 Ikhide G. Imumorin1,2*
Ikhide G. Imumorin1,2* Eun-Hee Kim3 Yun-Mi Lee3 Dirk-Jan De Koning4,5
Eun-Hee Kim3 Yun-Mi Lee3 Dirk-Jan De Koning4,5 Johan A. van Arendonk4
Johan A. van Arendonk4 Marcos De Donato1,2,6
Marcos De Donato1,2,6 Jeremy F. Taylor2,7 and Jong-Joo Kim2,3*
Jeremy F. Taylor2,7 and Jong-Joo Kim2,3*- 1 Department of Animal Science, Cornell University, Ithaca, NY, USA
- 2 Animal Genetics Section, Department of Animal Science, Texas A&M University, College Station, TX, USA
- 3 School of Biotechnology, Yeungnam University, Gyeongsan, South Korea
- 4 Animal Breeding and Genomics Center, Wageningen Institute of Animal Sciences, Wageningen University, Wageningen, Netherlands
- 5 Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, Uppsala, Sweden
- 6 Laboratorio Genética Molecular, Instituto de Investigaciones en Biomedicina y Ciencias Aplicadas, Universidad de Oriente, Cumaná, Venezuela
- 7 Division of Animal Sciences, University of Missouri, Columbia, MO, USA
Parent-of-origin effects (POE) such as genomic imprinting influence growth and body composition in livestock, rodents, and humans. Here, we report the results of a genome scan to detect quantitative trait loci (QTL) with POE on growth and carcass traits in Angus × Brahman cattle crossbreds. We identified 24 POE–QTL on 15 Bos taurus autosomes (BTAs) of which six were significant at 5% genome-wide (GW) level and 18 at the 5% chromosome-wide (CW) significance level. Six QTL were paternally expressed while 15 were maternally expressed. Three QTL influencing post-weaning growth map to the proximal end of BTA2 (linkage region of 0–9 cM; genomic region of 5.0–10.8 Mb), for which only one imprinted ortholog is known so far in the human and mouse genomes, and therefore may potentially represent a novel imprinted region. The detected QTL individually explained 1.4 ∼ 5.1% of each trait’s phenotypic variance. Comparative in silico analysis of bovine genomic locations show that 32 out of 1,442 known mammalian imprinted genes from human and mouse homologs map to the identified QTL regions. Although several of the 32 genes have been associated with quantitative traits in cattle, only two (GNAS and PEG3) have experimental proof of being imprinted in cattle. These results lend additional support to recent reports that POE on quantitative traits in mammals may be more common than previously thought, and strengthen the need to identify and experimentally validate cattle orthologs of imprinted genes so as to investigate their effects on quantitative traits.
Introduction
Applications of marker-based technologies to genetic evaluation as well as to quantitative trait loci (QTL) mapping often assume Mendelian inheritance of equal expression of parental alleles in progeny (Andersson, 2001). On the other hand, genomic imprinting is the expression of only one of two copies of a gene in the progeny based on parent-of-origin of the alleles (Barlow, 1995). Several studies have reported parent-of-origin effects (POE) on quantitative traits in mammalian livestock: pigs (Vries et al., 1994; De Koning et al., 1999, 2000; Van Laere et al., 2003; Thomsen et al., 2004; Boysen et al., 2010), sheep (Cockett et al., 1996; Garfield et al., 2003; Vuocolo et al., 2007; Fleming-Waddell et al., 2009; Matika et al., 2010), and in cattle (Kuehn et al., 2007; Allan et al., 2009; Neugebauer et al., 2010; Pausch et al., 2011). In addition, reciprocal differences have been detected in B. taurus × B. indicus (BT × BI) calves for growth and carcass characteristics (Thallman et al., 1993; Rohrer et al., 1994; Amen et al., 2007a,b). Furthermore, since the calves used by Thallman et al. (1993), Rohrer et al. (1994), and Amen et al. (2007a,b) were gestated and raised by unrelated recipient cows which were randomly assigned to embryos in a multiple ovulation and embryo transfer (MOET) program, the detected reciprocal effects are not due to the maternal effects of milk production, passive immunity or uterine environment (after day 7; Thallman et al., 1993; Rohrer et al., 1994) but appear to represent real genetic or epigenetic differences. The basis for this non-Mendelian effect on intrauterine growth remains unexplained, but increasing evidence strongly suggest that epigenetic inheritance involving imprinted genes is in play (Duselis et al., 2005; Jiang et al., 2007; Loschiavo et al., 2007; Cheverud et al., 2008; Hager et al., 2008; Wolf et al., 2008). Recently, a handful of known and putative imprinted genes have reportedly been associated with quantitative traits in beef cattle (Magee et al., 2010, 2011; Berkowicz et al., 2011; Chen et al., 2011; Sikora et al., 2011).
With the increasing role of epigenetic effects on complex traits of livestock, knowledge of the genomic regions harboring these loci will lead to the identification of the underlying causal genes. To date, only 14 of the roughly 142 genes previously known to be imprinted in mammals1 have been experimentally shown to be primarily imprinted in bovine embryos and fetuses (Dindot et al., 2004; Ruddock et al., 2004; Arnold et al., 2006; Zaitoun and Khatib, 2006; Kim et al., 2007; Tveden-Nyborg et al., 2008; Flisikowski et al., 2010). However, recent work in mouse has dramatically increased the number of mammalian imprinted genes to 1,300 (Gregg et al., 2010). Since only very few genome scans to identify POE has been published in cattle, in this paper we report the results of a genome scan to detect POE–QTL affecting seven growth and carcass traits in Angus × Brahman crossbred beef cattle.
Materials and Methods
Resource Family Structure and Phenotypes
Details about resource family structures and phenotypes of this study population have previously been described by Kim et al. (2003). The Angleton reference QTL population comprised 80 Brahman, Angus grandparents, and F1 parents with 14 Angus and 15 Brahman reciprocal backcross, and three F2 families. The average number of progeny per family was 19.1 ± 6.5, and steers and heifers (n = 602) were produced by MOET using randomly assigned multiparous Brahman × Hereford crossbred recipient dams. The progeny were raised under similar conditions from birth at the Texas A&M Agricultural Experiment Station in Angleton, TX, were weaned at approximately 7 months of age, backgrounded on pasture for an average of 215 days, and fed for approximately 170 days on a corn-based finishing diet. Cattle were then transported to the Rosenthal Meat Science and Technology Center in College Station, TX, USA where they were processed. Traits analyzed in this study were birth weight (BWT); weaning weight (WWT); yearling weight (YWT); slaughter weight (SWT); hot carcass weight (HCW), a measure of trimmed final carcass weight at approximately 20 months of age; adjusted subcutaneous fat thickness between the 12th and 13th ribs (ABF); and percentage of kidney, pelvic, and heart fat relative to carcass weight (KPH). The two carcass traits were evaluated according to United States Department of Agriculture specifications (USDA, 1989). Summary statistics for the traits in the population have previously been published (Kim et al., 2003).
Genetic Map Construction, Information Content, and Segregation Distortion
A total of 357 genetic markers, mainly microsatellites, were scored for the construction of linkage maps, and the best orders and map distances between markers were determined using CRI-MAP, version 2.4 (Green et al., 1990) as previously described (Kim et al., 2003). The list of primers have been described and published in Kim et al. (2003). Four QTL genotype probabilities for an offspring, e.g., P(QQ), P(Qq), P(qQ), P(qq; Q allele is inherited from Angus and q from Brahman grand-parental breed, and the first letter indicates an allele inherited from the progeny’s sire and the latter from its dam), were derived at a given chromosomal location by using the option of combined backcross and F2 crosses in QTL Express software (Seaton et al., 2002). Information content (IC) based on these genotype probabilities was obtained (Knott et al., 1998) as:
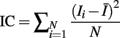
where, for paternal expression IC, Ii is [P(QQ) + P(Qq)] − [P(qQ) + P(qq)] for the ith individual, and I is the mean across all Ii values, and for maternal expression IC, Ii is [P(QQ) + P(qQ)] − [P(Qq) + P(qq)]. The more informative the marker region, the greater the value of IC which asymptotes at one.
The genotype probabilities can also be used to evaluate segregation distortion (SD) at a chromosomal location as:
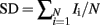
for which an excess of one parentally inherited allele, e.g., P(QQ + P(Qq), or P(qQ) + P(qq) for paternally inherited alleles, causes SD value to deviate from zero toward 1 or −1. Deviation of the SD value from “0,” i.e., under the null hypothesis condition of no SD can be tested by assuming a normal distribution under the Central Limit Theorem. It is then necessary to adjust the obtained comparison-wise P value for the test statistic to allow for multiple testing to a genome-wise significance level. As the average length of each chromosome was about 90 cM across the 29 autosomes, 45 cM was assumed to be the chromosome length unit for independence for each test, suggesting that 58 independent tests were performed across the whole genome. Consequently, the genome-wise P value was obtained by using the Bonferroni correction (Knott et al., 1998).
