


- 1 Department of Biology, University of Alabama at Birmingham, Birmingham, AL, USA
- 2 Center for Aging, University of Alabama Birmingham, Birmingham, AL, USA
- 3 Comprehensive Cancer Center, University of Alabama Birmingham, Birmingham, AL, USA
- 4 Nutrition Obesity Research Center, University of Alabama Birmingham, Birmingham, AL, USA
- 5 Comprehensive Diabetes Center, University of Alabama Birmingham, Birmingham, AL, USA
Ovarian cancer is a major cause of death among gynecological cancers and its etiology is still unclear. Currently, the two principle obstacles in treating this life threatening disease are lack of effective biomarkers for early detection and drug resistance after initial chemotherapy. Similar to other cancers, the initiation and development of ovarian cancer is characterized by disruption of oncogenes and tumor suppressor genes by both genetic and epigenetic mechanisms. While it is well known that it is challenging to treat ovarian cancer through a genetic strategy due in part to its heterogeneity, the reversibility of epigenetic mechanisms involved in ovarian cancer opens exciting new avenues for treatment. The epigenomics of ovarian cancer has therefore become a rapidly expanding field leading to intense investigation. A review on the current status of the field is thus warranted. In this analysis, we will evaluate the current status of epigenomics of ovarian cancer and will include epigenetic mechanisms involved in ovarian cancer development such as DNA methylation, histone modifications, and non-coding microRNA. Development of biomarkers, the epigenetic basis for drug resistance and improved chemotherapy for ovarian cancer will also be assessed. In addition, the potential use of natural compounds as epigenetic modulators in chemotherapy shows promise in moving to the forefront of ovarian cancer treatment strategies.
Introduction
Overview of Ovarian Cancer
Ovarian cancer has the highest mortality rate among reproductive cancers and is the leading cause of gynecologic cancer death. The ovaries contain three types of tissue: germ, stromal, and epithelial cells. Germ cells and stromal cells are inside of the ovary, while epithelial cells are on the surface of the ovary. Germ cells can make eggs, and stromal cells produce most of the female hormones (estrogen and progesterone). The epithelium is the tissue where most ovarian cancers arise. Ovarian cancer is classified into several stages according to the American Joint committee on Cancer/Tumor Node Metastasis (AJCC/TNM) and International Federation of Gynecology and Obstetrics (FIGO) staging systems which are based on how far the cancer has spread. In stages I and II, the tumor is confined to the ovaries, while there is local metastasis (usually lymph) in stage III and there is distal organ metastases in stage IV (Yarbro et al., 1999).
The etiology of ovarian cancer is still not clear. However, previous epidemiology studies indicate that for sporadic ovarian cancer, the younger the individuals are when they have their first child and the more children they have, the less the risk of suffering from ovarian cancer (Chen and Karlan, 1998). A hypothesis has been proposed based on these epidemiology studies, that is, too much wound repair after ovulation may contribute to the ovarian tumorigenesis processes (Roett and Evans, 2009).
Due to the atypical syndrome of the early stage of ovarian cancer, it is difficult to diagnose in its early stages. By the time most ovarian cancers are diagnosed, they are already at stage III or IV. The two most significant obstacles to the effective treatment of ovarian cancers are the lack of early diagnostic markers and the development of drug resistance after therapeutic treatment of advanced disease. Ovarian cancer screening with transvaginal ultrasound (TVU) and CA125 was evaluated in the Prostate, Lung, Colorectal, and Ovarian (PLCO) trial, however, it was revealed that the predictive value of both tests was relatively low (Buys et al., 2005). Increasing evidence indicates that epigenetic mechanisms may play a major role in the development of ovarian cancer.
Epigenetic Modifications in Cancer
The epigenome is defined as the mechanisms that regulate all the genes expression without alteration of the DNA sequence (Bernstein et al., 2007). These epigenetic mechanisms include DNA methylation, histone modification, and microRNA (miRNAs) expression which are known to regulate gene expression in cancer cells. DNA methylation is characterized by transferring of a methyl moiety to cytosine at the 5-carbon (C5) position by enzymes known as DNA methyltransferases (DNMTs). DNA methylation can regulate gene transcription by recruiting methyl CpG-binding proteins and therefore regulate chromatin status. Typically, DNA hypermethylation could silence genes (e.g., tumor suppressors) due to recruitment of repressors and further lead to chromatin condensation (Herceg, 2007).
