


- Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA
Tetrahydrofolates (THF) are a family of cofactors that function as one-carbon donors in folate-dependent one-carbon metabolism, a metabolic network required for the de novo synthesis of purines, thymidylate, and for the remethylation of homocysteine to methionine in the cytoplasm. 5-FormylTHF is not a cofactor in one-carbon metabolism, but serves as a storage form of THF cofactors. 5-formylTHF is mobilized back into the THF cofactor pool by methenyltetrahydrofolate synthetase (MTHFS), which catalyzes the irreversible and ATP-dependent conversion 5-formyltetrahydrofolate to 5,10-methenyltetrahydrofolate. Mthfs is not an essential gene in Arabidopsis, but MTHFS expression is elevated in animal tumors, enhances de novo purine synthesis, confers partial resistance to antifolate purine synthesis inhibitors and increases rates of folate catabolism in mammalian cell cultures. However, the mechanisms underlying these effects of MTHFS expression have yet to be established. The purpose of this study was to investigate the role and essentiality of MTHFS in mice. Mthfs was disrupted through the insertion of a gene trap vector between exons 1 and 2. Mthfsgt/+ mice were fertile and viable. No Mthfsgt/gt embryos were recovered from Mthfsgt/+ intercrosses, indicating Mthfs is an essential gene in mice. Tissue MTHFS protein levels are decreased in both Mthfsgt/+ and Mthfs+/+ mice placed on a folate and choline deficient diet, and mouse embryonic fibroblasts from Mthfsgt/+ embryos exhibit decreased capacity for de novo purine synthesis without impairment in de novo thymidylate synthesis. MTHFS was shown to co-localize with two enzymes of the de novo purine synthesis pathway in HeLa cells in a cell cycle-dependent manner, and to be modified by the small ubiquitin-like modifier (SUMO) protein. Mutation of the consensus SUMO modification sites on MTHFS eliminated co-localization of MTHFS with the de novo purine biosynthesis pathway under purine-deficient conditions. The results from this study indicate that MTHFS enhances purine biosynthesis by delivering 10-formylTHF to the purinosome in a SUMO-dependent fashion.
Introduction
Tetrahydrofolates (THF) are a family of cofactors that carry and chemically activate single carbons on N5, N10, or bridged between N5 and N10 (Shane, 1995). THF cofactors contain a glutamate polypeptide that contains 2–9 glutamate residues linked through γ-peptide linkages (Shane, 1995). The glutamate residues serve both to retain the co-factors within the cell and increase the affinity of folate-utilizing enzymes. THF cofactors donate or accept one-carbons in a network of interconnected pathways referred to as folate-mediated one-carbon metabolism (OCM). In the cytoplasm, folate-mediated OCM is required for the de novo synthesis of purines, thymidylate, and for the remethylation of homocysteine to methionine (Figure 1). Methionine can be converted to the methyl donor S-adenosylmethionine (AdoMet), which is a cofactor for numerous methylation reactions, including the methylation of lipids, proteins, DNA, and RNA. Alterations in folate metabolism are associated with several pathologies and developmental anomalies including certain cancers (Suh et al., 2001) and neural tube defects (Beaudin and Stover, 2009), but the mechanisms have not been established. Impairments in OCM can result from primary folate deficiency, polymorphisms in genes that encode folate-dependent enzymes, increased rates of folate degradation, and secondary nutrient deficiencies including vitamin B6 or vitamin B12. Biomarkers for impaired OCM include elevations in tissue and serum homocysteine and S-adenosylhomocysteine (AdoHcy) levels (Lindenbaum and Allen, 1995; Selhub, 1999; Ueland et al., 2000; Yi et al., 2000), increased rates of uracil incorporation into DNA which increases the incidence of DNA strand breaks (Blount et al., 1997; Melnyk et al., 1999), and DNA hypomethylation (Yi et al., 2000; Friso et al., 2002).
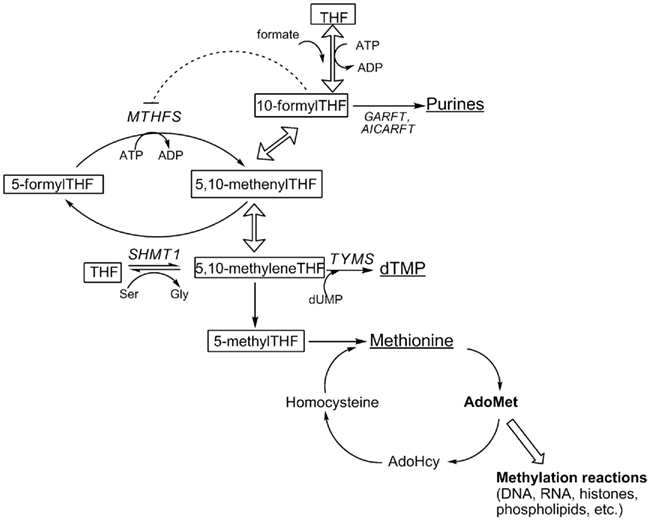
Figure 1. Folate-dependent one-carbon metabolism in the cytoplasm. Products of one-carbon metabolism are underlined; folate cofactors are boxed. THF, tetrahydrofolate; MTHFS, methenyltetrahydrofolate synthetase; SHMT1, serine hydroxymethyltransferase; GARFT, glycinamide ribonucleotide formyltransferase; AICARFT, phosphoribosylaminoimidazole carboxamide formyltransferase; TYMS, thymidylate synthase; dUMP, deoxyuridine monophsophate. Hollow arrows represent reactions catalyzed by MTHFD1.
There are three THF cofactors that carry single carbons at the oxidation level of formate: 10-formylTHF, 5-formylTHF, and 5,10-methenylTHF. These three forms of folate exist in chemical equilibrium and slowly interconvert (Stover and Schirch, 1992). In the cell, 10-formylTHF is synthesized through the ATP-dependent condensation of formate and THF catalyzed by the 10-formyltetrahydrofolate synthetase activity of the trifunctional enzyme C1-tetrahydrofolate synthase (referred to as MTHFD1, EC 1.5.1.5, 3.5.4.9, 6.3.4.3; Figure 1). 5,10-methenylTHF is synthesized from 10-formylTHF both non-enzymatically and through the cyclohydrolase activity of MTHFD1. 5-formylTHF concentrations are regulated by a futile cycle involving serine hydroxymethyltransferase (SHMT, EC 2.1.2.1) and 5,10-methenyltetrahydrofolate synthetase (MTHFS, EC 6.3.3.2; Stover and Schirch, 1993). SHMT catalyzes the formation of 5-formylTHF from 5, 10-methenylTHF, through a second catalytic activity, whereas 5-formylTHF is mobilized back into the THF cofactor pool through the activity of MTHFS, which catalyzes the ATP-dependent conversion of 5-formylTHF to 5,10-methenylTHF (Stover and Schirch, 1993; Figure 1).
The role of 5-formylTHF, and hence MTHFS, in mammalian OCM has not been fully established. 5-FormylTHF is not an enzyme cofactor, but is a potent inhibitor of several folate-dependent enzymes including aminoimidazolecarboxamide ribonucleotide formyltransferase (AICARFT, EC 2.1.2.3; Bertrand and Jolivet, 1989) and SHMT (Stover and Schirch, 1991). It has also been suggested to serve as a stable storage form of THF in spores (Kruschwitz et al., 1994), but is not known to accumulate in mammalian cells. Whereas all cells contain SHMT, certain prokaryotes lack MTHFS (Jeanguenin et al., 2010), and MTHFS is not an essential gene in Arabidopsis (Goyer et al., 2005). In bacteria, expression of prokaryotic glutamate formiminotransferases, which are involved in folate-dependent histidine catabolism, can functionally complement MTHFS activity by catabolizing 5-formylTHF (Jeanguenin et al., 2010). In mammalian cells, it is not known if MTHFS activity is essential, or if mammalian glutamate formiminotransferases are functionally redundant with MTHFS activity.
Both 5-formylTHF and MTHFS have been shown to regulate de novo purine biosynthesis. 10-formylTHF is the cofactor that supplies the number 2 and 8 carbons for the de novo synthesis of the purine ring, catalyzed by the enzymes AICARFT and glycinamide ribonucleotide formyltransferase (GARFT, EC 2.1.2.2), respectively (Figure 1). Recently, the enzymes that constitute the pathway for the de novo synthesis of purines were shown to form a multi-enzyme complex referred to as the “purinosome” (An et al., 2008).
Inhibition of MTHFS in MCF-7 cells impairs purine biosynthesis in cultured cells (Bertrand and Jolivet, 1989), whereas overexpression enhances rates of de novo purine biosynthesis (Field et al., 2006). MTHFS binds10-formylTHF tightly (Field et al., 2006); [6R,S]-10-FormylTHF tri-glutamates competitively inhibit recombinant mouse MTHFS enzymatic activity with Ki = 30 nM, suggesting that 10-formylTHF is an effective in vivo inhibitor of MTHFS activity (Field et al., 2006). Expression of MTHFS elevates cellular 10-formylTHF levels in cultured cells (Girgis et al., 1997), indicating that the MTHFS-10-formylTHF binary complex is available for purine biosynthesis, and suggests that MTHFS may function to deliver 10-formylTHF to the de novo purine synthesis pathway (Field et al., 2006). Furthermore, MTHFS overexpression in neuroblastoma confers resistance to the antifolate LY309887, a highly specific inhibitor that targets GARFT and 10-formylTHF-dependent purine biosynthesis (Field et al., 2009).
