


- 1 Department of Molecular Virology, Immunology and Medical Genetics, Ohio State University, Columbus, OH, USA
- 2 Comprehensive Cancer Center, Ohio State University, Columbus, OH, USA
- 3 Molecular Oncology Unit, Istituto Di Ricovero e Cura a Carattere Scientifico, Referral Cancer Center of Basilicata–Crob,Rionero in Vulture, Italy
- 4 Lautenberg Center for Immunology and Cancer Research, Institute for Medical Research Israel–Canada, Hebrew University–Hadassah Medical School, Jerusalem, Israel
MicroRNAs (miRNAs) are short non-coding RNAs that play critical roles in numerous cellular processes through post-transcriptional regulating functions. The aberrant role of miRNAs has been reported in a number of hematopoietic malignancies including multiple myeloma (MM). In this review we summarize the current knowledge on roles of miRNAs in the pathogenesis of MM.
Introduction
Multiple myeloma (MM) is a clonal B-cell malignancy characterized by the aberrant expansion of plasma cells (PCs) within the bone marrow, as well as into extramedullary sites (Bommert et al., 2006; Mahindra et al., 2010). The American Cancer Society estimates that 20,180 new cases of MM (11,170 in men and 9,010 in women) will be diagnosed while 10,650 deaths are expected to occur in the United States in 2010 (A.C.S., 2010). The lifetime risk of getting MM is 1 in 159 (0.63%) and the 5-year relative survival rate for MM is around 35%. Although the exact causes of MM remain elusive, our understanding of the cellular events underlying MM development is greatly advancing. This has been facilitated by extensive research that enabled thorough analysis of the changes during the pathogenesis of MM.
Multiple myeloma develops from a benign condition called monoclonal gammopathy of undetermined significance (MGUS) (Weiss et al., 2009). Individuals with MGUS often remain stable for years and do not require treatment. However, for unknown reasons, this benign condition can evolve into MM at a rate of ∼1% per year, with some MMs developing after many years (Kuehl and Bergsagel, 2002; Fonseca et al., 2009). Malignant PCs progressively infiltrate the bone marrow and produce monoclonal immunoglobulin (Ig) (M protein) or an Ig chain (M-component). It is well known that the bone marrow microenvironment plays a prominent role in the biology of MM; adhesion of MM cells to the bone marrow stroma triggers cytokine production, enhances cell proliferation and resistance to chemotherapy by activation of NFκB, PI3K/AKT, and STAT-3 pathways through IL-6 (Kastrinakis et al., 2000; Katz, 2010). The progression of MM is associated with osteolytic lesions, bone marrow failure, suppression of normal Ig production and renal insufficiency.
Multiple myeloma is characterized by various cytogenetic abnormalities some of which are of prognostic significance (Fonseca et al., 2004; Avet-Loiseau et al., 2007). Specifically, del(13), t(11;14), t(4;14), t(14;16), hyperdiploidy, MYC translocations, and del(17p) are among the most common cytogenetic changes in MM patients. Univariate statistical analyses revealed that del(13), t(4;14), non-hyperdiploidy, and del(17p) negatively impact both the event-free survival and the overall survival, whereas t(11;14) and MYC translocations are thought not to influence the prognosis. Nevertheless, cumulative evidence suggest that only t(4;14) and del(17p) retained prognostic value for both the event-free and overall survivals as assessed by multivariate analyses (Avet-Loiseau et al., 2007; Neben et al., 2010). These results have implications for risk-adapted management for patients with MM and may suggest that different treatment options might have variable success, depending on the underlying genetic nature of the clone. Despite these recent advances in cytogenetics, oncogenomics, and MM cell–stroma interactions (Avet-Loiseau et al., 2007; Katz, 2010; Neben et al., 2010; Anderson, 2011), further studies are required to better reveal critical players in MM development. The emerging role of microRNAs (miRNAs) in numerous types of human cancer led us and others to hypothesize that this group of non-coding genes might also be involved in the pathogenesis of MM.
MicroRNAs are a relatively recently identified class of regulatory non-coding RNAs, typically 19–25 nucleotides in length, that function primarily by targeting specific messenger RNAs (mRNAs) for degradation or inhibition of translation through base pairing to partially or fully complementary sites (Ambros, 2003; Bartel, 2004). MiRNAs are involved in a variety of biological processes, including development, differentiation, apoptosis, survival, senescence, and metabolism (Ambros, 2004; He and Hannon, 2004). Several hundred miRNAs have been identified and studied so far, but it is predicted that there are many more miRNAs whose significance and role in homeostasis and pathology is yet to be identified. In 2002, miR-15a and miR-16-1 located at 13q14.3, a region that is commonly deleted in chronic lymphocytic leukemia (CLL) and MM, were identified as potential cancer genes in the pathogenesis of CLL (Calin et al., 2002). This finding provided the first evidence that miRNA genes might be important for cancer development. Later on, the use of different approaches including microarrays (Calin et al., 2004; Volinia et al., 2006) and quantitative reverse transcription PCR (qRT-PCR; Schmittgen et al., 2004; Jiang et al., 2005), revealed that miRNAs are differentially expressed in cancer cells and identified significant miRNA genes that are of biological and clinical relevance in human diseases (Volinia et al., 2006; Liu et al., 2008). Several miRNAs have been identified as oncomirs (miRNAs that are amplified or overexpressed in cancer and were shown to have a promoting role in the development of primary tumors) or tumor suppressors (miRNAs that are deleted or reduced in cancer cells and their loss is associated with tumor development; reviewed in Calin and Croce, 2006; Ventura and Jacks, 2009). Various causes of deregulated miRNA expression in cancer have been identified including changes in gene copy number (CN), chromosomal translocation, mutations, transcriptional activation, epigenetic silencing and defective miRNA processing, and biogenesis (Calin and Croce, 2006). Collectively, it is realized that deregulation of, at least some, miRNAs might also contribute to tumorigenesis.
During the last 4 years, several studies investigated miRNAs expression and function in the pathogenesis of MM. In this review, we will discuss the classification of these miRNAs, their cause of deregulation, the mechanisms by which these genes exert their functions and the clinical relevance of their expression in MM.
