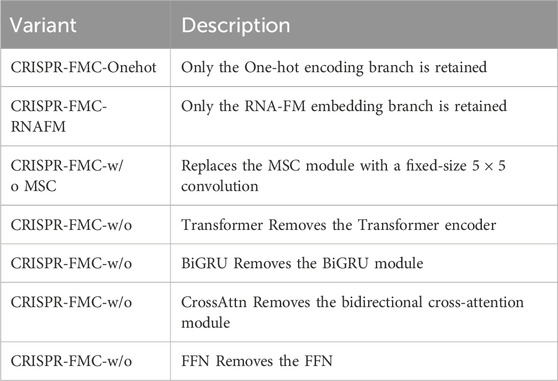
Table 2. Summary of CRISPR-FMC Variants in the Ablation Study. The average SCC and PCC values are computed across all nine CRISPR-Cas9 datasets to evaluate the impact of each architectural component.
As shown in Figure 4, the complete CRISPR-FMC model achieves the highest average SCC and PCC values of 0.709 and 0.716, respectively. Excluding either input encoding branch leads to notable performance degradation. Retaining only one-hot encoding still yields reduced performance (SCC: 0.654, PCC: 0.663), while relying solely on RNA-FM embeddings results in the most significant drop (SCC: 0.541, PCC: 0.553). These findings demonstrate that the two encoding schemes capture complementary aspects of sgRNA sequence information and jointly contribute to a robust feature representation.
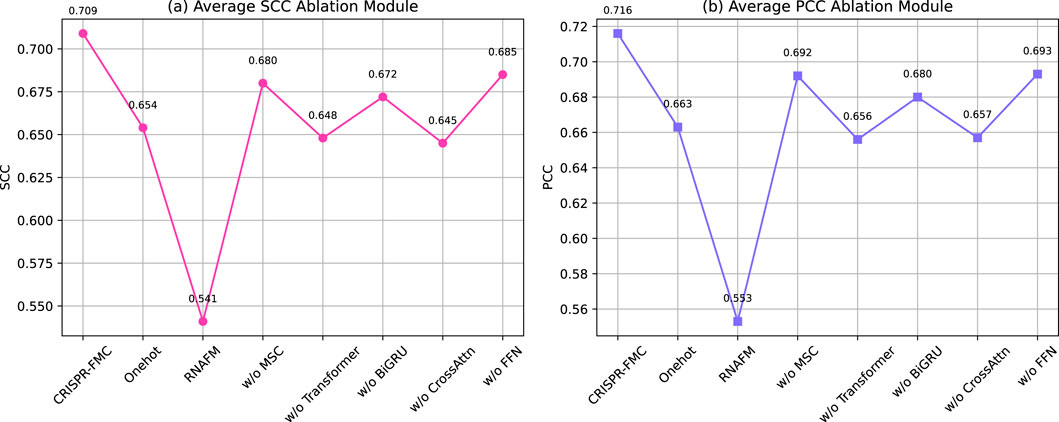
Figure 4. Ablation study evaluating the contribution of individual structural components to the predictive performance of the CRISPR-FMC model. The left panel displays the (a) Average SCC Ablation Module, while the right panel shows the (b) Average PCC Ablation Module. The results highlight the importance of multi-level sequence encoding and hierarchical feature extraction for accurate prediction.
In the sequence modeling pipeline, ablation of the MSC module (replaced with a fixed-size
The cross-modal interaction mechanism is further tested via an additional variant, oCrossAttn, which retains only the bidirectional cross-attention module while removing other fusion components. This version yields lower average scores (SCC: 0.645, PCC: 0.657), indicating that although cross-attention facilitates inter-modality alignment, it cannot fully replace the complete multimodal fusion framework. Lastly, eliminating the FFN module results in SCC and PCC scores of 0.685 and 0.693, respectively, suggesting its role in refining the fused representation through high-order nonlinear transformation.
Taken together, these ablation results validate the functional necessity and complementary contributions of each structural component in CRISPR-FMC. The observed performance declines confirm the value of multi-level encoding, hierarchical feature extraction, and semantic fusion.