In this study, the essentiality of MTHFS in mice was investigated. This study is the first examination of the effects of reduced MTHFS expression in mice. The results demonstrate that Mthfs is an essential gene in mice and that reduced MTHFS expression in mouse embryonic fibroblast (MEF) cells is associated with decreased capacity for de novo purine synthesis in these cells. MTHFS haploinsufficiency did not affect tumorigenesis in the Apcmin/+ model of sporadic intestinal tumor formation. Furthermore, MTHFS is shown to associate with the “purinosome” in mammalian cells, indicating that MTHFS enhances de novo purine biosynthesis by delivering 10-formylTHF cofactors to the purinosome.
Materials and Methods
Generation of Mthfsgt/+ Mice
All study protocols were approved by the Institutional Animal Care and Use Committee of Cornell University and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mouse embryonic stem cells containing a gene trap vector between exon 1 and 2 of the mouse Mthfs gene were purchased from Bay Genomics (San Fransisco, CA). Gene trap vector pGT1LXF contains the engrailed 2 (En2) intron located 5′ of a βgeo cassette. Integration of the gene trap vector was verified at Bay Genomics. Embryonic stem cells containing this vector were injected in to C57Bl/6 blastocysts at the Cornell University Transgenic Mouse Core Facility (Ithaca, NY). Germ line transmission of the Mthfsgt allele was confirmed by PCR using purified nuclear DNA, as described below. Mice were backcrossed for 10 generations and maintained on C57Bl/6 background under specific-pathogen free conditions.
Genotyping of MTHFS Deficient Mice
Genotyping was carried out using PCR from tail tissue nuclear DNA that was isolated using the DNeasy DNA purification kit (Qiagen) per manufacturer’s instructions. A duplex PCR reaction consisting of the following primers (priming sites shown in Figure 2A) detected for the wild-type Mthfs allele and the Mthfsgt allele: Mthfs forward, 5′-aggaatggcggcggtcacg-3′; Mthfs reverse, 5′-gtctccttcctctgctacatcc-3′; RGT 480, 5′-ggcctccagacaagtagat-3′. PCR conditions were 94°C for 2 min, followed by 30 cycles of 94°C for 30 s, 61°C for 30 s, 72°C for 30 s, and a final extension of 72°C for 7 min. The wild-type allele is detected as a 530 bp fragment and the gene trap allele is detected as a 400 bp fragment (Figure 2B).
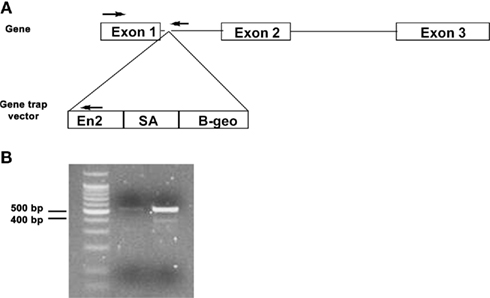
Figure 2. Schematic of gene trap vector and PCR genotyping of Mthfsgt allele. (A) Schematic of Mthfs gene, which consists of three exons. The gene trap vector was inserted between exon 1 and 2. PCR priming sites are shown with arrows. (B) PCR products (run on 2% agarose gel) for nuclear tail DNA of Mthfs+/+ and Mthfsgt/+. The wild-type allele produces a 530 bp product and the gene trap (gt) allele produces a 400 bp product.
Diet Studies
Male Mthfsgt/+ and Mthfs+/+ mice were randomly assigned to either the control (AIN-93G diet) or to the Modified AIN-3G diet lacking folate and choline (folate/choline deficient diet; Dyets, Inc., Bethlehem, PA) at weaning (3 weeks of age). Mice were maintained on the diet for 5 weeks. Mouse weight and food consumption were monitored weekly. Food was removed from the cage 24 h prior to sacrifice, after 12 h each mouse was given one pellet of food to synchronize intake. Mice were sacrificed another 12 h later by cervical dislocation.
In order to determine whether reduced MTHFS expression and a folate/choline deficient diet modifies intestinal cancer risk, Mthfsgt/+ mice were mated with C57Bl/6-Apcmin/+ mice. F1 Apcmin/+/Mthfs+/+ and Apcmin/+/Mthfsgt/+ male mice were randomly assigned to either the control or folate/choline deficient (FCD) diet. The mice remained on the diet for eleven weeks, and were monitored and sacrificed as in the five week diet study (described above). Genotyping of the Apcmin allele was carried out as previously described (Macfarlane et al., 2011).
Tissue Collection
Blood was collected by cardiac puncture into heparinized tubes. Plasma was separated from red blood cells by centrifugation. Brain, kidney and liver were removed, washed in cold phosphate-buffered saline and snap frozen in liquid nitrogen. The colon was removed, flushed with cold phosphate-buffered saline, opened longitudinally and cut into three sections and again washed in cold phosphate-buffered saline before being snap frozen in liquid nitrogen. All samples were stored at -80°C.
Mouse Embryonic Fibroblast Culture
Mouse embryonic fibroblasts were isolated from female Mthfsgt/+ mice bred to male Mthfsgt/+ mice at 10–14 days post coitus (dpc). Briefly, embryos were dissected from the uterus into cold sterile PBS. The heads were taken for genotyping and the eviscerated bodies were cut into 1–2 mm pieces and digested in 0.05% trypsin at 37±C. Cells were cultured in alpha-minimal essential medium (alpha-MEM; Hyclone Laboratories) supplemented with 10% fetal calf serum (Hyclone Laboratories), 0.1 mM non-essential amino acids (Gibco), 1 mM sodium pyruvate (Gibco) and penicillin/streptomycin (Gibco) and incubated at 37°C in a 5% CO2 atmosphere.
Western Blot
Snap-frozen tissues or cell pellets were lysed in 10 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 5 mM DTT, 1% Triton X-100, and Mammalian Protease Inhibitor Cocktail (Sigma). Tissue lysates were loaded onto 10% SDS-PAGE gels with 25 μg total protein/lane for MEF cells, kidney, liver, and colon samples and 50 μg total protein/lane for brain samples [protein concentrations were determined using the Lowry–Bensadoun method (Bensadoun and Weinstein, 1976)]. Proteins were then transferred to an immobilon-P PVDF membrane (Millipore). The membrane was blocked overnight at 4±C in phosphate-buffered saline with 10% non-fat dry milk and 1% NP40. The membrane was incubated at 4±C overnight in a purified antibody (1:5,000 dilution) raised against murine MTHFS (peptide sequence  AKRGLRAELKQRLR
AKRGLRAELKQRLR ). After four washes of 10 min each, the membrane was incubated for 2 h in 1:20,000 HRP-conjugated goat α-rabbit antibody (Pierce). After four washes of 10 min each, membranes were developed in SuperSignal West Pico Chemiluminescent Substrate (Pierce) and films exposed. As a loading control, membranes were also probed with an antibody to glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Novus Biologicals) used at 1:40,000 dilution; HRP-conjugated goat α-mouse secondary antibody (Pierce) was used at 1:10,000 dilution. Images were digitized and quantification of protein expression was performed using Image J software (NIH). For each given tissue, MTHFS expression was analyzed using a two-way ANOVA to assess differences by genotype and diet; in MEF cell lysates, expression was analyzed using a student’s t-test.
). After four washes of 10 min each, the membrane was incubated for 2 h in 1:20,000 HRP-conjugated goat α-rabbit antibody (Pierce). After four washes of 10 min each, membranes were developed in SuperSignal West Pico Chemiluminescent Substrate (Pierce) and films exposed. As a loading control, membranes were also probed with an antibody to glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Novus Biologicals) used at 1:40,000 dilution; HRP-conjugated goat α-mouse secondary antibody (Pierce) was used at 1:10,000 dilution. Images were digitized and quantification of protein expression was performed using Image J software (NIH). For each given tissue, MTHFS expression was analyzed using a two-way ANOVA to assess differences by genotype and diet; in MEF cell lysates, expression was analyzed using a student’s t-test.
Purine Biosynthesis Assay
This assay was carried out on eight MEF cell lines (n = 4 Mthfs+/+, n = 4 Mthfsgt/+) using methods detailed previously (Field et al., 2006).
Determination of Plasma and Tissue Folate Concentration
Plasma and tissue folate concentrations were determined using the Lactobacillus casei microbiological assay as previously described (Suh et al., 2000).
dU Suppression
This assay was carried out on four MEF cell lines (n = 2 Mthfs+/+, n = 2 Mthfsgt/+) using methods detailed previously (Macfarlane et al., 2011).