MiRNAs are Differentially Expressed in MM
Initially, it has been shown that miR-21 levels in MM cells are controlled in a STAT-3-IL-6 dependent manner (Loffler et al., 2007). Treatment of IL-6-dependent MM cells with IL-6 activated STAT-3, which in turn enhanced miR-21 transcription. Importantly, ectopic expression of miR-21 was sufficient to sustain growth of IL-6-dependent MM cells in the absence of IL-6. Subsequent studies showed that miR-21 levels are upregulated in MM and MGUS samples with respect to its levels in healthy PCs (Pichiorri et al., 2008; Zhou et al., 2010, see below). These results suggested that miRNAs levels might be implicated in MM pathogenesis.
In 2008, we reported a unique and comprehensive miRNA expression profiling of normal PCs, MGUS, and MM (Pichiorri et al., 2008). We utilized both a custom-made microarray chip and qRT-PCR of CD138+ normal PCs, MGUS, primary samples and MM cell lines. When compared with normal PCs, MGUS patients revealed a group of upregulated miRNAs with known oncogenic potential including miR-21 and the miR-106∼25 cluster. Although we are aware that the small set of MGUS samples (n = 10) used for these analyses could limit its significance, our study for the first time suggested that miRNAs deregulation could be associated with early stages of PC transformation. The fact that both miR-21 and the miR-106∼25 cluster were shown to target, among others, PTEN, BIM, and p21, tumor suppressors that inhibit survival and promote apoptosis, could suggest that these miRNAs predispose to secondary events that might ultimately lead to full blown myeloma.
To better shed light on the role of miRNAs in MM, we next analyzed their expression in a group of MM cell lines and normal PCs (Pichiorri et al., 2008). Our data revealed a signature comprised of upregulated miRNAs, including among others miR-32, miR-21, miR-17∼92, miR-106∼25, and miR-181a/b. The Myc related miR-17∼92 cluster were highly expressed only in MM patients, suggesting that these miRNAs are MM-specific and their expression could be related to Myc upregulation during MM progression (Chesi et al., 2008). To further investigate the potential involvement of miRNAs in MM, Zhou et al. (2010) performed integrative analyses of both miRNA expression profiles and protein coding gene expression profiles of myeloma cells. Their analyses reveal a global increase in miRNA expression in high-risk (poor prognosis) MM patients. This association was reinforced by the increase in viability of MM cells depleted of Argonaute 2 (AGO2), a master regulator of miRNA maturation and function (Liu et al., 2004; Diederichs and Haber, 2007). In fact, AGO2 is also involved in B-cell differentiation (O’carroll et al., 2007) and was reported as an important marker for MM disease prognosis (Shaughnessy et al., 2007). Nevertheless, expression of AGO2 did not significantly associate with global miRNA (Zhou et al., 2010) suggesting that other factors might contribute to the overall deregulation of miRNA in MM. Analysis of miRNA expression by another group revealed upregulation of miR-193b-365 among previously identified oncomiRs (Unno et al., 2009).
Intriguingly, many of the aforementioned miRNAs were previously identified (Pichiorri et al., 2008), though others were not or had an opposite trend. These differences could stem from the different platforms used for analyzing miRNA expression, number of cases analyzed and/or genetic and cytogenetic abnormalities in the different cases or cell lines. Furthermore, it is possible that miRNAs play opposing roles at different stages of the disease; i.e., in MGUS versus MM. Regardless of the disagreement among the different studies, the overall conclusions confirm that deregulation of miRNA expression adds a further level to our understanding of the biological and clinical variability of MM and warrants further investigations.
Mechanisms of miRNA Deregulation in MM
Additional studies further demonstrated deregulation of miRNA expression in the different subtypes of MM and shed light on the mechanism of this alteration. Lionetti et al.employed an integrative high-resolution microarray analysis of miRNAs and DNA CN or gene expression profiles in MM cell lines (Lionetti et al., 2009). These analyses revealed the deregulation of 16 miRNAs mapped to chromosomal regions frequently involved in allelic imbalances in MM. Among these were miR-22 at 17p13.3, miR-106b and miR-25 at 7q22.1, miR-15a at 13q14.3, miR-21 at 17q23.1, and miR-92b at 1q22. In another study, it was shown that expression of miR-15a/16-1 cluster at 13q14 display a range of expression patterns in MM cases independent of chromosome 13 status (Roccaro et al., 2009). Further, 32 intragenic miRNAs significantly correlated with that of their host genes of which some are known genes implicated in MM including miR-152-COPZ2, miR-335-MEST, and miR-342 3p-EVL(Ronchetti et al., 2008; Lionetti et al., 2009). Collectively, these data suggest that CN and frequent co-expression of intronic miRNAs with their respective host genes could be leading mechanisms to altered miRNAs expression in MM.
Since MM is characterized by very complex cytogenetic aberrations that affects prognosis and molecular profiling (Sawyer, 2011), it is likely that these aberrations might affect miRNA expression as well. Indeed, recent studies have shown, for example, that upregulation of miR-1 and miR-133a is correlated with t(14;16) MM cases suggesting that deregulation of miRNA expression could be associated with chromosomal aberrations (Gutierrez et al., 2010). Other examples are shown in Table 1.