Parent-of-Origin QTL Analysis
Least squares interval mapping models were used for parent-of-origin QTL detection on the autosomal chromosomes. The base model was a Mendelian line-cross model (Mend), which assumes a one QTL and single-trait model with alternate QTL alleles fixed in each of the grand-parental breeds (Haley et al., 1994) is the MENDELIAN MODEL:
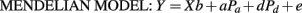
where Y is a vector of phenotypes of F2 or backcross individuals; X is a design matrix; b is a vector of fixed and covariate effects; a is the additive QTL effect, modeled as half of the difference between Angus and Brahman breed homozygotes; d is the dominance effect, modeled as the difference between the average of Angus and Brahman heterozygotes and the homozygote midpoint; Pa and Pd are vectors containing functions of genotype probabilities for each animal at the chromosomal position of the putative QTL conditional on flanking marker genotypes. The genotype probabilities were calculated differently according to the cross type of each animal. For example, the element of Pa was P(QmatQpat) − P(qmatqpat), P(QmatQpat), or −P(qmatqpat) for the F2, Angus backcross and Brahman backcross types, respectively, and e is a vector of uncorrelated residuals with constant variance. The second model was the FULL IMPRINTING MODEL:
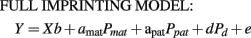
where Y, X, b, and e are as defined previously, and apat, amat, and d are the paternally inherited, maternally inherited, and dominance QTL coefficients, respectively. Vector Ppat contains probabilities of inheriting an Angus allele, Q vs. Brahman allele, q from the sire, Pmat probabilities of inheriting an Angus allele, Q vs. Brahman allele, q from the dam, and Pd probabilities of being heterozygous. The parent-of-origin genotype probabilities were also derived differently according to the cross type of each animal. For example, elements of Ppat were P(QmatQpat) + P(Qpatqmat) and P(QmatQpat) − P(qpatQmat) for type 1 (Angus × F1) and type 2 (F1 × Angus) Angus backcross progeny, respectively, and −P(qmatQpat) − P(qpatqmat) and P(Qmatqpat) − P(qpatqmat) for type 1 (Brahman × F1) and type 2 (F1 × Brahman) Brahman backcross progeny, respectively, and P(QmatQpat) + P(Qmatqpat)–P(qmatQpat) − P(qmatqpat) for F2 individuals. The following define the paternal (Pat), maternal (Mat) expression models, and null model:

where all terms are as previously defined. All models were fitted at 1 cM increments along each of the chromosomes, similar to De Koning et al. (2000).
To define a QTL as being Mendelian, paternal, maternal, or partial expression QTL, the following decision tree (Figure 1), based on Kim et al. (2003), Thomsen et al. (2004), and McElroy et al. (2006), was used with some minor modifications for the specific tests:
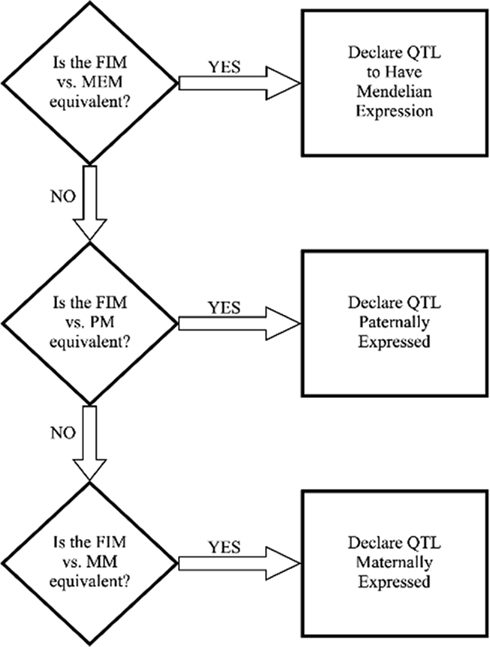
Figure 1. Decision tree to compare different QTL models. FIM, full imprinting model; MEM, Mendelian model; MM, maternal model; PM, paternal model.
If the MENDELIAN MODEL vs. the NULL MODEL was significant:
(1) The FULL IMPRINTING MODEL was tested against the MENDELIAN MODEL at the most likely QTL position detected in the full model around the region where the QTL was detected in the MENDELIAN MODEL. If this F-test was not significant, then the QTL was classified as Mend.
(2) If the FULL IMPRINTING MODEL vs. the MENDELIAN MODEL was significant, then the FULL IMPRINTING MODEL was tested against the PATERNAL and MATERNAL MODELS.
(a) If the FULL IMPRINTING MODEL vs. the PATERNAL MODEL was not significant and the FULL IMPRINTING MODEL vs. the MATERNAL MODEL was significant at the most likely QTL position under the PATERNAL MODEL, then the QTL was classified as paternally expressed.
(b) If the FULL IMPRINTING MODEL vs. the PATERNAL MODEL was significant and the FULL IMPRINTING MODEL vs. the MATERNAL MODEL was not significant at the most likely QTL position under the MATERNAL MODEL, then the QTL was classified as maternally expressed.
(c) If the FULL IMPRINTING MODEL vs. the PATERNAL MODEL and the FULL IMPRINTING MODEL vs. the MATERNAL MODEL were both significant or both not significant, then the QTL was classified as partially expressed.
If the MENDELIAN MODEL vs. the NULL MODEL was not significant:
(1) The FULL IMPRINTING MODEL was tested against the NULL MODEL. If this test was significant, then the FULL IMPRINTING MODEL was tested against the MATERNAL MODEL and PATERNAL MODEL as described in step 2 above.
(2) If the FULL IMPRINTING MODEL vs. the NULL MODEL was not significant, then the PATERNAL MODEL and MATERNAL MODEL were tested against the NULL MODEL. If the PATERNAL MODEL vs. the NULL MODEL was significant, then the QTL was classified as paternally expressed. If the MATERNAL MODEL vs. the NULL MODEL was significant, then the QTL was classified as maternally expressed. It is unlikely that tests of both the PATERNAL and MATERNAL MODELS vs. the NULL MODEL will be significant if the test of the FULL IMPRINTING MODEL vs. the NULL MODEL is not significant.
A paternally (maternally) expressed QTL is one that shows a significant allelic effect when inherited from the sires (dams) of progeny without showing a significant allelic effect when inherited from the dams (sires) of progeny. A partially expressed QTL shows unequal allelic effect in progeny, conditioned on the sex of the parent from which it was inherited. For all models, the estimated proportion of phenotypic variance explained by a detected QTL was calculated by comparing the reduction of the residual sums of squares with and without fitting the QTL in the model (Kim et al., 2003). For all models, fixed effects were included for year season of birth, gender, cross type (two double reciprocal backcrosses and F2). Covariates were weaning age for WWT, yearling age for YWT, and days on feed and age at slaughter for post-slaughter measures. For the QTL detected at the 5% GW level, 95% confidence intervals for the QTL location were obtained by using 10,000 bootstrap samples according to Visscher et al. (1996).
Permutation Tests
Significance thresholds to determine the presence of QTL, i.e., Full, Mend, Pat, or Mat model vs. Null model, were based on single-trait analysis under one QTL model. Permutation tests were performed with 10,000 replicates to empirically determine P values at the CW-significance level. Permutation of the phenotypes, fixed factors, and covariates to marker genotypes were restricted to within each of the five cross types. For a QTL detected at 5% CW-significance level, the P value for a GW significance level was then obtained using the Bonferroni correction:
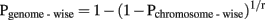
where r is the proportion of total genome length attributed to the chromosome (De Koning et al., 2001). Significance thresholds to determine type of QTL, i.e., Full model vs. Mend, Pat or Mat model were determined at the 5% comparison-wise level. The overall significance level reached by a QTL was determined using the model that corresponded to the classification of the QTL, i.e., Mend, Full (partial), Pat, or Mat.
Comparative Analysis of Genomic Regions in Human and Mouse Homologs
A list of all known imprinted genes in the human and mouse genomes was compiled from the Catalog of parent-of-origin Effects Database2, the MRC Mouse Imprinting Map Database3 and from a structured query of publications in PubMed/MEDLINE to yield a total of 1,442 genes for analyses. These were cross-referenced with NCBI resources including OMIM, UniGene, and LocusLink. Bovine orthologs are either known or putative based on the most significant alignments produced by BLAST analyses against Bta genome sequence version 5.2. The positions of imprinted genes in the mapped QTL regions were inferred from the MARC linkage map (Ihara et al., 2004), and ILTX radiation hybrid (RH) map (Everts-van der Wind et al., 2005) because the microsatellite markers used in our study are found on these maps and can be localized in the bovine reference genome sequence. In addition, we cross-referenced the public database of the human and mouse genomes for comparisons with cattle positions based on published comparative maps (Band et al., 2000; Everts-van der Wind et al., 2005). This was followed by presenting the genes to the gene ontology analysis database and we selected candidate genes by (1) its presence in matched syntenic regions between cattle vs. mouse and human genomes, (2) the most frequent gene ontology terms or by the terms that most closely related the gene to a quantitative trait of interest where available according to the methods described by Silva et al. (2007) slightly modified for our purposes. Briefly, we compiled a list of known and putative genes within 10 Mb of each mapped QTL region by using the NCBI Map Viewer of the bovine genome and compared them to the comprehensive list of all known and predicted imprinted genes in reported in mouse (Nikaido et al., 2003; Gregg et al., 2010). Some genes were selected to be associated with at least one growth-related phenotype, confirmed by citations on the NCBI PubMed browser4, whereas others were included if they mapped to the closest genomic coordinates of the linkage and RH maps and the genome sequence of orthologous genes in cattle.