To further validate these findings, we conducted a detailed per-dataset ablation analysis and found that the contribution of each encoding branch varies with dataset size. Specifically, the one-hot branch dominates performance on large datasets (e.g., WT, ESP, HF), where abundant data allows the model to learn directly from sequence. In contrast, the RNA-FM branch is more effective on small datasets (e.g., HCT116, HELA, HL60), acting as a regularizer and injecting prior biological knowledge. Detailed SCC and PCC results are visualized in Supplementary Figures 6, 7, and the encoding contribution trend is highlighted in Supplementary Figure 6.
3.3 Generalization capability of the model
To evaluate the transferability and generalization capability of the CRISPR-FMC model across different data distributions, we conducted a cross-dataset migration experiment. Specifically, the model was independently trained on two large-scale datasets, WT and ESP, and subsequently evaluated on a small-scale dataset (HL60). The performance was assessed based on SCC and PCC. This setup simulates real-world application scenarios in which the model must generalize from well-annotated source data to novel cellular contexts characterized by limited annotations or high noise levels (Wang et al., 2020).
As shown in Table 3, CRISPR-FMC achieves superior and stable cross-dataset generalization under both training settings. When trained on the WT dataset, the model attains an SCC of 0.468 and a PCC of 0.459 on HL60, outperforming competing models such as CrnnCrispr (SCC: 0.456, PCC: 0.438) and TransCrispr (SCC: 0.448, PCC: 0.432). When trained on the ESP dataset, CRISPR-FMC still maintains top performance with an SCC of 0.448 and a PCC of 0.432, again surpassing baselines such as CRISPR-ONT (SCC: 0.432, PCC: 0.421) and C-RNNCrispr (SCC: 0.434, PCC: 0.429).
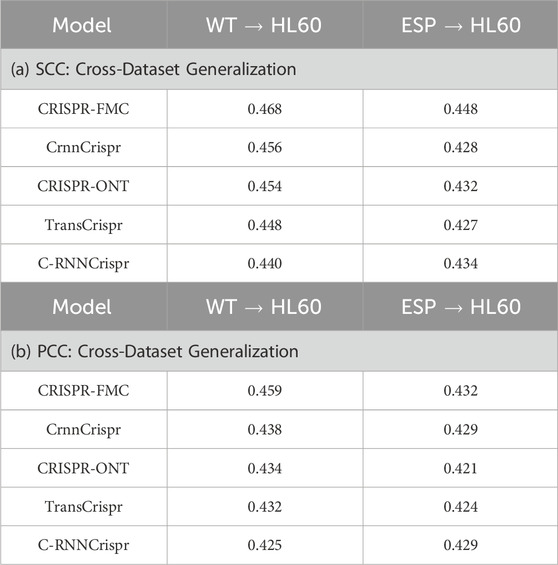
Table 3. Cross-dataset migration performance of different models trained on the WT or ESP datasets and tested on HL60. (a) SCC: Cross-Dataset Generalization; (b) PCC: Cross-Dataset Generalization.
Overall, CRISPR-FMC consistently achieves the highest SCC and PCC values on HL60 across both training sources, highlighting its robustness in cross-cell line migration tasks (Wang C. et al., 2014). Notably, HL60 represents a challenging test case due to its small sample size and high noise levels, yet CRISPR-FMC maintains strong predictive accuracy and correlation.
This strong generalization performance can be attributed to the architectural design of CRISPR-FMC. The dual-branch encoding scheme—combining one-hot and RNA-FM representations—enables the model to simultaneously capture local structural patterns and global semantic context. The integration of multi-scale convolution (MSC), BiGRU modules, and a bidirectional cross-attention mechanism further enhances feature expressiveness and semantic alignment, collectively improving the model’s adaptability to distributional shifts and noise.
3.4 Model interpretability
To gain a deeper understanding of the decision-making process of the CRISPR-FMC model, we conducted a base substitution experiment to analyze the positional sensitivity of the model’s predicted cleavage efficiency. In this experiment, the original nucleotide at each position was sequentially replaced with each of the other three bases (A, T, C, G), and the resulting changes in the model’s predictions were recorded. The impact of each substitution was quantified by calculating the Z-score (Ma et al., 2016), which reflects the magnitude of change in the predicted output.
