
- 1 Center for Integrated Animal Genomics, Department of Animal Science, Iowa State University, Ames, IA, USA
- 2 The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Cambridge, UK
The newly available pig genome sequence has provided new information to fine map quantitative trait loci (QTL) in order to eventually identify causal variants. With targeted genomic sequencing efforts, we were able to obtain high quality BAC sequences that cover a region on pig chromosome 17 where a number of meat quality QTL have been previously discovered. Sequences from 70 BAC clones were assembled to form an 8-Mbp contig. Subsequently, we successfully mapped five previously identified QTL, three for meat color and two for lactate related traits, to the contig. With an additional 25 genetic markers that were identified by sequence comparison, we were able to carry out further linkage disequilibrium analysis to narrow down the genomic locations of these QTL, which allowed identification of the chromosomal regions that likely contain the causative variants. This research has provided one practical approach to combine genetic and molecular information for QTL mining.
Introduction
A large number of quantitative trait loci (QTL) for economically important traits has been identified in pigs over the past 15+ years. More than 6,300 pig QTL have been deposited in the Animal QTLdb (http://www.animalgenome.org/QTLdb/) as of January 1, 2011. Despite the large number of QTL reported, the screening of QTL for causal mutations still suffers from the fact that QTL often span large chromosomal intervals, which makes their practical use in pig breeding schemes very limited. In essence, the causal variant(s) for any given QTL are likely in strong linkage disequilibrium (LD) with other genetic markers, which makes identification difficult. However, this may or may not always be the case. Previously, only a limited number of causal or presumed variants for QTL have been discovered in pigs (Milan et al., 2000; Ciobanu et al., 2001; Van Laere et al., 2003).
Sequencing of the pig genome has provided a new approach for QTL examinations. As part of the Swine Genome Sequencing Consortium (SGSC), Iowa State University allocated funds toward targeted sequencing of pig chromosome 17. The sequencing was carried out at the Wellcome Trust Sanger Institute (Hinxton, UK) and generated 70 high quality BACs ordered by overlapping tile path (Hart et al., 2007). Due to limitations using known publicly available software to assemble them for their relatively large clone sizes (>200 kbp), we have taken an ad hoc approach to combine information from several sources including the BAC finger printed clones (FPC) tiling path, comparative human maps, and overlapping BAC-end sequence blast evidence, to assemble the BAC sequences in alignment with the known linkage map. This resulted in a ∼8-Mbp chromosomal contig that harbors 19 genes or open reading frames (ORFs), which were identified by comparative synteny alignment to the human genome.
We have previously identified five meat quality QTL on pig chromosome 17 in a genome scan using an F2 population derived from a Berkshire × Yorkshire (BY) cross (Malek et al., 2001a). In order to increase the marker density under the QTL region on SSC17, we have previously added 21 new markers to the SSC17 linkage map (Ramos et al., 2006). We have added more markers in this study to facilitate the fine mapping of QTL. The objectives of the current study were to use the genome sequence information to fine map the SSC17 QTL region, identify the chromosomal region(s) most likely to contain the causative variant(s) responsible for the observed SSC17 meat quality QTL and to identify potential causative variants.
Materials and Methods
Animals and Phenotype Data
Resource population: two Berkshire sires were crossed with nine Yorkshire dams to produce nine F1 litters. From these litters, 8 sires and 26 dams were selected and crossed to generate 515 F2 individuals (Malek et al., 2001b). Growth, carcass composition and meat quality data were collected in the F2 individuals. Traits and procedures to collect the trait data were as described previously (Malek et al., 2001b).
Sequencing of Individual Genes and Addition of New Markers to the Linkage Map
Pooled DNA from BY founder animals were used to sequence 15 selected genes in the chromosomal region (correspond to 54–64 Mbp on pig assembly-10) of interest: Melanocortin 3 receptor (MC3R), Aurora kinase A (AURKA), Cleavage stimulation factor 3′ pre-RNA, subunit 1 (CSTF1), Transcription factor AP-2 gamma (TFAP2C), Bone morphogenetic protein 7 (BMP7), Protein phosphatase 4, Regulatory subunit 1-like (PPP4R1L), RAB22A member RAS oncogene family (RAB22A), vesicle-associated membrane protein (VAMP), associated protein B and C (VAPB), Phosphoenolpyruvate carboxykinase 1 (PCK1), Chromosome 20 ORFs 108 (C20orf108), 32 (C20orf32), 43 (C20orf43), 106 (C20orf106), 174 (C20orf174). The entire coding regions and the 5′ and 3′ UTR regions of the 15 genes were sequenced. A computer program, Expeditor (Hu et al., 2005) was used to design 114 sets of primers based on the completed pig SSC17 sequence.