Results
Overall QTL Analyses
The average marker POE IC derived from the simultaneous use of flanking markers was 83% (84%) under paternal (maternal) expression models and was higher than the 78% for the Mendelian model (Kim et al., 2003) in this same population. The chromosomal distribution of IC under POE and Mendelian models were generally consistent (results not shown). We also tested for SD at 1-cM intervals throughout the genome to ensure that there was no over-representation of Angus or Brahman alleles at any locus. We found only one position (7 cM on BTA2) at which there was evidence for an excess of Brahman alleles transmitted through F1 sires (GW P = 0.049). Since no paternally expressed QTL were detected in this region (Table 1), we conclude that the SD may either be a chance event or may be due to a nearby locus that promotes meiotic drive of Brahman alleles to cause distortion of allelic segregation away from Mendelian expected ratios. The comparison-wise −log10 P-value corresponding to a 5% CW threshold in the gender-averaged QTL models was 2.24 ± 0.17 when averaged across models and traits. However, the −log10 P-values for the 5% CW thresholds were higher for the gender-specific QTL models (3.14 ± 1.15).
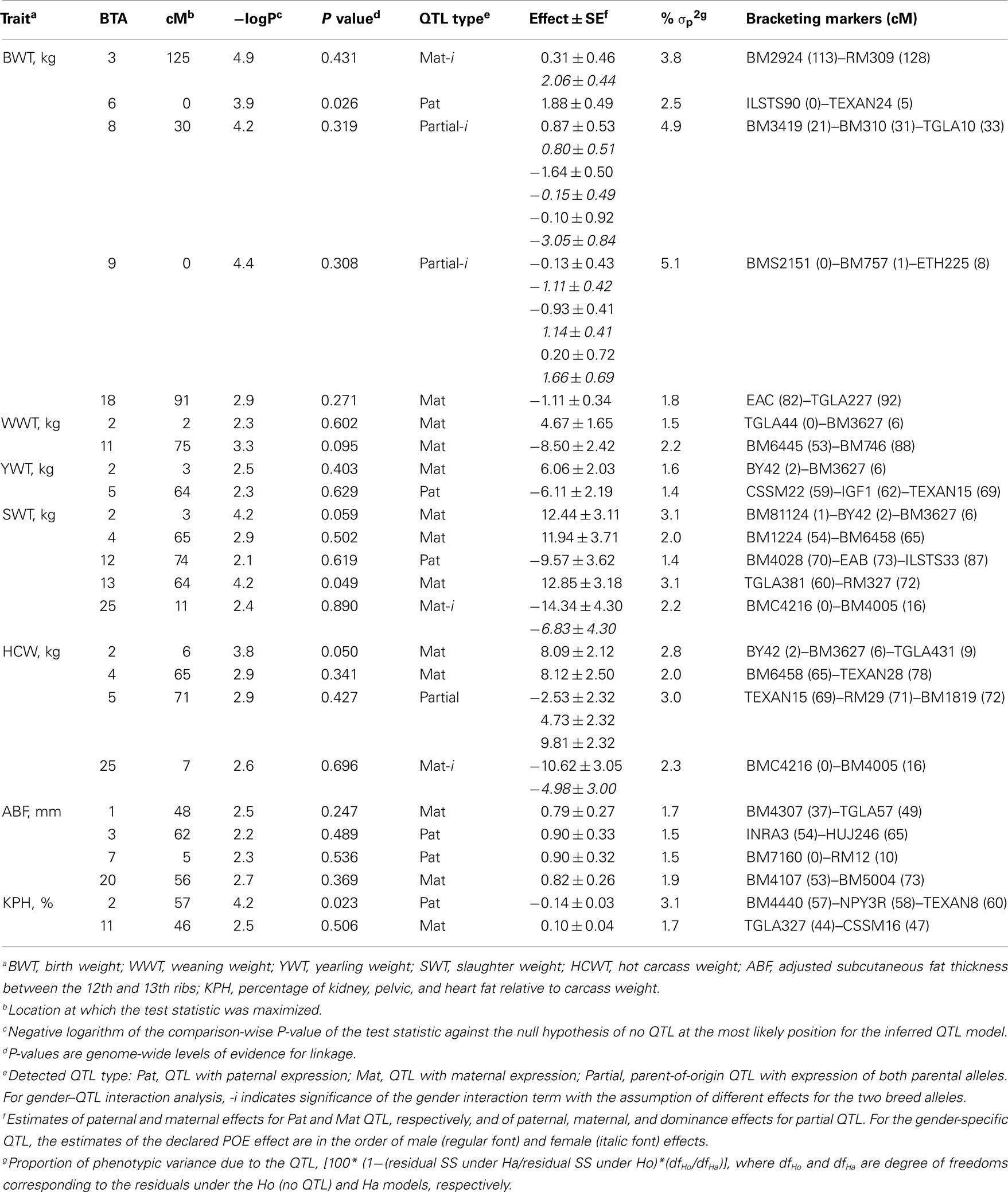
Table 1. Most likely position, test statistic values and estimated effects of parent-of-origin QTL for growth and beef carcass traits that were detected with at least 5% chromosome-wise evidence for linkage.
Twenty four POE–QTL were found on 15 autosomes with six QTL detected at the 5% GW-significance level and 18 at the 5% CW-significance level (Table 1). Six of the POE–QTL showed paternal expression. Three QTL were partially expressed and the remaining 15 QTL were maternally expressed. Five QTL had gender-specific effects for BWT on BTA3, 8, and 9, and for SWT and HCWT on BTA25. All of the detected QTL individually explained small portions of each trait’s phenotypic variance ranging from 1.4% for the SWT QTL on BTA12 and YWT QTL on BTA2 to 5.1% for the BWT QTL on BTA9 (Table 1).
Parent-of-Origin QTL Analysis for Growth and Carcass Traits
Eighteen POE–QTL or 75% of the total QTL detected on 11 chromosomes influenced growth traits compared to six or 25% of POE–QTL which influenced carcass traits on six chromosomes (Table 1). Several maternally expressed QTL affecting post-weaning growth traits were detected in the proximal region of BTA2: WWT, YWT, SWT, and HCWT (Figure 2). For all four QTL, the Angus allele conferred a weight advantage over the Brahman allele (Table 1). Two QTL affecting YWT and HCWT were detected at 64 and 71 cM on BTA5 with paternal and partial expression modes of inheritance, respectively. Significant interactions between progeny gender and POE–QTL were detected for BWT on BTA3, 8, and 9 (Table 1).
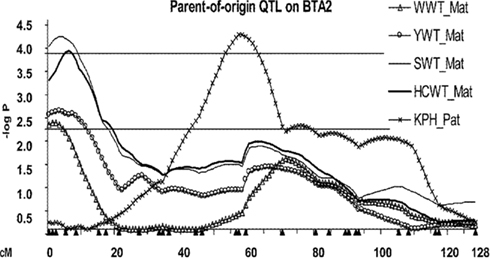
Figure 2. Profile of POE–QTL on BTA2 for growth and carcass traits (WWT, weaning weight; YWT, yearling weight; SWT, slaughter weight; HCWT, hot carcass weight; KPH, percentage of kidney, pelvic, and heart fat relative to carcass weight). The negative of the logarithm (base 10) of the comparison-wise significance value is shown for the inferred QTL models against the chromosomal position. Upper and lower lines indicate the average –log10 values of 2.24 and 3.85 across models, traits, and chromosomes for the detected QTL, representing 5% chromosome and 5% genome-wise significance thresholds, respectively. Filled triangles under the X-axis are marker positions. Mat, maternally expressed. Pat, paternally expressed.
For the QTL on BTA3, the maternal expression was only significant in female progeny. For the partially expressed BWT QTL on BTA8, the maternally expressed and dominance effects were only significant in male and female progeny, respectively. For the partially expressed BWT QTL on BTA9, paternally expressed and dominance effects were only significant in female progeny, while the effects of maternal expression were similar between male and female progeny (Table 1). There were also significant interactions between progeny gender and maternally expressed QTL for SWT and HCWT on BTA25, for which the QTL effects were only significant in male progeny (Table 1). Since the proportions of phenotypic variance due to QTL were very small at only 1.4 ∼ 5%, and the linkage map was relatively sparse, the observed confidence intervals were relatively large (results not shown).