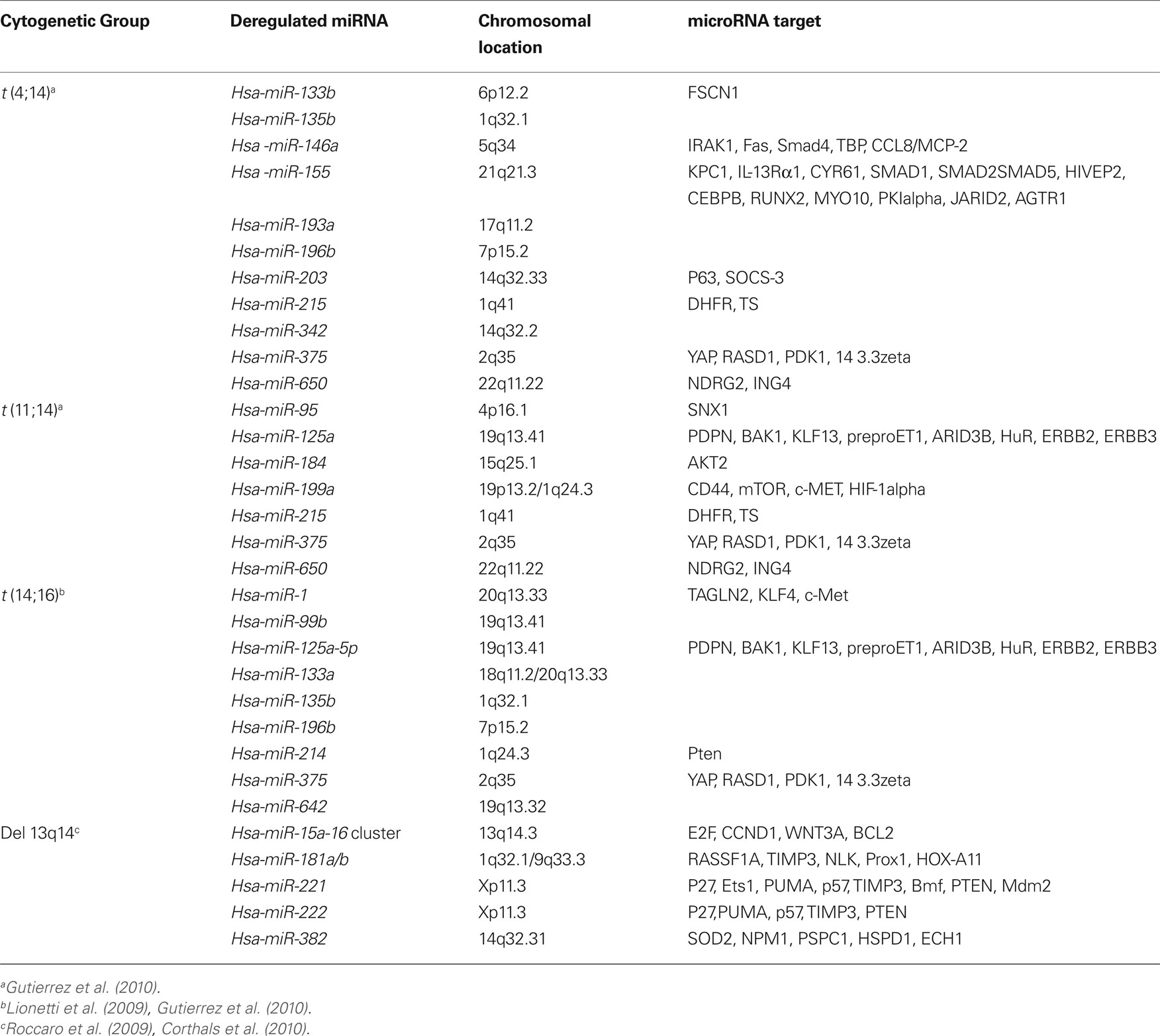
Table 1. miRNA genes deregulated in specific cytogenetic aberrations in multiple myeloma.
Impaired processing of miRNAs was also reported to be associated with high-risk MM. The findings of Zhou et al. (2010) clearly suggest that depletion of AGO2, involved in miRNA genesis and functionality, is associated with growth arrest and apoptosis in MM cells. Altered expression of AGO2 might lead to impaired miRNAs genesis thereby affecting their functions in MM pathogenesis. In agreement, it was recently shown that altered expression of Dicer, but not Drosha, might be associated with progression and prognosis of MM. In particular, it was suggested that MGUS premalignant cases display higher levels of Dicer than do SMM and MM cases and are very similar to normal PCs (Sarasquete et al., 2010). Moreover, higher expression of Dicer was associated with improved progression-free survival in symptomatic MM cases. Whether upregulation of Dicer in MGUS and MM is associated with impaired miRNA expression and function is still to be determined. These findings may shed light on the potential prognostic and therapeutic strategies in which regulators for both miRNA maturation and function, such as AGO2 and Dicer, might be used as prognostic tools and/or treatments of MM.
Deregulation of miRNAs Regulates Critical Genes Associated with MM
Deregulation of miRNAs was shown to be associated with targeting coding genes that are implicated in MM. For example, miR-17∼92 cluster located at 13q31-32, a region frequently amplified in malignant B-cell lymphomas (He et al., 2005), and overexpressed in 65% of the B-cell lymphoma patients (Ota et al., 2004), has been shown to contribute to B-cell lymphoma by targeting PTEN, E2F1, and BIM. By overexpressing miR-17∼92 in lymphoid progenitor cells from mice that carry the MYC transgene, He et al. reported an accelerated lymphomagenesis by the cluster (He et al., 2005). A different group generated mice with high miR-17-92 lymphocytic expression and observed a higher rate of lymphoproliferative disorders, autoimmunity, and premature death in the transgenic mice (Xiao et al., 2008). Further, the work of Ventura et al. (2008) demonstrated that this cluster is essential for B-cell development and that its deficiency leads to increased levels of the pro-apoptotic protein BIM thereby inhibiting the transition of pro-B to pre-B stage. Therefore, it is likely that upregulation of the miR-17∼92 cluster negatively regulates these critical tumor suppressor genes contributing to PCs transformation and MM progression.
On the other hand, the miR-106∼25 cluster together with miR-32 were shown to target PCAF (Pichiorri et al., 2008), a p53 positive regulator (Schiltz and Nakatani, 2000; Linares et al., 2007). Downregulation of PCAF could keep p53 at low level via its histone acetyltransferase function contributing to MM progression. Additionally, miR-106a/b, miR-17-5, and miR-20b were shown to target the CDKN1A1/p21 cell cycle regulator implicated in MM (Chen et al., 1999; Lavelle et al., 2001). Another central pathway that is deregulated in MM pathogenesis is the STAT-3/IL-6 pathway (Bommert et al., 2006). Targeting of this pathway by miR-19a/b was demonstrated in MM cells (Pichiorri et al., 2008). MiR-19a/bdirectly targets a negative regulator of IL-6, SOCS1, contributing to its frequent downregulation in MM cells. Importantly, xenograft studies using human MM cell lines treated with miR-19a/b and miR-181a/b antagonists (antimiRs) resulted in significant suppression of tumor growth in immunocompromised mice (Pichiorri et al., 2008).