Polymorphic sites were identified by sequence comparisons to develop PCR–RFLP tests for genotyping and subsequently mapping them. The methods used for sequencing, PCR–RFLP testing and linkage analysis were as previously described (Ramos et al., 2006).
QTL Scan
Ab initio least-squares regression interval mapping analysis was performed using an F2 model by QTL Express (Seaton et al., 2002). The analysis used 41 SSC17 markers for all meat quality traits collected from the BY resource population. The regression models for each trait included sex and slaughter date as fixed effects. Chromosome-wide significance thresholds for each individual trait were determined by random permutation of 5,000 times. In order to assess significance of QTL at the genome level, we used a genome-wide significance threshold previously determined by Malek et al. (2001a).
QTL Fine Mapping and Analysis
The QXPAK software (Perez-Enciso and Misztal, 2004), containing packages for LD association analysis, QTL segment analysis, multi-trait QTL analysis, and a multi-QTL analysis, was used to conduct detailed QTL analysis in the F2 population. We have divided the SSC17 distal region into 32 small segments, each flanked by two markers, to estimate the genetic variance of a trait explained by each segment. We tested hypothesis for all possible combinations of the significant QTL traits for multi-traits (pleiotropy), multi-QTL for the refinement of the chromosome genetic architecture. Significance threshold correction for multiple comparisons was determined based on the correlation and dependence among SNPs to estimate the number of independent tests within a gene (Cheverud, 2001). A value of P < 0.001 was therefore considered significant for the single QTL test.
Association Analyses
Association analyses were performed using a mixed model method. All models included sex, slaughter date, and marker genotype as fixed effects, while dam was fitted as a random effect. Least-squares means and SE were estimated for different genotype effects. All association analyses were performed such that a single marker was fitted at a time. The PROC MIXED procedure of SAS package was used to perform all analyses.
Additional association analyses that combined information from more than one marker at a time were also performed. The combined genotype analysis was done by grouping animals that shared common genotypes with different markers. A gene effect was declared to be significant when significant P-values were reached (P < 0.05) in both analysis of variance of the gene and the least-squares means analysis for all markers within the gene.
Results
Sequence Assembly, Candidate Gene Search, and Molecular Dissection
Sequencing of 70 selected BACs was carried out at the Wellcome Trust Sanger Institute (Hart et al., 2007). The order of the BACs was based on the minimum tiling path and best BAC-end sequence blast overlaps (Hu et al., 2006). The finished sequence of all clones comprised 7,792,673 bp that were confirmed by Hart et al. (2007). Because of an extensive conservation between SSC17 and HSA20 (Lahbib-Mansais et al., 2005), 15 candidate genes, or ORFs were selected from the homologous region of the human genome. The coding sequences of the selected genes were localized to SSC17 by blast analysis to confirm their candidacy.
We used pooled DNA to sequence exons of all candidate genes in order to detect polymorphisms by hybrid peaks on sequencing chromatograms. In total, 53 exonic and 146 intronic polymorphisms were identified. Non-synonymous SNPs were validated by additional sequence analysis of individual founder animal or by PCR–RFLP tests. Fourteen exonic polymorphisms resulted in amino acid changes. The experimental details of the 30 mapped markers are listed in Appendix.
Linkage and QTL Mapping
All genes were linked to markers previously mapped to SSC17. In Table 1, polymorphism information used to map each of the 30 genes/markers is reported. The new SSC17 linkage map for the BY population contained 41 markers and was 122.2 cM in length, which is 2.9 cM longer than previously published SSC17 map (Ramos et al., 2006).