Expression of Fluorescent Constructs in HeLa Cells
The human MTHFS cDNA (described in Girgis et al., 1997) was amplified by PCR using forward primer 5′-aatggcggcggcagcggtgag-3′and reverse primer 5′-gagctgttgacgagtct tcgtaaaggac-3′, gel purified using a High Pure PCR Product Purification Kit (Roche) and inserted into the pcDNA3.1/CT-GFP-TOPO® (Invitrogen) per manufacturer’s instructions. The resulting vector, expressing human MTHFS-GFP (MTHFS with a C-terminal GFP tag) was transfected into HeLa cells using Lipofectamine 2000 (Invitrogen) per manufacturer’s instructions. The MTHFS-K140R, K190R-GFP point mutants were made using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) according to manufacturers’ instructions using primers 5′-ccagtgaatgaaaacgacatgcgcgtagatgaagtcctttacg-3′ and 5′-cgtaaaggacttcatctacgcgcatgtcgttttcattcactgg-3′ (for K190R) and primers 5′-ggccttgggtttgacaggcatggcaaccgaact-3′ and 5′-agttcggttgccatgcctgtcaaacccaaggcc-3′ (for K140R). The sites of the lysine to arginine mutations are underlined. Plasmids expressing formylglycinamidine ribonucleotide fused to orange fluorescent protein (hFGAMS-OFP) and the trifunctional protein TrifGART fused to orange fluorescent protein (hTrifGART-OFP) were a generous gift from Stephen Benkovic of The Pennsylvania State University and have been described elsewhere (An et al., 2008).
HeLa cells were maintained in either purine-rich minimal essential media alpha-modification (αMEM, Hyclone) supplemented with 10% fetal bovine serum (FBS, Hyclone) for “purine-rich” conditions or in Roswell Park Memorial Media (RPMI 1640, Hyclone) supplemented with 5% dialyzed FBS for “purine-depleted” conditions. FBS was dialyzed against phosphate-buffered saline with four buffer changes for a minimum of 24 h at 4°C, and then charcoal treated to remove any residual small molecules.
Cell-Cycle Arrest
HeLa cells maintained in purine-depleted media at 50% confluence were treated with either 60 μM mevinolin (also known as Lovastatin, Sigma) or 120 ng/mL nocodazole (Sigma) 24 h after transfection with MTHFS-GFP (as described above). After another 24 h, cells were analyzed by confocal microscopy. To confirm cell-cycle arrest, cells were treated with Lipofectamine 2000 alone (“mock” transfected) then treated 24 h later with cell cycle blocking agents. Cells were analyzed by fluorescence activated cell sorting (FACS) 24 h after arrest.
Fluorescence Microscopy of Live Cells
HeLa cells were maintained in either purine-rich αMEM or purine-depleted RPMI 1640 and transfected 48 h prior to visualization. Confocal fluorescence microscopy (Leica TCS SP2 system) was used to image all cells at the Cornell Microscope and Imaging Facility. Draq5 (Biostatus Limited) was used as a nuclear marker. To visualize MTHFS-GFP a 488 nm laser line and optical filter were used. For OFP and Draq5 the 543nm and 633nm laser lines, respectively, were used with a 543 nm/633 nm filter set. To ensure there was no bleed over between channels, the images for each color were taken sequentially and then merged in silico using Image J software.
In vitro Sumoylation Assay
Purified recombinant mouse MTHFS was SUMOylated using the SUMOlink SUMO-1 kit (Active Motif) according to manufacturer’s instructions. Reactions were analyzed via Western Blot using either α-SUMO-1 (Active Motif) or α-MTHFS (described above).
Sumo Immunoprecipitation
MTHFS-GFP was transfected into HeLa cells maintained in purine-deficient RPMI-1640 using Lipofectamine 2000 (Invitrogen), as described above. Cells were trypsinized and collected via centrifugation. Cells were lysed in Lysis Buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1% NP-40, 5 mM EDTA) to which 5 mM DTT, protease inhibitor cocktail (Sigma) and 1 mM SUMO protease inhibitor N-ethylmaleimide (NEM) were added. Cell extracts (180 μg/sample) were incubated with 4 μg α-SUMO-1 antibody (Active Motif) overnight at 4°C. Dynabeads Protein G (Invitrogen) were used to pull down protein complexes, which were eluted using 1 × SDS-PAGE sample buffer. Western Blot was then performed as previously described.
Statistical Methods
For animals studies in which mice of both Mthfs+/+ and Mthfsgt/+ were placed onto either of two diets (control or folate/choline deficient) results were analyzed using a two-way ANOVA to assess differences between genotype and diet and any possible gene-diet interactions. For cell culture studies (dU suppression and purine biosynthesis assay), a two-tailed student’s t-test was used to assess differences between genotypes.
Results
MTHFS Null Mice are not Viable
Mthfsgt/+ mice were backcrossed onto a C57Bl/6 background for 10 generations, and were viable and fertile. Mthfsgt/+ intercrosses yielded the expected frequency of Mthfs+/+ and Mthfsgt/+ pups, but did not yield Mthfsgt/gt pups in more than 100 live births (data not shown). To determine if the lethality could be rescued by maternal dietary purines, the drinking water of the Mthfsgt/+ females was supplemented with 500 μM hypoxanthine during mating, and throughout pregnancy. No Mthfsgt/gt embryos were observed at day 9.5 dpc in seven litters containing 42 fetuses (Table 1), indicating that embryos homozygous for the Mthfsgt allele did not survive early embryogenesis and that Mthfs is an essential gene in C57Bl/6 mice.
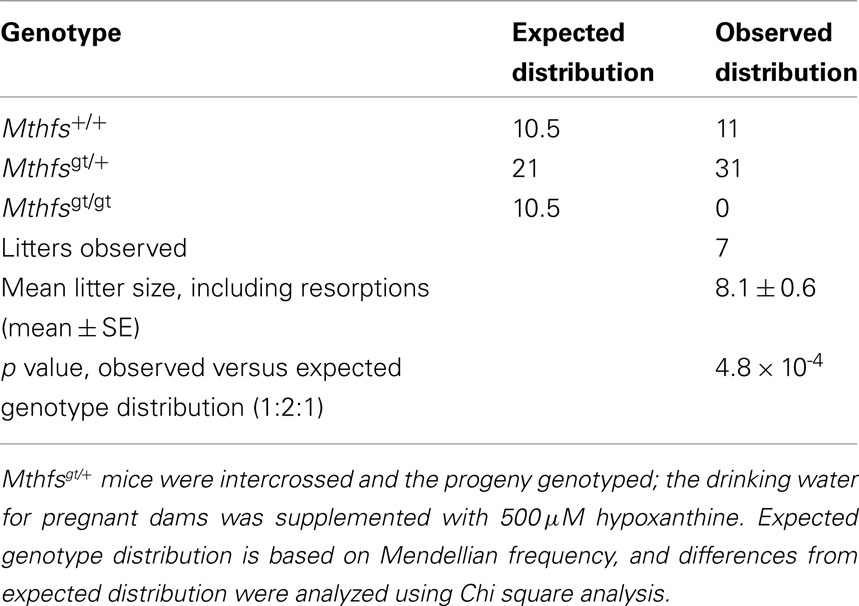
Table 1. Mthfs null mice are not viable and lethality is not rescued with 500 μM hypoxanthine supplementation to pregnant dams.
The effect of Mthfs deletion on growth and food consumption was determined over a 5 week period. There were no significant differences in weight or food intake between Mthfsgt/+ and their Mthfs+/+ littermates when fed the standard AIN93G diet (control diet), or a modified AIN93G diet lacking folate and choline (FCD diet; Figure 3).
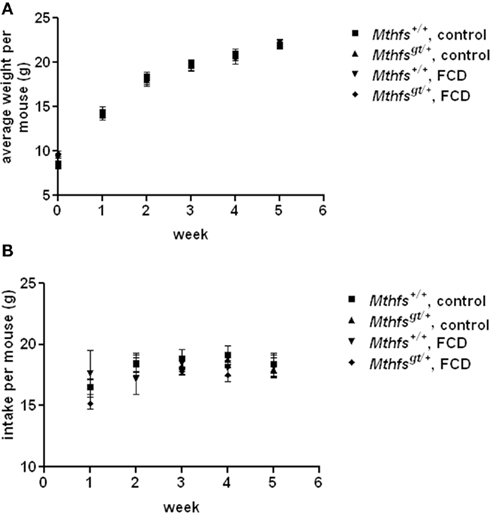
Figure 3. Weight gain and food intake in Mthfs+/+ and Mthfsgt/+ mice fed either control diet (AIN-93G) or folate-choline deficient diet (FCD; AIN-93G lacking folate and choline) for five weeks. In both (A) weight gain per mouse and (B) intake per mouse Mthfs+/+ on control diet is shown by squares; Mthfsgt/+ on control diet shown by triangles; Mthfs+/+ on FCD shown by inverted triangles; Mthfsgt/+ on FCD shown by diamonds. Data is shown as the mean and standard deviation with n = 10 per group.
MTHFS Protein Levels in Mthfsgt/+ Mice
Male Mthfsgt/+ mice were crossed to female C57Bl/6 mice to generate Mthfsgt/+ and Mthfs+/+ male mice which were randomly assigned to either control or FCD diet for 5-weeks. MTHFS protein levels are slightly, but not statistically significantly, reduced in the liver, kidney, and colon in Mthfsgt/+ animals placed on control diet compared to wild-type animals on the same diet (Figures 4A,B). Both Mthfsgt/+ and Mthfs+/+ mice placed on FCD diet exhibited decreased MTHFS levels in these tissues compared to animals on the control diet. Interestingly, MTHFS levels in both the kidney and liver were reduced by more than 60% in animals placed on the FCD relative to control diet (p < 0.01 for liver, and p = 0.02 for kidney) when densitomety values were analyzed using a two-way ANOVA (data not shown). MTHFS protein levels in all tissues were not significantly different between Mthfsgt/+ and Mthfs+/+ mice placed on FCD diet.