MiRNAs 15a/16-1 Function as Tumor Suppressors in MM
As mentioned above, miR-15a and miR-16-1 are located at 13q14.3, a region that is commonly deleted in CLL and MM among other malignancies (Aqeilan et al., 2010). Deletion of this region occurs in more than 50% of cases and is believed to take place at an early stage of the disease suggesting that it might be associated with MM pathogenesis (Fonseca et al., 2004). MiRNA expression profiling of bone marrow derived CD138 + MM cells versus their normal cellular counterparts identified an MM-specific miRNA signature characterized by significant downregulation of miR-15a/16 (Roccaro et al., 2009; Corthals et al., 2010). Similar to CLL, it was hypothesized that minimal deletion in 13q14.3 containing the miR-15a/16 cluster might have an oncosuppressor effect on MM pathogenesis. Indeed, the functional role of miR-15a/16 in MM cells revealed their regulation of cell proliferation, survival, and cell cycle progression in vitro and in vivo (Roccaro et al., 2009). Recent studies from our laboratory have shown that the expression of the DLEU-2 gene, located at 13q14.3 and containing miR-15a/16 clusters, is completely absent in MM cells as well as in normal PCs. Nevertheless, miR-15a and miR-16 are indeed downregulated. This might strongly suggest that the expression of these miRNAs could be driven from their related cluster at chromosome 3 (q25.33). Alternatively, an internal promoter unrelated to the host gene could drive their expression in PCs.
Although at the genetic level it is still considered an open question, at the molecular level miR-15a/16 are shown to regulate cell cycle proteins including cyclinD1, cyclinD2, and CDC25A expression. Moreover, miR-15a/16 target BCL2 expression (Aqeilan et al., 2010) and their overexpression in MM cells induces apoptosis as indicated by an increased percentage of sub-G1 population. MiR-15a/16 were also shown to inhibit AKT3 and NFκB pathways in MM cells (Roccaro et al., 2009). Moreover, ectopic expression of miR-15a/16 displayed antiangiogenic activity through regulation of VEGF. Most recently, Gatt et al. further investigated the tumor suppressor role of miR-15a/16 in MM (Gatt et al. 2010). Using “sponge” lentiviral vectors directed toward the mature form of miR-15a/16, it was shown that inhibition of miR-16 enhanced proliferative and invasive ability of MMS1 cells in vitro. Further, depletion of miR-16 decreased animal survival in a xenograft model of MM through increasing tumor load and host angiogenesis. Gene expression profiling of miR-16- depelted MM1S cells revealed a large number of miR-16 target genes including FGFR1, VEGFa, and MDM4. Importantly, miR-16 levels correlated with 13q14 deletion in MM samples and with those having larger blood vessels (Gatt et al. 2010). These observations clearly support the fact that a single miRNA, for example miR-16, can have far-reaching effects on the overall function of the cell. Altogether, it is becoming obvious that miR-15a/16 are critical regulators of MM pathogenesis through targeting of multiple pathways implicated in clonal PCs and bone marrow neoangiogenesis.
MiRNA Functional Cross-Talk with P53 in Multiple Myeloma
The tumor suppressor p53 is frequently inactivated by mutations or deletions in cancer. p53 acts as a potent transcription factor and can be activated in response to diverse stresses, leading to induction of cell cycle arrest, apoptosis, or senescence (Xue et al., 2007; Junttila and Evan, 2009). Although regulation of the p53 pathway is not fully understood at the molecular level, it has been well established that activated p53 suppresses cancer progression, underscoring why cancer cells have developed multiple mechanisms to disable p53 function (Danovi et al., 2004; Ventura et al., 2007). In human tumors that retain wild-type (WT) p53 (Lane, 2001; Junttila and Evan, 2009), p53 can be antagonized by murine double minute 2 (MDM2), a negative regulator of p53 that is overexpressed in many human tumors, offering a therapeutic strategy (Brown et al., 2009; Dickens et al., 2010). Indeed, it has been reported that inhibiting MDM2 expression can reactivate p53 in cancer cells, leading to their demise (Dickens et al., 2010; Saha et al., 2010a,b).
TP53 mutation is rarely detected at diagnosis in many hematological cancers including MM, CLL, acute myeloid leukemia, and Hodgkin’s disease. Thus, numerous reports have shown that therapeutic induction of p53 might be particularly suitable for the treatment of hematological malignancies (Saha et al., 2010b) including MM (Kuehl and Bergsagel, 2002; Fonseca et al., 2009). Mutations of p53 are rare in untreated MM and the tumors appear to have intact, if perhaps suppressed, p53 function, suggesting that therapeutic modulation of the p53/MDM2 pathway holds promise to help the majority of patients. Several reports (Quesnel et al., 1994; Teoh et al., 1997) and our data suggest that MDM2 overexpression in MMs, not its gene amplification (Quesnel et al., 1994), could be responsible for p53 inactivation in cells retaining functional p53 pathways. This supports the idea that induction of p53 in this setting might be a suitable treatment for MM. Interestingly, in MM cells, expression of p53 protein levels can be rescued by antagonizing MDM2. Several reports have focused on the p53-mediated apoptotic pathway, upon endogenous p53 protein re-expression by the small-molecule MDM2 antagonists (Nutlins) and target genes which may be involved in p53-dependent apoptosis in MM cells which have been identified (Stuhmer and Bargou, 2006). We recently showed that alteration in miRNA expression during the progression from MGUS to newly diagnosed MM could partially be responsible for p53 inactivation in MM patients (Pichiorri et al., 2010). We found that, among others, miR-192, 194, and 215, which are downregulated in a subset of newly diagnosed MMs, can be transcriptionally activated by p53 and then modulate MDM2 expression (Table 2). Furthermore, ectopic re-expression of these miRNAs in MM cells increases the therapeutic action of MDM2 inhibitors in vitro and in vivo by enhancing their p53-activating effects. In addition, miR-192 and 215 target the IGF pathway, preventing enhanced migration of PCs into bone marrow.