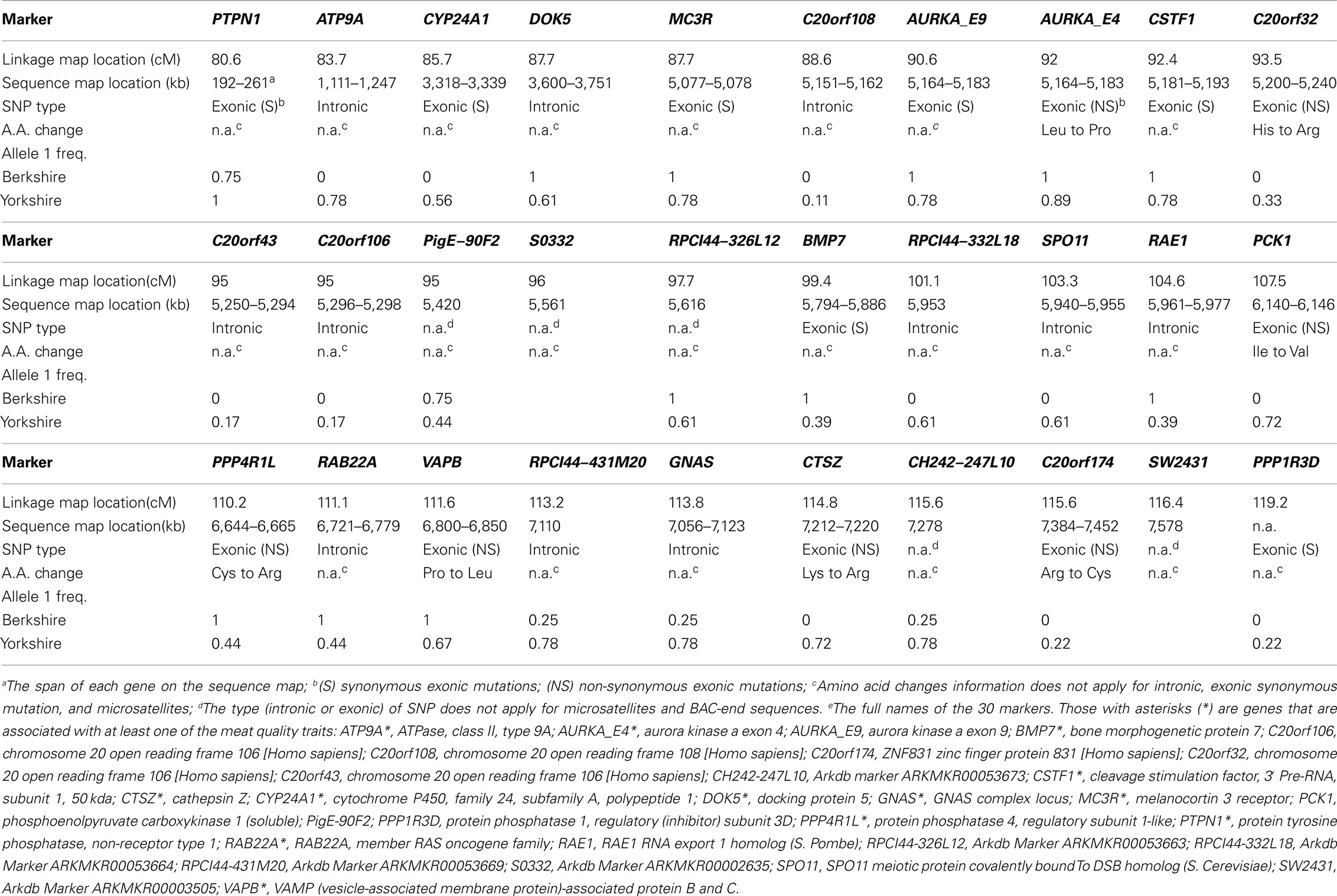
Table 1. Molecular information of 30 genetically mapped markers over the interested region of SSC17.
Quantitative trait loci analysis with QTL Express confirmed five significant meat quality QTL (Figures 1 and 2) that have been previously reported by Malek et al. (2001a). Notably, while the QTL reported by Malek et al. (2001a) were at 5% genome-wide level, several QTL, including Minolta L scores (LABLM) and Hunter L score (LABLH), and color score, are detected at 1% genome-wide level. This improvement may be due to the increased marker density used in this study. In addition, a new significant QTL was detected for average drip percentage (AVDRIP).
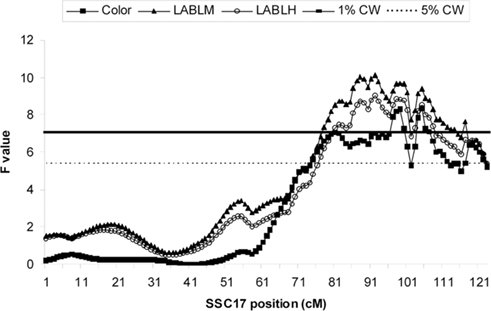
Figure 1. F-statistic curves from univariate F2 QTL analysis from QTL Express. QTL position estimates for color, 48 h Minolta L score (LABLM) and 48 h Hunter L score (LABLH) are shown. The 1 and 5% chromosome-wide significance levels were estimated to be 7.08 (solid line) and 5.38 (dashed line) respectively, while the 1 and 5% genome-wide significance levels used were 9.96 and 8.22 respectively.