Comparative Genomic Analysis of Imprinted Regions
Table 2 summarizes our results of known bovine orthologs of genes imprinted in human and mouse that map to the bovine chromosomal regions detected as harboring imprinted QTL in this study. This was defined as a region within 10 Mb of peak cM of estimated QTL position due to a relatively sparse linkage map and relatively large 95% confidence intervals. Our in silico comparative genomics analysis indicate that 32 out of 1,442 imprinted genes have been reported on the human and mouse homologs of bovine chromosomes 1, 2, 3, 4, 5, 6, 7, 8, 9, 11, 12, 13, and 18, and none on BTA20 and 25. Of these, two genes (GNAS and PEG3) have experimental support for imprinting status in cattle, although the demonstrated parental allele for GNAS was consistent with maternally expressed QTL for SWT on BTA13 while the paternally expressed status of ZIM2 and PEG3 are opposite of the QTL found in this study for BWT on BTA18. Although the largest number of POE–QTL mapped to BTA2, we only found one gene (IWS1 homolog) which recruits the HYPB/Setd2 histone methyltransferase to the RNA polymerase II elongation complex and is required for H3K36 trimethylation (H3K36me3), thereby affecting the histone modification state of active genes (Yoh et al., 2008) on the corresponding homologous human and mouse chromosomes. This may represent a novel region in the bovine genome that harbors imprinted genes affecting postnatal growth and development which are yet to be discovered in human and mouse.
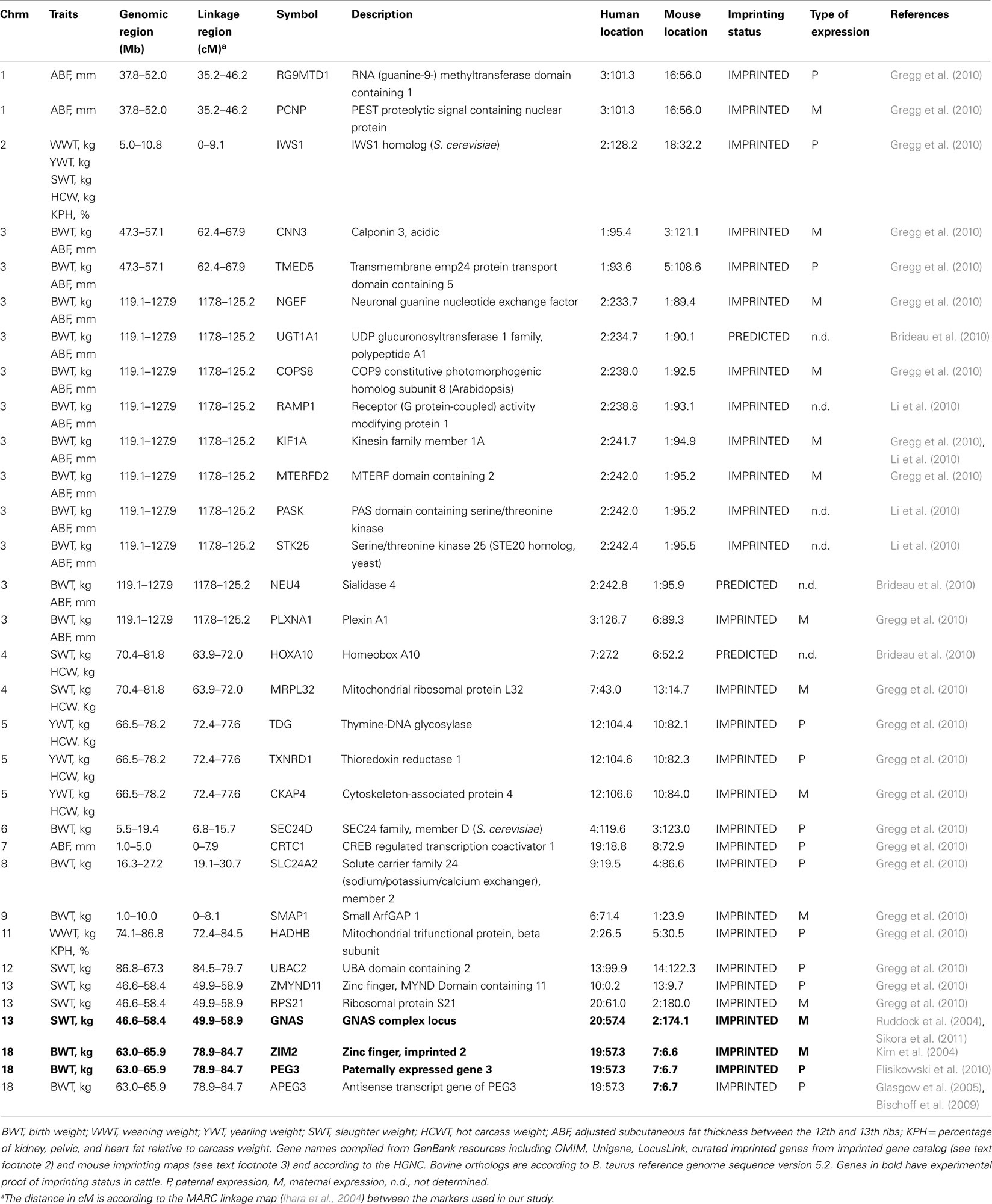
Table 2. Bovine orthologs of imprinted mammalian genes in human and mouse inside the regions harboring POE–QTL.
Discussion
Parent-of-origin QTL Analysis and Effects
Of the 15 chromosomes identified in our study as harboring POE–QTL, 13 coincide with regions of imprinted gene locations or clusters in human and mouse species (Table 2) and BTA2 also point to a possible novel region in the bovine genome that require further exploration (Table 1). Recently, it has been shown that the common assumption of line-fixed QTL made in QTL analyses face several problems in this specific design and model assumption for detecting POE–QTL in out-crossing species like livestock (De Koning et al., 2002; Sandor and Georges, 2008). It appears that demonstrating genuine imprinting by comparing QTL allele substitution effect of a proven identity-by-descent (IBD) pair of alleles upon maternal vs. paternal transmission is difficult to achieve in livestock as it is very difficult to have a sufficiently large number of F1 dams that have a genotype that is unambiguously IBD with that of one or more F1 sires (Sandor and Georges, 2008). Several remedies prescribed by Sandor and Georges (2008) may not solve the inherent intractable problems of accurately estimating parental imprinting due to the unique scenarios presented by livestock breeds and breeding. Sandor and Georges (2008) argued that the ultimate proof to test parental imprinting hypothesis is the use of thousands or even millions of single nucleotide polymorphisms (SNPs) that will allow the recognition of haplotypes that are known with virtual certainty to be IBD even in the absence of pedigree data. However, our results would appear to suggest that our methodology was robust enough to identify regions with imprinted genes that may potentially underlie genuine imprinting effects in cattle.
Our study showed a putative cluster of growth-related POE–QTL on BTA2 influencing weights between weaning and slaughter. One of the reasons may be long term implementation of selection on growth in the US beef cattle during the last several decades (Koots et al., 1994a,b) causing dramatic changes of allele frequencies for growth genes between breeds, which would enable detection of more growth QTL compared with QTL influencing composition in the breed-cross QTL models applied in this study. In addition, the identified imprinted regions may represent effects at the extreme ends of a spectrum of POE effects with potential allelic imbalance in gene expression, although more research is needed to ascertain the magnitude and direction of such effects.
Growth traits, notably BWT showed a large number of imprinted QTL, which is consistent with the known effects of genomic imprinting on growth, particularly embryonic and fetal growth (Cui et al., 2004; Constancia et al., 2005; Isles and Holland, 2005; Jiang et al., 2007; Charalambous et al., 2010). Carcass composition traits related to adiposity including ABF and KPH, had POE–QTL mapped to bovine chromosomes with relatively few known imprinted genes on their human and mouse homologs (Table 2). Obtaining significant results for body composition traits is consistent with imprinting effects on human adult obesity and body composition (Georges et al., 2003; Gorlova et al., 2003; Dong et al., 2005) and on adult obesity and body composition in mice (Casellas et al., 2009). It is now known that imprinting marks, such as DNA methylation and histone configurations, often persist into adulthood (Gorlova et al., 2003; Christensen et al., 2009; Trowbridge and Orkin, 2010; Woodfine et al., 2011; Wu et al., 2011), and that imprinting may play a physiological role in metabolism and body composition throughout life, thereby contributing both to normal variation and the architecture of complex traits rather than being restricted to prenatal and neonatal effects (Rance et al., 2005; Smith et al., 2006; Cheverud et al., 2008; Casellas et al., 2009; Hager et al., 2009; Garfield et al., 2011). Our current understanding of the function of imprinted genes is overwhelmingly biased toward growth and development (Constancia et al., 2005; Abu-Amero et al., 2006; Delaval et al., 2006; Fowden et al., 2006; Fradin et al., 2006; Smith et al., 2006, 2007; Wu et al., 2006; Jiang et al., 2007; Charalambous et al., 2010) and only recently have we begun to gain a better understanding in mice of the effects of genomic imprinting on physiological traits expressed long after embryogenesis and fetal development (Rance et al., 2005; Cheverud et al., 2008; Casellas et al., 2009; Hager et al., 2009; Garfield et al., 2011). The effects of imprinting on fetal and early postnatal development have been well characterized in mice (Cheverud et al., 2008; Garfield et al., 2011) but just beginning to get attention in other mammals. Wolf et al. (2008) recently reported that the effects of imprinted QTL in mice were mostly restricted to traits expressed after weaning, and they also show that the imprinting pattern of a locus can vary over ontogenetic time and, in contrast to current dogma, may often be stronger at later stages in life. This latter view is supported by recent analysis of Grb10 gene in adult mouse where epigenetic effects began early (Charalambous et al., 2010) and persisted into late adulthood (Garfield et al., 2011).