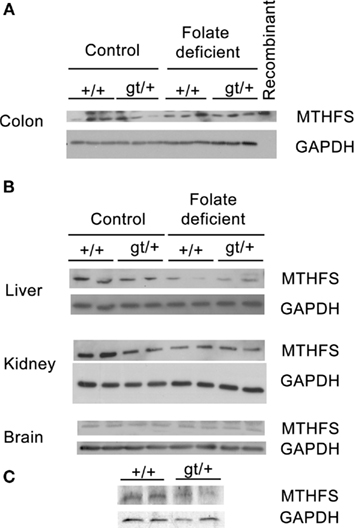
Figure 4. Western blot of Mthfs+/+ and Mthfsgt/+ tissues. Tissue lysates from liver, kidney, colon (25 μg total protein per lane), and brain (50 μg total protein per lane) were separated on a 10% SDS-PAGE gel. Membranes were probed with sheep anti-mouse MTHFS antibody (described in section Materials and methods). Glycerol aldehyde phosphate dehydrogenase (GAPDH) was used as a loading control. (A) Three samples per genotype and diet were run for colon, and this blot includes mouse recombinant MTHFS (purified as described previously Anguera and Stover, 2006). (B) Two samples were run per genotype per diet for liver, kidney and brain. (C) Cell lysates from Mthfs+/+ and Mthfsgt/+ MEF cells, n = 2 cell lines per genotype.
Mthfsgt/+ MEFs Demonstrate Reduced de novo Purine Synthesis
The effect of Mthfs disruption on de novo purine biosynthesis relative to purine synthesis through the folate-independent salvage pathway was determined in murine embryonic fibroblasts isolated from Mthfsgt/+ and Mthfs+/+ embryos. In this assay [14C]-formate is incorporated into purines via the folate-dependent de novo purine synthesis pathway and [3H]-hypoxanthine is incorporated via the purine salvage pathway, in which hypoxanthine is converted to inosine monophosphate (IMP) and then to adenosine and guanosine monophosphate in a folate-independent manner (Yamaoka et al., 1997). Therefore, the ratio of 14C/3H in nuclear DNA serves as a measure of the ability of de novo purine synthesis to suppress contributions from the salvage pathway. As shown in Figure 5, Mthfs+/+ MEFs displayed approximately two-fold greater 14C/3H than Mthfsgt/+ cells (p < 0.05), indicating that cells with 50% reduced MTHFS expression (p < 0.05, Figure 4C) have reduced de novo purine synthesis. Therefore, these data support the previous finding that MTHFS expression enhances de novo purine synthesis (Field et al., 2006).
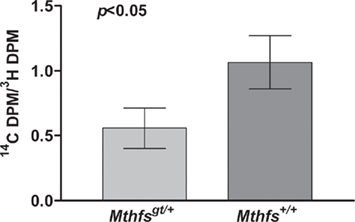
Figure 5. Mthfsgt/+ mouse embryonic fibroblasts (MEFs) exhibit less efficient de novo purine biosynthesis. MEFs were isolated as described in Section “Materials and methods” and cultured in the presence of [3H]-hypoxanthine and [14C]-formate. [3H]-Hypoxanthine is converted to purines via the folate-independent salvage pathway, whereas [14C]-formate is incorporated into purines via the de novo pathway. The 14C/3H DPM ratio was determined in nuclear DNA from two independent cell lines for both Mthfs+/+ and Mthfsgt/+ genotypes. The 14C and 3H content (dpm) in resulting fractions were quantified on a Beckman Coulter LS6500 Scintillation Counter. Variation is expressed as standard deviation of the mean (two cell lines per genotype), data were analysed using a student’s t-test.
Mthfsgt/+ MEFs do not Demonstrate Altered de novo dTMP Synthesis.
The effect of Mthfs disruption on de novo thymidylate biosynthesis relative to thymidine synthesis through the folate-independent salvage pathway was determined in murine embryonic fibroblasts isolated from Mthfsgt/+ and Mthfs+/+ embryos. This assay is similar to the formate suppression assay with the exception that Mthfsgt/+ and Mthfs+/+ MEFs were incubated with [14C]-deoxyuridine and [3H]-thymidine; deoxyuridine is incorporated in thymidylate via the folate-dependent de novo synthesis pathway and thymidine is incorporated via the salvage pathway (Oppenheim et al., 2001; Macfarlane et al., 2011). As shown in Figure 6, there is no difference in de novo thymidylate synthesis capacity in cells with reduced MTHFS expression.
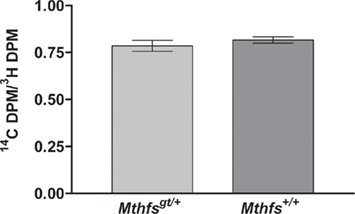
Figure 6. Mthfsgt/+ mouse embryonic fibroblasts (MEFs) do not exhibit altered de novo thymidylate biosynthesis. MEFs were isolated as described in Section “Materials and methods” and cultured in the presence of [3H]-thymidine and [14C]-dUMP. [3H]-thymidine is converted to thymidylate via the folate-independent salvage pathway, whereas [14C]-dUMP is incorporated into thmidylate via the de novo pathway. The 14C/3H DPM ratio was determined in nuclear DNA from two independent cell lines for both Mthfs+/+ and Mthfsgt/+ genotypes. The 14C and 3H content (dpm) in resulting fractions were quantified on a Beckman Coulter LS6500 Scintillation Counter. Variation is expressed as standard deviation of the mean (two cell lines per genotype), data were analysed using a student’s t-test. dUMP, deoxyuridine monophosphate.
Plasma of Mthfsgt/+ Mice Exhibits Altered Folate Levels
The effect of MTHFS depletion on plasma and tissue folate concentrations were determined for Mthfsgt/+ and Mthfs+/+ mice. Plasma folate levels were lower for animals on FCD at both time points compared to animals fed the control diet (p < 0.001; Tables 2 and 3). Mthfsgt/+ male mice exhibited a 14% reduction in plasma folate compared to Mthfs+/+ mice on the control diet at 5 weeks (p < 0.001; Table 2). Folate levels in liver, kidney, and colon were lower in animals on FCD at 5-weeks (Table 2) and in liver and colon after 11 weeks (Table 3); there was no effect on brain folate with the FCD, nor did Mthfs genotype influence folate levels in liver, kidney, or colon (Tables 2 and 3).
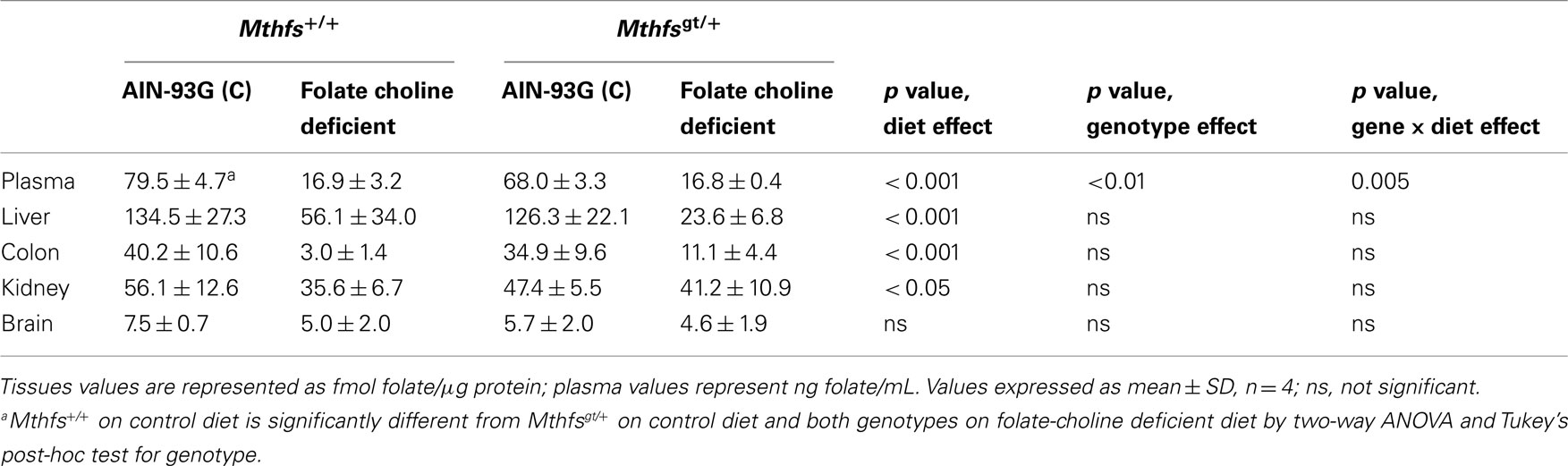
Table 2. Folate levels of Mthfs+/+ and Mthfsgt/+ tissues after 5 weeks on AIN-93G and AIN-93G lacking folate and choline.