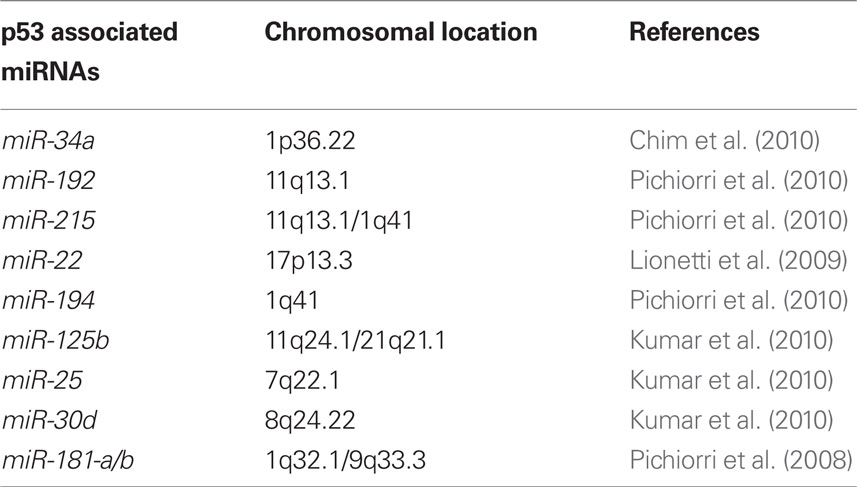
Table 2. miRNAs deregulation associated with p53 expression in multiple myeloma.
In seeking an explanation for the lack of expression of these miRNAs in MM, we have noted that the genes for these miRNAs are located in chromosomal regions in MM that are normally characterized by chromosome gain and translocations rather than deletions (Fonseca et al., 2009). This led us to hypothesize that other mechanisms are responsible for the observed downregulation of these miRNAs. Indeed, it turns out that epigenetic cross-talk between methylation and histone acetylation (Vaissiere et al., 2008; Walker et al., 2011) in the promoter of the miR-194-2-192 is r esponsible for this silencing in MM cell lines. This could support the hypothesis that the transition from MGUS to MM could be favored by clonal selection of cells with the epigenetic inactivation not only of p53 but principally of its direct targets. This could be associated with a decreasing ability of p53 to down-modulate MDM2 expression thus tipping the regulatory balance in favor of MDM2 in MM cells. Whether methylation of the miR-194-2-192 promoter in primary patients and in MGUS is responsible for this cluster inactivation is currently under investigation.
We and others also have shown that the upregulation of miR-25 and miR-181a in MM cells of a specific set of miRNAs could negatively regulate the expression of the tumor suppressor gene p53 emphasizing the important cross-talk between p53 and aberrant microRNA expression in MM cells (Kumar et al., 2010; Pichiorri et al., 2010). Of note, monoallelic deletion of TP53 in MM, which often seems to occur without mutation on the other allele, is associated with an extremely poor prognosis (Chng et al., 2007). This supports the idea that a decrease in TP53 gene content is associated with tumor progression, which supports the hypothesis that a partial lack of expression of these miRNAs in MMs could create a p53 imbalance with direct biological consequences. The deletion of one copy of p53 by FISH has been uniformly found to be an adverse prognostic factor with all therapies used in the treatment of MM: alkylating agents, proteasome inhibitors, and immune modulator-based therapies (Lode et al., 2010). Due to the emerging role of epigenetic modifications in MM progression and the key role of p53 regulation by miRNAs it should be relevant to know if deacetylase inhibitors (DACi) could affect a set of miRNA expression in MM cells able to affect p53 re-expression making the cells more sensitive to further therapies.
Conclusions and Future Perspectives
Genetic alterations in MM have been identified and were shown to be associated with different biologic features and heterogeneity in clinical outcome. Recent research work highlights the possible contribution of a new class of non-coding genes, miRNA (Figure 1). Cumulative evidence indicates that miRNAs function as “fine” regulators of cell commitment and differentiation during human normal hematopoiesis (Fabbri et al., 2009). In parallel, several research groups are studying whether and how the miRNome differs in hematological malignancies (i.e., MM) with respect to the normal hematologic counterpart (i.e., CD138+ PCs). The study of miRNAs deregulation and their functional consequences in MM shed light on the molecular mechanisms of MM and B-cell malignancies in general. Intriguingly, the unique signature of miRNA gene expression in MM differentiates it from CD138+ B-cells and MGUS suggesting that miRNAs can be used in diagnosis and, probably, in prognosis and treatment of these tumors. Moreover, by revealing the molecular effectors of miRNAs, it is becoming evident that the use of miRNA and/or anti-miRNA molecules together with chemotherapeutic regimens might increase the success rate in treating MM. The interplay of miRNA genes and the bone marrow microenvironment and their role in MM pathogenesis and drug resistance is another interesting topic that would warrant investigation in the future.
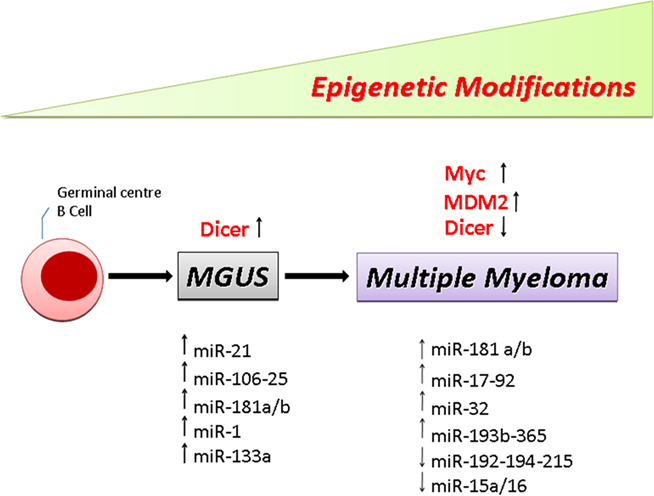
Figure 1. Schematic drawing showing the multistep molecular process of PC transformation. Representative list of the miRNAs and genes significantly deregulated in MGUS and MM patients versus normal PCs.
Although for many years no single mouse model seemed capable of reproducing all facets of a specific human multiple myeloma, the Vk*MYC mice (Chesi et al., 2008) fulfill many of the biologic and genetic criteria of an ideal mouse model, showing a high degree of homology to the clinical phenotype of human MM. The Vk*MYC model relies upon a sporadic, yet precisely timed, physiologic mechanism for oncogenic activation, providing a model of tumorigenesis with high penetrance. It should be really interesting to study the miRNAs changes during myeloma development from MGUS to active MM in this mouse model. Whether MYC upregulation in early stages of the disease could lead to epigenetic modification in the genome that is related to p53 downregulation through MDM2 upregulation and higher chromosomal instability is another open question. While many answers are still pending, the need for further analysis of this new class of players in MM, and more general in cancer, is therefore required to better decipher miRNAs’ role in this heterogeneous and incurable disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to apologize for the many colleagues that we could not cite their work due to space limitation. We are grateful for Mrs. Aliza Forman for critical reading of the manuscript. Flavia Pichiorri is a Kimmel Scholar and Rami I. Aqeilan is a winner of the Ma’of Fellowship.