Positional Candidates in Identified POE–QTL Regions
Given the relatively large confidence intervals, it is possible that our 10 Mb intervals flanking the peak centimorgan may have underestimated the number of possible orthologs. But our analysis focused only on putative imprinted genes with experimental proof in other species, making the size of the interval less critical in our selection of positional candidate genes. Altogether, 32 orthologous bovine genes were identified in the QTL regions homologous to human and mouse chromosomes from a pool of 1,442 known imprinted genes (Table 2). Although there is the possibility that one or more of these genes may be genuine positional candidates directly responsible for the imprinted QTL effects, another possibility is they are close to novel imprinted genes that are yet to be identified in human and mouse or may indicate underlying genes that are bovine-specific since imprinting status of genes are not necessarily conserved across mammalian species. The maternally expressed QTL location on BTA2 may be just one QTL manifesting at different time points on different stages of growth and development (Cheverud et al., 2008; Wolf et al., 2008; Brideau et al., 2010; Garfield et al., 2011). It is noteworthy that this region does not appear to possess a correspondingly large number of known imprinted orthologs in human and mouse (see text footnotes 1 and 2) although significant contribution of maternal effect QTL to early growth in mice appears consistent with our findings (Wolf et al., 2002; Casellas et al., 2009). Recent scans for GW imprinted QTL in mice mapped them to novel locations that have not previously been associated with imprinting effects nor previously known to harbor imprinted genes at all (Cheverud et al., 2008; Wolf et al., 2008). This is due to the fact that hitherto, most known imprinted genes were biased in location toward regions of the genome with chromosomal aberrations and biased to loci with large phenotypic effects. Therefore, only a few years after it was suggested that as many as 600 genes may be imprinted in mammals (Luedi et al., 2005), more recent research with next generation sequencing of mRNA libraries analyzed for parent-of-origin bias in expression overcame that previous bottleneck and suggest as many as 1,300 imprinted genes in mouse (Gregg et al., 2010).
Of the 32 genes mapped to the bovine QTL locations, only PEG3 (Flisikowski et al., 2010) and GNAS (Khatib, 2004; Ruddock et al., 2004; Sikora et al., 2011) have experimental proof of being imprinted in cattle, while ZIM2 found in the same domain as PEG3 was biallelically expressed in testis (Kim et al., 2004). It is possible however, that the bovine ZIM2 may be imprinted in other tissues of metabolic and developmental importance other than testis or even at other stages yet to be investigated. The paternally expressed PEG3 gene (Flisikowski et al., 2010) on BTA18 lies within a QTL region identified in this study that is homologous to a well known imprinting domain. A novel mutation in this imprinting domain causing a 110-kb microdeletion results in the loss of paternal MIMT1 expression leading to late term abortion and stillbirth in cattle (Flisikowski et al., 2010). Recently SNPs within this gene has been associated with traits related to calving, calf performance and gestation length suggesting that this domain on chromosome 18 plays a key role in regulating prenatal growth, development and fertility (Magee et al., 2010).
Parent-of-Origin Effects and Longitudinal Growth and Development
Phenotypic selection for growth in beef cattle has historically been very successful due to the ease of phenotype measurement and the moderate to high trait heritabilities (Koots et al., 1994a). However, because of the moderate to high genetic correlations among growth traits at different ages (Koots et al., 1994b), selection to increase postnatal growth usually results in increased BWT and calving difficulty. This presents a special problem for understanding the genetic architecture of growth in mammalian livestock because even though phenotypic measurements represent a single point in time, phenotypes are highly correlated over an animal’s life span from birth, through weaning to slaughter. From an epigenetic standpoint, imprinted genes underlying POE may change between birth and adulthood from mono-allelic to bi-allelic expression concomitant with aging (Bennett-Baker et al., 2003; Delaval et al., 2006; Fowden et al., 2006; Smith et al., 2006; Wolf et al., 2008), even switch which parental allele is expressed (Garfield et al., 2011), or the imprinting mechanism itself could exhibit polymorphism within a population (Xu et al., 1993). None of these possible mechanisms for the regulation of gene expression have been studied in cattle.
Bos belongs to the tribe Bovini and taurine and indicine cattle are considered subspecies within the Bos genus (Lenstra and Bradley, 1999). Crossbreeding between taurine and zebu-type cattle breeds is widely practiced to take advantage of combinations of dissimilar genotypes for traits in the crossbred progeny by exploiting heterosis. Speciation within the extant Bovini tribes began about one million years ago and is not yet complete since fertile offspring are produced by B. taurus × B. indicus crosses reflecting divergence time of only 100,000–200,000 years (Bradley et al., 1996). These may underlie significant reciprocal differences in pre- and post-weaning traits observed between B. taurus × B. indicus and B. indicus × B. taurus crosses. Reciprocal differences observed in interspecies hybrids of Mus musculus × M. spretus and Peromyscus polionotus × P. maniculatus (Vrana et al., 2000; Shi et al., 2005) which are somewhat analogous to B. taurus × B. indicus crosses result in altered embryonic and placental growth, which in Mus musculus × M. spretus has been attributed to loss of imprinting (LOI; Shi et al., 2004). This LOI suggests that an imbalance in the expression of imprinted genes could underlie the divergent growth phenotypes in reciprocal mammalian hybrids (Shi et al., 2004). This has led to the hypothesis that abnormal reprogramming after fertilization and during preimplantation development in utero may partly be responsible for hybrid dysgenesis, for which a strong epigenetic basis has been demonstrated (Wolf et al., 2002; Cui et al., 2004; Duselis et al., 2005; Wu et al., 2006). This may help to partly explain our results in which a preponderance of maternally expressed QTL were found to affect growth. For now, it is unclear whether these differences result from strain-specific genetic variation, sex-specific gene expression, tissue-specific gene expression, or combinations of these effects. The observation that the most severe overgrowth is accompanied by widespread relaxation of imprinting of mostly paternally expressed genes (Shi et al., 2004) will be interesting to test in B. taurus × B. indicus crosses. The on-going speciation among Bos may involve subtle effects of LOI that may be amenable to molecular analysis in B. taurus × B. indicus crosses across multiple generations to ascertain transgenerational effects of imprinted genes.
Our inability to confirm some previous QTL influencing growth in this population (Kim et al., 2003), under the current POE model may partly be due to (1) different methods of estimating QTL breed-of-origin; (2) exclusion of the fixed effect of families nested within cross and of the random effect for recipient dams that were fitted in the previous study (Kim et al., 2003) which may have changed significance thresholds for QTL detection. Some previous Mendelian QTL detected in this population were re-classified as POE–QTL in this study, for instance QTL for BWT on BTA3, and QTL for post-weaning growth on BTA2 with increased statistical evidence for linkage (Table 1; Kim et al., 2003). This appears to be largely consistent with the predictions of simulations reported by Cui (2007), Cui et al. (2007), and De Koning et al. (2002) that imprinted QTL that may otherwise remain undetected when analyzing the genome with Mendelian models may become significant when tested against a Mendelian QTL using the null hypothesis of an imprinted QTL such as the methods used in this study. The underlying challenge of segregating QTL in founder lines leading to spurious imprinted QTL detection, especially for designs with a small number of F1 sires appear to be extenuated by the large number of imprinted genes coincident with the identified bovine QTL regions with 32 imprinted orthologs found in homologs of human and mouse. It will be worthwhile in a follow up study to see if all 32 genes are also imprinted in cattle, determine if they underlie the identified POE–QTL and ascertain whether they exert any influence on these quantitative traits.
Phenotypic Variance and Parent-of-Origin QTL Effects
Phenotypic variance explained by the detected POE–QTL in our study are consistent with an average of 1–4% reported in mice and may underscore the subtle effects of imprinting (Hager et al., 2009). Although these effects are individually small and not as common as additive genotypic effects, they are about as frequent as QTL with dominance effects in intercross populations (Cheverud et al., 2008), and our results appear to demonstrate that this may be typical for other similar populations. QTL found for HCWT at 71 cM on BTA5 and for BWT at BTA8 and BTA9 showed partial (unequal) expression of POE–QTL with the involvement of both parental alleles. Similar partial imprinting has been reported for imprinted QTL in mice where the difference between the two homozygotes is larger than the difference between the two heterozygotes (Wolf et al., 2008). This observation may reflect subtle differences resulting from complex molecular events underlying complex traits (Allis et al., 2007), because even in the simple case of monoallelic expression, patterns of effects on higher-order phenotypes, such as body weight and obesity, may not correspond to mRNA levels in a simple linear fashion (Cheverud et al., 2008). In addition, imprinted QTL effects may reflect strict mono-allelic expression during one stage of development/life and Mendelian expression at another stage (Bennett-Baker et al., 2003; Wolf et al., 2008) or as recently shown in mouse, a later stage switch of which parental allele becomes transcriptionally active (Garfield et al., 2011).
Conclusion
In conclusion, our results further demonstrate that non-Mendelian growth and carcass QTL subject to POE are not rare phenomena in livestock species and indicate the need to perform POE analyses to better understand the genetic architecture of complex traits. POE remain a complex and intriguing phenomenon to study in mammalian livestock and more methodological work is required to characterize how imprinted genes interact with the growth trajectory over an animal’s lifespan. The current paucity of known imprinted genes in the bovine genome strengthens the need to identify imprinted genes in cattle. This will enable and improve our ability to evaluate them as candidate genes underlying POE–QTL, for possible use in breeding programs under specific selection regimes such as in crossbreeding systems with specialized sire and dam lines (Vries et al., 1994; Neugebauer et al., 2010).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was financially supported by the Texas Agricultural Experiment Station, a grant from the Beef Industry Council of the National Livestock and Meat Board and by National Research Initiative Competitive Grant Nos. 94-37205-1224 and 95-37205-2273 from USDA National Institute of Food and Agriculture to Jeremy Francis Taylor and Grant No. 2006-35205-16864 to Ikhide G. Imumorin. Jong-Joo Kim was partly supported by TDPAF, Ministry of Agriculture and Forestry, Korea.
Footnotes
References
Abu-Amero, S., Monk, D., Apostolidou, S., Stanier, P., and Moore, G. (2006). Imprinted genes and their role in human fetal growth. Cytogenet. Genome Res. 113, 262–270.
Allan, M. F., Kuehn, L. A., Cushman, R. A., Snelling, W. M., Echternkamp, S. E., and Thallman, R. M. (2009). Confirmation of quantitative trait loci using a low-density single nucleotide polymorphism map for twinning and ovulation rate on bovine chromosome 5. J. Anim. Sci. 87, 46–56.
Allis, C. D., Jenuwein, T., and Reinberg, D. (2007). Epigenetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Amen, T. S., Herring, A. D., Sanders, J. O., and Gill, C. A. (2007a). Evaluation of reciprocal differences in Bos indicus x Bos taurus backcross calves produced through embryo transfer: I. Birth and weaning traits. J. Anim. Sci. 85, 365–372.
Amen, T. S., Herring, A. D., Sanders, J. O., and Gill, C. A. (2007b). Evaluation of reciprocal differences in Bos indicus x Bos taurus backcross calves produced through embryo transfer: II. Postweaning, carcass, and meat traits. J. Anim. Sci. 85, 373–379.
Andersson, L. (2001). Genetic dissection of phenotypic diversity in farm animals. Nat. Rev. Genet. 2, 130–138.
Arnold, D. R., Lefebvre, R., and Smith, L. C. (2006). Characterization of the placenta specific bovine mammalian achaete scute-like homologue 2 (Mash2) gene. Placenta 27, 1124–1131.
Band, M. R., Larson, J. H., Rebeiz, M., Green, C. A., Heyen, D. W., Donovan, J., Windish, R., Steining, C., Mahyuddin, P., Womack, J. E., and Lewin, H. A. (2000). An ordered comparative map of the cattle and human genomes. Genome Res. 10, 1359–1368.
Bennett-Baker, P. E., Wilkowski, J., and Burke, D. T. (2003). Age-associated activation of epigenetically repressed genes in the mouse. Genetics 165, 2055–2062.
Berkowicz, E. W., Magee, D. A., Sikora, K. M., Berry, D. P., Howard, D. J., Mullen, M. P., Evans, R. D., Spillane, C., and MacHugh, D. E.. (2011). Single nucleotide polymorphisms at the imprinted bovine insulin-like growth factor 2 (IGF2) locus are associated with dairy performance in Irish Holstein-Friesian cattle. J. Dairy Res. 78, 1–8.
Bischoff, S. R., Tsai, S., Hardison, N., Motsinger-Reif, A. A., Freking, B. A., Nonneman, D., Rohrer, G., and Piedrahita, J. A. (2009). Characterization of conserved and nonconserved imprinted genes in swine. Biol. Reprod. 81, 906–920.
Boysen, T. J., Tetens, J., and Thaller, G. (2010). Detection of a quantitative trait locus for ham weight with polar overdominance near the ortholog of the callipyge locus in an experimental pig F2 population. J. Anim. Sci. 88, 3167–3172.
Bradley, D. G., MacHugh, D. E., Cunningham, P., and Loftus, R. T. (1996). Mitochondrial diversity and the origins of African and European cattle. Proc. Natl. Acad. Sci. U.S.A. 93, 5131–5135.
Brideau, C. M., Kauppinen, K. P., Holmes, R., and Soloway, P. D. (2010). A non-coding RNA within the Rasgrf1 locus in mouse is imprinted and regulated by its homologous chromosome in trans. PLoS ONE 5, e13784. doi: 10.1371/journal.pone.0013784
Casellas, J., Farber, C. R., Gularte, R. J., Haus, K. A., Warden, C. H., and Medrano, J. F. (2009). Evidence of maternal QTL affecting growth and obesity in adult mice. Mamm. Genome 20, 269–280.
Charalambous, M., Cowley, M., Geoghegan, F., Smith, F. M., Radford, E. J., Marlow, B. P., Graham, C. F., Hurst, L. D., and Ward, A. (2010). Maternally-inherited Grb10 reduces placental size and efficiency. Dev. Biol. 337, 1–8.
Chen, F. Y., Niu, H., Wang, J. Q., Lei, C. Z., Lan, X. Y., Zhang, C. L., Li, M. J., Hua, L. S., Wang, J., and Chen, H. (2011). Polymorphism of DLK1 and CLPG gene and their association with phenotypic traits in Chinese cattle. Mol. Biol. Rep. 38, 243–248.
Cheverud, J. M., Hager, R., Roseman, C., Fawcett, G., Wang, B., and Wolf, J. B. (2008). Genomic imprinting effects on adult body composition in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 4253–4258.
Christensen, B. C., Houseman, E. A., Marsit, C. J., Zheng, S., Wrensch, M. R., Wiemels, J. L., Nelson, H. H., Karagas, M. R., Padbury, J. F., Bueno, R., Sugarbaker, D. J., Yeh, R. F., Wiencke, J. K., and Kelsey, K. T. (2009). Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 5, e1000602, doi: 10.1371/journal.pgen.1000602
Cockett, N. E., Jackson, S. P., Shay, T. L., Farnir, F., Berghmans, S., Snowder, G. D., Nielsen, D. M., and Georges, M. (1996). Polar overdominance at the ovine callipyge locus. Science 273, 236–238.
Constancia, M., Angiolini, E., Sandovici, I., Smith, P., Smith, R., Kelsey, G., Dean, W., Ferguson-Smith, A., Sibley, C. P., Reik, W., and Fowden, A. (2005). Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc. Natl. Acad. Sci. U.S.A. 102, 19219–19224.
Cui, Y. (2007). A statistical framework for genome-wide scanning and testing of imprinted quantitative trait loci. J. Theor. Biol. 244, 115–126.
Cui, Y., Casella, G., and Wu, R. (2004). Mapping quantitative trait loci interactions from the maternal and offspring genomes. Genetics 167, 1017–1726.
Cui, Y., Cheverud, J. M., and Wu, R. (2007). A statistical model for dissecting genomic imprinting through genetic mapping. Genetica 130, 227–239.
De Koning, D.-J., Bovenhuis, H., and van Arendonk, J. A. M. (2002). On the detection of imprinted quantitative trait loci in experimental crosses of outbred species. Genetics 161, 931–938.
De Koning, D.-J., Harlizius, B., Rattink, A. P., Groenen, M. A. M., Brascamp, E. W., and van Arendonk, J. A. M. (2001). Detection and characterization of quantitative trait loci for meat quality traits in pigs. J. Anim. Sci. 79, 2812–2819.
De Koning, D.-J., Janss, L. L. G., Rattink, A. P., van Oers, P. A. M., de Vries, B. J., Groenen, M. A. M., van der Poel, J. J., de Groot, P. N., Brascamp, E. W., and van Arendonk, J. A. M. (1999). Detection of quantitative trait loci for backfat thickness and intramuscular fat content in pigs (Sus scrofa). Genetics 152, 1679–1690.
De Koning, D.-J., Rattink, A. P., Harlizius, B., van Arendonk, J. A. M., Brascamp, E. W., and Groenen, M. A. M. (2000). Genome-wide scan for body composition in pigs reveals important role of imprinting. Proc. Natl. Acad. Sci. U.S.A. 97, 7947–7950.
Delaval, K., Wagschal, A., and Feil, R. (2006). Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. Bioessays 28, 453–459.
Dindot, S. V., Kent, K. C., Evers, B., Loskutoff, N., Womack, J., and Piedrahita, J. A. (2004). Conservation of genomic imprinting at the XIST, IGF2, and GTL2 loci in the bovine. Mamm. Genome 15, 966–974.
Dong, C., Li, W. D., Geller, F., Lei, L., Li, D., Gorlova, O. Y., Hebebrand, J., Amos, C. I., Nicholls, R. D., and Price, R. A. (2005). Possible genomic imprinting of three human obesity-related genetic loci. Am. J. Hum. Genet. 76, 427–437.
Duselis, A. R., Wiley, C. D., O’Neill, M. J., and Vrana, P. B. (2005). Genetic evidence for a maternal effect locus controlling genomic imprinting and growth. Genesis 43, 155–165.
Everts-van der Wind, A., Larkin, D. M., Green, C. A., Elliott, J. S., Olmstead, C. A., Chiu, R., Schein, J. E., Marra, M. A., Womack, J. E., and Lewin, H. A. (2005). A high-resolution whole-genome cattle-human comparative map reveals details of mammalian chromosome evolution. Proc. Natl. Acad. Sci. U.S.A. 102, 18526–18531.
Fleming-Waddell, J. N., Olbricht, G. R., Taxis, T. M., White, J. D., Vuocolo, T., Craig, B. A., Tellam, R. L., Neary, M. K., Cockett, N. E., and Bidwell, C. A. (2009). Effect of DLK1 and RTL1 but not MEG3 or MEG8 on muscle gene expression in callipyge lambs. PLoS ONE 4, e7399. doi: 10.1371/journal.pone.0007399
Flisikowski, K., Venhoranta, H., Nowacka-Woszuk, J., McKay, S. D., Flyckt, A., Taponen, J., Schnabel, R., Schwarzenbacher, H., Szczerbal, I., Lohi, H., Fries, R., Taylor, J. F., Switonski, M., and Andersson, M. (2010). A novel mutation in the maternally imprinted PEG3 domain results in a loss of MIMT1 expression and causes abortions and stillbirths in cattle (Bos taurus). PLoS ONE 5, e15116. doi: 10.1371/journal.pone.0015116
Fowden, A. L., Sibley, C., Reik, W., and Constancia, M. (2006). Imprinted genes, placental development and fetal growth. Horm. Res. 65(Suppl. 3), 50–58.
Fradin, D., Boileau, P., Lepercq, J., and Bougneres, P. (2006). Non-Mendelian’ genetics of fetal growth. J. Endocrinol. Invest. 29(1 Suppl.), 11–15.
Garfield, A. S., Cowley, M., Smith, F. M., Moorwood, K., Stewart-Cox, J. E., Gilroy, K., Baker, S., Georges, M., Charlier, C., and Cockett, N. (2003). The callipyge locus: evidence for the trans interaction of reciprocally imprinted genes. Trends Genet. 19, 248–252.
Garfield, A. S., Cowley, M., Smith, F. M., Moorwood, K., Stewart-Cox, J. E., Gilroy, K., Baker, S., Xia, J., Dalley, J. W., Hurst, L. D., Wilkinson, L. S., Isles, A. R., and Ward, A. (2011). Distinct physiological and behavioural functions for parental alleles of imprinted Grb10. Nature 469, 534–538.
Georges, M., Charlier, C., and Cockett, N. (2003). The callipyge locus: evidence for the trans interaction of reciprocally imprinted genes. Trends Genet. 19, 248–252.
Glasgow, E., Ryu, S. L., Yamashita, M., Zhang, B. J., Mutsuga, N., and Gainer, H. (2005). APeg3, a novel paternally expressed gene 3 antisense RNA transcript specifically expressed in vasopressinergic magnocellular neurons in the rat supraoptic nucleus. Brain Res. Mol. Brain Res. 137, 143–151.
Gorlova, O. Y., Amos, C. I., Wang, N. W., Shete, S., Turner, S. T., and Boerwinkle, E. (2003). Genetic linkage and imprinting effects on body mass index in children and young adults. Eur. J. Hum. Genet. 11, 425–432.
Green, P., Falls, K., and Crooks, S. (1990). Documentation for CRI-MAP, version 2.4. St. Louis, MO: Washington University School of Medicine.
Gregg, C., Zhang, J., Weissbourd, B., Luo, S., Schroth, G. P., Haig, D., and Dulac, C. (2010). High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329, 643–648.
Hager, R., Cheverud, J. M., Leamy, L. J., and Wolf, J. B. (2008). Sex dependent imprinting effects on complex traits in mice. BMC Evol. Biol. 8, 303. doi: 10.1186/1471-2148-8-303
Hager, R., Cheverud, J. M., and Wolf, J. B. (2009). Relative contribution of additive, dominance, and imprinting effects to phenotypic variation in body size and growth between divergent selection lines of mice. Evolution 63, 1118–1128.
Haley, C. S., Knott, S. A., and Elsen, J.-M. (1994). Mapping quantitative trait loci in crosses between outbred lines using least squares. Genetics 136, 1195–1207.
Ihara, N., Takasuga, A., Mizoshita, K., Takeda, H., Sugimoto, M., Mizoguchi, Y., Hirano, T., Itoh, T., Watanabe, T., Reed, K. M., Snelling, W. M., Kappes, S. M., Beattie, C. W., Bennett, G. L., and Sugimoto, Y. (2004). A comprehensive genetic map of the cattle genome based on 3802 Microsatellites. Genome Res. 14, 1987–1998.
Isles, A. R., and Holland, A. J. (2005). Imprinted genes and mother-offspring interactions. Early Hum. Dev. 81, 73–77.
Jiang, L., Jobst, P., Lai, L., Samuel, M., Ayares, D., Prather, R. S., and Tian, X. C. (2007). Expression levels of growth-regulating imprinted genes in cloned piglets. Cloning Stem Cells 9, 97–106.
Kim, J., Bergmann, A., Choo, J. H., and Stubbs, L. (2007). Genomic organization and imprinting of the Peg3 domain in bovine. Genomics 90, 85–92.
Kim, J., Bergmann, A., Lucas, S., Stone, R., and Stubbs, L. (2004). Lineage-specific imprinting and evolution of the zinc-finger gene ZIM2. Genomics 84, 47–58.
Kim, J.-J., Farnir, F., Savell, J., and Taylor, J. F. (2003). Detection of quantitative trait loci for growth and beef carcass fatness traits in a cross between Bos taurus (Angus) and Bos indicus (Brahman) cattle. J. Anim. Sci. 81, 1933–1942.
Knott, S. A., Marklund, L., Haley, C. S., Andersson, K., Davis, W., Ellegren, H., Fredholm, M., Hansson, I., Hoyheim, B., Lundstrom, K., Moller, M., and Andersson, L. (1998). Multiple marker mapping of quantitative trait loci in a cross between outbred wild boar and large white pigs. Genetics 149, 1069–1080.
Koots, K. R., Gibson, J. P., Smith, C., and Wilton, J. W. (1994a). Analyses of published genetic parameter estimates for beef production traits. 1. Heritability. Anim. Breed. Abstr. 62, 309–338.
Koots, K. R., Gibson, J. P., Smith, C., and Wilton, J. W. (1994b). Analyses of published genetic parameter estimates for beef production traits. 2. Phenotypic and genotypic correlations. Anim. Breed. Abstr. 62, 825–853.
Kuehn, C., Edel, C., Weikard, R., and Thaller, G. (2007). Dominance and parent-of-origin effects of coding and non-coding alleles at the acylCoA-diacylglycerol-acyltransferase (DGAT1) gene on milk production traits in German Holstein cows. BMC Genet. 8, 62.
Lenstra, J. A., and Bradley, D. G. (1999). “Systematics and phylogeny of cattle,” in Genetics of Cattle, eds R. Fries and A. Ruvinsky (London: CAB International), 1–14.
Li, Y., Zhu, J., Tian, G., Li, N., Li, Q., Ye, M., Zheng, H., Yu, J., Wu, H., Sun, J., Zhang, H., Chen, Q., Luo, R., Chen, M., He, Y., Jin, X., Zhang, Q., Yu, C., Zhou, G., Sun, J., Huang, Y., Zheng, H., Cao, H., Zhou, X., Guo, S., Hu, X., Li, X., Kristiansen, K., Bolund, L., Xu, J., Wang, W., Yang, H., Wang, J., Li, R., Beck, S., Wang, J., and Zhang, X. (2010). The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 8, e1000533. doi: 10.1371/journal.pbio.1000533
Loschiavo, M., Nguyen, Q. K., Duselis, A. R., and Vrana, P. B. (2007). Mapping and identification of candidate loci responsible for Peromyscus hybrid overgrowth. Mamm. Genome 18, 75–85.
Luedi, P. P., Hartemink, A. J., and Jirtle, R. L. (2005). Genome-wide prediction of imprinted murine genes. Genome Res. 15, 875–884.
Magee, D. A., Berry, D. P., Berkowicz, E. W., Sikora, K. M., Howard, D. J., Mullen, M. P., Evans, R. D., Spillane, C., and MacHugh, D. E. (2011). Single nucleotide polymorphisms within the bovine DLK1- DIO3 imprinted domain are associated with economically important production traits in cattle. J. Hered. 102, 94–101.
Magee, D. A., Sikora, K. M., Berkowicz, E. W., Berry, D. P., Howard, D. J., Mullen, M. P., Evans, R. D., Spillane, C., and MacHugh, D. E. (2010). DNA sequence polymorphisms in a panel of eight candidate bovine imprinted genes and their association with performance traits in Irish Holstein-Friesian cattle. BMC Genet. 11, 93. doi: 10.1186/1471-2156-11-93
Matika, O., Sechi, S., Pong-Wong, R., Houston, R. D., Clop, A., Woolliams, J. A., and Bishop, S. C. (2010). Characterization of OAR1 and OAR18 QTL associated with muscle depth in British commercial terminal sire sheep. Anim. Genet. 42, 172–180.
McElroy, J. P., Kim, J.-J., Harry, D. E., Brown, S., Dekkers, J. C. M., and Lamont, S. J. (2006). Trait loci affecting white meat percent and other growth and carcass traits in commercial broiler chickens. J. Poultry Sci. 85, 593–605.
Nikaido, I., Saito, C., Mizuno, Y., Meguro, M., Bono, H., Kadomura, M., Kono, T., Morris, G. A., Lyons, P. A., Oshimura, M., Hayashizaki, Y., and Okazaki, Y., RIKEN GER Group and GSL Members. (2003). Discovery of imprinted transcripts in the mouse transcriptome using large-scale expression profiling. Genome Res. 13, 1402–1409.
Neugebauer, N., Räder, I., Schild, H. J., Zimmer, D., and Reinsch, N. (2010). Evidence for parent-of-origin effects on genetic variability of beef traits. J. Anim. Sci. 88, 523–532.
Pausch, H., Flisikowski, K., Jung, S., Emmerling, R., Edel, C., Götz, K. U., and Fries, R. (2011). Genome-wide association study identifies two major loci affecting calving ease and growth-related traits in cattle. Genetics 187, 289–297.
Rance, K. A., Fustin, J. M., Dalgleish, G., Hambly, C., Bunger, L., and Speakman, J. R. (2005). A paternally imprinted QTL for mature body mass on mouse chromosome 8. Mamm. Genome 16, 567–577.
Rohrer, G. A., Taylor, J. F., Sanders, J. O., and Thallman, R. M. (1994). Evaluation of line and breed of cytoplasm effects on performance of purebred Brangus cattle. J. Anim. Sci. 72, 2798–2803.
Ruddock, N. T., Wilson, K. J., Cooney, M. A., Korfiatis, N. A., Tecirlioglu, R. T., and French, A. J. (2004). Analysis of imprinted messenger RNA expression during bovine preimplantation development. Biol. Reprod. 70, 1131–1135.
Sandor, C., and Georges, M. (2008). On the detection of imprinted quantitative trait loci in line crosses: effect of linkage disequilibrium. Genetics 180, 1167–1175.
Seaton, G., Haley, C. S., Knott, S. A., Kearsey, M., and Visscher, P. M. (2002). QTL Express: mapping quantitative trait loci in simple and complex pedigrees. Bioinformatics 18, 339–340.
Shi, W., Krella, A., Orth, A., Yu, Y., and Fundele, R. (2005). Widespread disruption of genomic imprinting in adult interspecies mouse (Mus) hybrids. Genesis 43, 100–108.
Shi, W., Lefebvre, L., Yu, Y., Otto, S., Krella, A., Orth, A., and Fundele, R. (2004). Loss-of-imprinting of Peg1 in mouse interspecies hybrids is correlated with altered growth. Genesis 39, 65–72.
Sikora, K. M., Magee, D. A., Berkowicz, E. W., Berry, D. P., Howard, D. J., Mullen, M. P., Evans, R. D., MacHugh, D. E., and Spillane, C. (2011). DNA sequence polymorphisms within the bovine guanine nucleotide-binding protein Gs subunit alpha (Gsα)-encoding (GNAS) genomic imprinting domain are associated with performance traits. BMC Genet. 12, 4.
Silva, G. J., Pereira, A. C., Krieger, E. M., and Krieger, J. E. (2007). Genetic mapping of a new heart rate QTL on chromosome 8 of spontaneously hypertensive rats. BMC Med. Genet. 8, 17. doi: 10.1186/1471-2350-8-17
Smith, A. C., Choufani, S., Ferreira, J. C., and Weksberg, R. (2007). Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr. Res. 61(5 Pt 2), 43R–47R.
Smith, F. M., Garfield, A. S., and Ward, A. (2006). Regulation of growth and metabolism by imprinted genes. Cytogenet. Genome Res. 113, 279–291.
Thallman, R. M., Sanders, J. O., and Taylor, J. F. (1993). Non-Mendelian genetic effects in reciprocal cross Brahman x Simmental F1 calves produced by embryo transfer. PR-5053. Beef Cattle Research in Texas. College Station: Texas A&M University, 8–14.
Thomsen, H., Lee, H. K., Rothschild, M. F., Malek, M., and Dekkers, J. C. M. (2004). Characterization of quantitative trait loci for growth and meat quality in a cross between commercial breeds of swine. J. Anim. Sci. 82, 2213–2228.
Trowbridge, J. J., and Orkin, S. H. (2010). DNA methylation in adult stem cells: new insights into self-renewal. Epigenetics 5, 189–193.
Tveden-Nyborg, P. Y., Alexopoulos, N. I., Cooney, M. A., French, A. J., Tecirlioglu, R. T., Holland, M. K., Thomsen, P. D., and D’Cruz, N. T. (2008). Analysis of the expression of putatively imprinted genes in bovine peri-implantation embryos. Theriogenology 70, 1119–1128.
USDA. (1989). Official United States standards for grades of carcass beef. Washington, DC: United States Department of Agriculture Marketing Services.
Van Laere, A. S., Nguyen, M., Braunschweig, M., Nezer, C., Collette, C., Moreau, L., Archibald, A. L., Haley, C. S., Buys, N., Tally, M., Andersson, G., Georges, M., and Andersson, L. (2003). A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425, 832–836.
Visscher, P. M., Thompson, R., and Haley, C. S. (1996). Confidence intervals in QTL mapping by bootstrapping. Genetics 143, 1013–1020.
Vrana, P. B., Fossella, J. A., Matteson, P., del Rio, T., O’Neill, M. J., and Tilghman, S. M. (2000). Genetic and epigenetic incompatibilities underlie hybrid dysgenesis in Peromyscus. Nat. Genet. 25, 120–124.
Vries, A. G., Kerr, R., Tier, B., Long, T., and Meuwissen, T. H. E. (1994). Gametic imprinting effects on rate and composition of pig growth. Theor. Appl. Genet. 88, 1037–1042.
Vuocolo, T., Byrne, K., White, J., McWilliam, S., Reverter, A., Cockett, N. E., and Tellam, R. L. (2007). Identification of a gene network contributing to hypertrophy in callipyge skeletal muscle. Physiol. Genomics 28, 253–272.
Wolf, J. B., Cheverud, J. M., Roseman, C., and Hager, R. (2008). Genome-wide analysis reveals a complex pattern of genomic imprinting in mice. PLoS Genet. 4, e1000091. doi: 10.1371/journal.pgen.1000091
Wolf, J. B., Vaughn, T. T., Pletscher, L. S., and Cheverud, J. M. (2002). Contribution of maternal effect QTL to genetic architecture of early growth in mice. Heredity 89, 300–310.
Woodfine, K., Huddleston, J. E., and Murrell, A. (2011). Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenetics Chromatin 4, 1.
Wu, G., Bazer, F. W., Wallace, J. M., and Spencer, T. E. (2006). Board-invited review: intrauterine growth retardation: implications for the animal sciences. J. Anim. Sci. 84, 2316–2337.
Wu, H. C., John, E. M., Ferris, J. S., Keegan, T. H., Chung, W. K., Andrulis, I., Delgado-Cruzata, L., Kappil, M., Gonzalez, K., Santella, R. M., and Terry, M. B. (2011). Global DNA methylation levels in girls with and without a family history of breast cancer. Epigenetics 6, 29–33.
Xu, Y., Goodyer, C. G., Deal, C., and Polychronakos, C. (1993). Functional polymorphism in the parental imprinting of the human IGF2R gene. Biochem. Biophys. Res. Commun. 197, 747–754.
Yoh, S. M., Lucas, J. S., and Jones, K. A. (2008). The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 22, 3422–3434.
Keywords: parent-of-origin, imprinting, QTL, growth traits, carcass traits, comparative genomics, beef cattle, BTA2
Citation: Imumorin IG, Kim E-H, Lee Y-M, De Koning D-J, van Arendonk JA, De Donato M, Taylor JF and Kim J-J (2011) Genome scan for parent-of-origin QTL effects on bovine growth and carcass traits. Front. Gene. 2:44. doi: 10.3389/fgene.2011.00044
Received: 23 March 2011; Paper pending published: 14 April 2011;
Accepted: 25 June 2011; Published online: 12 July 2011.
Edited by:
Peter Dovč, University of Ljubljana, SloveniaReviewed by:
Peter Dovč, University of Ljubljana, SloveniaIno Čurik, University of Zagreb, Croatia
Simon Horvat, University of Ljubljana, Slovenia
Copyright: © 2011 Imumorin, Kim, Lee, De Koning, van Arendonk, De Donato, Taylor and Kim. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ikhide G. Imumorin, Department of Animal Science, Cornell University, 267 Morrison Hall, Ithaca, NY 14853, USA. e-mail:aWdpMkBjb3JuZWxsLmVkdQ==; Jong-Joo Kim, School of Biotechnology, Yeungnam University, Gyeongsan, Gyeongbuk 712749, South Korea. e-mail:a2ltampAeXVtYWlsLmFjLmty