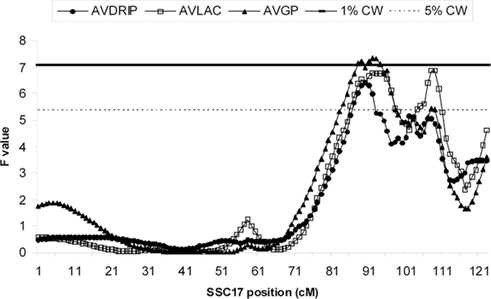
Figure 2. F-statistic curves from an univariate F2 QTL analysis from QTL Express. QTL position estimates for average drip percentage (AVDRIP), average lactate (AVLAC), and average glycolytic potential (AVGP) are shown. The 1 and 5% chromosome-wide significance levels were estimated to be 7.08 (solid line) and 5.38 (dashed line) respectively, while the 1 and 5% genome-wide significance levels used were 9.96 and 8.22 respectively.
Previously Malek et al. (2001a) reported that five QTL were located in this genome region, but each had only one single QTL peak while in this study multiple significant closely positioned QTL peaks for all traits were observed (Figures 1 and 2).
Segment Analysis, Association Analysis, and QTL Fit
Quantitative trait loci segment analysis was used to complement the classical QTL scans and was done for all significant QTL traits from the original analysis (Figure 3). The LD and QTL segment mapping analyses in the F2 population identified significant QTL peaks that were either on the same or in very nearby positions to the markers. Results combined from these analyses showed strong agreement between different approaches used to refine the QTL locations.
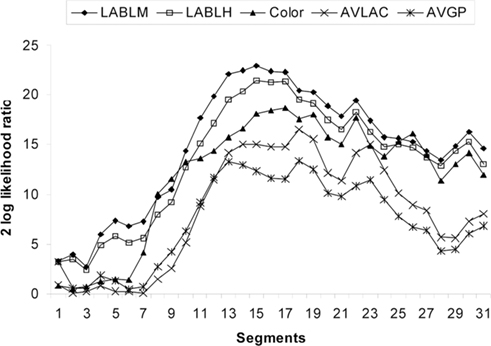
Figure 3. Log likelihood profiles of the QTL segment mapping analysis with QXPAK for 48 h Minolta L score (LABLM), 48 h Hunter L score (LABLH), color, average lactate (AVLAC), and average glycolytic potential (AVGP). Shown on the x axis are the chromosomal segments, each is flanked with 2 markers: 1 (SW335 – SWR1004); 2 (SWR1004 – SW2441); 3 (SW2441 – SIGLEC1); 4 (SIGLEC1 – MYLK2); 5 (MYLK2 – ASIP); 6 (ASIP – S0292); 7 (S0292 – S0359); 8 (S0359 – PKIG); 9 (PKIG – MMP9); 10 (MMP9 – PTPN1); 11 (PTPN1 – ATP9A); 12 (ATP9A – CYP24A1); 13 (CYP24A1 – MC3R/DOK5); 14 (MC3R/DOK5 – AURKA); 15 (AURKA – CSTF1); 16 (CSTF1 – C20orf43); 17 (C20orf43 – PigE-90F2); 18 (PigE-90F2 – S0332); 19 (S0332 – RPCI44-326L12); 20 (RPCI44-326L12 – RPCI44-332L18); 21 (RPCI44-332L18 – SPO11); 22 (SPO11 – RAE1); 23 (RAE1 – PCK1); 24 (PCK1 – RAB22A); 25 (RAB22A – RPCI44-431M20); 26 (RPCI44-431M20 – GNAS); 27 (GNAS – CTSZ); 28 (CTSZ – CH242-247L10); 29 (CH242-247L10 – SW2431); 30 (SW2431 – PPP1R3D); 31 (PPP1R3D – SW2427). The y axis shows the log likelihood values.
Linkage disequilibrium association analysis for all markers and traits on SSC17 indicated that microsatellite S0332 was significantly associated with all traits analyzed. Based on the 33 marker SSC17 linkage map, this region spanned 6 cM and included seven genes (MC3R, C20orf108, AURKA, CSTF1, C20orf32, C20orf43, and C20orf106). With the exception of MC3R, all genes are located in one BAC clone of approximately 200 kb, which further narrowed down the region.
Our multi-trait QTL analyses provided strong evidence of pleiotropy between LABLM and LABLH. This may be partly due to the fact that these biological traits/events are highly correlated. For the combination of remaining traits, results consistently supported the linkage (one QTL) hypothesis. In contrast, although the multi-QTL analyses for each trait supported the hypothesis of only one QTL per trait for all traits, the profiles from the LD association showed multiple peaks above the significance threshold. While it is possible that more than one QTL may exist for the meat quality traits on SSC17, it is of interest in the future to carry out further analyses.