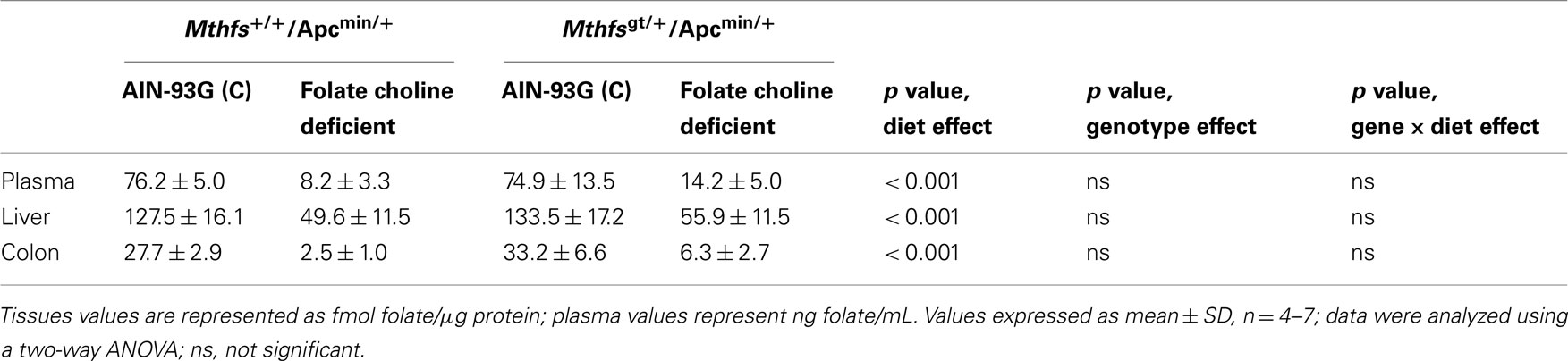
Table 3. Folate levels of Mthfs+/+/Apcmin/+ and Mthfsgt/+/Apcmin/+ tissues after 11 weeks on AIN-93G and AIN-93G lacking folate and choline.
Mthfs Disruption does not Modify Tumor Development in the Apcmin/+ Model
The effect of MTHFS disruption on intestinal tumor incidence was determined by crossing Mthfsgt/+ and Mthfs+/+ mice to the intestinal carcinogenesis model Apcmin/+ mice. Apcmin/+, Mthfsgt/+ and Apcmin/+, Mthfs+/+ mice were maintained on either the control or FCD diet for 11 weeks. As shown in Table 4, there were no differences in tumor number in the small intestine nor the colon in Mthfsgt/+/Apcmin/+ mice compared to Mthfs+/+/Apcmin/+ controls. The folate/choline deficient diet did not modify total tumor number, and there were no genotype x diet interactions (Table 4). Similarly, no genotype, diet, or genotype x diet effects on tumor size (tumor area) were observed in either the small intestine or colon (data not shown).
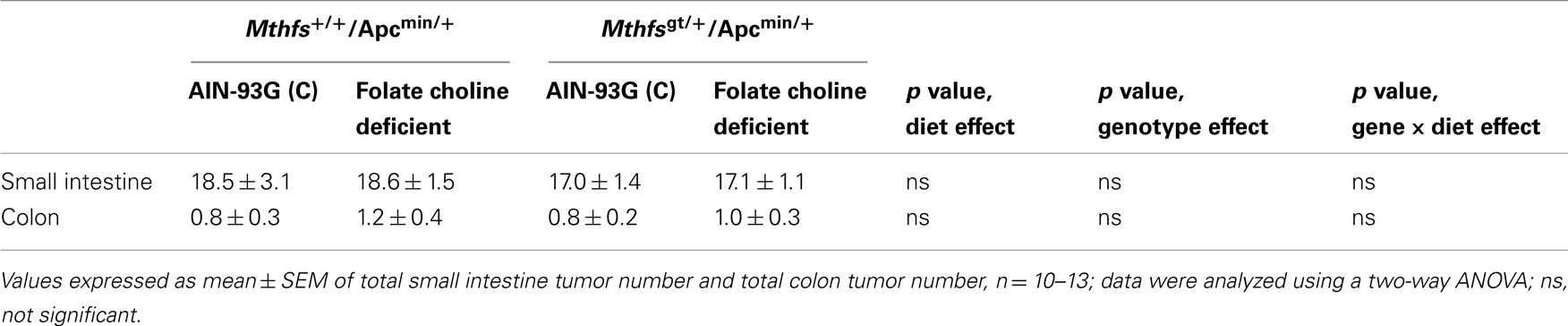
Table 4. Tumor number in small intestine and colon of and Mthfs+/+/Apcmin/+Mthfsgt/+/Apcmin/+ after 11 weeks on AIN-93G and AIN-93G lacking folate and choline.
MTHFS co-localizes with Enzymes of de novo Purine Synthesis
Recently, all the enzymes necessary for de novo purine synthesis were found to co-localize to a multi-enzyme “purinosome” complex under purine-depleted conditions (An et al., 2008). Two of the enzymes in this complex, AICARFT and GARFT, require the folate cofactor 10-formylTHF. Interestingly, the enzyme responsible for the synthesis of 10-formylTHF, C1-THF synthase, is not a part of the “purinosome,” suggesting that another protein is required to deliver 10-formylTHF to the complex. To test our hypothesis that MTHFS functions to provide 10-formylTHF for de novo purine biosynthesis (Field et al., 2006, 2009), the localization of MTHFS within the “purinosome” was investigated in HeLa cells. As shown in Figures 7A–C, MTHFS-GFP expression is diffuse throughout the cytoplasm and nucleus of cells maintained in purine-rich αMEM. However, in cells maintained in purine-depleted media (Figures 7D–F), MTHFS-GFP expression is clustered in the cytoplasm of approximately 50% of observed cells. Since this clustering mimicked the clustering of the “purinosome,” MTHFS-GFP was co-expressed with hFGAMS-OFP or hTrifGART-OFP (GARFT) under purine-depleted conditions. As shown in Figure 8, MTHFS-GFP is found in the same cytoplasmic cluster as both hFGAMS-OFP and hTrifGART-OFP. These data demonstrate that MTHFS is a component of the purinosome and may function to deliver 10-formylTHF to this complex.
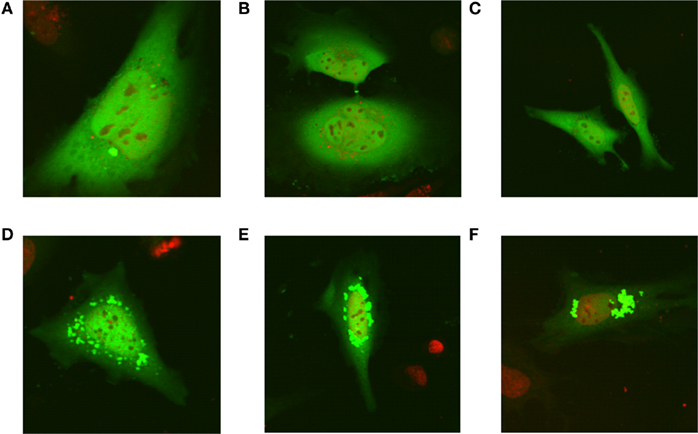
Figure 7. MTHFS-GFP forms clusters in purine-depleted media. The MTHFS-GFP fusion protein was expressed in HeLa cells maintained in either purine-rich αMEM medium (A–C) or purine-depleted RPMI-1640 medium (D–F) as described in Section “Materials and methods.” Nuclear DNA stain in red (DRAQ5) and MTHFS-GFP is green.
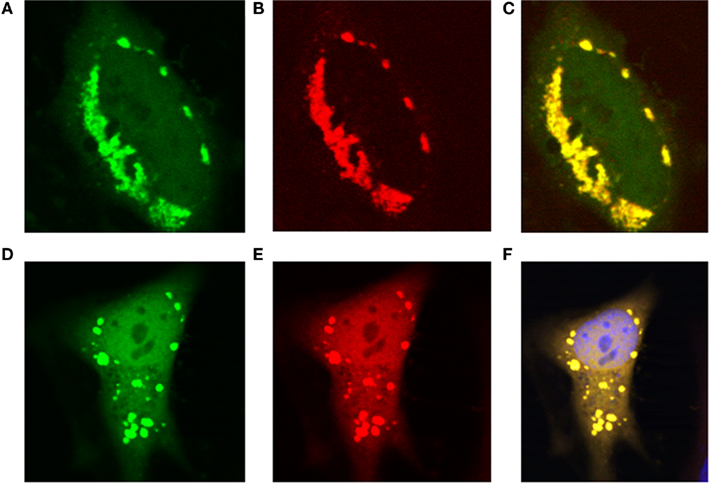
Figure 8. MTHFS-GFP co-localizes with hTrifGART-OFP and hFGAMS-OFP under purine-depleted conditions. Fluorescent fusion proteins were expressed in HeLa cells maintained in purine-deficient RPMI as described in Section “Materials and methods.” (A) hMTHFS-GFP fusion protein, (B) hFGAMS-OFP fusion protein, (C) merge of (A) and (B). (D) hMTHFS-GFP fusion protein, (E) hTrifGART-OFP fusion protein, (F) merge of (D) and (E); nuclear stain (DRAQ5) shown in blue.
Sumo Modification Targets MTHFS to the “Purinosome” Complex
The MTHFS primary sequence contains two conserved SUMO modification sites at residues K140 and K190. Recombinant murine MTHFS was shown to be a substrate for sumoylation in vitro using the SUMOlink SUMO-1 kit (Active Motif; Figure 9). To determine if MTHFS is sumoylated in vivo, an immunoprecipitation with α-SUMO1 antibody was performed in extracts from cells transfected with constructs expressing either MTHFS-GFP or MTHFS-K140R, K190R-GFP. Significantly less MTHFS-K140R, K190R-GFP protein was immunoprecipitated compared to MTHFS-GFP protein (Figure 10), indicating that K140 and/or K190 are SUMO modified in HeLa cells, but that additional MTHFS lysine(s) are SUMO modified other than K140 and K190.