Figure 5 illustrates the effect of different base substitutions on the Z-score. The model exhibits the highest sensitivity in the PAM-proximal region (positions 16–20), where substitutions consistently lead to large fluctuations in the Z-score. For instance, positions 18 and 20 show particularly strong effects. Other positions, such as position three and 14, also demonstrate notable sensitivity, indicating their contribution to the model’s predictions. This observation aligns with established biological mechanisms, especially regarding the binding affinity of Cas9 to the target DNA. Previous studies, including those by Doench et al. (2016) and Duan et al. (2014), have reported that mutations in the PAM-proximal region can substantially influence Cas9 cleavage activity, reflecting the sequence-specific binding nature of the Cas9 protein.
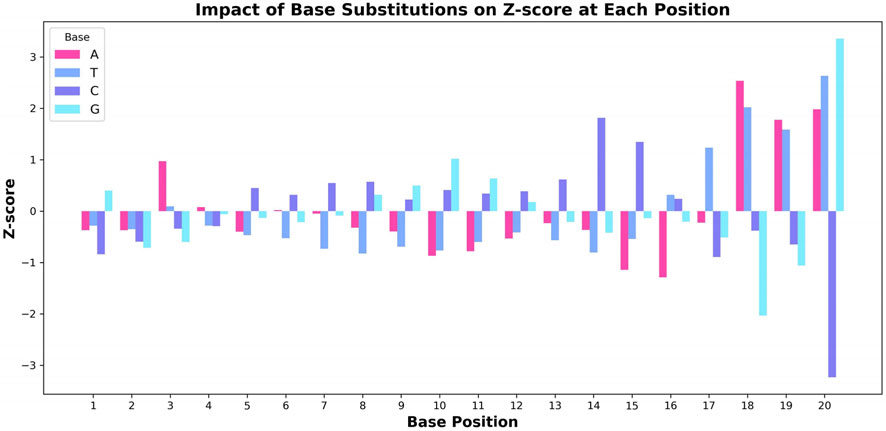
Figure 5. Effect of base substitution on the Z-score at each nucleotide position. The changes in Z-score resulting from substituting each base (A, T, C, G) in the sgRNA sequence are visualized, highlighting the pronounced impact of positions associated with the PAM region on predicted editing efficiency.
Through this analysis, we have gained insights into how the CRISPR-FMC model prioritizes specific nucleotide positions when predicting cleavage efficiency. This understanding provides valuable guidance for optimizing sgRNA sequence design and offers a targeted strategy for enhancing CRISPR editing performance.
4 Contribution and discussion
This study presents CRISPR-FMC, a dual-branch deep neural network designed to accurately predict sgRNA cleavage efficiency in the CRISPR-Cas9 system. The model integrates semantic embeddings from the pre-trained RNA language model RNA-FM with conventional One-hot encoding, alongside a combination of MSC, BiGRU, and Transformer-based feature extraction modules. A bidirectional cross-attention mechanism and a FFN are incorporated to enable multimodal semantic alignment and global feature fusion. Extensive evaluations on nine publicly available datasets demonstrate that CRISPR-FMC consistently achieves higher performance scores than state-of-the-art models in both SCC and PCC metrics, exhibiting particular strengths in low-data settings and cross-dataset generalization.
The effectiveness of CRISPR-FMC stems from a synergistic and, as highlighted by our ablation studies, context-dependent integration of its components. The dual-branch encoding strategy demonstrates complementary strengths that are crucial for the model’s versatility. On large- and medium-scale datasets, performance is heavily driven by the One-hot encoding branch, which excels at capturing explicit nucleotide patterns when sufficient data is available. In contrast, on small datasets, the contribution of the RNA-FM branch becomes vital. The pre-trained RNA-FM embeddings provide rich contextual priors learned from millions of sequences, acting as an effective regularizer that guides the model toward generalizable features and prevents overfitting in data-scarce scenarios. Therefore, the architecture does not merely combine features, but adaptively leverages them—relying on robust pattern extraction in data-rich environments while harnessing pre-trained biological knowledge to ensure reliability when data is limited. This adaptability to varying data scales is a key strength, enhancing the model’s practical utility for a wide range of bioinformatics applications. Despite its promising performance, several limitations remain. A key limitation is our use of RNA-FM for sgRNA embeddings. We acknowledge the important biological nuance that the sgRNA functions within a Cas9-sgRNA ribonucleoprotein complex to target DNA, rather than as a free, unbound non-coding RNA. The target recognition process is therefore critically dependent on the properties of the target DNA sequence and its local chromatin context. This is consistent with our ablation study results, where the performance gains from RNA-FM were more modest on large-scale datasets, suggesting that DNA-level features are likely stronger determinants of cleavage efficiency. Furthermore, the scope of this study is confined to on-target activity, while predicting and minimizing off-target effects remains an equally critical challenge for safe and effective genome editing. In addition, the current web platform lacks support for high-throughput input and batch prediction, highlighting opportunities for improving deployment scalability.