Meat Color QTL on SSC17
There were 12 markers detected to be significantly (P < 0.05) associated with color, LABLM, and LABLH (Table 2). Each marker was represented by one preferred genotype and was associated with darker meat color for each of the three color traits.
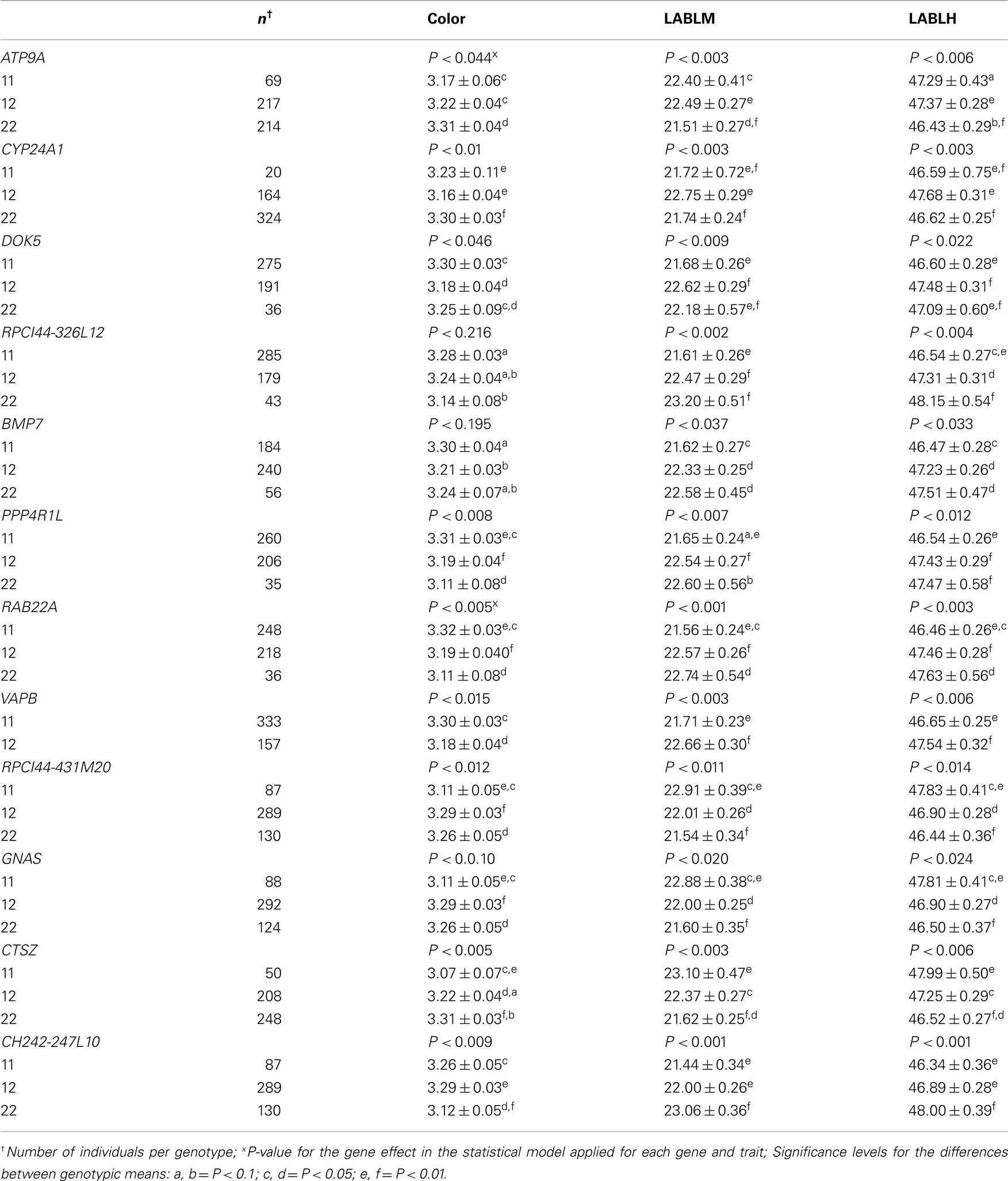
Table 2. Least-squares means and SE for the association analysis of 12 markers with meat color traits [color score; 48 h Minolta L score (LABLM); and 48 h Hunter L score (LABLH)] in F2 Berkshire × Yorkshire population.
The most significant QTL peaks for LABLM and LABLH were detected at 87 and 91 cM (Figure 1). Significant associations with the meat color traits analyzed were detected for DOK5, a gene that has the same position as MC3R in the linkage map (87.7 cM). On the linkage map, this region is collapsed to a very narrow distance due to lack of polymorphic markers. However, as it is revealed by sequence map, this region spans about 1.5 Mbp, where a gene cerebellin 4 precursor (CBLN4) was found between DOK5 and MC3R. It is yet unknown how this gene is related with the LABLM/LABLH QTL in the region.