References
A.C.S. (2010). Multiple Myeloma. Available at http://www.cancer.org/Cancer/MultipleMyeloma/index
Ambros, V. (2003). MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell 113, 673–676.
Anderson, K. C. (2011). Oncogenomics to target myeloma in the bone marrow microenvironment. Clin. Cancer Res. 17, 1225–1233.
Aqeilan, R. I., Calin, G. A., and Croce, C. M. (2010). miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 17, 215–220.
Avet-Loiseau, H., Attal, M., Moreau, P., Charbonnel, C., Garban, F., Hulin, C., Leyvraz, S., Michallet, M., Yakoub-Agha, I., Garderet, L., Marit, G., Michaux, L., Voillat, L., Renaud, M., Grosbois, B., Guillerm, G., Benboubker, L., Monconduit, M., Thieblemont, C., Casassus, P., Caillot, D., Stoppa, A. M., Sotto, J. J., Wetterwald, M., Dumontet, C., Fuzibet, J. G., Azais, I., Dorvaux, V., Zandecki, M., Bataille, R., Minvielle, S., Harousseau, J. L., Facon, T., and Mathiot, C. (2007). Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood 109, 3489–3495.
Bommert, K., Bargou, R. C., and Stuhmer, T. (2006). Signalling and survival pathways in multiple myeloma. Eur. J. Cancer 42, 1574–1580.
Brown, C. J., Lain, S., Verma, C. S., Fersht, A. R., and Lane, D. P. (2009). Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 9, 862–873.
Calin, G. A., and Croce, C. M. (2006). MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 66, 7390–7394.
Calin, G. A., Dumitru, C. D., Shimizu, M., Bichi, R., Zupo, S., Noch, E., Aldler, H., Rattan, S., Keating, M., Rai, K., Rassenti, L., Kipps, T., Negrini, M., Bullrich, F., and Croce, C. M. (2002). Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 99, 15524–15529.
Calin, G. A., Liu, C. G., Sevignani, C., Ferracin, M., Felli, N., Dumitru, C. D., Shimizu, M., Cimmino, A., Zupo, S., Dono, M., Dell’aquila, M. L., Alder, H., Rassenti, L., Kipps, T. J., Bullrich, F., Negrini, M., and Croce, C. M. (2004). MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. U.S.A. 101, 11755–11760.
Chen, Y. H., Lavelle, D., Desimone, J., Uddin, S., Platanias, L. C., and Hankewych, M. (1999). Growth inhibition of a human myeloma cell line by all-trans retinoic acid is not mediated through downregulation of interleukin-6 receptors but through upregulation of p21(WAF1). Blood 94, 251–259.
Chesi, M., Robbiani, D. F., Sebag, M., Chng, W. J., Affer, M., Tiedemann, R., Valdez, R., Palmer, S. E., Haas, S. S., Stewart, A. K., Fonseca, R., Kremer, R., Cattoretti, G., and Bergsagel, P. L. (2008). AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 13, 167–180.
Chim, C. S., Wong, K. Y., Qi, Y., Loong, F., Lam, W. L., Wong, L. G., Jin, D. Y., Costello, J. F., and Liang, R. (2010). Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis 31, 745–750.
Chng, W. J., Glebov, O., Bergsagel, P. L., and Kuehl, W. M. (2007). Genetic events in the pathogenesis of multiple myeloma. Best Pract. Res. Clin. Haematol. 20, 571–596.
Corthals, S. L., Jongen-Lavrencic, M., De Knegt, Y., Peeters, J. K., Beverloo, H. B., Lokhorst, H. M., and Sonneveld, P. (2010). Micro-RNA-15a and micro-RNA-16 expression and chromosome 13 deletions in multiple myeloma. Leuk. Res. 34, 677–681.
Danovi, D., Meulmeester, E., Pasini, D., Migliorini, D., Capra, M., Frenk, R., De Graaf, P., Francoz, S., Gasparini, P., Gobbi, A., Helin, K., Pelicci, P. G., Jochemsen, A. G., and Marine, J. C. (2004). Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell. Biol. 24, 5835–5843.
Dickens, M. P., Fitzgerald, R., and Fischer, P. M. (2010). Small-molecule inhibitors of MDM2 as new anticancer therapeutics. Semin. Cancer Biol. 20, 10–18.
Diederichs, S., and Haber, D. A. (2007). Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131, 1097–1108.
Fabbri, M., Croce, C. M., and Calin, G. A. (2009). MicroRNAs in the ontogeny of leukemias and lymphomas. Leuk. Lymphoma 50, 160–170.
Fonseca, R., Barlogie, B., Bataille, R., Bastard, C., Bergsagel, P. L., Chesi, M., Davies, F. E., Drach, J., Greipp, P. R., Kirsch, I. R., Kuehl, W. M., Hernandez, J. M., Minvielle, S., Pilarski, L. M., Shaughnessy, J. D. Jr., Stewart, A. K., and Avet-Loiseau, H. (2004). Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 64, 1546–1558.
Fonseca, R., Bergsagel, P. L., Drach, J., Shaughnessy, J., Gutierrez, N., Stewart, A. K., Morgan, G., Van Ness, B., Chesi, M., Minvielle, S., Neri, A., Barlogie, B., Kuehl, W. M., Liebisch, P., Davies, F., Chen-Kiang, S., Durie, B. G., Carrasco, R., Sezer, O., Reiman, T., Pilarski, L., and Avet-Loiseau, H. (2009). International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia 23, 2210–2221.
Gatt, M. E., Zhao, J. J., Ebert, M. S., Zhang, Y., Chu, Z., Mani, M., Gazit, R., Carrasco, D. E., Dutta-Simmons, J., Adamia, S., Minvielle, S., Tai, Y. T., Munshi, N. C., Avet-Loiseau, H., Anderson, K. C., and Carrasco, D. R. (2010). MicroRNAs 15a/16-1 function as tumor suppressor genes in multiple myeloma. Blood (in press).