Therefore, a significant future direction will be to shift the modeling focus toward the biological context of the DNA target site. Future work will focus on integrating DNA-specific language models (e.g., Nucleotide Transformer, HyenaDNA) to generate embeddings from the target DNA sequence, including flanking genomic regions. This approach would allow the model to capture features more central to the biological mechanism, such as DNA accessibility and sequence complementarity, potentially leading to substantial gains in predictive power. Beyond this, the dual-branch architecture of CRISPR-FMC could be adapted to address off-target predictions. By training the model on datasets of guide-mismatch sequence pairs, the framework could learn to predict cleavage efficiency at unintended sites, representing another promising extension of this work. We will also continue to explore incorporating other multi-omics data and uncertainty quantification techniques to improve prediction robustness. These extensions aim to provide more comprehensive and reliable decision support for sgRNA design across diverse biological contexts.
Data availability statement
The datasets and source code generated for this study are publicly available at https://github.com/xx0220/CRISPR-FMC. A lightweight web interface is also included to allow users to test CRISPR-Cas9 activity predictions by inputting sequences and selecting target datasets. Further inquiries can be directed to the corresponding authors.
Author contributions
CL: Investigation, Methodology, Writing – original draft, Conceptualization. JL: Writing – review and editing, Formal Analysis, Visualization. QZ: Resources, Data curation, Writing – review and editing, Supervision. HF: Writing – review and editing, Supervision, Conceptualization, Funding acquisition, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the National Natural Science Foundation of China (32471860); the scientific research project of the education department of Zhejiang province (Y202352570).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgeed.2025.1643888/full#supplementary-material
References
Alzubaidi, L., Zhang, J., Humaidi, A. J., Al-Dujaili, A., Duan, Y., Al-Shamma, O., et al. (2021). Review of deep learning: concepts, cnn architectures, challenges, applications, future directions. J. big Data 8, 53–74. doi:10.1186/s40537-021-00444-8
Ates, G. C., Mohan, P., and Celik, E. (2023). Dual cross-attention for medical image segmentation. Eng. Appl. Artif. Intell. 126, 107139. doi:10.1016/j.engappai.2023.107139
Bao, C., and Liu, F. (2024). “Prediction of crispr on-target effects via deep learning,” in Proceedings of the 1st workshop on NLP for science (NLP4Science), 9–15.
Benesty, J., Chen, J., and Huang, Y. (2008). On the importance of the pearson correlation coefficient in noise reduction. IEEE Trans. Audio, Speech, Lang. Process. 16, 757–765. doi:10.1109/tasl.2008.919072
Bochman, M. L., Paeschke, K., and Zakian, V. A. (2012). Dna secondary structures: stability and function of g-quadruplex structures. Nat. Rev. Genet. 13, 770–780. doi:10.1038/nrg3296
Chai, T., and Draxler, R. R. (2014). Root mean square error (rmse) or mean absolute error (mae)? arguments against avoiding rmse in the literature. Geosci. model Dev. 7, 1247–1250. doi:10.5194/gmd-7-1247-2014
Chen, C.-F. R., Fan, Q., and Panda, R. (2021). “Crossvit: cross-attention multi-scale vision transformer for image classification,” in Proceedings of the IEEE/CVF international conference on computer vision, 357–366. doi:10.48550/arXiv.2204.00300
Chen, J., Hu, Z., Sun, S., Tan, Q., Wang, Y., Yu, Q., et al. (2022). Interpretable rna foundation model from unannotated data for highly accurate rna structure and function predictions. arXiv Prepr. arXiv:2204.00300. doi:10.48550/arXiv.2204.00300
Chuai, G., Ma, H., Yan, J., Chen, M., Hong, N., Xue, D., et al. (2018). Deepcrispr: optimized crispr guide rna design by deep learning. Genome Biol. 19, 80–18. doi:10.1186/s13059-018-1459-4
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using crispr/cas systems. Science 339, 819–823. doi:10.1126/science.1231143
Dhruv, P., and Naskar, S. (2020). “Image classification using convolutional neural network (cnn) and recurrent neural network (rnn): a review,” in Proceedings of ICMLIP 2019, 367–381. doi:10.1007/978-981-15-1884-3_34
Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., et al. (2016). Optimized sgrna design to maximize activity and minimize off-target effects of crispr-cas9. Nat. Biotechnol. 34, 184–191. doi:10.1038/nbt.3437
Doudna, J. A., and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. doi:10.1126/science.1258096
Duan, J., Lu, G., Xie, Z., Lou, M., Luo, J., Guo, L., et al. (2014). Genome-wide identification of crispr/cas9 off-targets in human genome. Cell Res. 24, 1009–1012. doi:10.1038/cr.2014.87
Elkayam, S., and Orenstein, Y. (2022). Deepcristl: deep transfer learning to predict crispr/cas9 functional and endogenous on-target editing efficiency. Bioinformatics 38, i161–i168. doi:10.1093/bioinformatics/btac218
Haeussler, M., Schönig, K., Eckert, H., Eschstruth, A., Mianné, J., Renaud, J.-B., et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide rna selection tool crispor. Genome Biol. 17, 148–12. doi:10.1186/s13059-016-1012-2
Han, K., Xiao, A., Wu, E., Guo, J., Xu, C., and Wang, Y. (2021). Transformer in transformer. Adv. neural Inf. Process. Syst. 34, 15908–15919. Available online at: https://proceedings.neurips.cc/paper_files/paper/2021/file/854d9fca60b4bd07f9bb215d59ef5561-Paper.pdf
Hart, T., Chandrashekhar, M., Aregger, M., Steinhart, Z., Brown, K., Macleod, G., et al. (2015). High-Resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526. doi:10.1016/j.cell.2015.11.015
Hosmer Jr, D. W., Lemeshow, S., and Sturdivant, R. X. (2013). Applied logistic regression. John Wiley and Sons.
Huang, Z., Liang, M., Qin, J., Zhong, S., and Lin, L. (2023). “Understanding self-attention mechanism via dynamical system perspective,” in Proceedings of the IEEE/CVF international conference on computer vision, 1412–1422.
Jin, Y., Qin, C., Zhang, Z., Tao, J., and Liu, C. (2022). A multi-scale convolutional neural network for bearing compound fault diagnosis under various noise conditions. Sci. China Technol. Sci. 65, 2551–2563. doi:10.1007/s11431-022-2109-4
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-rna–guided dna endonuclease in adaptive bacterial immunity. science 337, 816–821. doi:10.1126/science.1225829
Kim, H. K., Kim, Y., Lee, S., Min, S., Bae, J. Y., Choi, J. W., et al. (2019). Spcas9 activity prediction by deepspcas9, a deep learning–based model with high generalization performance. Sci. Adv. 5, eaax9249. doi:10.1126/sciadv.aax9249
Kim, N., Kim, H. K., Lee, S., Seo, J. H., Choi, J. W., Park, J., et al. (2020). Prediction of the sequence-specific cleavage activity of cas9 variants. Nat. Biotechnol. 38, 1328–1336. doi:10.1038/s41587-020-0537-9
Li, C., Zou, Q., Li, J., and Feng, H. (2025). Prediction of crispr-cas9 on-target activity based on a hybrid neural network. Comput. Struct. Biotechnol. J. 27, 2098–2106. doi:10.1016/j.csbj.2025.05.001
Liu, Y., Guan, Q., Zhao, X., and Cao, Y. (2018). “Image forgery localization based on multi-scale convolutional neural networks,” in Proceedings of the 6th ACM workshop on information hiding and multimedia security, 85–90.
Liu, Q., He, D., and Xie, L. (2019). Prediction of off-target specificity and cell-specific fitness of crispr-cas system using attention boosted deep learning and network-based gene feature. PLoS Comput. Biol. 15, e1007480. doi:10.1371/journal.pcbi.1007480
Luo, Y., Chen, Y., Xie, H., Zhu, W., and Zhang, G. (2024). Interpretable crispr/cas9 off-target activities with mismatches and indels prediction using bert. Comput. Biol. Med. 169, 107932. doi:10.1016/j.compbiomed.2024.107932
Ma, J., Köster, J., Qin, Q., Hu, S., Li, W., Chen, C., et al. (2016). Crispr-do for genome-wide crispr design and optimization. Bioinformatics 32, 3336–3338. doi:10.1093/bioinformatics/btw476
Marmolin, H. (1986). Subjective mse measures. IEEE Trans. Syst. man, Cybern. 16, 486–489. doi:10.1109/tsmc.1986.4308985
Meier, J. A., Zhang, F., and Sanjana, N. E. (2017). Guides: sgrna design for loss-of-function screens. Nat. methods 14, 831–832. doi:10.1038/nmeth.4423
Moreno-Mateos, M. A., Vejnar, C. E., Beaudoin, J.-D., Fernandez, J. P., Mis, E. K., Khokha, M. K., et al. (2015). Crisprscan: designing highly efficient sgrnas for crispr-cas9 targeting in vivo. Nat. methods 12, 982–988. doi:10.1038/nmeth.3543
Qin, H., and Song, Y. (2022). “Reinforced cross-modal alignment for radiology report generation,” in Findings of the Association for Computational Linguistics: ACL 2022, 448–458.
Roberts, A., and Barrangou, R. (2020). Applications of crispr-cas systems in lactic acid bacteria. FEMS Microbiol. Rev. 44, 523–537. doi:10.1093/femsre/fuaa016
Saini, A., Stoica, P., Babu, P., and Arora, A. (2024). Min-max framework for majorization-minimization algorithms in signal processing applications: an overview. Found. Trends® Signal Process. 18, 310–389. doi:10.1561/2000000129
Sermanet, P., and LeCun, Y. (2011). “Traffic sign recognition with multi-scale convolutional networks,” in The 2011 international joint conference on neural networks (IEEE), 2809–2813.
Shaheen, Z., Wohlgenannt, G., and Filtz, E. (2020). Large scale legal text classification using transformer models. arXiv Prepr. arXiv:2010.12871. doi:10.48550/arXiv.2010.12871
Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., et al. (2014). Genome-scale crispr-cas9 knockout screening in human cells. Science 343, 84–87. doi:10.1126/science.1247005
Song, X., Hua, Z., and Li, J. (2024a). Context spatial awareness remote sensing image change detection network based on graph and convolution interaction. IEEE Trans. Geoscience Remote Sens. 62, 1–16. doi:10.1109/tgrs.2024.3357524
Song, Z., Lu, Q., Xu, H., Zhu, H., Buckeridge, D., and Li, Y. (2024b). “Timelygpt: extrapolatable transformer pre-training for long-term time-series forecasting in healthcare,” in Proceedings of the 15th ACM international conference on bioinformatics, computational biology and health informatics, 1–10.
Sun, J., Guo, J., and Liu, J. (2024). Crispr-m: predicting sgrna off-target effect using a multi-view deep learning network. PLOS Comput. Biol. 20, e1011972. doi:10.1371/journal.pcbi.1011972
Taud, H., and Mas, J.-F. (2017). “Multilayer perceptron (mlp),” in Geomatic approaches for modeling land change scenarios (Springer), 451–455.
Vishwanathan, S., and Murty, M. N. (2002). Ssvm: a simple svm algorithm. Proc. 2002 Int. Jt. Conf. Neural Netw. IJCNN’02 (Cat. No. 02CH37290) 3, 2393–2398. doi:10.1109/ijcnn.2002.1007516
Wan, Y., and Jiang, Z. (2022). Transcrispr: transformer based hybrid model for predicting crispr/cas9 single guide rna cleavage efficiency. IEEE/ACM Trans. Comput. Biol. Bioinforma. 20, 1518–1528. doi:10.1109/TCBB.2022.3201631
Wang, C., Chowdhury, S., Driscoll, M., Parent, C. A., Gupta, S., and Losert, W. (2014a). The interplay of cell–cell and cell–substrate adhesion in collective cell migration. J. R. Soc. Interface 11, 20140684. doi:10.1098/rsif.2014.0684
Wang, T., Wei, J. J., Sabatini, D. M., and Lander, E. S. (2014b). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. doi:10.1126/science.1246981
Wang, D., Zhang, C., Wang, B., Li, B., Wang, Q., Liu, D., et al. (2019). Optimized crispr guide rna design for two high-fidelity cas9 variants by deep learning. Nat. Commun. 10, 4284. doi:10.1038/s41467-019-12281-8
Wang, J., Xiang, X., Bolund, L., Zhang, X., Cheng, L., and Luo, Y. (2020). Gnl-scorer: a generalized model for predicting crispr on-target activity by machine learning and featurization. J. Mol. cell Biol. 12, 909–911. doi:10.1093/jmcb/mjz116
Wong, N., Liu, W., and Wang, X. (2015). Wu-crispr: characteristics of functional guide rnas for the crispr/cas9 system. Genome Biol. 16, 218–8. doi:10.1186/s13059-015-0784-0
Xiang, X., Corsi, G. I., Anthon, C., Qu, K., Pan, X., Liang, X., et al. (2021). Enhancing crispr-cas9 grna efficiency prediction by data integration and deep learning. Nat. Commun. 12, 3238. doi:10.1038/s41467-021-23576-0
Xie, J., Liu, M., and Zhou, L. (2023a). Crispr-ote: prediction of crispr on-target efficiency based on multi-dimensional feature fusion. IRBM 44, 100732. doi:10.1016/j.irbm.2022.07.003
Xie, S., Zhang, H., Guo, J., Tan, X., Bian, J., Awadalla, H. H., et al. (2023b). Residual: transformer with dual residual connections. arXiv Prepr. arXiv:2304.14802. doi:10.48550/arXiv.2304.14802
Xu, X., Li, J., Yang, Y., and Shen, F. (2020). Toward effective intrusion detection using log-cosh conditional variational autoencoder. IEEE Internet Things J. 8, 6187–6196. doi:10.1109/jiot.2020.3034621
Yan, J., Liu, J., Yu, Y., and Xu, H. (2021). Water quality prediction in the luan river based on 1-drcnn and BiGRU hybrid neural network model. Water 13, 1273. doi:10.3390/w13091273
Yu, H., Yang, H., Sun, W., Yan, Z., Yang, X., Zhang, H., et al. (2024). Plantrna-fm: an interpretable rna foundation model for exploration functional rna motifs in plants. bioRxiv 2024–06. doi:10.1101/2024.06.24.600509
Zar, J. H. (2005). “Spearman rank correlation,” in Encyclopedia of biostatistics. 7th Edn. Oxford, England: Wiley.
Zhang, G., Dai, Z., and Dai, X. (2020). C-rnncrispr: prediction of crispr/cas9 sgrna activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 18, 344–354. doi:10.1016/j.csbj.2020.01.013
Zhang, G., Zeng, T., Dai, Z., and Dai, X. (2021). Prediction of crispr/cas9 single guide rna cleavage efficiency and specificity by attention-based convolutional neural networks. Comput. Struct. Biotechnol. J. 19, 1445–1457. doi:10.1016/j.csbj.2021.03.001
Zheng, Y., Zou, Q., Li, J., and Yang, Y. (2025). Crispr-mfh: a lightweight hybrid deep learning framework with multi-feature encoding for improved crispr-cas9 off-target prediction. Genes 16, 387. doi:10.3390/genes16040387
Keywords: CRISPR-Cas9, RNA-FM, deep learning, on-target activity, SgRNA
Citation: Li C, Li J, Zou Q and Feng H (2025) CRISPR-FMC: a dual-branch hybrid network for predicting CRISPR-Cas9 on-target activity. Front. Genome Ed. 7:1643888. doi: 10.3389/fgeed.2025.1643888
Received: 09 June 2025; Accepted: 08 August 2025;
Published: 29 August 2025.
Edited by:
Ji-Ping Wang, Northwestern University, United StatesReviewed by:
Qingyang Zhang, University of Arkansas, United StatesAdriano Setti, Sapienza University of Rome, Italy
Copyright © 2025 Li, Li, Zou and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Li, bGlqaWFuMDEyMEB6YWZ1LmVkdS5jbg==; Hailin Feng, aGxmZW5nQHphZnUuZWR1LmNu