In a significant QTL peak between 98 and 99 cM for color, LABLM, and LABLH (Figure 1), there is a polymorphic site in BMP7 that was significantly associated with these two color traits. The favorable allele analysis shows that allele 1 was fixed in the Berkshire sires while its frequency in the Yorkshire dams was only 0.39. In addition, haplotype analysis for S0332, RPCI44-326L12, and BMP7 indicated that they were significantly associated with color (P < 0.004), LABLM (P < 0.003), and LABLH (0.003). While no synonymous mutations within BMP7 were found, our analysis indicates that BMP7 maybe a plausible candidate gene for meat color QTL.
The most significant QTL peak for color, LABLM, and LABLH was near 104 cM (Figure 1) where RAE1 located. Favorable allele analysis of PPP4R1L and RAB22A showed that genotype 11 were significantly associated with color (P < 0.02), LABLM (P < 0.004), and LABLH (P < 0.008). This is in agreement with LD association analyses in which RAB22A is found to be significantly associated with all color traits. However we were not able to pinpoint the association to any specific mutation at this time.
The fourth QTL peak for color traits was found near 116–117 cM (Figure 1). Several genes displayed significant associations with all meat color traits are RPCI44-431M20 (located in GNAS intron 3), GNAS (on its intron 8), CTSZ, and CH242-247L10. Association analysis, favorable allele analysis and genotype analysis all show that animals carrying the favorable 22–22 genotypes for CTSZ and CH242-247L10 were significantly associated with color (P < 0.007), LABLM (P < 0.02), and LABLH (P < 0.03).
Average Lactate and Average Glycolytic Potential QTL on SSC17
There were eight markers associated with average lactate (AVLAC) and average glycolytic potential (AVGP; Table 3). QTL peaks for AVLAC and AVGP were near 91 cM where AURKA is found. Among the mutations found in AURKA gene, mutations in exons 4 and 5 both caused amino acid changes (Valine → Alanine, Leucine → Proline substitutions respectively) and both are in complete LD in the BY population. However, other mutations (one in exon 9 and a second one in exon 4) in the same gene are not in complete LD. Interestingly, the mutation in exon 4 was associated with both traits while the mutation in exon 9 was not. More biochemistry investigation and a better understanding of the underlying LD may be needed to determine if AURKA is a candidate gene that contributes to the AVLAC and AVGP trait variations.
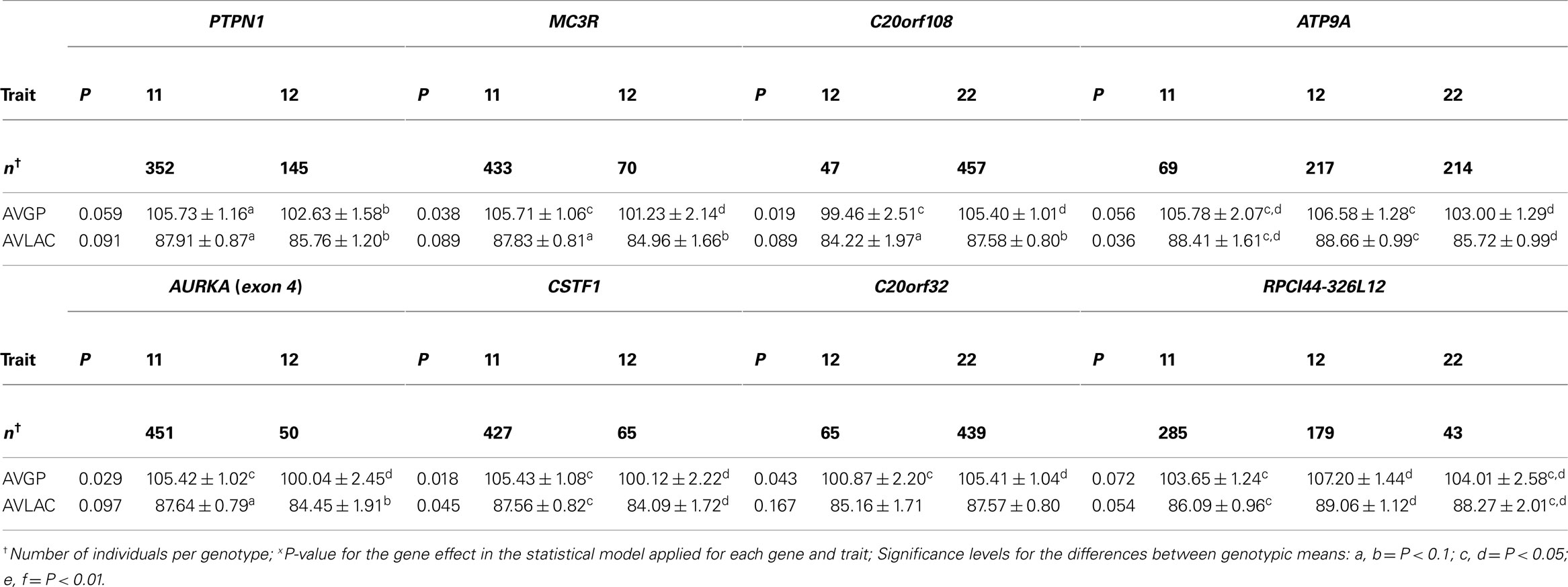
Table 3. Least-Squares means and SE for eight markers with the average glycolytic potential (AVGP) and average lactate (AVLAC) traits in F2 Berkshire × Yorkshire population.