Gutierrez, N. C., Sarasquete, M. E., Misiewicz-Krzeminska, I., Delgado, M., De Las Rivas, J., Ticona, F. V., Ferminan, E., Martin-Jimenez, P., Chillon, C., Risueno, A., Hernandez, J. M., Garcia-Sanz, R., Gonzalez, M., and San Miguel, J. F. (2010). Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 24, 629–637.
He, L., and Hannon, G. J. (2004). MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531.
He, L., Thomson, J. M., Hemann, M. T., Hernando-Monge, E., Mu, D., Goodson, S., Powers, S., Cordon-Cardo, C., Lowe, S. W., Hannon, G. J., and Hammond, S. M. (2005). A microRNA polycistron as a potential human oncogene. Nature 435, 828–833.
Jiang, J., Lee, E. J., Gusev, Y., and Schmittgen, T. D. (2005). Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 33, 5394–5403.
Junttila, M. R., and Evan, G. I. (2009). p53 – a Jack of all trades but master of none. Nat. Rev. Cancer 9, 821–829.
Kastrinakis, N. G., Gorgoulis, V. G., Foukas, P. G., Dimopoulos, M. A., and Kittas, C. (2000). Molecular aspects of multiple myeloma. Ann. Oncol. 11, 1217–1228.
Katz, B. Z. (2010). Adhesion molecules – The lifelines of multiple myeloma cells. Semin. Cancer Biol. 20, 186–195.
Kuehl, W. M., and Bergsagel, P. L. (2002). Multiple myeloma: evolving genetic events and host interactions. Nat. Rev. Cancer 2, 175–187.
Kumar, M., Lu, Z., Takwi, A. A., Chen, W., Callander, N. S., Ramos, K. S., Young, K. H., and Li, Y. (2010). Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene 30, 843–853.
Lavelle, D., Chen, Y. H., Hankewych, M., and Desimone, J. (2001). Histone deacetylase inhibitors increase p21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am. J. Hematol. 68, 170–178.
Linares, L. K., Kiernan, R., Triboulet, R., Chable-Bessia, C., Latreille, D., Cuvier, O., Lacroix, M., Le Cam, L., Coux, O., and Benkirane, M. (2007). Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat. Cell Biol. 9, 331–338.
Lionetti, M., Agnelli, L., Mosca, L., Fabris, S., Andronache, A., Todoerti, K., Ronchetti, D., Deliliers, G. L., and Neri, A. (2009). Integrative high-resolution microarray analysis of human myeloma cell lines reveals deregulated miRNA expression associated with allelic imbalances and gene expression profiles. Genes Chromosomes Cancer 48, 521–531.
Liu, C. G., Spizzo, R., Calin, G. A., and Croce, C. M. (2008). Expression profiling of microRNA using oligo DNA arrays. Methods 44, 22–30.
Liu, J., Carmell, M. A., Rivas, F. V., Marsden, C. G., Thomson, J. M., Song, J. J., Hammond, S. M., Joshua-Tor, L., and Hannon, G. J. (2004). Argonaute2 is the catalytic engine of mammalian RNAi. Science 305, 1437–1441.
Lode, L., Eveillard, M., Trichet, V., Soussi, T., Wuilleme, S., Richebourg, S., Magrangeas, F., Ifrah, N., Campion, L., Traulle, C., Guilhot, F., Caillot, D., Marit, G., Mathiot, C., Facon, T., Attal, M., Harousseau, J. L., Moreau, P., Minvielle, S., and Avet-Loiseau, H. (2010). Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 95, 1973–1976.
Loffler, D., Brocke-Heidrich, K., Pfeifer, G., Stocsits, C., Hackermuller, J., Kretzschmar, A. K., Burger, R., Gramatzki, M., Blumert, C., Bauer, K., Cvijic, H., Ullmann, A. K., Stadler, P. F., and Horn, F. (2007). Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 110, 1330–1333.
Mahindra, A., Hideshima, T., and Anderson, K. C. (2010). Multiple myeloma: biology of the disease. Blood Rev. 24(Suppl. 1), S5–S11.
Neben, K., Jauch, A., Bertsch, U., Heiss, C., Hielscher, T., Seckinger, A., Mors, T., Müller, N. Z., Hillengass, J., Raab, M. S., Ho, A. D., Hose, D., and Goldschmidt, H. (2010). Combining information regarding chromosomal aberrations t(4;14) and del(17p13) with the International Staging System classification allows stratification of myeloma patients undergoing autologous stem cell transplantation. Haematologica 95, 1150–1157.
O’carroll, D., Mecklenbrauker, I., Das, P. P., Santana, A., Koenig, U., Enright, A. J., Miska, E. A., and Tarakhovsky, A. (2007). A slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev. 21, 1999–2004.
Ota, A., Tagawa, H., Karnan, S., Tsuzuki, S., Karpas, A., Kira, S., Yoshida, Y., and Seto, M. (2004). Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 64, 3087–3095.
Pichiorri, F., Suh, S. S., Ladetto, M., Kuehl, M., Palumbo, T., Drandi, D., Taccioli, C., Zanesi, N., Alder, H., Hagan, J. P., Munker, R., Volinia, S., Boccadoro, M., Garzon, R., Palumbo, A., Aqeilan, R. I., and Croce, C. M. (2008). MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 12885–12890.
Pichiorri, F., Suh, S. S., Rocci, A., De Luca, L., Taccioli, C., Santhanam, R., Zhou, W., Benson, D. M. Jr., Hofmainster, C., Alder, H., Garofalo, M., Di Leva, G., Volinia, S., Lin, H. J., Perrotti, D., Kuehl, M., Aqeilan, R. I., Palumbo, A., and Croce, C. M. (2010). Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 18, 367–381.
Quesnel, B., Preudhomme, C., Oscier, D., Lepelley, P., Collyn-D’hooghe, M., Facon, T., Zandecki, M., and Fenaux, P. (1994). Over-expression of the MDM2 gene is found in some cases of haematological malignancies. Br. J. Haematol. 88, 415–418.
Roccaro, A. M., Sacco, A., Thompson, B., Leleu, X., Azab, A. K., Azab, F., Runnels, J., Jia, X., Ngo, H. T., Melhem, M. R., Lin, C. P., Ribatti, D., Rollins, B. J., Witzig, T. E., Anderson, K. C., and Ghobrial, I. M. (2009). MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood 113, 6669–6680.