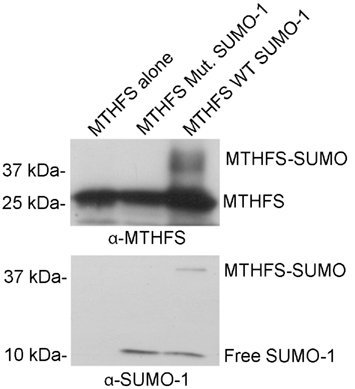
Figure 9. MTHFS is sumoylated in vitro. Purified recombinant mouse MTHFS was SUMOylated using the SUMOlink SUMO-1 kit (Active Motif) according to manufacturer’s instructions. Membranes were probed with 1:5,000 dilution of sheep anti-mouse MTHFS antibody (described in section Materials and methods) or 1:10,000 α-SUMO1 antibody (ActiveMotif).
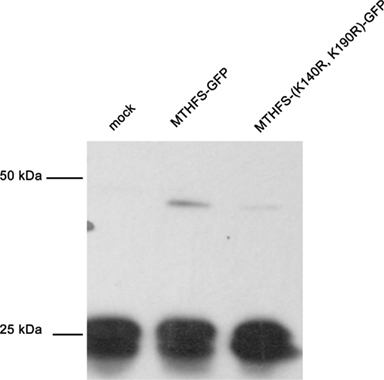
Figure 10. Sumo Immunoprecipitation. The MTHFS-GFP fusion protein was expressed in HeLa cells maintained in purine-deficient RPMI-1640 using Lipofectamine 2000 (Invitrogen), as described above. Mock transfection contained only Lipofectamine 2000. Cell extracts (180 μg/sample) were incubated with 4 μg α-SUMO-1 antibody (Active Motif) overnight at 4°C. Dynabeads Protein G (Invitrogen) were used to pull down protein complexes, which were eluted using 1 × SDS-PAGE sample buffer and resolved on a 12% SDS-PAGE acrylamide gel. Membranes were probed with 1:5,000 dilution of sheep anti-mouse MTHFS antibody (described in section Materials and methods).
To determine if MTHFS sumoylation was essential for its co-localization with the purinosome, the MTHFS-K140R, K190R-GFP fusion protein was expressed in Hela cells maintained in both purine-rich and purine-depleted media. Mutation of K140 and K190 ablated the clustering of MTHFS-GFP under purine-depleted conditions (Figures 11C,D). MTHFS-K140R, K190R-GFP remained diffuse throughout the cytoplasm and nucleus under both purine-rich (Figures 11A,B) and purine-depleted conditions (Figures 11C,D), whereas the control MTHFS-GFP formed punctuate clusters under purine-deficient conditions (Figures 11E,F). This suggests that sumoylation of MTHFS at K140 and/or K190 is required for the formation of the punctuate clusters and localization of MTHFS to the “purinosome.”
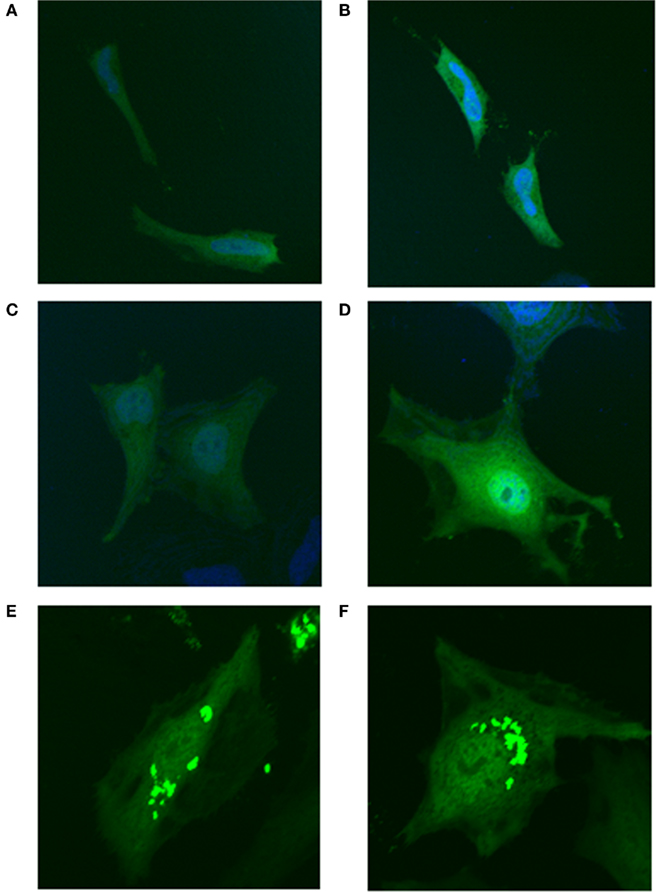
Figure 11. Sumoylation of MTHFS at K140 and/or K190 is required for clustering under purine-deficient conditions. MTHFS-K140R, K190R-GFP fusion proteins were expressed in HeLa cells and visualized by confocal microscopy as described in Section “Materials and methods.” (A, B) The MTHFS-K140R, K190R-GFP fusion protein in purine-rich αMEM; (C, D) The MTHFS-K140R, K190R-GFP fusion protein in purine-depleted RPMI-1640; (E, F) The MTHFS-GFP fusion protein in “purine-depleted” RPMI-1640. Nuclear stain (DRAQ5) shown in blue.
Formation of “Purinosome” Clusters is Abolished by Addition of Adenosine or Guanosine to Depleted Media
The original experiments describing the identification of the “purinosome” were carried out in “purine-depleted RMPI-1640” media (An et al., 2008) that lacked not only purine bases, but all nucleosides and nucleotides. To ensure that the formation of these clusters was purine-specific, the “purine-depleted” media was supplemented with either 10 mg/L adenosine or 10mg/L guanosine (concentration of nucleosides in the control purine replete media αMEM). Addition of either purine base resulted in diffuse localization of MTHFS-GFP throughout the cells (Figure 12). Addition of cytosine and uridine to the culture medium at a concentration of 10 mg/L did not perturb “purinosome” cluster formation (Figure 12).
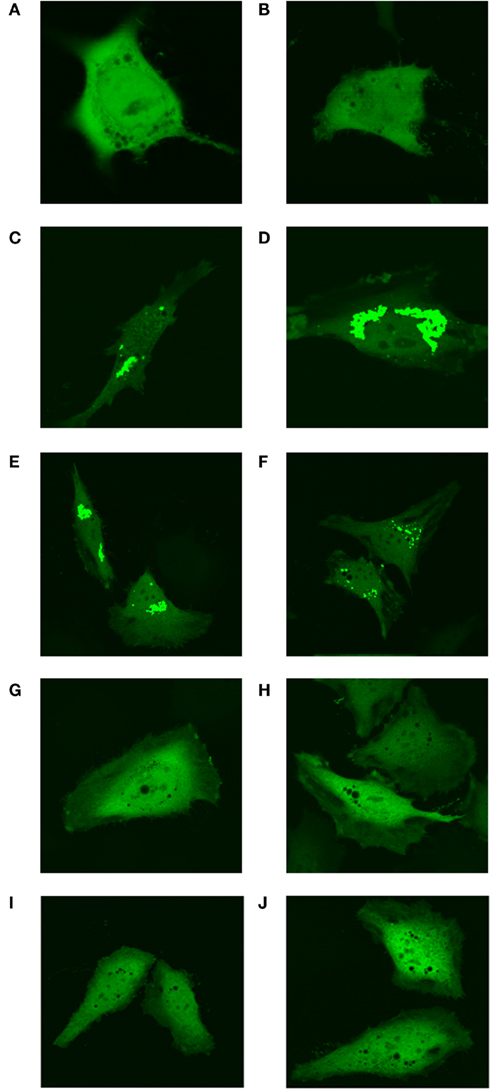
Figure 12. Formation of punctuate “purinosome” clusters is abolished by addition of adenosine and guanosine to media. The MTHFS-GFP protein was expressed in HeLa cells and visualized by confocal microscopy as described in Section “Materials and methods.” Cells were cultured with either purine-rich α-MEM (A, B), purine-depleted RPMI-1640 (C, D), or purine-depleted RPMI-1640 supplemented with individual nucleosides. The nucleosides were added at 10 mg/L, the concentration at which they are present in the purine-rich α-MEM. Pyrimidine nucleosides uridine and cytosine were added together (E, F). Purine nucleosides adenosine (G, H) and guanosine (I, J) were added individually.
The “Purinosome” forms during G1 Phase of the Cell Cycle
In purine-depleted media, roughly 50% of the cells on a given plate exhibit “purinsome” cluster formation. To determine whether “purinosome” formation was cell cycle regulated, we arrested cells maintained in purine-depleted media at G1-phase with lovastatin or at G2/M with nocodazol 24 h after transfection with MTHFS-GFP. Cell-cycle arrest was confirmed by FACS (data not shown). In cells arrested in G1-phase, nearly 90% of observed cells exhibited “purinosome” formation (Figures 13A,B). We were unable to identify any cells with clustered MTHFS-GFP in the G2/M arrested cells (Figures 13C,D). These observations suggest that the “purinosome” forms primarily during G1-phase of the cell cycle, as has been suggested previously (An et al., 2008). Since nocodazole disrupts microtubule polymerization, these data are consistent with the observation that “purinosome” formation requires intact microtubules (An et al., 2010a).
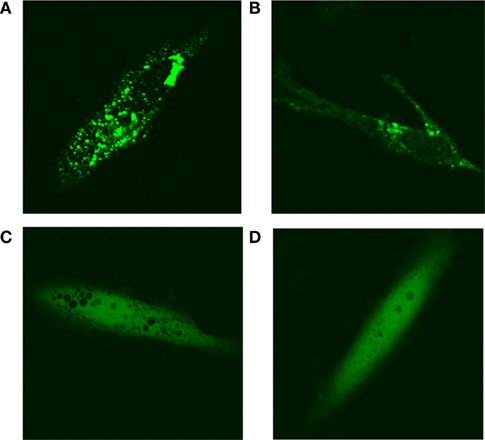
Figure 13. MTHFS-GFP forms clusters in purine-depleted media during G1-phase of the cell cycle. The MTHFS-GFP fusion protein (shown in green) was expressed in HeLa cells maintained in either purine-depleted RPMI-1640 medium as described in Section “Materials and methods.” 24 h prior to visualization, cells were treated with 60 μM mevinolin (lovastatin) to arrest in G1-phase (A, B) or 120 ng/mL nocodazole to arrest at G2/M (C, D). Cell-cycle arrest was confirmed by FACS.
Discussion
Homozygous disruption of Mthfs is embryonic lethal prior to embryonic day 9.5 (Table 1), indicating that MTHFS is an essential gene in mice. Mthfsgt/+ mice are viable and fertile and do not show any difference in weight gain nor food intake, compared to wild-type littermates (Figure 3). This result contrasts what was reported for Arabidopsis, where MTHFS is not essential (Goyer et al., 2005). The differences in the essentiality of MTHFS between species may result from differences in the ability of glutamate formiminotransferase to functionally compensate for MTHFS activity. Alternatively, MTHFS may only be essential in organisms, like mammals, that are not capable of folate biosynthesis and therefore cannot tolerate the accumulation of a single folate vitamer, including 5-formylTHF.
MTHFS enzymatically converts 5-formylTHF, a stable storage form of folate, to 5,10-methenylTHF, which is a metabolically active form of folate (Figure 1). Interestingly, MTHFS protein levels were more than 60% lower in liver and kidney tissues of animals placed on the FCD diet (Figure 4). Given that tissue folate levels were reduced significantly when animals were placed on the FCD diet (Tables 2 and 3) and that elevated MTHFS expression in cell culture models has been shown to increase rates of folate catabolism (Anguera et al., 2003), we hypothesize that MTHFS expression may be downregulated in response to folate deficiency to protect cellular folate pools. Furthermore, MTHFS protein levels were not significantly decreased in tissues from Mthfsgt/+ animals relative to wild-type littermates, suggesting that MTHFS levels may be upregulated in a post-transcriptional or post-translational fashion. A similar phenomenon was observed in N-ethyl-N-nitrosourea (ENU) mutagenized mice with an amino acid substitution in the thymidylate synthase (TYMS) gene (Ching et al., 2010).
Mthfsgt/+ mice are not more susceptible than Mthfs+/+ mice to tumorigenesis when when crossed to C57Bl/6-Apcmin/+ mice (Table 4), a model of spontaneous intestinal neoplasia (Taketo and Edelmann, 2009). Neoplastic tissues cannot effectively salvage purines, and therefore rely more heavily than normal tissue on the de novo purine synthesis pathway (Jackson and Harkrader, 1981; Habeck et al., 1994; McGuire, 2003), and Mthfsgt/+ MEFs exhibited decreased de novo purine synthesis capacity relative to the salvage pathway (Figure 5). However, because MTHFS protein levels did not differ between Mthfsgt/+ and Mthfs+/+ on the FCD diet, we cannot make conclusions regarding the role of MTHFS activity as a modifier of carcinogenesis in the Apcmin/+ model.
Mthfsgt/+ MEFs exhibit a 50% decrease in the efficiency of de novo purine biosynthesis relative to wild-type mice, suggesting that MTHFS may be essential for de novo purine biosynthesis. This finding is consistent with previous results of cell culture models that demonstrate increased MTHFS expression enhances de novo purine biosynthesis relative to the purine salvage pathway (Field et al., 2006). Furthermore, MTHFS expression in neuroblastoma has been shown to decrease the cytotoxicity of the purine-specific chemotherapeutic antifolate LY309887 (Field et al., 2009). However, the mechanism whereby MTHFS stimulates de novo purine biosynthesis and protects the pathway from antifolate inhibitors has remained elusive. The recent discovery that all six enzymes required for de novo purine synthesis co-localize during purine depletion, and the observation in this study that MTHFS-GFP co-localizes with two fluorescently tagged enzymes of the de novo purine synthesis pathway, hFGAMS-OFP and hTrifGART-OFP under purine-deficient conditions (Figures 7 and 8) suggests that MTHFS delivers the chemically labile 10-formylTHF to this “purinosome” complex and protects the complex from antifolate inhibitors. This observation is particularly interesting given that the enzyme responsible for the synthesis of 10-formylTHF, C1-THF Synthase, is not part of the “purinosome” (An et al., 2008, 2010b). The co-localization of MTHFS with the “purinosome” sheds light onto the mechanism by which the tight binding of 10-formylTHF to MTHFS results in enhanced de novo purine synthesis; the 50% reduction in the efficiency of de novo purine biosynthesis in Mthfsgt/+ MEFs indicates that MTHFS and 10-formylTHF may be rate limiting for de novo purine biosynthesis.
These data also demonstrate that MTHFS localization to the “purinosome” complex is dependent on MTHFS sumoylation. The small ubiquitin-related modifier (SUMO, NP_001005781) is a 10 kDa protein that covalently attaches to lysine residues primarily, but not exclusively, in ΨKxE motifs (where Ψ is a hydrophobic amino acid, K is the lysine that is modified and x is any amino acid). This process is facilitated by several enzymes: AOS1/UBA2, the E1 activating enzyme; UBC9, the E2 conjugating enzyme, which recognizes the acceptor site on the target protein; E3 ligase. Sumoylation has been shown to both enhance and inhibit protein-protein interactions and plays an essential role in the intracellular targeting of proteins (Geiss-Friedlander and Melchior, 2007). For instance, the folate-dependent enzymes of the thymidylate cycle have been shown to localize to the nucleus in a sumoylation-dependent fashion during S-phase of the cell cycle (Woeller et al., 2007). In this study, MTHFS is shown to be sumoylated in both an in vitro sumoylation assay (Figure 9) and in HeLa cell transfected with MTHFS-GFP (Figure 10). Mutation of lysine to arginine on the two potential SUMO modification sites results in impaired sumoylation in HeLa cells (Figure 10). Interestingly, MTHFS did not form clusters under purine-deficient conditions when K140 and K190 were mutated to arginine (Figure 11), demonstrating that sumoylation at either or both of these sites is required for complex formation and may enhance the protein-protein interactions underlying the assembly of this complex.
MTHFS-GFP was shown to localize with the “purinosome” in nucleotide/nucleoside-deficient RPMI-1640 media as in the original reports identifying the “purinosome” (An et al., 2008, 2010b). Nucleotide supplementation demonstrated that MTHFS-GFP was only associated with the purinosome when exogenous purines were not available; MTHFS-GFP did not form clusters when purine nucleosides (adenosine or guanosine) were added to the media (Figure 12). Conversely, addition of pyrimidines (uridine and cytidine) to the culture medium did not impair formation of “purinosome” with MTHFS-GFP (Figure 12). Furthermore, localization of MTHFS-GFP to “purinosome” clusters occurs in cells arrested in G1-phase of the cell cycle (Figures 13A,B), but not in cells arrested at the G2/M transition (Figures 13C,D). This is consistent with the observation that “purinosomes” formation is reversible and responds to cellular demands for nucleotides required to meet energetic needs and to maintain balanced nucleotide pools for DNA replication during S-phase and for the G1/S transition (Kondo et al., 2000; Mathews, 2006; An et al., 2008). Taken together, the data presented further support the suggestion that MTHFS responds to reduced intracellular purine levels presumably by delivering 10-formylTHF to the enzymes of de novo purine synthesis that require this co-factor, GARFT and AICARFT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Amanda MacFarlane, Anna Beaudin, Cheryll Perry, Rachel Slater, and Qinghui Ai for technical assistance. We thank Songon An and Stephen Benkovic of The Pennsylvania State University for the generous gift of hFGAMS-OFP and hTrifGART-OFP plasmids. This work was supported by Public Health Service grant DK58144.
References
An, S., Deng, Y. J., Tomsho, J. W., Kyoung, M., and Benkovic, S. J. (2010a). Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc. Natl. Acad. Sci. U.S.A. 107, 12872–12876.
An, S., Kyoung, M., Allen, J. J., Shokat, K. M., and Benkovic, S. J. (2010b). Dynamic regulation of a metabolic multi-enzyme complex by protein kinase CK2. J. Biol. Chem. 285, 11093–11099.
An, S. G., Kumar, R., Sheets, E. D., and Benkovic, S. J. (2008). Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320, 103–106.
Anguera, M. C., and Stover, P. J. (2006). Methenyltetrahydrofolate synthetase is a high-affinity catecholamine-binding protein. Arch. Biochem. Biophys. 455, 175–187.
Anguera, M. C., Suh, J. R., Ghandour, H., Nasrallah, I. M., Selhub, J., and Stover, P. J. (2003). Methenyltetrahydrofolate synthetase regulates folate turnover and accumulation. J. Biol. Chem. 278, 29856–29862.
Beaudin, A. E., and Stover, P. J. (2009). Insights into metabolic mechanisms underlying folate-responsive neural tube defects: a minireview. Birth Defects Res. A Clin. Mol. Teratol. 85, 274–284.
Bensadoun, A., and Weinstein, D. (1976). Assay of proteins in the presence of interfering materials. Anal. Biochem. 70, 241–250.
Bertrand, R., and Jolivet, J. (1989). Methenyltetrahydrofolate synthetase prevents the inhibition of phosphoribosyl 5-aminoimidazole 4-carboxamide ribonucleotide formyltransferase by 5-formyltetrahydrofolate polyglutamates. J. Biol. Chem. 264, 8843–8846.
Blount, B. C., Mack, M. M., Wehr, C. M., Macgregor, J. T., Hiatt, R. A., Wang, G., Wickramasinghe, S. N., Everson, R. B., and Ames, B. N. (1997). Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. U.S.A. 94, 3290–3295.
Ching, Y. H., Munroe, R. J., Moran, J. L., Barker, A. K., Mauceli, E., Fennell, T., Dipalma, F., Lindblad-Toh, K., Abcunas, L.M., Gilmour, J. F., Harris, T. P., Kloet, S. L., Luo, Y., Mcelwee, J. L., Mu, W., Park, H. K., Rogal, D. L., Schimenti, K. J., Shen, L., Shindo, M., Shou, J. Y., Stenson, E. K., Stover, P. J., and Schimenti, J. C. (2010). High resolution mapping and positional cloning of ENU-induced mutations in the Rw region of mouse chromosome 5. BMC Genet. 11, 106.
Field, M. S., Anguera, M. C., Page, R., and Stover, P. J. (2009). 5,10-Methenyltetrahydrofolate synthetase activity is increased in tumors and modifies the efficacy of antipurine LY309887. Arch. Biochem. Biophys. 481, 145–150.
Field, M. S., Szebenyi, D. M., and Stover, P. J. (2006). Regulation of de novo purine biosynthesis by methenyltetrahydrofolate synthetase in neuroblastoma. J. Biol. Chem. 281, 4215–4221.
Friso, S., Choi, S. W., Girelli, D., Mason, J. B., Dolnikowski, G. G., Bagley, P. J., Olivieri, O., Jacques, P. F., Rosenberg, I. H., Corrocher, R., and Selhub, J. (2002). A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci U.S.A. 99, 5606–5611.
Geiss-Friedlander, R., and Melchior, F. (2007). Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956.
Girgis, S., Suh, J. R., Jolivet, J., and Stover, P. J. (1997). 5-Formyltetrahydrofolate regulates homocysteine remethylation in human neuroblastoma. J. Biol. Chem. 272, 4729–4734.
Goyer, A., Collakova, E., De La Garza, R. D., Quinlivan, E. P., Williamson, J., Gregory, J. F., Shachar-Hill, Y., and Hanson, A. D. (2005). 5-Formyltetrahydrofolate is an inhibitory but well tolerated metabolite in Arabidopsis leaves. J. Biol. Chem. 280, 26137–26142.
Habeck, L. L., Leitner, T. A., Shackelford, K. A., Gossett, L. S., Schultz, R. M., Andis, S. L., Shih, C., Grindey, G. B., and Mendelsohn, L. G. (1994). A novel class of monoglutamated antifolates exhibits tight-binding inhibition of human glycinamide ribonucleotide formyltransferase and potent activity against solid tumors. Cancer Res. 54, 1021–1026.
Jackson, R. C., and Harkrader, R. J. (1981). "The contributions of de novo and salvage pathways of nucleotide biosynthesis in normal and malignant cells," in Nucleosides and Cancer Treatment, eds M. N. H. Tattersall and R. M. Fox (Sydney: Academic Press), 18–31.
Jeanguenin, L., Lara-Nunez, A., Pribat, A., Mageroy, M. H., Gregory, J. F. III, Rice, K. C., De Crecy-Lagard, V., and Hanson, A. D. (2010). Moonlighting glutamate formiminotransferases can functionally replace 5-formyltetrahydrofolate cycloligase. J. Biol. Chem. 285, 41557–41566.
Kondo, M., Yamaoka, T., Honda, S., Miwa, Y., Katashima, R., Moritani, M., Yoshimoto, K., Hayashi, Y., and Itakura, M. (2000). The rate of cell growth is regulated by purine biosynthesis via ATP production and G(1) to S phase transition. J. Biochem. 128, 57–64.
Kruschwitz, H. L., Mcdonald, D., Cossins, E. A., and Schirch, V. (1994). 5-Formyltetrahydropteroylpolyglutamates are the major folate derivatives in Neurospora crassa conidiospores. J. Biol. Chem. 269, 28757–28763.
Lindenbaum, J., and Allen, R. H. (1995). “Clinical spectrum and diagnosis of folate deficiency,” in Folate in Health and Disease, ed. L. B. Bailey (New York: Marcel Dekker, Inc.), 43–73.
Macfarlane, A. J., Perry, C. A., Mcentee, M. F., Lin, D. M., and Stover, P. J. (2011). Shmt1 heterozygosity impairs folate-dependent thymidylate synthesis capacity and modifies risk of Apcmin-mediated intestinal cancer risk. Cancer Res. 71, 2098–2107.
McGuire, J. J. (2003). Anticancer antifolates: current status and future directions. Curr. Pharm. Des. 9, 2593–2613.
Melnyk, S., Pogribna, M., Miller, B. J., Basnakian, A. G., Pogribny, I. P., and James, S. J. (1999). Uracil misincorporation, DNA strand breaks, and gene amplification are associated with tumorigenic cell transformation in folate deficient/repleted Chinese hamster ovary cells. Cancer Lett. 146, 35–44.
Oppenheim, E. W., Adelman, C., Liu, X., and Stover, P. J. (2001). Heavy chain ferritin enhances serine hydroxymethyltransferase expression and de novo thymidine biosynthesis. J. Biol. Chem. 276, 19855–19861.
Shane, B. (1995). “Folate chemistry and metabolism,” in Folate in Health and Disease, ed. L. B. Bailey (New York: Marcel Dekker, Inc.), 1–22.
Stover, P., and Schirch, V. (1991). 5-Formyltetrahydrofolate polyglutamates are slow tight binding inhibitors of serine hydroxymethyltransferase. J. Biol. Chem. 266, 1543–1550.
Stover, P., and Schirch, V. (1992). Evidence for the accumulation of a stable intermediate in the nonenzymatic hydrolysis of 5,10-methenyltetrahydropteroylglutamate to 5-formyltetrahydropteroylglutamate. Biochemistry 31, 2148–2155.
Stover, P., and Schirch, V. (1993). The metabolic role of leucovorin. Trends Biochem. Sci. 18, 102–106.
Suh, J. R., Herbig, A. K., and Stover, P. J. (2001). New perspectives on folate catabolism. Annu. Rev. Nutr. 21, 255–282.
Suh, J. R., Oppenheim, E. W., Girgis, S., and Stover, P. J. (2000). Purification and properties of a folate-catabolizing enzyme. J. Biol. Chem. 275, 35646–35655.
Taketo, M. M., and Edelmann, W. (2009). Mouse models of colon cancer. Gastroenterology 136, 780–798.
Ueland, P. M., Refsum, H., Beresford, S. A., and Vollset, S. E. (2000). The controversy over homocysteine and cardiovascular risk. Am. J. Clin. Nutr. 72, 324–332.
Woeller, C. F., Anderson, D. D., Szebenyi, D. M., and Stover, P. J. (2007). Evidence for small ubiquitin-like modifier-dependent nuclear import of the thymidylate biosynthesis pathway. J. Biol. Chem. 282, 17623–17631.
Yamaoka, T., Kondo, M., Honda, S., Iwahana, H., Moritani, M., Ii, S., Yoshimoto, K., and Itakura, M. (1997). Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. J. Biol. Chem. 272, 17719–17725.
Keywords: folate, MTHFS, purinosome, SHMT, 5-formyltetrahydrofolate, purine biosynthesis, leucovorin, SUMO
Citation: Field MS, Anderson DD and Stover PJ (2011) Mthfs is an essential gene in mice and a component of the purinosome. Front. Gene. 2:36. doi: 10.3389/fgene.2011.00036
Received: 04 March 2011; Paper pending published: 03 April 2011;
Accepted: 08 June 2011; Published online: 20 June 2011.
Edited by:
Michael Felix Fenech, Commonwealth Scientific and Industrial Research Organisation, AustraliaReviewed by:
Dilip Ghosh, Neptune Bio-Innovations Pty Ltd, AustraliaSharon Ross, National Cancer Institute, USA
Copyright: © 2011 Field, Anderson and Stover. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Patrick J. Stover, Division of Nutritional Sciences, Cornell University, 315 Savage Hall, Ithaca, NY 14853, USA. e-mail:UEpTMTNAY29ybmVsbC5lZHU=