Quantitative trait loci for AVLAC and AVGP were also detected in the 107–108 cM region where PCK1 was mapped. This gene catalyzes the conversion of oxaloacetate to phosphoenolpyruvate, the rate-limiting step in the gluconeogenesis, hence an excellent candidate among the causative factors for AVLAC and AVGP variations. However several mutations in this gene were not significantly associated with AVGP and AVLAC by association segment analyses. Further segregation analysis with a breeding scheme specifically designed for loci in this gene might help to dissect the genetic architecture in which the QTL may be pinpointed.
Discussion
The distal region of the long arm on SSC17 has been of interest since several meat quality QTL were confirmed. In this study, we have attempted to use genome sequence information to enrich the promising chromosome region with information from comparative genomics, which turned out to be very efficient for candidate gene searches by using conserved synteny across species. However, the molecular mining of candidate genes for causative variants has not been very straight forward.
First of all, identification of variants responsible for complex traits in livestock species remains a challenge due to a number of factors contributing to the difficulty in detecting, localizing, and resolving trait variations to relatively small chromosomal segments where many polymorphic markers are also available for genotyping. In this study, we combined a variety of different approaches in an attempt to dissect and rectify the QTL for meat quality QTL region on SSC17 looking for causal mutations.
The availability of genome sequence dramatically changes the extent to which genome regions can be interrogated with respect to identification of polymorphisms responsible for QTL. We see that, by going through the process of bringing the genome sequence and linkage information together, the power of genome sequence information has been limited in terms of resolving QTL imparted by LD. We have significantly improved the resolution of several overlapping meat quality QTL on SSC17. However, the final outcome has not been as we wished for in terms of resolving QTL to causal mutations. For example, the LD among multiple SNPs on AURKA gene impairs the ways to analyze the gene as a genetic unit. In contrast, haplotype analysis of S0332, RPCI44-326L12, and BMP7 helped to gain more detection power. Therefore, how to properly use the marker information to gain detection power presents a challenge. In addition, we attempted to use gene information from orthologs to aid the comparative QTL mining but this has not been fruitful.
While this study has illustrated some of the limitations of using F2 populations for fine QTL mapping, we want to realize that the expectation for causal mutations under a QTL to exist may very well be an over simplification of genetic mechanisms in which quantitative trait variations are controlled. In fact, genetic factors (QTL) for a trait may exist on several chromosomes, each of which may control the same or different part of an expression pathway in which a trait is finally formed. The multiple factors (QTL) interactions may happen in different ways, levels, or manners. As such, the success rates for traits controlled by several genes may be greatly vary in hunting for causal genes/mutations depending on the resource population used, genetic architecture of a QTL, or molecular/quantitative analysis tools available. Therefore, the ultimate success of future QTL mining may lie in system biology approaches or a more complete genetic architecture analysis involving biochemical/physiology pathways.
Conclusion
In this study, we were able to carry out LD analysis with an additional 25 new genetic markers that were identified by sequence comparison. This has helped to narrow down the genomic locations of these QTL to more confined regions that likely contain the causative variants. This research has also provided one practical approach to combine genetic and molecular information for QTL mining.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by Sygen International and the Iowa Agriculture and Home Economics Experimental Station, State of Iowa, and Hatch funds. Financial support from Iowa State University and the Iowa Pork Producers Association for the SSC17 sequencing is highly appreciated. Financial support for Antonio Marcos Ramos was provided by FCT Fellowship BD-6877-2001. Useful contributions and discussions from Dr. Graham Plastow is valued. The authors would like to also thank Dr. Miguel Perez-Enciso’s assistance with the QXPAK analyses, Dr. Hauke Thomsen and Jong Joo Kim on the initial QTL analyses.
References
Cheverud, J. M. (2001). A simple correction for multiple comparisons in interval mapping genome scans. Heredity 87, 52–58.
Ciobanu, D. C., Bastiaansen, J., Malek, M., Helm, J., Woollard, J., Plastow, G. S., and Rothschild, M. F. (2001). Evidence for new alleles in the protein kinase AMP-activated, subunit gene associated with low glycogen content in pig skeletal muscle and improved meat quality. Genetics 159, 1151–1162.
Hart, E. A., Caccamo, M., Harrow, J. L., Humphray, S., Gilbert, J. G. R., Trevanion, S., Hubbard, T., Rogers, J., and Rothschild, M. F. (2007). Lessons learned from the initial sequencing of the pig genome: comparative analysis of an 8 MB region of pig chromosome 17. Genome Biol. 8, R168.
Hu, Z.-L., Glenn, K., Ramos, A. M., Otieno, C. J., Reecy, J. M., and Rothschild, M. F. (2005). Expeditor: a pipeline for designing primers using human gene structure and livestock animal EST information. J. Hered. 96, 80–82.
Hu, Z.-L., Humphray, S., Scott, C., Rogers, J., Ramos, A. M., Reecy, J. M., and Rothschild, M. F. (2006). “From genome scan to fine mapping to sequence information: steps towards the clarification of the mechanisms controlling porcine chromosome 17 QTL for meat quality,” in Proceeding of XIV PAG, San Diego, CA, 246.
Lahbib-Mansais, Y., Karlskov-Mortensen, P., Mompart, F., Milan, D., Jorgensen, C. B., Cirera, S., Gorodkin, J., Faraut, T., Yerle, M., and Fredholm, M. (2005). A high-resolution comparative map between pig chromosome 17 and human chromosomes 4, 8, and 20: identification of synteny breakpoints. Genomics 86, 405–413.
Malek, M., Dekkers, J. C., Lee, H. K., Baas, T. J., Prusa, K., Huff-Lonergan, E., and Rothschild, M. F. (2001a). A molecular genome scan analysis to identify chromosomal regions influencing economic traits in the pig. II. Meat and muscle composition. Mamm. Genome 12, 637–645.
Malek, M., Dekkers, J. C., Lee, H. K., Baas, T. J., and Rothschild, M. F. (2001b). A molecular genome scan analysis to identify chromosomal regions influencing economic traits in the pig. I. Growth and body composition. Mamm. Genome 12, 630–636.
Milan, D., Jeon, J. T., Looft, C., Amarger, V., Robic, A., Thelander, M., Rogel-Gaillard, C., Paul, S., Iannuccelli, N., Rask, L., Ronne, H., Lundstrom, K., Reinsch, N., Gellin, J., Kalm, E., Le Roy, P., Chardon, P., and Andersson, L. (2000). A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288, 1248–1251.
Perez-Enciso, M., and Misztal, I. (2004). QXPAK: a versatile mixed model application for genetical genomics and QTL analyses. Bioinformatics 20, 2792–2798.
Ramos, A. M., Helm, J., Sherwood, J., Rocha, D., and Rothschild, M. F. (2006). Mapping of 21 genetic markers to a QTL region for meat quality on pig chromosome 17. Anim. Genet. 37, 296–297.
Seaton, G., Haley, C. S., Knott, S. A., Kearsey, M., and Visscher, P. M. (2002). QTL express: mapping quantitative trait loci in simple and complex pedigrees. Bioinformatics 18, 339–340.
Keywords: meat quality QTL, pig chromosome 17, integrated analysis
Citation: Hu Z-L, Ramos AM, Humphray SJ, Rogers J, Reecy JM and Rothschild MF (2011) Use of genome sequence information for meat quality trait QTL mining for causal genes and mutations on pig chromosome 17. Front. Gene. 2:43. doi: 10.3389/fgene.2011.00043
Received: 30 March 2011; Accepted: 24 June 2011;
Published online: 14 July 2011.
Edited by:
Johan Van Arendonk, Wageningen University, NetherlandsReviewed by:
Shu-Hong Zhao, Huazhong Agricultural University, ChinaMerete Fredholm, University of Copenhagen, Denmark
Copyright: © 2011 Hu, Ramos, Humphray, Rogers, Reecy and Rothschild. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Max F. Rothschild, Department of Animal Science, Iowa State University, 2255 Kildee Hall, Ames, IA 50011, USA. e-mail:bWZyb3Roc2NAaWFzdGF0ZS5lZHU=