Ronchetti, D., Lionetti, M., Mosca, L., Agnelli, L., Andronache, A., Fabris, S., Deliliers, G. L., and Neri, A. (2008). An integrative genomic approach reveals coordinated expression of intronic miR-335, miR-342, and miR-561 with deregulated host genes in multiple myeloma. BMC Med. Genomics 1, 37. doi: 10.1186/1755-8794-1-37
Saha, M. N., Jiang, H., Jayakar, J., Reece, D., Branch, D. R., and Chang, H. (2010a). MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol. Ther. 9, 936–944.
Saha, M. N., Micallef, J., Qiu, L., and Chang, H. (2010b). Pharmacological activation of the p53 pathway in haematological malignancies. J. Clin. Pathol. 63, 204–209.
Sarasquete, M. E., Gutierrez, N. C., Misiewicz-Krzeminska, I., Paiva, B., Chillon, M. C., Alcoceba, M., Garcia-Sanz, R., Hernandez-Rivas, J. M., Gonzalez, M., and San Miguel, J. F. (2010). Up-regulation of dicer is more frequent in monoclonal gammopathies of undetermined significance than in multiple myeloma patients and is associated with longer survival in symptomatic myeloma patients. Haematologica 96, 468–471.
Sawyer, J. R. (2011). The prognostic significance of cytogenetics and molecular profiling in multile myleoma. Cancer Genet. 204, 3–12.
Schiltz, R. L., and Nakatani, Y. (2000). The PCAF acetylase complex as a potential tumor suppressor. Biochim. Biophys. Acta 1470, M37–M53.
Schmittgen, T. D., Jiang, J., Liu, Q., and Yang, L. (2004). A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 32, e43.
Shaughnessy, J. D. Jr., Zhan, F., Burington, B. E., Huang, Y., Colla, S., Hanamura, I., Stewart, J. P., Kordsmeier, B., Randolph, C., Williams, D. R., Xiao, Y., Xu, H., Epstein, J., Anaissie, E., Krishna, S. G., Cottler-Fox, M., Hollmig, K., Mohiuddin, A., Pineda-Roman, M., Tricot, G., Van Rhee, F., Sawyer, J., Alsayed, Y., Walker, R., Zangari, M., Crowley, J., and Barlogie, B. (2007). A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 109, 2276–2284.
Stuhmer, T., and Bargou, R. C. (2006). Selective pharmacologic activation of the p53-dependent pathway as a therapeutic strategy for hematologic malignancies. Cell Cycle 5, 39–42.
Teoh, G., Urashima, M., Ogata, A., Chauhan, D., Decaprio, J. A., Treon, S. P., Schlossman, R. L., and Anderson, K. C. (1997). MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood 90, 1982–1992.
Unno, K., Zhou, Y., Zimmerman, T., Platanias, L. C., and Wickrema, A. (2009). Identification of a novel microRNA cluster miR-193b-365 in multiple myeloma. Leuk. Lymphoma 50, 1865–1871.
Vaissiere, T., Sawan, C., and Herceg, Z. (2008). Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 659, 40–48.
Ventura, A., and Jacks, T. (2009). MicroRNAs and cancer: short RNAs go a long way. Cell 136, 586–591.
Ventura, A., Kirsch, D. G., Mclaughlin, M. E., Tuveson, D. A., Grimm, J., Lintault, L., Newman, J., Reczek, E. E., Weissleder, R., and Jacks, T. (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665.
Ventura, A., Young, A. G., Winslow, M. M., Lintault, L., Meissner, A., Erkeland, S. J., Newman, J., Bronson, R. T., Crowley, D., Stone, J. R., Jaenisch, R., Sharp, P. A., and Jacks, T. (2008). Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132, 875–886.
Volinia, S., Calin, G. A., Liu, C. G., Ambs, S., Cimmino, A., Petrocca, F., Visone, R., Iorio, M., Roldo, C., Ferracin, M., Prueitt, R. L., Yanaihara, N., Lanza, G., Scarpa, A., Vecchione, A., Negrini, M., Harris, C. C., and Croce, C. M. (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 103, 2257–2261.
Walker, B. A., Wardell, C. P., Chiecchio, L., Smith, E. M., Boyd, K. D., Neri, A., Davies, F. E., Ross, F. M., and Morgan, G. J. (2011). Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 117, 553–562.
Weiss, B. M., Abadie, J., Verma, P., Howard, R. S., and Kuehl, W. M. (2009). A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 113, 5418–5422.
Xiao, C., Srinivasan, L., Calado, D. P., Patterson, H. C., Zhang, B., Wang, J., Henderson, J. M., Kutok, J. L., and Rajewsky, K. (2008). Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 9, 405–414.
Xue, W., Zender, L., Miething, C., Dickins, R. A., Hernando, E., Krizhanovsky, V., Cordon-Cardo, C., and Lowe, S. W. (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660.
Zhou, Y., Chen, L., Barlogie, B., Stephens, O., Wu, X., Williams, D. R., Cartron, M. A., Van Rhee, F., Nair, B., Waheed, S., Pineda-Roman, M., Alsayed, Y., Anaissie, E., and Shaughnessy, J. D. Jr. (2010). High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proc. Natl. Acad. Sci. U.S.A. 107, 7904–7909.
Keywords: miRNAs, multiple myeloma, microarray, profiling, p53, MDM2
Citation: Pichiorri F, De Luca L and Aqeilan RI (2011) MicroRNAs: new players in multiple myeloma. Front. Gene. 2:22. doi: 10.3389/fgene.2011.00022
Received: 20 February 2011;
Paper pending published: 22 March 2011;
Accepted: 06 May 2011;
Published online: 24 May 2011.
Edited by:
Michael Rossbach, Genome Institute of Singapore, SingaporeReviewed by:
Vladimir Benes, European Molecular Biology Laboratory, GermanyAmelia Cimmino, Consiglio Nazionale delle Ricerche, Italy
Copyright: © 2011 Pichiorri, De Luca and Aqeilan. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Rami I. Aqeilan, Hebrew University–Hadassah Medical School, Ein-Karem Campus, P.O. Box 12272, Jerusalem 91120, Israel. e-mail:YXFlaWxhbkBjYy5odWppLmFjLmls; Flavia Pichiorri, Ohio State University, Biomedical Research Tower room #1060, 460 W. 12th Avenue, Columbus, OH 43210.e-mail:ZmxhdmlhLnBpY2hpb3JyaUBvc3VtYy5lZHU=