Histone modifications are characterized by covalent modification of the N-terminal tails of core of histones (H2A, H2B, H3, and H4) including acetylation, methylation, phosphorylation, biotinylation, and ubiquitination (Feinberg et al., 2006; Hassan and Zempleni, 2006; Doi et al., 2009). Based on studies focused on the biological significance of these modifications, concepts like “histone code” and “histone language” have been proposed (Oliver and Denu, 2011). Acetylation has been the most studied in cancer among all these histone modifications. Histone acetyltransferases (HATs) are responsible for the acetylation of the lysine residues of histones (Turner, 2000), while histone deacetylases (HDACs) can remove the acetyl moieties (Mottet and Castronovo, 2008; Choudhuri et al., 2010). Histones acetylation generates an open conformation of chromatin which is accessible to transcription regulators, leading to the promotion of gene expression. Moreover HDACs can remove acetyl moieties from acetylated sites in non-histone proteins thereby adding another layer of regulation on cellular activities (Marson, 2009).
Non-coding miRNAs are regulatory RNAs that are 20–30 nucleotides in length that match the 3′ untranslated regions of target mRNAs. miRNAs can inhibit mRNA translation or degrade mRNA (Ducasse and Brown, 2006; Guil and Esteller, 2009; Lujambio et al., 2010). miRNAs target multiple protein-coding transcripts and are known to cause posttrancriptional gene down-regulation and regulate a number of genes (Jones and Baylin, 2007). While miRNAs are tightly regulated in normal cells, in cancer cells miRNAs are vastly deregulated (Veeck and Esteller, 2010). Additionally, miRNA profiles are being used to classify cancers and the misexpression of miRNAs is associated with cancer development (Iorio et al., 2010; Veeck and Esteller, 2010). A global down-regulation of miRNAs in multiple tumor tissues has also been observed (Lu et al., 2005).
Epigenomic Mechanisms on the Development of Ovarian Cancer
Role of DNA Methylation in the Development of Ovarian Cancer
The genome contains a wealth of CpG dinucleotides and the cytosine in CpG dinucleotides can be methylated by DNMTs. Some of the methylated CpGs are located in the centromere and subtelomeric regions and can also be found in repeated sequences, such as Alu and LINEs sequences. Many CpGs in euchromatin are located in the promoters of genes and when present are termed CpG islands (CGIs). The methylation status of CGIs is often under strict regulation during cell differentiation, which constitutes one of the basic concepts of epigenetics. The abnormal methylation status can cause disorders in gene expression, thus leading to certain diseases, including cancer. Hypermethylation is usually associated with gene silencing, while hypomethylation is generally associated with gene expression. In cancer cells, CGI methylation has been identified as a major cause of silencing of tumor suppressor genes (TSGs), while CGI demethylation has been recognized as a common mechanism involved in the activation of oncogenes. CGI hypomethylation in the centromere and subtelomeric regions have also been implicated in causing genomic instability (GI) leading to chromosomal translocations and gene disruption through the reactivation of endoparasitic sequences (Walsh et al., 1998; Gaudet et al., 2003; Esteller and Almouzni, 2005). In ovarian cancer, satellite DNA hypomethylation is increased compared to normal tissues and this is more pronounced in advanced stages than in early stages of ovarian cancer (Widschwendter et al., 2004). TSGs include DNA mismatch repair genes such as hMLH1, cell cycle-related genes such as p16, p21, and p27, and genes involved in signal transduction (e.g., PTEN). Typically, epigenetic change of certain genes involved in DNA mismatch repair could lead to GI. This would further trigger abnormal expression of other tumor-related genes through mutation or epigenetic mechanisms, which is consistent with the “drivers” and “passengers” mutation theory. In the theory, “drivers” refers to those genes after mutation that can confer advantageous biological phenotypes, while “passengers” refers to those mutations that reflect the general increased mutability of tumor cell genomes due to mutation of “drivers” (Weinberg, 2008).
In ovarian cancer, a number of TSGs have been found to undergo hypermethylation (Table 1). As one of the genes that is involved in inherited ovarian cancer, the promoter of BRCA1 is hypermethylated in sporadic cases of ovarian cancer (Baldwin et al., 2000), which leads to the silencing of this gene in these tumors (Wilcox et al., 2005). Consistent with the aforementioned “drivers” and “passengers” mutation theory, silencing of BRCA1 together with other DNA repair genes, such as hMLH1 (Strathdee et al., 1999), increase the chance of mutation of other genes related to ovarian cancer cell proliferation by both genetic and epigenetic approaches (Esteller, 2000), which further confers a growth advantage to cancer cells. Other genes that can be silenced by promoter hypermethylation in ovarian cancer include genes involved in cell cycle regulation and genes that promote apoptosis such as LOT1, DAPK, TMS1/ASC, and PAR-4.

Table 1. Tumor suppressor genes (TSGs) regulated by epigenetic mechanisms in ovarian cancer.
In a recent study conducted by Matsumura et al. (2011) microarray analysis of genome-wide changes in gene expression was applied to 39 cell lines and 17 cultured primary ovarian cancer specimens treated with DNA hypomethylating agents. In total, 378 candidate methylated genes contributing to ovarian cancer were identified and a number of genes involved in the TGF-beta pathway were found to be regulated by methylation in ovarian cancer (Matsumura et al., 2011). A distinct gene cluster strongly correlated with TGF-beta pathway activity that discriminates patients based on age. This was generated by using the 378 genes through hierarchical clustering of ovarian cancers, which suggests that suppression of TGF-beta signaling is caused by accumulation of age-related epigenetic modifications.
Studies in this regard not only reveal gene regulation networks, but also provide information on the potential cause of complicating diseases, such as cancer. Clinically, it can lead to discovery of novel biomarkers and therapeutic targets; it can also lay the foundation for personalized medicine that is tailored to each individual’s genome and epigenomic status (Figure 1).
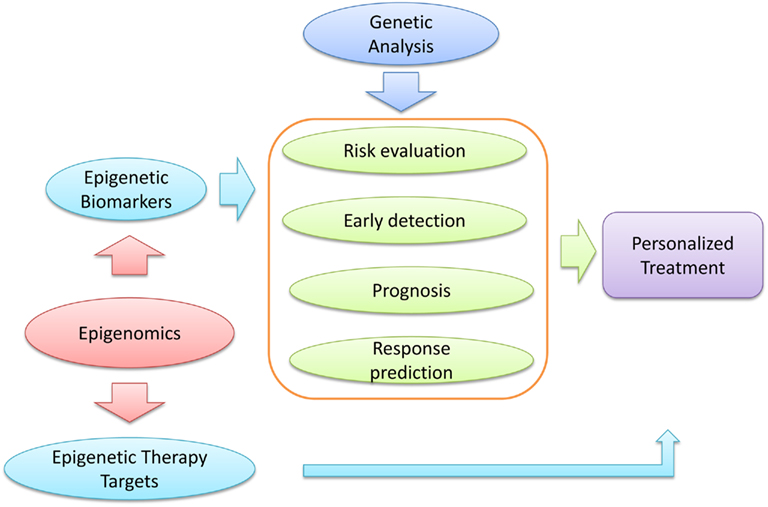
Figure 1. Function of epigenomic studies in cancer prevention. Epigenomic analyses of cancer could provide novel epigenetic biomarkers which can serve for cancer risk evaluation, early detection, prognosis, and chemotherapy response prediction. Studies of this nature could also provide novel epigenetic therapy targets. In addition, genetic analysis could contribute to these processes. Taken together, epigenomic and genetic analyses could promote a personalized treatment for the optimal benefits to cancer patients.
Role of Histone Modifications in the Development of Ovarian Cancer
Another major epigenetic mechanism involved in gene expression is histone modification. These modifications include acetylation, methylation, and phosphorylation. Genes involved in cell cycle regulation, such as cyclinB1 (Valls et al., 2005), p21 (Richon et al., 2000), and ADAM19 (Chan et al., 2008) have been indicated to be regulated by histone modification. Overexpression of the cancer-promoting genes claudin-3 and claudin-4 in ovarian cancer is associated with loss of trimethylated histone 3 lysine 27 (H3K27me3) (Kwon et al., 2010). A protein, osteoprotegerin (OPG), reportedly can inhibit cancer cell growth through apoptotic mechanisms, and its gene may be silenced in ovarian cancer. The loss of OPG gene expression correlates with reduced histone 3 lysine 4 trimethylation (H3K4me3) and increased H3K27me3 (Lu et al., 2009). GATA4 and GATA6 gene silencing also correlated with hypoacetylation of histones H3 and H4 and loss of H3K4me3 at their promoters in five epithelial ovarian carcinoma cell lines (Caslini et al., 2006) and only treatment with the HDAC inhibitor (HDACIs), trichostatin A (TSA) can re-express these genes in the ovarian cancer cells.
Role of miRNAs in the Development of Ovarian Cancer
microRNA is another epigenetic mechanism that often contributes to regulation of gene expression and miRNAs can participate in cancer progression by regulating expression of oncogenes or TSGs. In ovarian cancer, miR-187 has been shown to inhibit its progression by down-regulating Dab2, which may promote tumor progression in advanced cancers through epithelial-to-mesenchymal transition (EMT) (Chao et al., 2011). Additionally, ovarian tumors were found to have down-regulated miR-100 and miRNA let-7i and up-regulated miR-199a, miR-200a, and miR-214 (Iorio et al., 2007; Yang et al., 2008a,c). Specifically, up-regulation of miR-214 in ovarian cancer can induce the degradation of PTEN mRNA which leads to activation of Akt pathway (Yang et al., 2008b). miRNAs have also been shown to regulate the apoptosis pathway in ovarian cancer (Mezzanzanica et al., 2011).
Ovarian Cancer Biomarker Identification at the Epigenomic Level
Biomarkers are those biological molecules that could be used as markers for diagnostic, prognostic, or therapeutic purposes. Biomarkers that can represent the early stages of ovarian cancer are of significant importance for early detection of the disease due to its atypical syndrome. Also, biomarkers can be used for better stratification of ovarian cancer subtypes for individualized treatment. So far only two biomarkers of protein origin (CA125 and HE4) have been approved by the FDA for monitoring ovarian cancer (Moore et al., 2010). However, benign gynecological and medical conditions such as endometriosis, congestive heart failure, and cirrhosis can also have elevated CA125 levels, and elevated serum HE4 level was only related to the advanced stage of epithelial ovarian cancer. More specific and early detection biomarkers are sorely needed. As one of the basic elements of epigenetic mechanisms, DNA methylation has been recognized as a potential ideal biomarker due to its stability compared with RNA and protein, sensitivity of detection by PCR, the possibility of localization to a specific gene region and potential of development as a high-throughput screening method specific for cancer detection (Ahluwalia et al., 2001; Laird, 2003; Anglim et al., 2008).
Epigenetic mechanisms as main players in cancer development are emerging as attractive targets for characterizing reliable biomarkers of ovarian cancer. Due to inaccessibility of ovaries through a non-invasive strategy, association between methylation of certain genes in DNA isolated from blood and cancer on ovaries can improve the diagnosis of ovarian cancer in a non-invasive manner. Hypermethylation of a number of TSG promoters has been detected in the serum or peritoneal fluid of a group of ovarian cancer patients in stage I. These TSGs include RASSF1A, BRCA1, APC, CDKN2A, and DAPK (Ibanez De Caceres et al., 2004; Table 1). Especially for DAPK methylation, there is a tight association of the methylation status between DNA isolated from the peripheral blood and primary tumor (Collins et al., 2006).
Furthermore, epigenetic biomarkers can be applied to stratify patients according to their epigenetic status of ovarian cancer. Recently, 302 loci spanning 189 promoter CGIs at 137 genes in the Wnt signaling pathway were analyzed by differential methylation hybridization. Several genes (FZD4, DVL1, NKD1, ROCK1, AXIN1, LRP5, and NFATC3) involved in the Wnt pathway were found to be significantly associated with progression-free survival (PFS). Furthermore, NKD1 and DVL1 were shown to associate with two groups of patients with distinct PFS (Dai et al., 2011).
More recently, an integrated genomic analysis of ovarian carcinoma was performed by the Network (2011). Four ovarian cancer transcriptional subtypes, three miRNAs subtypes, four promoter methylation subtypes, and a transcriptional signature associated with survival duration were identified in 489 high-grade serous ovarian adenocarcinomas.
Genes that are specifically methylated in ovarian cancer still wait to be discovered that have the potential to distinguish ovarian cancer from other cancers and to therefore serve diagnostic purposes. Epigenomics studies consisting of methylomic analysis may hold the key in this regard. However, multicenter-conducted studies and well-controlled clinical trials will eventually be needed to further validate these biomarkers.
Epigenomic Studies on the Chemoprevention of Ovarian Cancer
Chemotherapy and Drug Resistance of Ovarian Cancer
Currently, the gold standard treatment for ovarian cancer is surgical excision followed by cytotoxic chemotherapy. Cisplatin and carboplatin have often been mainstays for treatment of ovarian cancer. Cisplatin can form interstrand and intrastrand adducts once it bound to DNA double strands. The adducts are characterized by DNA bending toward the major groove, unwinding of the double helix, and alterations of the grooves and of the puckering of the sugars (Takahara et al., 1995). Adducts can further be recognized by several proteins of the DNA damage repair system of the cell and eventually leads to apoptosis. Although most ovarian cancers respond to first-line chemotherapy, recurrence of ovarian cancer occurs in up to 75% of the patients (Agarwal and Kaye, 2003). This resistance can be intrinsic or acquired. Intrinsic resistance means that the resistant cell already exists in the tumor. The hypothesis is that the pretreatment tumors contain certain gene expression signatures that lead to the resistance. By contrast, acquired resistance occurs in tumors that generate resistance after chemotherapy by gene mutation or epigenetic modulation. The nature of cisplatin-mediated chemotherapy (targeted at DNA) is based on the potential of acquired resistance (caused by gene mutation) occurring. Cisplatin resistance could be caused by multiple factors. Generally, those factors could be pharmacokinetics-related, including cisplatin uptake and efflux, inactivation by thiol-containing molecules, such as glutathione (gamma-glutamylcysteinylglycine, GSH), or could be caused by the sensitivity of the target cell to DNA damage caused by cisplatin, including an aberrant DNA repair system, change in apoptosis pathway, or hyperactivation of oncogenes (Borst et al., 2008; Figure 2).
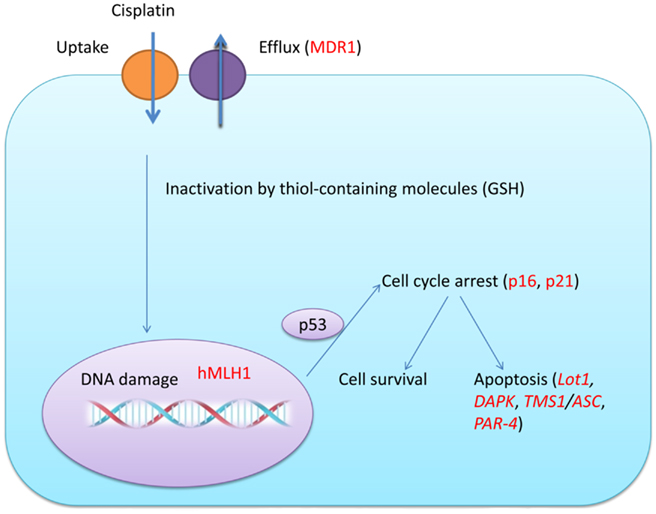
Figure 2. Potential pathways involved in cisplatin resistance in ovarian cancer cells. Cisplatin resistance can occur due to the decrease of proteins that are responsible for its uptake and an increase of proteins that can transport cisplatin to the outside of a cell. Cisplatin in the cell can also be inactivated by thiol-containing molecules (GSH) and cisplatin could cause DNA damage. The DNA repair system (hMLH1) can then work together with p53 and cell cycle regulating proteins (p16, p21) to engage G1 and G2 cell cycle arrest. Depending on the result of DNA repair and the balance between pro-apoptosis and anti-apoptosis proteins, the cell can either survive or undergo apoptosis. Targets that could be regulated by epigenetic mechanisms had been highlighted in red.
Apparently, genetic and epigenetic mechanisms both play essential roles in these processes. Due to the reversible nature of epigenetic modulation, it has become a promising target for cancer treatment. With the consideration that multiple factors may be involved in the resistance of ovarian cancer cells to cisplatin treatment, a strategy of targeting those multiple factors simultaneously may hold the key for this refractory disease. Potential targets for epigenetic mechanisms on chemotherapy possibly include most of the key genes involved in the metabolism pathway of chemotherapy drugs and apoptosis pathway. Epigenomic studies thus may provide us valuable information on the aberrant epigenetic status of those genes which could guide specialized therapy.
Several studies had indicated that epigenetic inactivation of genes involved in trigging an apoptotic response would result in drug resistance. For instance, MLH1 can recognize certain damages in DNA which can further activate a cascade response that can induce apoptosis. Methylation of MLH1 and further transcriptional silencing occurs in cisplatin-resistant ovarian cancer (Gifford et al., 2004). DAPK, which is a gene involved in apoptosis, has also been indicated to be silenced in drug resistant cancer due to methylation (Lehmann et al., 2002). In a recent study by Li et al. (2009) DNA methylation and gene expression were integrated together to analyze the pathways involved in platinum resistance in ovarian cancer. This study indicates disruption of tumor-suppressive functions by hypermethylation, such as cell adhesion molecules, tight junction formation, PPAR signaling, and leukocyte transendothelial migration pathways. Significant up-regulation of tumor-promoting cascades by hypomethylation, such as PIK3K, PDGFRA, E2F1, and TGFBR2, which represents PI3K/Akt, cell cycle progression, and TGF-beta pathways, was also reported.
Epigenomic Studies on the Resensitization of Ovarian Cancer
A number of studies have been performed to investigate the ability of epigenetic modulating drugs on resensitizing resistant ovarian cancer to chemotherapy. DNMT inhibitors (DNMTIs) are analogs of deoxycytosine with different substitutions at their 5-carbons. They can prevent transfer of a methyl moiety by trapping the methyltransferases once they are incorporated into DNA (Lyko and Brown, 2005). These inhibitors have been used to induce re-expression of silenced TSGs caused by hypermethylation. It is hypothesized that reactivation of those genes could sensitize the apoptotic pathway mediated by chemotherapy. RASSF1A and hMLH1 up-regulation by decitabine and zebularine, two DNMTIs, has contributed to the resensitization of cisplatin-resistant ovarian cancer cells to platinum (Balch et al., 2005).
Histone deacetylation is another recognized mechanism that contributes to silencing of TSGs in cancer. Histone deacetylation could lead to a compact conformation of DNA which is not accessible by DNA transcription factors. HDACIs could inhibit deacetylation by HDACs, thereby releasing genes from epigenetic repression. HDACIs have been indicated to induce cell cycle arrest, differentiation, or apoptosis in cancer cells (Secrist et al., 2003).
The combination of a DNMTI and HDACI to re-sensitize resistant cancer cells has also been investigated. It is hypothesized that there should be an additive or synergestic effect on silenced TSGs expression through this approach (Karpf and Jones, 2002). In studies conducted by Steele et al. (2009) it was shown that a combination of decitabine (a DNMTI) with belinostat (an HDACI) is more effective in resensitizing ovarian cancer xenografts than decitabine alone.
Several studies have also demonstrated that miRNAs act as potential targets for resensitization of ovarian cancer. Liang et al. (2010) showed miRNA-related genetic polymorphisms may impact ovarian cancer predisposition and clinical outcome both individually and jointly. Others showed that low tumoral miR-200 expression was significantly associated with high β-tubulin III protein content, a trend toward poor PFS (Leskelä et al., 2011). In a more recent study, Boyerinas found that Let-7 can sensitize ovarian cancer with acquired resistance to Taxanes via targeting IMP-1, a protein known to stabilize mRNAs of a number of genes, which can lead to degradation of the drug resistance gene MDR1 (Boyerinas et al., 2011).
Natural Compounds as Potential Epigenomic Modulators to Re-Sensitize Resistant Ovarian Cancer Cells
Although conventional epigenetic drugs are promising in treating cancer, their non-specific effect could be detrimental to cancer prevention. DNMTIs could lead to genomic hypomethylation which could result in GI. They could also activate potential oncogenes or drug resistant genes due to their demethylation effect on the CGIs of these genes. HDACIs could have a similar effect due to their non-specific targets.
A number of natural compounds extracted from plants, including epigallocatechin-3-gallate (EGCG) from green tea, sulforaphane (SFN) from cruciferous vegetables, and genistein from soybean have been demonstrated to effectively inhibit proliferation of cancer cells via their epigenetic modulating effects. EGCG has been found to function as a DNMTI to reactivate TSGs (Fang et al., 2003). It has also been shown to inhibit telomerase expression in cancer cells through epigenetic remodeling of the hTERT promoter (Mittal et al., 2004; Berletch et al., 2008; Chen et al., 2009; Meeran et al., 2011). The advantage of these natural compounds compared with conventional epigenetic modulators is their low toxic effect to normal cells. Furthermore, a combination of these natural compounds may exert additive or synergistic effects in killing cancer cells which would allow yet lower and safer doses to be applied. Epigenomic studies on the epigenetic alterations of genes in response to treatment with natural compounds may reveal potential mechanisms for how these compounds exert their extra benefits and which could guide design of future anticancer drugs.
Summary
Epigenetic regulation of gene expression plays a fundamental role in normal tissue as well as diseases. Previous epigenetic studies focused on individual genes have provided us with a wealth of insights into the mechanisms of ovarian cancer. In this review, we summarized most of the related progress on epigenomics of ovarian cancer, including its development, potential biomarker, chemoresistance, and possible treatment on its resensitization. To understand this complex disease comprehensively, there is an urgent need to reveal the epigenetic status genome-wide (epigenomics) in ovarian cancer. Numerous pathways involved in basic cell metabolism have been demonstrated to be aberrantly regulated by epigenetic mechanisms during the progression of ovarian cancer, such as Wnt, TGF-beta, cell cycle regulation, and the apoptosis pathway. Although considerable progress has been made on the understanding of the nature of ovarian cancer epigenomics, especially of DNA methylation, more effort is still needed, specifically on histone modifications and miRNA-related epigenomics underlying the development of different stages and chemoresistance of ovarian cancer. Knowledge in this regard will undoubtedly promote the development of novel biomarkers and lead to the design of more effective epigenetically based chemotherapy. As ovarian cancer patients have already begun to benefit from studies in this field, continued efforts in this direction would be justified.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported in part by grants from the National Cancer Institute (RO1 CA129415), the American Institute for Cancer Research, and the Norma Livingston Foundation.
References
Abdollahi, A., Pisarcik, D., Roberts, D., Weinstein, J., Cairns, P., and Hamilton, T. C. (2003). LOT1 (PLAGL1/ZAC1), the candidate tumor suppressor gene at chromosome 6q24-25, is epigenetically regulated in cancer. J. Biol. Chem. 278, 6041–6049.
Abou-Zeid, A. A., Azzam, A. Z., and Kamel, N. A. (2011). Methylation status of the gene promoter of cyclin-dependent kinase inhibitor 2A (CDKN2A) in ovarian cancer. Scand. J. Clin. Lab. Invest. doi: 10.3109/00365513.2011.590224. [Epub ahead of print].
Agarwal, R., and Kaye, S. B. (2003). Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 3, 502–516.
Ahluwalia, A., Hurteau, J. A., Bigsby, R. M., and Nephew, K. P. (2001). DNA methylation in ovarian cancer. II. Expression of DNA methyltransferases in ovarian cancer cell lines and normal ovarian epithelial cells. Gynecol. Oncol. 82, 299–304.
Anglim, P. P., Alonzo, T. A., and Laird-Offringa, I. A. (2008). DNA methylation-based biomarkers for early detection of non-small cell lung cancer: an update. Mol. Cancer 7, 81.
Balch, C., Yan, P., Craft, T., Young, S., Skalnik, D. G., Huang, T. H., and Nephew, K. P. (2005). Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Mol. Cancer Ther. 4, 1505–1514.
Baldwin, R. L., Nemeth, E., Tran, H., Shvartsman, H., Cass, I., Narod, S., and Karlan, B. Y. (2000). BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. 60, 5329–5333.
Berletch, J. B., Liu, C., Love, W. K., Andrews, L. G., Katiyar, S. K., and Tollefsbol, T. O. (2008). Epigenetic and genetic mechanisms contribute to telomerase inhibition by EGCG. J. Cell. Biochem. 103, 509–519.
Bernstein, B. E., Meissner, A., and Lander, E. S. (2007). The mammalian epigenome. Cell 128, 669–681.
Borst, P., Rottenberg, S., and Jonkers, J. (2008). How do real tumors become resistant to cisplatin? Cell Cycle 7, 1353–1359.
Boyerinas, B., Park, S. M., Murmann, A. E., Gwin, K., Montag, A. G., Zillardt, M. R., Hua, Y. J., Lengyel, E., and Peter, M. E. (2011). Let-7 modulates acquired resistance of ovarian cancer to taxanes via IMP-1-mediated stabilization of MDR1. Int. J. Cancer. doi: 10.1002/ijc.26190. [Epub ahead of print].
Buys, S. S., Partridge, E., Greene, M. H., Prorok, P. C., Reding, D., Riley, T. L., Hartge, P., Fagerstrom, R. M., Ragard, L. R., Chia, D., Izmirlian, G., Fouad, M., Johnson, C. C., Gohagan, J. K., and Team, P. P. (2005). Ovarian cancer screening in the prostate, lung, colorectal and ovarian (PLCO) cancer screening trial: findings from the initial screen of a randomized trial. Am. J. Obstet. Gynecol. 193, 1630–1639.
Caslini, C., Capo-Chichi, C. D., Roland, I. H., Nicolas, E., Yeung, A. T., and Xu, X. X. (2006). Histone modifications silence the GATA transcription factor genes in ovarian cancer. Oncogene 25, 5446–5461.
Chan, M. W., Huang, Y. W., Hartman-Frey, C., Kuo, C. T., Deatherage, D., Qin, H., Cheng, A. S., Yan, P. S., Davuluri, R. V., Huang, T. H., Nephew, K. P., and Lin, H. J. (2008). Aberrant transforming growth factor beta 1 signaling and SMAD4 nuclear translocation confer epigenetic repression of ADAM19 in ovarian cancer. Neoplasia 10, 908–919.
Chao, A., Lin, C. Y., Lee, Y. S., Tsai, C. L., Wei, P. C., Hsueh, S., Wu, T. I., Tsai, C. N., Wang, C. J., Chao, A. S., Wang, T. H., and Lai, C. H. (2011). Regulation of ovarian cancer progression by microRNA-187 through targeting disabled homolog-2. Oncogene. doi: 10.1038/onc.2011.269. [Epub ahead of print].
Chen, H., Li, Y., and Tollefsbol, T. O. (2009). Strategies targeting telomerase inhibition. Mol. Biotechnol. 41, 194–199.
Chen, L. M., and Karlan, B. Y. (1998). Early detection and risk reduction for familial gynecologic cancers. Clin. Obstet. Gynecol. 41, 200–214.
Choudhuri, S., Cui, Y., and Klaassen, C. D. (2010). Molecular targets of epigenetic regulation and effectors of environmental influences. Toxicol. Appl. Pharmacol. 245, 378–393.
Collins, Y., Dicioccio, R., Keitz, B., Lele, S., and Odunsi, K. (2006). Methylation of death-associated protein kinase in ovarian carcinomas. Int. J. Gynecol. Cancer 16(Suppl. 1), 195–199.
Dai, W., Teodoridis, J. M., Zeller, C., Graham, J., Hersey, J., Flanagan, J. M., Stronach, E., Millan, D. W., Siddiqui, N., Paul, J., and Brown, R. (2011). Systematic CpG islands methylation profiling of genes in the wnt pathway in epithelial ovarian cancer identifies biomarkers of progression-free survival. Clin. Cancer Res. 17, 4052–4062.
Doi, A., Park, I.-H., Wen, B., Murakami, P., Aryee, M. J., Irizarry, R., Herb, B., Ladd-Acosta, C., Rho, J., Loewer, S., Miller, J., Schlaeger, T., Daley, G. Q., and Feinberg, A. P. (2009). Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 41, 1350–1353.
Esteller, M. (2000). Epigenetic lesions causing genetic lesions in human cancer: promoter hypermethylation of DNA repair genes. Eur. J. Cancer 36, 2294–2300.
Esteller, M., and Almouzni, G. (2005). How epigenetics integrates nuclear functions. Workshop on epigenetics and chromatin: transcriptional regulation and beyond. EMBO Rep. 6, 624–628.
Fang, M. Z., Wang, Y., Ai, N., Hou, Z., Sun, Y., Lu, H., Welsh, W., and Yang, C. S. (2003). Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 63, 7563–7570.
Feinberg, A. P., Ohlsson, R., and Henikoff, S. (2006). The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33.
Gaudet, F., