 Hyungjun Park
Hyungjun Park Takeshi Kuroha
Takeshi Kuroha Hiroaki Saika
Hiroaki Saika Masaharu Kuroda
Masaharu Kuroda Hitoshi Yoshida
Hitoshi Yoshida- Institute of Agrobiological Science, National Agriculture and Food Research Organization (NARO), Tsukuba, Ibaraki, Japan
Introduction: Copy number variation (CNV) is one of the crucial elements among genomic structural variations that span plant breeding. However, its impact on agricultural traits has remained elusive.
Methods: We modulated CNVs using two genome-editing technologies, CRISPR/Cas9 and Cas3, along with their verification methods in rice to elucidate the effect of CNVs and further harness to improve relevant agronomic traits.
Results: The addition of cytosine extension to the conventional single-guide RNA and its combination with Cas9 generated a frameshift mutation in parts of the OsGA20ox1 gene copies, substantially modifying its CNV. Phenotypes of the copy number variants revealed OsGA20ox1 copy number as a determinant of seedling vigor in rice. The Cas3 nuclease, which induces large-scale deletions, effectively decreased the copy number of the OsMTD1 gene. We verified the copy number of each gene by combining droplet digital polymerase chain reaction (ddPCR), Sanger sequencing, and bioinformatics tools.
Discussion: Altogether, the two technologies are expected to lay the foundation for new approaches to plant breeding by controlling CNV.
1 Introduction
Long-read DNA sequencing technologies have been recently applied to construct high-quality genome sequences and databases and identify structural and copy number variations (CNVs) from multiple perspectives (Fuentes et al., 2019; Qin et al., 2021; Wei et al., 2021; Shang et al., 2022; Wang et al., 2023; Yu et al., 2023). CNV refers to a functional gene with different numbers between cultivars; thus, CNVs represent a form of structural variation denoting specific DNA sequences arising from deletions, duplications, or amplifications. The CNVs have provided insights into plant breeding, including domestication, adaptation, and evolution (Lye and Purugganan, 2019; Wang et al., 2023; Wilson et al., 2025). However, the impact of CNV on agronomic traits remains unclear.
Studies have reported that certain genes, including OsGA20ox1 and OsMTD1, which are possibly associated with agronomic traits, harbor CNVs (Alonge et al., 2020; Liu et al., 2020; Qin et al., 2021; Wang et al., 2023; Wilson et al., 2025). OsGA20ox1 encodes gibberellin (GA) 20-oxidase, a crucial enzyme involved in the penultimate stage of GA production in rice; it is associated with plant stature as well (Oikawa et al., 2004). This gene is a major quantitative locus affecting the phenotypes such as seedling vigor and grain number per panicle (Abe et al., 2012; Yano et al., 2012; Wu et al., 2016). In addition, it contributes to rice’s tolerance to alkali soil and heat stress and is therefore significant for the next-generation Green Revolution (Guo et al., 2025). OsGA20ox1 possesses CNVs, particularly in japonica rice (Qin et al., 2021; Wang et al., 2023), which have been implicated in several agronomic traits significant for rice breeding. In addition, OsMTD1, a gene potentially linked to rice plant architecture and tiller number, has CNVs (Liu et al., 2015; Liu et al., 2020).
Agricultural traits are influenced by a wide range of factors, making it challenging to evaluate the effect of CNVs of a single gene on agronomic traits using cultivars with different genetic backgrounds. The role of CNVs of OsGA20ox1 on agronomic traits, depending on trait comparison of cultivars with varying genetic backgrounds and overexpression experiments has been previously reported (Wang et al., 2023). Studying agricultural traits and CNVs in the same genetic background can reveal the mechanism by which CNVs influence traits. Establishing NILs with different CNVs is time-consuming, whereas using genome editing for CNV variants with the same genetic background could be beneficial. However, precise modification of CNVs through genome editing remains challenging for the following reasons. Because multiple copies of genes with CNVs arise from the duplication of the genomic region, these copies share mostly identical nucleotide sequences. In particular, a length of 10–20 kb before and after the gene is mostly identical (herein referred to as “gene block”) (Lye and Purugganan, 2019; Qin et al., 2021). This makes it difficult to independently edit one of the duplicated genes using the existing genome editing method with Cas9 nuclease due to its high efficacy and targeted specificity (Wang and Doudna, 2023). Several Cas9 nuclease-independent genome editing technologies have been developed recently (Liu G. et al., 2022). Thus, controlling CNV using different genome-editing technologies is highly anticipated (Shen et al., 2017).
We report technical strategies on two model cases to modify the CNVs and overcome the current limitations of the existing Cas9 nuclease. We modified CNVs by complementing the existing Cas9 method, adding cytosine (Kawamata et al., 2023), and using Cas3 (Dolan et al., 2019; Morisaka et al., 2019), a completely different type of nuclease. Furthermore, we propose genotype detection guides for each approach and effective progeny selection strategies in later generations. We successfully produced dosage-reduced mutant alleles in rice using these strategies. We believe our findings will set practical examples for the effective utilization of the increasingly intensive CNV data from different crops.
2 Materials and methods
2.1 Target gene selection and CNV identification
A total of 1,003 genes showing CNVs between cv. Nipponbare (NIP) and cv. Koshihikari (KOSH) were extracted from the Rice Resource Center database (https://ricerc.sicau.edu.cn/) (Ricerc, 2019; Qin et al., 2021; Supplementary Table S1). We investigated the function of extracted genes using the Rice Gene Index database (https://riceome.hzau.edu.cn/) (Riceome, 2021; Yu et al., 2023) and existing studies. We selected genes OsGA20ox1 (Os03g0856700 or LOC_Os03g63970) and OsMTD1 (Os08g0441300 or LOC_Os08g34249), which are possibly closely related to rice agronomic traits, to confirm the effect of CNV modification in rice (Supplementary Table S2). Next, nucleotide information from each variety was extracted from the Rice Resource Center, annotated, and compared using CLC Main Workbench 25 (Qiagen) and GenomeMatcher to determine the CNV structure of the two genes (Ohtsubo et al., 2008).
2.2 Plant materials and culture conditions
Oryza sativa L. cv. Nipponbare and cv. Koshihikari were used to generate genome-edited rice. All rice seeds were surface sterilized in 70% ethanol for 5 min and in a 50% sodium hypochlorite solution for 30 min, rinsed with sterile water, placed on Petri dishes with half-strength solid Murashige and Skoog (MS) medium, and subsequently cultured for at least 14 days in a growth chamber. Afterward, the seedlings were moved to soil in black vinyl pots (9 cm in diameter) until they reached maturity. Plants were grown in the greenhouse under LD conditions, which included 10 h of darkness at 25 °C and 14 h of light at 28 °C.
2.3 Generation of genome-edited rice
Genome editing was conducted using the vector pZNH2GTRU6 (manuscript in progress) through recognition of the NGG as the PAM sequence. This vector, derived from plasmid pZ2028 (Kuroda et al., 2010), consists of single guide RNAs (sgRNAs) for editing target genes driven by the rice U6-2 noncoding RNA promoter, Streptococcus pyogenes Cas9 driven by the modified rice polyubiquitin promoter, and the hygromycin phosphotransferase (HPT) gene driven by the nopaline synthase promoter. The sgRNAs were inserted into the vectors using the BbsI site by ligation.
The vector pZD202-Cas3 (Cas758+Cas1163+hpt) (Saika et al., 2025) was used for genome editing through recognition of AAG as the PAM sequence. This vector contained the HPT gene driven by the nopaline synthase promoter, Cas3 and other Cas genes driven by the modified maize polyubiquitin promoter and CaMV 35S promoter, and CRISPR RNAs (crRNAs) for editing target genes driven by the rice U6-2 noncoding RNA promoter. The crRNAs were inserted into the vectors using the SpeI site by In-Fusion HD cloning (TaKaRa Bio Inc., Shiga, Japan). The vector pOsU6Cas3gRNA rev was used for the crRNA expression vector and is resistant to kanamycin. The crRNAs were inserted into the vectors using the BsaI site by ligation.
The target sequences for sgRNA were selected using CHOPCHOP v3 (Labun et al., 2019) and DeepSpCas9 (Kim et al., 2019). The crRNA sequences were designed manually; sgRNA and crRNA sequences are listed in Supplementary Table S3. Primers and oligonucleotides for construction are listed in Supplementary Table S4.
Agrobacterium tumefaciens-mediated transformation was used to introduce transgenes into rice cv. Nipponbare or cv. Koshihikari (Toki, 1997; Ozawa, 2012). Hygromycin-resistant plants were transferred to soil in black plastic pots (9 cm in diameter) until maturity. The genomic DNA was extracted from elongated leaves of all material plants using the Qiagen DNeasy® Plant Mini Kit (Qiagen, Hilden, Germany) to verify genome editing.
2.4 PCR, droplet digital PCR (ddPCR), and long-range PCR (LR-PCR)
Polymerase chain reactions (PCRs) were performed with a total volume of 10 μL to investigate the Cas9-induced mutation on OsGA20ox1 in both Nipponbare and Koshihikari. The PCR mixture contained 5 μL of Emerald Amp® PCR Master Mix (Takara), 1 μL of forward primer, 1 of μL reverse primer, 1 μL of genomic DNA, and 2 μL of betaine. The mixture was thermally cycled in the following conditions: pre-denaturing at 94 °C for 1 min, 40 cycles of PCR at 98 °C for 10 s, annealing at 64 °C for 30 s, and extension at 72 °C for 1 min, and final extension at 72 °C for 5 min, following which the reaction was stopped at 4 °C. The reaction solution was used for direct Sanger sequencing after confirming the band size using gel electrophoresis.
Cycle sequencing PCR was conducted with a total volume of 10 μL using the BigDyeTM Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA, United States), following the manufacturer’s instructions. After the PCR, all samples were purified by the ethylenediaminetetraacetic acid (EDTA)/ethanol precipitation method. Next, the pellet was dissolved in Hi-diTM Formamide (Thermo Fisher Scientific) and incubated at 95 °C for 2 min. The reaction solutions were used for the sequence analysis by ABI PRISM® 3130xl genetic analyzer (Thermo Fisher Scientific).
Droplet digital PCR (ddPCR) was used to detect the Cas3-induced CNV change in OsMTD1 in Nipponbare and the Cas9-mediated CNV change in OsGA20ox1 in Koshihikari. It was performed in a reaction volume of 22 μL using a QX200 Auto DG Droplet Digital PCR platform (Bio-Rad, Hercules, CA, United States). The PCR mixture contained 11 μL of the ddPCR Supermix for probes with no dUTP, 1 μL of upstream primer, 1 μL of downstream primer, 0.6 μL of the probe, 1.1 μL of HEX (OsACT1, PrimePCR Probe Assay: ACT1), 6.3 μL of ddH2O, and 1 μL of DNA (20 ng treated with Hind III). The PCR mixture and Droplet generation oil were mixed, and a droplet was generated. The droplet emulsion was thermally cycled in the following conditions: pre-denaturing at 94 °C for 5 min, 40 cycles of PCR at 94 °C for 30 s, and at 55 or 59 °C for 1 min, and melting at 98 °C for 10 min, and finally, the reaction was finished at 4 °C. The completed reaction was used for ddPCR absolute quantification analysis. QuantaLife software was used to determine the nucleic acid copy number. The ratio of the target gene to the reference gene was calculated, and each ratio was converted as a relative value based on the value of the wild-type with one copy as 1.
Next, long-range PCR was performed with a total volume of 20 μL to identify the Cas3-induced deletion size on OsMTD1 in Nipponbare. The PCR mixture contained 10 μL of KOD One® PCR Master Mix (TOYOBO, Osaka, Japan), 1 μL of the forward primer, 1 μL of the reverse primer, 1 μL of the genomic DNA, and 7 μL of distilled water. The mixture was thermally cycled in the following conditions: pre-denaturation at 94 °C for 5 min, 35 cycles of PCR at 98 °C for 10 s, annealing at 60 °C for 5 s, and extension at 68 °C for 2 min 10 s. The reaction was finished at 4 °C. The reaction solution was used for direct Sanger sequencing after confirming the band size by gel electrophoresis. Primers and probes for all PCR types are listed in Supplementary Table S4.
2.5 Genotyping by sanger sequencing and TIDE
The genotyping of genome-edited rice lines of the OsGA20ox1 gene was confirmed by Sanger sequencing. Sanger sequencing chromatograms were analyzed visually and using the TIDE software (Brinkman et al., 2014) to determine the mutation patterns at each target site (Supplementary Figure S1).
2.6 Allelic variant identification via TA cloning and chi-square goodness-of-fit test
Allelic variants were identified to verify the copy number of OsGA20ox1 PCR products amplified using primers GA20ox1-F, R (Supplementary Table S4) were purified using QIAquick PCR Purification Kit (Qiagen) and subsequently cloned into a pGEM®-T Easy Vector Systems (Promega, WI, United States). In total, 117 independent plasmid clones were selected and sequenced according to the manufacturer’s instructions. The allele ratios of the sequence results were used as the observed values (Oi), and the expected allele ratios for each CNV were used as the expected values (Ei). A chi-square goodness of fit test (χ2) was performed to verify the most appropriate CNV.
3 Results
3.1 Genome editing strategies for CNV modification
Two rice genes (OsGA20ox1 and OsMTD1) and two cultivars (Nipponbare and Koshihikari) were selected as models for CNV modification. Re-annotation was performed after acquiring the nucleotide information of each gene from each cultivar to verify the information of CNVs for these genes from the database and previous studies (Liu et al., 2020; Qin et al., 2021; Wang et al., 2023). In addition, we confirmed the multiplication status of each gene block using PCR analysis following the method described by Liu et al. (2020) to cross-validate sequence data from the database (Supplementary Figure S2).
Re-annotation and sequence comparison revealed that both the genes of interest and the genomic region including the genes (gene blocks) about 13–37 kb were multiplicated. Specifically, the length of the gene block, including OsGA20ox1, was approximately 37 kb, and the nucleotide sequences of individual OsGA20ox1 copies from each block were completely identical. In addition, the nucleotide sequences of gene blocks, including OsGA20ox1, shared a similarity of 99.94%. Moreover, gene blocks, including OsMTD1, which was 13 kb long, shared completely identical sequences (Supplementary Figure S3; Supplementary Table S5).
We proposed two strategies for modifying CNVs of these two model genes with substantial nucleotide similarity between gene blocks (Figure 1), i.e., the addition of cytosine extension to sgRNA in CRISPR/Cas9 (Strategy I) and CRISPR/Cas3 (Strategy II). Because the native CRISPR/Cas9 system introduces mutations at high efficiency in rice, most of the genome-edited plants possess bi-allelic mutations for each gene copy (complete knock-outs), making it challenging to obtain partially mutated plants. This implies that native CRISPR/Cas9 could not be efficiently used to obtain genome-edited plants with partially reduced CNVs. Therefore, we employed the first strategy to reduce Cas9 efficiency by adding 30 cytosine bases to the 5′-end of the sgRNA (Figure 2A), because the results of previous studies reported that adding 30 cytosines produced the most dramatic effect (Kawamata et al., 2023). This method affected the concentration of ideal sgRNA-Cas9 complexes and enabled the partial cause of mutations in CNVs by intentionally reducing editing efficiency without affecting target specificity. The second tactic induced large-scale deletions by recruiting Cas3 to the flanking region of the target gene block. Because the deletion orientation of Cas3 is mostly guided from the crRNAs to the PAM (Dolan et al., 2019; Morisaka et al., 2019), we designed crRNAs that caused Cas3-induced deletion toward the target CNV.
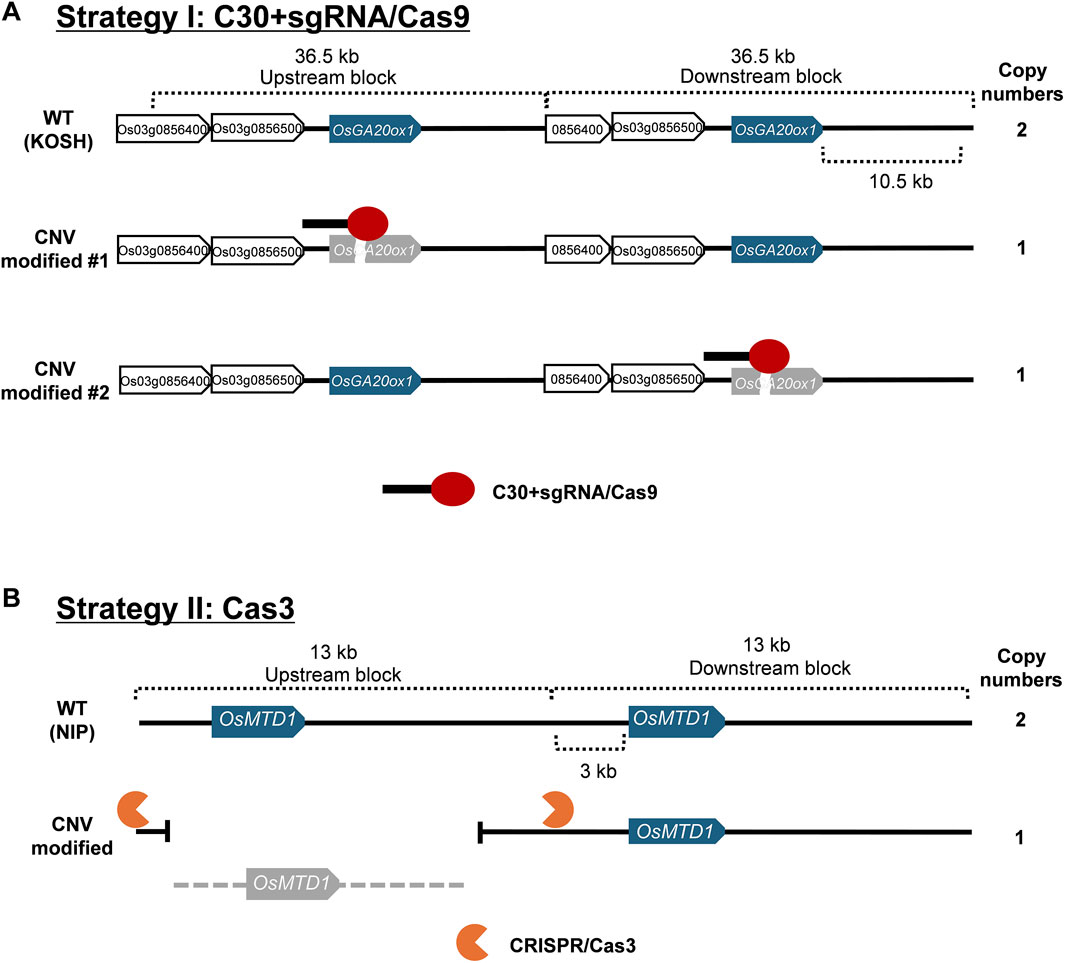
Figure 1. Schematic strategies for genome editing-mediated CNV modification. Information on nucleotide sequences at the expected multiplicated structures was extracted from the Rice Resource Center and analyzed manually. (A) Genome editing strategy I uses C30+sgRNA and Cas9 (red ovals with solid black bars). The gene block including OsGA20ox1 is approximately 36.5 kb in length. Blue boxes indicate the target OsGA20ox1 genes. White boxes show the other annotated genes in gene blocks. Gray boxes refer to genome-edited dysfunctional genes. (B) Genome editing strategy II using CRISPR/Cas3. The gene block including OsMTD1 is approximately 13 kb in length. Blue boxes indicate the target OsMTD1 gene. Gray boxes refer to dysfunctional genes due to a large-scale deletion.
![Diagram illustrating a gene editing experiment. Panel A shows the structure of [C30+sgRNA] with 30 cytosine extensions. Panels B and C display bar charts of the number of functional alleles in NIP and KOSH lines at generation T0. Panel D shows allele configurations across generations, indicating mutant and functional alleles in different configurations. Homozygous lines with reduced copy numbers are highlighted. Panel E presents bar charts for KOSH T1 lines (CR1 to CR4), indicating the ratio of lines with varying numbers of functional alleles. Blue represents functional alleles; gray, mutant alleles.](https://www.frontiersin.org/files/Articles/1652950/fgeed-07-1652950-HTML-r1/image_m/fgeed-07-1652950-g002.jpg)
Figure 2. Visualization of genome editing by C30+sgRNA/Cas9. (A) Schematic diagram of adding cytosine base extensions to sgRNA. (B,C) Comparison of genome-editing efficiency targeted to the OsGA20ox1 gene using sgRNA and C30+sgRNA in Nipponbare (B) and Koshihikari (C) T0 generation. Different colors imply the number of functional alleles after genome editing. Orange boxes imply partially modified mutant lines. (D) Strategy for efficient selection of progenies with desired copy numbers. The blue box indicates a functional allele. The gray box depicts a mutant allele generated by genome editing. The red rectangles show the efficient region of progeny selection. The orange box indicates the homozygous CNV-modified genotypes. (E) Results of progeny selection in Koshihikari T1 generation, according to the selection strategy. Different colors refer to the numbers of functional alleles and genotypes. Orange boxes refer to homozygous lines.
3.2 Genome editing strategy I: use of sgRNA with 30 cytosine extensions
Genome editing targeting the OsGA20ox1 exon was performed in Nipponbare to determine whether the sgRNA with 30 additional cytosine nucleotides (designated as “C30+sgRNA”) could effectively reduce genome editing in plants (Figure 2A). Because Nipponbare has one copy of the OsGA20ox1 gene, and bi-allelic mutations were predominantly induced using the native CRISPR/Cas9 system in rice, we examined whether mono-allelic mutations were induced using the C30+sgRNA by genotyping the T0 generation plants. The genome editing efficiency was reduced when using the C30+sgRNA/Cas9 compared to cases using the normal sgRNA (Figure 2B), which was consistent with the results of a previous study in mammalian cells (Kawamata et al., 2023). Specifically, the proportion of bi-allelic mutant lines decreased from 70% to 40%, and the proportion of mono-allelic and non-mutated lines increased, accordingly.
Next, genome editing with C30+sgRNA/Cas9 was applied to Koshihikari, which contains two copies of the OsGA20ox1 gene (Qin et al., 2021; Wang et al., 2023), using the same vectors. The genotyping of genome-edited lines revealed that the CR19 line exhibited a segregation pattern of −3, −1, and +1 bp indel mutations in the ratio of 1:2:3, as determined using TIDE analysis (Supplementary Figure S4A). This ratio was unexpected in genomes containing two CNV copies in Koshihikari. In addition, we identified other T0 line harboring more than four mutant alleles (Supplementary Figure S4A). The maximum number of distinct mutant alleles is expected to be four when the CNV is two in Koshihikari, which is inconsistent with the above results. Therefore, the CNV of OsGA20ox1 was thought to be three or more in Koshihikari. Next, we performed a quantitative analysis using ddPCR to re-examine the copy numbers of OsGA20ox1 in the Koshihikari and Nipponbare genomes. The relative value was approximateley threefold higher in Koshihikari than in Nipponbare (Supplementary Figure S4B).
For a more accurate verification of the TIDE results, we performed allelic variant identifications using CR19. The target region of OsGA20ox1 was amplified, and resulting PCR product was cloned. We sequenced 117 independent clones, the proportion of which was consistent with that of TIDE (Supplementary Figure S4C). C in the “+1 insertion” tab in Supplementary Figure S4A was not detected, possibly because of misinterpretation by TIDE. Next, we applied the chi-square goodness-of-fit test to compare multiple CNV hypotheses (CNV = 2, 3, and 4) against the observed allele frequencies derived from cloning and sequencing. The p-value (p = 0.8568) when the CNV of OsGA20ox1 was 3 was the most plausible (Supplementary Table S6).
Subsequently, the TIDE results were re-analyzed assuming that Koshihikari had three copies of the target gene, resulting in a consistent interpretation. Similarly, the genome editing efficiency of C30+sgRNA/Cas9 was reduced in Koshihikari (Figure 2C). Compared to the normal sgRNA, the percentage of C30+sgRNA/Cas9-mediated genome editing lines with bi-allelic mutations in all three copies of the target genes significantly decreased from 50% to 10%. Consequently, the diversity of lines with different numbers of functional alleles was increased, indicating the effectiveness of this strategy for modifying CNVs in rice.
3.3 Efficient screening of CNV-controlled lines by allele type diversification
The presence of multiple functional allele types complicated the genotyping of subsequent generations in genome editing of genes with more than three copies of CNV. The goal in such a scenario is to obtain progeny lines with one or two CNVs reduced (having two or four functional alleles). Thus, we proposed an effective progeny screening method for such cases (Figure 2D). First, the lines in the middle of the mutation patterns (having two to four functional alleles) were excluded from further screening. Because it is unknown how they will be inherited in future generations, further verification of the next generations is required. The most efficient method to obtain CNV-modified lines is to use lines with one or five functional alleles. These lines can be possibly used to obtain homozygous CNV-modified lines in the next-generation with a theoretical segregation ratio of approximately 25%. We screened T1 according to this strategy and obtained the expected rate (22%–44%) of CNV-modified homozygote lines (Figure 2E).
Next, we compared the seedling vigor of T1 homozygote lines with different copy numbers to examine the effects of copy number differences on OsGA20ox1. The leaf sheath length and the copy number of OsGA20ox1 were positively correlated (Figure 3). Thus, the CNV of OsGA20ox1 may be a determinant of the seedling vigor in rice. Considering previous reports demonstrating a correlation between the copy number and gene expression (Qin et al., 2021), this phenotype might have been caused by an altered OsGA20ox1 expression through its CNV modifications.
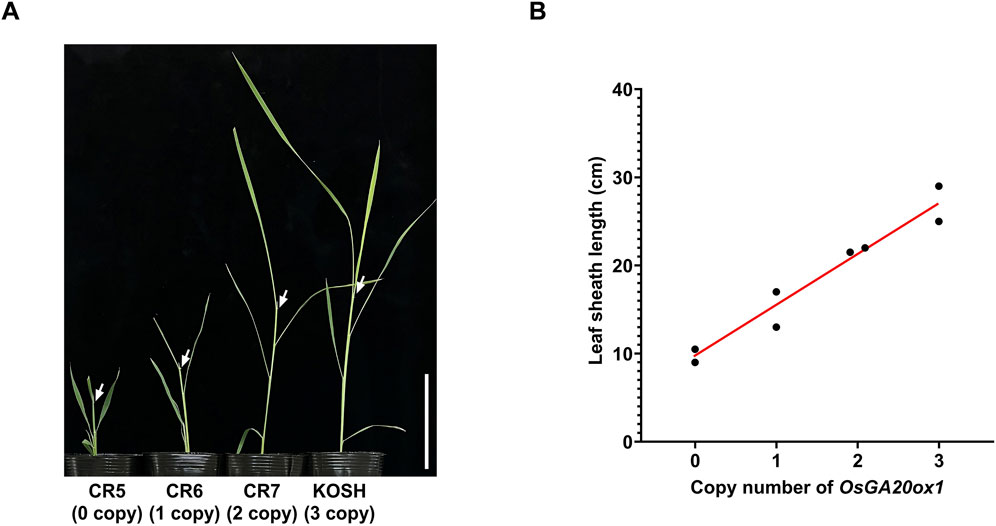
Figure 3. Seedling vigor of OsGA20ox1 CNV-modified T1 homozygous lines. (A) Representative phenotypes of each CNV-modified line 5 weeks after sowing. CR5: All the copies are dysfunctional, CR6: One CNV is functional, CR7: Two CNVs are functional, KOSH: Three CNVs are functional. White arrows refer to the leaf sheath location of each plant. Scale bar (white solid bar) = 10 cm. (B) Scatter plot of copy numbers (X-axis) and length of leaf sheath (Y-axis) of each plant. Leaf sheath length was measured 5 weeks after sowing. The red line is a trend line between copy numbers and leaf sheath length.
3.4 Establishment of a quantification method for screening CNV-modified lines by mismatching primer
Although TIDE-based genotype evaluation, using Sanger sequencing results, is rapid, cost-effective, and simple, it is susceptible to the amplification pattern of PCR or the accuracy of sequence result wavelength data. Therefore, we established a method to more accurately quantify genome-editing patterns. Because the SpCas9-induced mutation pattern is predominantly limited to small-scale mutations around 3 bp upstream of the PAM sequence, ddPCR technology using mismatch primers was designed. The forward primer was designed to overlap with the DNA cleavage site in the target sequence with an intentional mismatch in the third base from the 3′-end of the primer (Supplementary Figures S5A–E).
A general PCR conducted before ddPCR revealed no band amplified using the mismatch primers in the 0-functional allele line. A faint band appeared in the low-mutant allele number lines (Supplementary Figure S5F). Each plant with a distinct number of functional alleles, as inferred from the TIDE analysis, was further analyzed using ddPCR. Thus, the stepwise relative value according to each functional allele was detected in ddPCR (Figure 4). The wild-type Koshihikari and T0 plants recovered with six functional alleles in the T1 generation (CR8 and CR9) demonstrated around thrice the relative value of Nipponbare, or T0 plants having two functional alleles (CR16 and CR6). T0 plants having 0-functional alleles (CR19 and CR20) did not react in ddPCR.
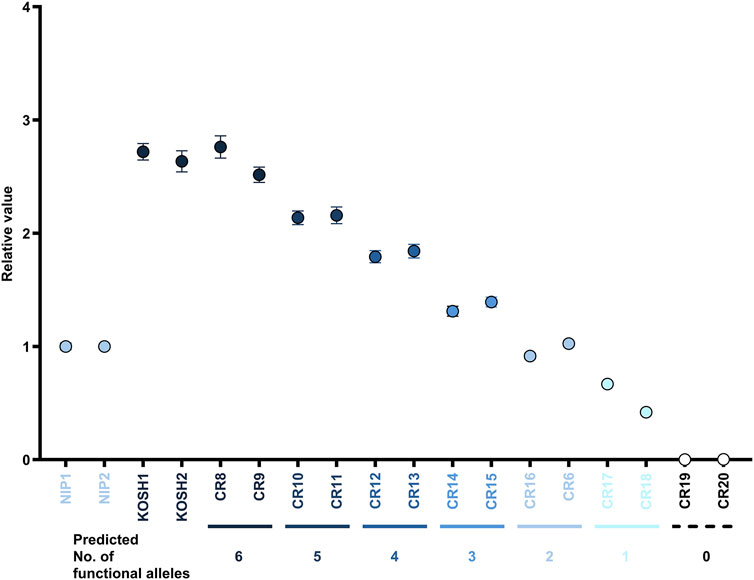
Figure 4. Quantification of unedited functional alleles in C30+sgRNA/Cas9-mediated T0 and T1 genome-edited lines using mismatch primer ddPCR. The expected number of functional alleles by interpretation of TIDE is shown below the X-axis. The Y-axis denotes the relative copy numbers in each sample corresponding to that of NIP. Error bars are calculated by Poisson’s law.
3.5 Genome editing strategy II: induction of large deletions via Cas3 nuclease
Next, we wanted to determine whether inducing Cas3 nuclease-mediated large-scale deletions would be effective in modifying CNV, we designed experiments in which a single sequence on the outside of a gene block was used to induce deletions of the upstream block inward (Figure 1B). Cas3-mediated genome editing was conducted on Nipponbare, which has two copies of the target gene OsMTD1, and CNV was surveyed using ddPCR. Nipponbare and IR64 cultivars were used as positive controls with two copies and one copy of OsMTD1, respectively. Among the 46 T0 generation lines, seven lines, accounted for 15.2%, demonstrating a decreased relative value of the target gene compared with Nipponbare (Figure 5A). The ddPCR results demonstrated that the T0 generation was divided into three groups containing 1. no deletions reaching the OsMTD1 region or no genome editing occurrence (blue), 2. heterozygous deletions occurrence (green), and 3. homozygous deletion occurrence (purple).
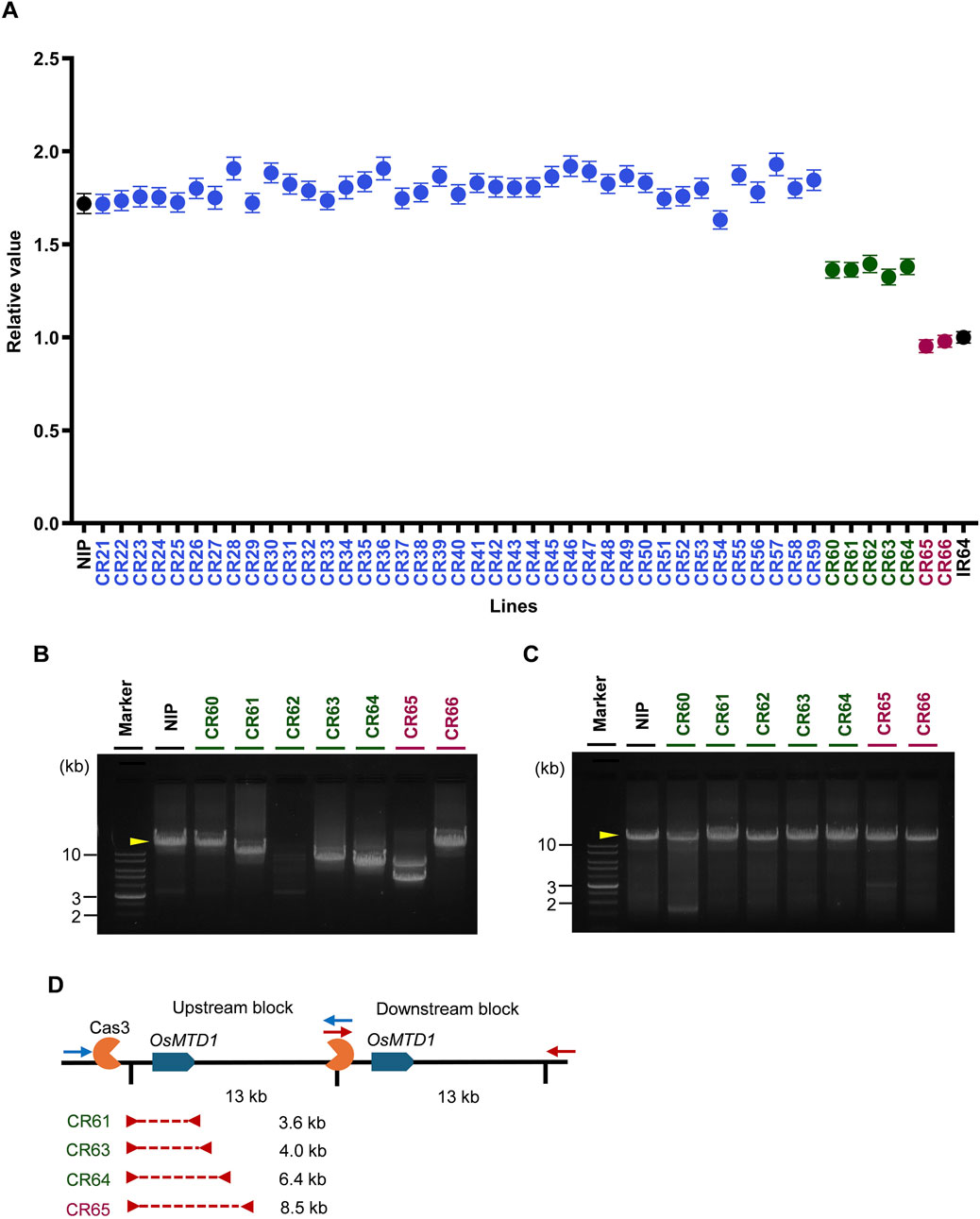
Figure 5. Evaluation of genome editing generated by CRISPR/Cas3. (A) Quantification of the numbers of OsMTD1 alleles using ddPCR in Cas3-introduced Nipponbare lines at T0 generation. The X-axis shows sample names. The Y-axis denotes the relative values of each sample corresponding to those of IR64. Error bars are calculated by the Poisson’s law. Black: Nipponbare (two copies) and IR64 (one copy). Blue: No genome editing occurred, or deletions did not reach the probe position in the OsMTD1 region. Green: CNV was modified heterozygously. Purple: CNV was modified homozygously. (B) Gel electrophoresis analysis of the OsMTD1 upstream block in CNV-modified T0 lines. Yellow arrowhead, fragment size amplified in wild-type. (C) Gel electrophoresis analysis of OsMTD1 downstream block in CNV-modified T0 lines. Yellow arrowhead, fragment size amplified in wild-type. (D) Schematic diagram showing the deletion size in the OsMTD1 CNV-modified T0 lines. Blue boxes indicate the target OsMTD1 genes. Blue and red arrows denote long-range PCR primers for upstream and downstream blocks. The Red dashed line refers to the deletion site.
Next, the deletion size was investigated by long-range PCR in each T0 CNV-modified line. We first conducted long-range PCR to detect the deletions in the OsMTD1 upstream block (Figure 5B). We could not detect deletion at least on gel electrophoresis in lines CR60, CR62, and CR66 because of almost the same size of PCR products (CR60 and CR66) or no amplification (CR62). In contrast, a decrease in the PCR product size was detected in lines CR61, CR63, CR64, and CR65 on gel electrophoresis. No deletion was observed in the OsMTD1 downstream block of all lines (Figure 5C). Sanger sequencing of PCR products revealed that CR61 and CR63 had 3.6 and 4.0 kb deletions that barely reached the OsMTD1 region in the upstream block. CR64 had a slightly longer deletion of 6.4 kb, whereas CR65 had the longest deletion of 8.5 kb (Figure 5D).
Subsequently, T1 generation lines presumed to have a decreased CNV in the T0 generation were genotyped to confirm the homozygosity. First, CR62, one of the heterozygous (green) T0 groups, was segregated into three groups in the next-generation; we obtained approximately 30% of the CNV-modified lines (purple) (Figure 6A). The target band was not observed in the LR-PCR results from the CR62 T1 individuals (Supplementary Figure S6), indicating that the binding site of the forward primer was deleted (Supplementary Figure S6B). Next, CR65, a CNV-decreased homozygous T0 group (purple), was confirmed to have a fixed CNV-decreased homozygous genotype in all the lines (Figure 6B). Finally, we could not obtain CNV-modified homozygous lines with the desired number of CNV (CNV = 1) for CR66, which was another CNV-decreased homozygous group (purple), unlike CR65 (Figure 6C). Taken together, homozygous genome-edited lines with desired CNVs can be selected using ddPCR-based genotyping of T1-segregating individuals. No clear phenotypic changes were observed in the OsMTD1 CNV modified lines of the T0 and T1 generations, including the homozygous knockout lines (CR66-17/18/19/20).
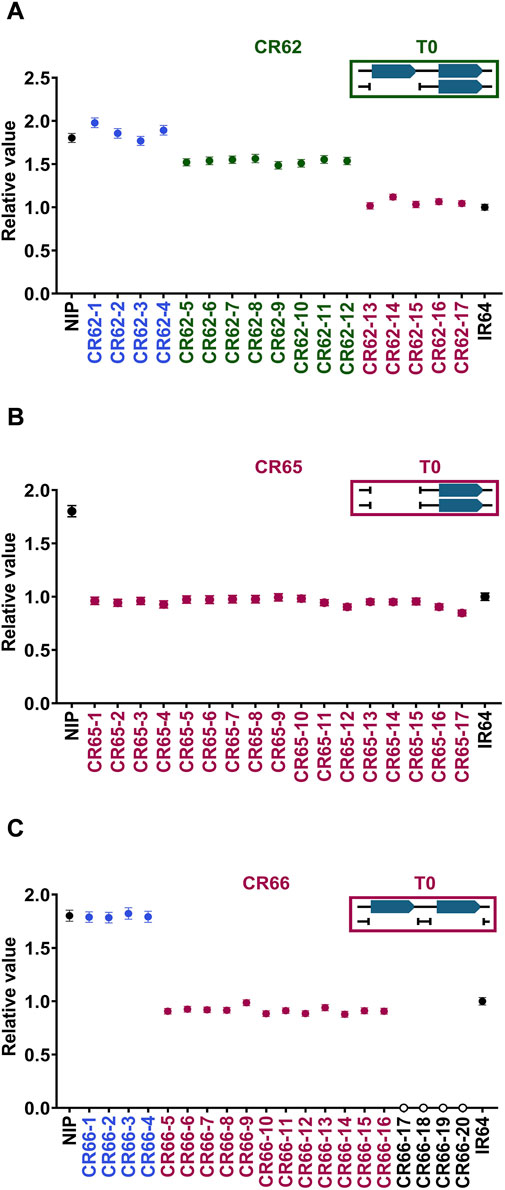
Figure 6. Segregation patterns of OsMTD1 CNV-modified T1 individuals analyzed by ddPCR. Segregation of T1 progenies of CR62 (A), CR65 (B), and CR66 (C) T0 lines, respectively. Black: Nipponbare (two copies) and IR64 (one copy). Blue: no genome editing occurred, or deletions did not reach the probe position in the OsMTD1 region. Green: CNVs were modified heterozygously. Purple: CNVs were modified homozygously. Yellow: Two CNV sites were deleted homozygously. Boxes in the upper right are schematic diagrams of the expected genotypes of each line at T0 generation.
4 Discussion
Despite the growing interest in CNVs, the mechanism controlling their numbers has rarely been studied (Li et al., 2017; Shen et al., 2017). We demonstrated that CNV modification of each target gene, especially by partial reduction of functional gene copies, can be successfully achieved with genome editing technologies. We proposed two independent strategies, i.e., I. controlling Cas9 activity by adding 30 cytosine extensions to sgRNA, and II. inducing large-scale deletions using Cas3 nuclease. The findings provide significant insights into future research directions for modulating and optimizing CNV through genome editing.
4.1 CNV modification using emerging genome-editing technologies
The SpCas9 in rice displays high activity, predominantly causing bi-allelic mutations rather than mono-allelic mutations for each target gene. This is also the case for other plants (Li et al., 2018; Naim et al., 2018; Wolabu et al., 2020; Lu and Tian, 2022; Yuan et al., 2024). Because several CNVs are caused by the multiplication of genome regions including the numbers of genes with almost identical nucleotide sequences, obtaining genome-edited plants with a partially reduced copy number of the target gene is challenging. This problem was solved by reducing and optimizing the activity of Cas9. Kawamata et al. (2023) reported that the addition of cytosine between 15 and 30 to the 5′-end of sgRNA caused mono-allelic mutations, instead of bi-allelic mutations, in animal cells. We demonstrated the effect of controlling the genome-editing frequency of Cas9 by adding 30 cytosines to sgRNA in plants, and its application to modify CNV in rice. This technique would be possible in other plants as well. Moreover, such mosaic editing may be useful in studying genes causing lethality or sterility as recessive homozygous (bi-allelic) mutations. A previous report on rice suggested that editing efficiency can be controlled by adjusting the number of nucleotides in the sgRNA, and this approach is also expected to facilitate the efficient modification of CNV (Liu X. J. et al., 2022).
Cas3 is characterized by easy induction of large-scale deletions compared to Cas9 (Dolan et al., 2019; Morisaka et al., 2019). We successfully modified CNV using Cas3 in the second strategy, targeting OsMTD1, which has a 13 kb gene block (Figure 5). Although it did not delete the entire 13 kb gene block, the ddPCR experiment demonstrated a reduced copy number of OsMTD1, being a good example for modulating the CNV of the target gene. Approximately 15% of the Cas3-mediated genome-edited plants displayed altered CNVs (Figure 5A), which was similar to the efficiency obtained previously (Li et al., 2023). This appears to be sufficient efficiency to obtain enough numbers of CNV-modified lines.
Each strategy has its advantages and limitations in several aspects (Table 1). An appropriate strategy depending on the purpose of the study and the structure of the CNV of the target gene should be adopted (Figure 7).
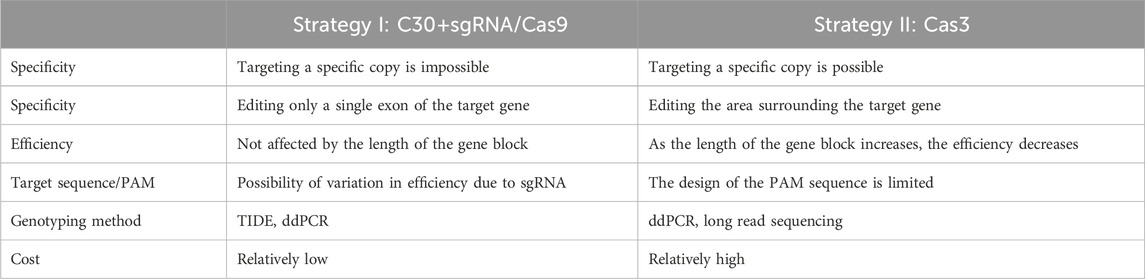
Table 1. Advantages and limitations of each strategy.
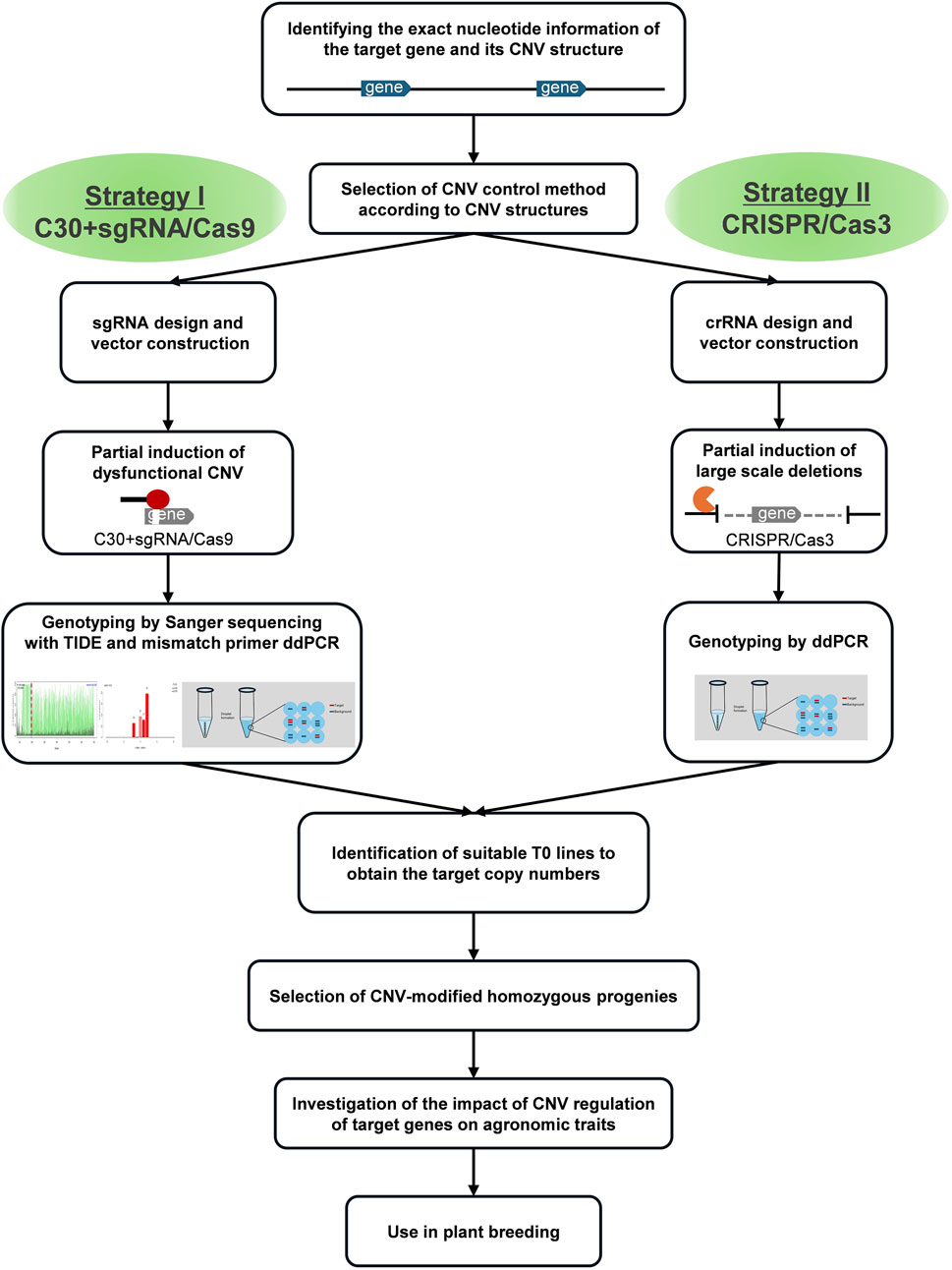
Figure 7. Flowchart of genome editing-mediated CNV modification in plant breeding.
4.2 Copy number evaluation in genome-edited plants
Because ddPCR is a useful technique to determine the copy number of target genes (Collier et al., 2017), we used it to evaluate the CNVs of genome-edited lines. We quantified the copy number of the target gene in Cas3-mediated genome-edited plants compared to that in the wild-type.
We successfully applied the TIDE software to evaluate the mutation pattern and CNV caused by the C30+sgRNA/Cas9 system (Supplementary Figures S1, S4A). Despite being an intuitive, rapid, and cost-effective software, TIDE has the drawback that the quality of the sequence reads and PCR product purity determine the reliability of the results (Brinkman et al., 2014). A previous study measured the efficiency of Cas9-induced genome editing using ddPCR (Peng et al., 2020). We developed a ddPCR method with a deliberately mismatched primer that could more accurately verify multiple copy numbers in genome-edited plants generated by the C30+sgRNA/Cas9 system (Figure 4; Supplementary Figure S5). Primers utilizing intentional mismatches have been successfully used for gene or cultivar screening using the SNP data (Hayashi et al., 2004; Park et al., 2020). A single base mismatch at the 3′ end is usually insufficient for accurate discrimination, and intentional addition of the mismatch in the PCR primer can resolve this problem (Hayashi et al., 2004). Because the predominant sites of Cas9-induced mutations are limited, and the actual mutation patterns can be identified by Sanger sequencing, we could distinguish the wild-type and edited mutant alleles by using such mismatch primers (Figure 4; Supplementary Figure S5). Regarding the stability of this technique, even for the same samples or lines assumed to have the same number of functional alleles, slight fluctuations in the ddPCR values were occasionally observed, as previously reported (Collier et al., 2017). Therefore, cross-validation of two methods, TIDE and ddPCR using mismatch primer, is recommended to accurately determine reduced CNV.
4.3 Impact of CNV modification on agronomic traits
The genotyping and ddPCR results demonstrated that Koshihikari may have three copies of the OsGA20ox1 gene, rather than two. These results are inconsistent with those of previous studies reporting a copy number of two of OsGA20ox1 in Koshihikari (Qin et al., 2021; Wang et al., 2023). Moreover, the phenotypes of CNV-modified lines support the hypothesis that Koshihikari possesses three copies of OsGA20ox1 (Figure 3). Such a misestimation of CNV has been reported in Arabidopsis using a probe-based technique, which differs from the results of previous whole-genome sequencing study (Samelak-Czajka et al., 2017). These inconsistencies may be due to technical limitations, accumulation of genomic data, interpretation errors due to computational interpretation bias, and genetic complexity. Therefore, cross-analysis using different types of experimental techniques, such as probe- or sequence-based techniques, is necessary to correctly identify CNVs. We comprehensively utilized three analytical techniques in this study. First, using TIDE, a software that interprets data based on sequencing wavelengths, we found that the proportion of alleles did not match existing data. Second, we used two different probes in ddPCR technology, which enabled absolute quantification. Finally, we isolated genetic fragments from CR19 and performed Sanger sequencing and statistical analyses, demonstrating that three CNVs were the most persuasive.
Previous studies reported that the expression of OsGA20ox1 controls the seedling vigor in rice; however, the genetic basis for its function has been unclear (Oikawa et al., 2004; Abe et al., 2012; Yano et al., 2012). The leaf sheath length of CNV-modified lines was measured to evaluate the regulatory roles for CNV of OsGA20ox1 in agronomic traits. Our results demonstrated stepwise differences in leaf sheath length depended on the copy number of OsGA20ox1 (Figure 3), and supported the idea that the difference in CNVs, not SNPs, of OsGA20ox1 affected the seedling vigor. In addition, the change in the expression of SlKLUH, a major gene related to increased tomato fruit weight, was previously assumed to be caused by a nucleotide alteration in the promoter region. However, Alonge et al. (2020) revealed that the CNV of SlKLUH is the direct cause of the change in the expression and agronomic traits. These findings are consistent with those of previous reports that the gene expression change depends on differences in CNV (Chiang et al., 2017; Lye and Purugganan, 2019; Qin et al., 2021; Wang et al., 2023).
Several recent studies reported that both simple functional knockout of target genes and fine-tuning of the gene expression through genome editing of cis-regulatory regions would be promising approaches to developing applicable genome-edited crops (Rodriguez-Leal et al., 2017; Wang et al., 2021; Ludwig et al., 2023; Zhou et al., 2024; Kuroha et al., 2025; Lanctot et al., 2025; Mou et al., 2025). Because the OsGA20ox1 is a crucial gene associated with several agronomic traits in rice, fine-tuning of this gene through CNV modification may have the potential to optimize appropriate seedling vigor and plant height, such as the SD1/OsGA20ox2 gene (Monna et al., 2002; Sasaki et al., 2002). In addition, our results suggest that fine-tuning of CNVs in agronomically important genes can contribute to the genetic improvement of multiple crops, including polyploid species.
4.4 Application of genome-editing-mediated CNV modification in breeding
We proposed a strategy to efficiently screen CNV-modified lines (Figure 2D) and successfully obtain CNV-modified homozygous lines at T1 generation (Figure 2E). This strategy could be applied to cases even when we aim to obtain lines with the desired copy number of the gene whose CNV is more than three. We designed a model for the increased copy number of the target gene to determine the number of functional alleles in T0 individuals suitable for the efficient selection of the progenies with the desired number of copies (Table 2). Here, we defined “the number of functional alleles at T0 generation that was suitable for efficient selection of progenies with desired CNV” as “functional allele numbers for selection (FAS)”. Next, based on the designed model, we proposed a formula to easily calculate FAS as a function of the number of desired gene copies “x.”

Table 2. Example of an efficient number of functional alleles to obtain homozygous lines in T1 generation when CNV is more than three.
Finally, as depicted in Table 3, we simulated the usefulness of the FAS in CNV-modified line screening. We assumed that the CNV of the wild-type genome was eight, the desired CNV was five, and FAS (5) was calculated as 9 or 11. Thus, we estimated that the probability of obtaining a homozygous line with the target copy number (five) in the T1 generation was the highest (17.1%–18.0%) when the two values (9 or 11) of FAS obtained through the formula were taken. They were about thrice higher than the probability calculated in not FAS (7 or 13). Homozygous genome-edited progenies with the desired CNV could be selected following this strategy, using a variety of edited allele types of T0 individuals.
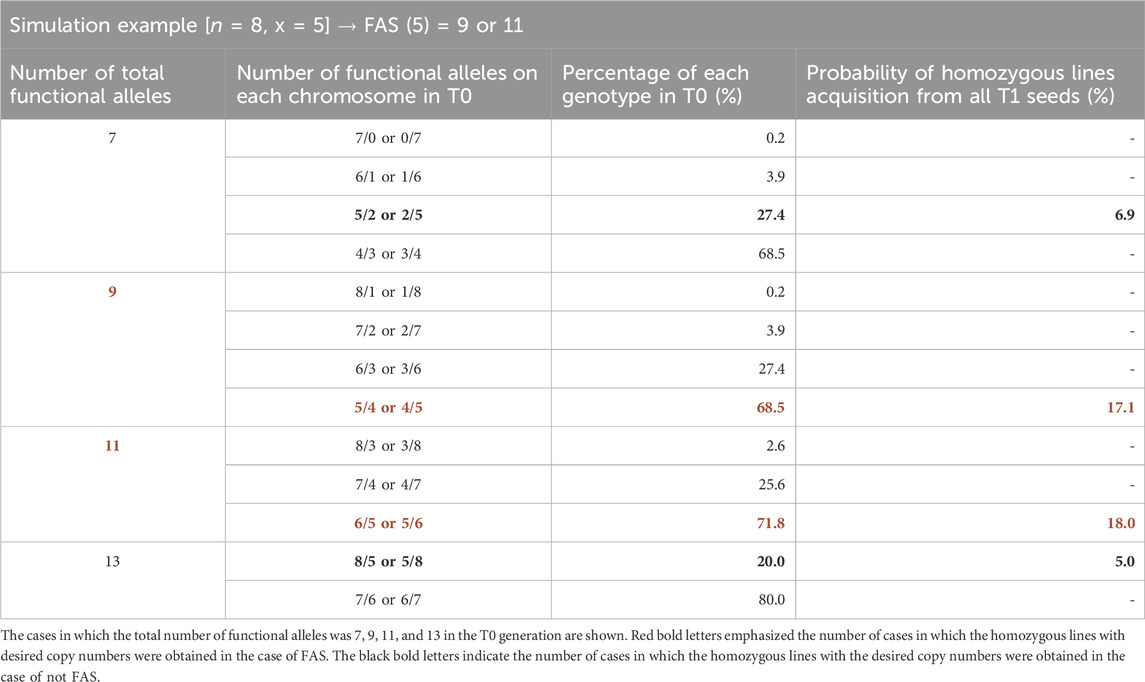
Table 3. Simulation example when the total CNV was eight and the desired number of copies was five.
In conclusion, we successfully modulated CNV using two technologies, CRISPR/Cas9 and Cas3, in both model cases. Our approach provides a solid foundation for future application of these techniques in agriculture by modifying CNVs related to agronomic traits. This strategy may not only reinforce the effectiveness of genetic modifications in different crops but also open new avenues for optimizing plant breeding for several purposes.
Data availability statement
The datasets presented in this article are not readily available because No restriction. Requests to access the datasets should be directed to Hitoshi Yoshida, eW9zaGlkYS5oaXRvc2hpOTIwQG5hcm8uZ28uanA=.
Author contributions
HP: Formal Analysis, Conceptualization, Methodology, Writing – original draft, Investigation. TK: Writing – review and editing, Methodology, Conceptualization. HS: Methodology, Writing – review and editing. MK: Methodology, Writing – review and editing, Supervision. HY: Methodology, Writing – review and editing, Supervision, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Cross-ministerial Strategic Innovation Promotion Program (SIP), “Building a Resilient and Nourishing Food Supply Chain Management for a Sustainable Future” (Grant number: JPJ012287; funding agency: Bio-oriented Technology Research Advancement Institution), and JSPS KAKENHI (Grant number: 22H00373).
Acknowledgments
We sincerely thank the staff and members of our laboratory for their assistance. We would like to thank Editage (www.editage.jp) for the English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgeed.2025.1652950/full#supplementary-material
References
Abe, A., Takagi, H., Fujibe, T., Aya, K., Kojima, M., Sakakibara, H., et al. (2012). OsGA20ox1, a candidate gene for a major QTL controlling seedling vigor in rice. Theor. Appl. Genet. 125 (4), 647–657. doi:10.1007/s00122-012-1857-z
Alonge, M., Wang, X., Benoit, M., Soyk, S., Pereira, L., Zhang, L., et al. (2020). Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182 (1), 145–161. doi:10.1016/j.cell.2020.05.021
Brinkman, E. K., Chen, T., Amendola, M., and van Steensel, B. (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42 (22), e168. doi:10.1093/nar/gku936
Chiang, C., Scott, A. J., Davis, J. R., Tsang, E. K., Li, X., Kim, Y., et al. (2017). The impact of structural variation on human gene expression. Nat. Genet. 49 (5), 692–699. doi:10.1038/ng.3834
Collier, R., Dasgupta, K., Xing, Y. P., Hernandez, B. T., Shao, M., Rohozinski, D., et al. (2017). Accurate measurement of transgene copy number in crop plants using droplet digital PCR. Plant J. 90 (5), 1014–1025. doi:10.1111/tpj.13517
Dolan, A. E., Hou, Z., Xiao, Y., Gramelspacher, M. J., Heo, J., Howden, S. E., et al. (2019). Introducing a spectrum of long-range genomic deletions in human embryonic stem cells using type I CRISPR-cas. Mol. Cell 74 (5), 936–950. doi:10.1016/j.molcel.2019.03.014
Fuentes, R. R., Chebotarov, D., Duitama, J., Smith, S., De la Hoz, J. F., Mohiyuddin, M., et al. (2019). Structural variants in 3000 rice genomes. Genome Res. 29 (5), 870–880. doi:10.1101/gr.241240.118
Guo, S. Q., Chen, Y. X., Ju, Y. L., Pan, C. Y., Shan, J. X., Ye, W. W., et al. (2025). Fine-tuning gibberellin improves rice alkali-thermal tolerance and yield. Nature 639 (8053), 162–171. doi:10.1038/s41586-024-08486-7
Hayashi, K., Hashimoto, N., Daigen, M., and Ashikawa, I. (2004). Development of PCR-based SNP markers for rice blast resistance genes at the Piz locus. Theor. Appl. Genet. 108 (7), 1212–1220. doi:10.1007/s00122-003-1553-0
Kawamata, M., Suzuki, H. I., Kimura, R., and Suzuki, A. (2023). Optimization of Cas9 activity through the addition of cytosine extensions to single-guide RNAs. Nat. Biomed. Eng. 7 (5), 672–691. doi:10.1038/s41551-023-01011-7
Kim, H. K., Kim, Y., Lee, S., Min, S., Bae, J. Y., Choi, J. W., et al. (2019). SpCas9 activity prediction by DeepSpCas9, a deep learning-based model with high generalization performance. Sci. Adv. 5 (11), eaax9249. doi:10.1126/sciadv.aax9249
Kuroda, M., Kimizu, M., and Mikami, C. (2010). A simple set of plasmids for the production of transgenic plants. Biosci. Biotechnol. Biochem. 74 (11), 2348–2351. doi:10.1271/bbb.100465
Kuroha, T., Lombardo, F., Iwasaki, W. M., Chechetka, S., Kawahara, Y., Yoshida, A., et al. (2025). Modification of TAWAWA1-mediated panicle architecture by genome editing of a downstream conserved noncoding sequence in rice. Plant Biotechnol. J. 23, 2667–2669. doi:10.1111/pbi.70043
Labun, K., Montague, T. G., Krause, M., Torres Cleuren, Y. N., Tjeldnes, H., and Valen, E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47 (W1), W171–W174. doi:10.1093/nar/gkz365
Lanctot, A., Hendelman, A., Udilovich, P., Robitaille, G. M., and Lippman, Z. B. (2025). Antagonizing cis-regulatory elements of a conserved flowering gene mediate developmental robustness. Proc. Natl. Acad. Sci. U. S. A. 122 (8), e2421990122. doi:10.1073/pnas.2421990122
Li, X., Xie, Y., Zhu, Q., and Liu, Y. G. (2017). Targeted genome editing in genes and cis-regulatory regions improves qualitative and quantitative traits in crops. Mol. Plant. 10 (11), 1368–1370. doi:10.1016/j.molp.2017.10.009
Li, X., Wang, Y., Chen, S., Tian, H., Fu, D., Zhu, B., et al. (2018). Lycopene is enriched in tomato fruit by CRISPR/Cas9-Mediated multiplex genome editing. Front. Plant. Sci. 9, 559. doi:10.3389/fpls.2018.00559
Li, Y., Huang, B., Chen, J., Huang, L., Xu, J., Wang, Y., et al. (2023). Targeted large fragment deletion in plants using paired crRNAs with type I CRISPR system. Plant Biotechnol. J. 21 (11), 2196–2208. doi:10.1111/pbi.14122
Liu, Q., Shen, G., Peng, K., Huang, Z., Tong, J., Kabir, M. H., et al. (2015). The alteration in the architecture of a T-DNA insertion rice mutant osmtd1 is caused by up-regulation of MicroRNA156f. J. Integr. Plant Biol. 57 (10), 819–829. doi:10.1111/jipb.12340
Liu, Q., Xu, J., Zhu, Y., Mo, Y., Yao, X. F., Wang, R., et al. (2020). The copy number variation of OsMTD1 regulates rice plant architecture. Front. Plant Sci. 11, 620282. doi:10.3389/fpls.2020.620282
Liu, G., Lin, Q., Jin, S., and Gao, C. (2022a). The CRISPR-cas toolbox and gene editing technologies. Mol. Cell 82 (2), 333–347. doi:10.1016/j.molcel.2021.12.002
Liu, X. J., Yang, J. T., Song, Y. Y., Zhang, X. C., Wang, X. J., and Wang, Z. X. (2022b). Effects of sgRNA length and number on gene editing efficiency and predicted mutations generated in rice. Crop J. 10 (2), 577–581. doi:10.1016/j.cj.2021.05.015
Lu, Q. S. M., and Tian, L. (2022). An efficient and specific CRISPR-Cas9 genome editing system targeting soybean phytoene desaturase genes. BMC Biotechnol. 22 (1), 7. doi:10.1186/s12896-022-00737-7
Ludwig, Y., Duenas, C., Arcillas, E., Macalalad-Cabral, R. J., Kohli, A., Reinke, R., et al. (2023). CRISPR-mediated promoter editing of a cis-regulatory element of OsNAS2 increases zn uptake/translocation and plant yield in rice. Front. Genome 5, 1308228. doi:10.3389/fgeed.2023.1308228
Lye, Z. N., and Purugganan, M. D. (2019). Copy number variation in domestication. Trends Plant Sci. 24 (4), 352–365. doi:10.1016/j.tplants.2019.01.003
Monna, L., Kitazawa, N., Yoshino, R., Suzuki, J., Masuda, H., Maehara, Y., et al. (2002). Positional cloning of rice semidwarfing gene, sd-1: rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 9 (1), 11–17. doi:10.1093/dnares/9.1.11
Morisaka, H., Yoshimi, K., Okuzaki, Y., Gee, P., Kunihiro, Y., Sonpho, E., et al. (2019). CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat. Commun. 10 (1), 5302. doi:10.1038/s41467-019-13226-x
Mou, R., Niu, R., Yang, R., and Xu, G. (2025). Engineering crop performance with upstream open reading frames. Trends Plant Sci. 30 (3), 311–323. doi:10.1016/j.tplants.2024.10.005
Naim, F., Dugdale, B., Kleidon, J., Brinin, A., Shand, K., Waterhouse, P., et al. (2018). Gene editing the phytoene desaturase alleles of Cavendish banana using CRISPR/Cas9. Transgenic Res. 27 (5), 451–460. doi:10.1007/s11248-018-0083-0
Ohtsubo, Y., Ikeda-Ohtsubo, W., Nagata, Y., and Tsuda, M. (2008). GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinforma. 9, 376. doi:10.1186/1471-2105-9-376
Oikawa, T., Koshioka, M., Kojima, K., Yoshida, H., and Kawata, M. (2004). A role of OsGA20ox1, encoding an isoform of gibberellin 20-oxidase, for regulation of plant stature in rice. Plant Mol. Biol. 55 (5), 687–700. doi:10.1007/s11103-004-1692-y
Ozawa, K. (2012). A high-efficiency agrobacterium-mediated transformation system of rice (Oryza sativa L.). Methods Mol. Biol. 847, 51–57. doi:10.1007/978-1-61779-558-9_5
Park, H., Kim, S., Nie, H., Kim, J., Lee, J., and Kim, S. (2020). Molecular identification of sweet potato accessions using ARMS-PCR based on SNPs. J. Plant Biotechnol. 47 (2), 124–130. doi:10.5010/jpb.2020.47.2.124
Peng, C., Zheng, M., Ding, L., Chen, X., Wang, X., Feng, X., et al. (2020). Accurate detection and evaluation of the gene-editing frequency in plants using droplet digital PCR. Front. Plant. Sci. 11, 610790. doi:10.3389/fpls.2020.610790
Qin, P., Lu, H., Du, H., Wang, H., Chen, W., Chen, Z., et al. (2021). Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184 (13), 3542–3558.e16. doi:10.1016/j.cell.2021.04.046
Riceome (2021). Rice gene index. Available online at: https://riceome.hzau.edu.cn/ (Accessed October 11, 2023).
Ricerc (2019). Rice resource center. Available online at: https://ricerc.sicau.edu.cn/ (Accessed November 27, 2023).
Rodriguez-Leal, D., Lemmon, Z. H., Man, J., Bartlett, M. E., and Lippman, Z. B. (2017). Engineering quantitative trait variation for crop improvement by genome editing. Cell 171 (2), 470–480. doi:10.1016/j.cell.2017.08.030
Saika, H., Hara, N., Yasumoto, S., Muranaka, T., Yoshimi, K., Mashimo, T., et al. (2025). Heritable large deletions using type I-E CRISPR-Cas3 in rice. bioRxiv. doi:10.1101/2025.06.05.658196
Samelak-Czajka, A., Marszalek-Zenczak, M., Marcinkowska-Swojak, M., Kozlowski, P., Figlerowicz, M., and Zmienko, A. (2017). MLPA-based analysis of copy number variation in plant populations. Front. plant Sci. 8, 222. doi:10.3389/fpls.2017.00222
Sasaki, A., Ashikari, M., Ueguchi-Tanaka, M., Itoh, H., Nishimura, A., Swapan, D., et al. (2002). Green revolution: a mutant gibberellin-synthesis gene in rice. Nature 416 (6882), 701–702. doi:10.1038/416701a
Shang, L., Li, X., He, H., Yuan, Q., Song, Y., Wei, Z., et al. (2022). A super pan-genomic landscape of rice. Cell Res. 32 (10), 878–896. doi:10.1038/s41422-022-00685-z
Shen, R., Wang, L., Liu, X., Wu, J., Jin, W., Zhao, X., et al. (2017). Genomic structural variation-mediated allelic suppression causes hybrid male sterility in rice. Nat. Commun. 8 (1), 1310. doi:10.1038/s41467-017-01400-y
Toki, S. (1997). Rapid and efficient agrobacterium-mediated transformation in rice. Plant Mol. Biol. Rep. 15 (1), 16–21. doi:10.1007/BF02772109
Wang, J. Y., and Doudna, J. A. (2023). CRISPR technology: a decade of genome editing is only the beginning. Science 379 (6629), eadd8643. doi:10.1126/science.add8643
Wang, X., Aguirre, L., Rodriguez-Leal, D., Hendelman, A., Benoit, M., and Lippman, Z. B. (2021). Dissecting cis-regulatory control of quantitative trait variation in a plant stem cell circuit. Nat. Plants 7 (4), 419–427. doi:10.1038/s41477-021-00898-x
Wang, Y., Li, F., Zhang, F., Wu, L., Xu, N., Sun, Q., et al. (2023). Time-ordering japonica/geng genomes analysis indicates the importance of large structural variants in rice breeding. Plant Biotechnol. J. 21 (1), 202–218. doi:10.1111/pbi.13938
Wei, X., Qiu, J., Yong, K., Fan, J., Zhang, Q., Hua, H., et al. (2021). A quantitative genomics map of rice provides genetic insights and guides breeding. Nat. Genet. 53 (2), 243–253. doi:10.1038/s41588-020-00769-9
Wilson, J., Bieker, V. C., Boheemen, L. V., Connallon, T., Martin, M. D., Battlay, P., et al. (2025). Copy number variation contributes to parallel local adaptation in an invasive plant. Proc. Natl. Acad. Sci. U. S. A. 122 (10), e2413587122. doi:10.1073/pnas.2413587122
Wolabu, T. W., Park, J. J., Chen, M., Cong, L., Ge, Y., Jiang, Q., et al. (2020). Improving the genome editing efficiency of CRISPR/Cas9 in arabidopsis and Medicago truncatula. Planta 252 (2), 15. doi:10.1007/s00425-020-03415-0
Wu, Y., Wang, Y., Mi, X. F., Shan, J. X., Li, X. M., Xu, J. L., et al. (2016). The QTL GNP1 encodes GA20ox1, which increases grain number and yield by increasing cytokinin activity in rice panicle meristems. PLoS Genet. 12 (10), e1006386. doi:10.1371/journal.pgen.1006386
Yano, K., Takashi, T., Nagamatsu, S., Kojima, M., Sakakibara, H., Kitano, H., et al. (2012). Efficacy of microarray profiling data combined with QTL mapping for the identification of a QTL gene controlling the initial growth rate in rice. Plant Cell Physiol. 53 (4), 729–739. doi:10.1093/pcp/pcs027
Yu, Z., Chen, Y., Zhou, Y., Zhang, Y., Li, M., Ouyang, Y., et al. (2023). Rice gene index: a comprehensive pan-genome database for comparative and functional genomics of Asian rice. Mol. Plant. 16 (5), 798–801. doi:10.1016/j.molp.2023.03.012
Yuan, C., Zeng, J., Liu, Y., Yu, H., Tong, Z., Zhang, J., et al. (2024). Establishment and application of Agrobacterium-delivered CRISPR/Cas9 system for wild tobacco (Nicotiana alata) genome editing. Front. Plant. Sci. 15, 1329697. doi:10.3389/fpls.2024.1329697
Keywords: copy number variation, cytosine extension, genome editing, OsGA20ox1, phenotype, plant breeding, rice, type-I enzyme
Citation: Park H, Kuroha T, Saika H, Kuroda M and Yoshida H (2025) CRISPR/Cas9- and Cas3-mediated modification of copy number variation in rice. Front. Genome Ed. 7:1652950. doi: 10.3389/fgeed.2025.1652950
Received: 24 June 2025; Accepted: 22 September 2025;
Published: 07 October 2025.
Edited by:
Hidetaka Kaya, Ehime University, JapanReviewed by:
Shigeo S. Sugano, National Institute of Advanced Industrial Science and Technology (AIST), JapanKeiji Nishida, Kobe University, Japan
Copyright © 2025 Park, Kuroha, Saika, Kuroda and Yoshida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takeshi Kuroha, a3Vyb2hhLnRha2VzaGk0ODRAbmFyby5nby5qcA==; Hitoshi Yoshida, eW9zaGlkYS5oaXRvc2hpOTIwQG5hcm8uZ28uanA=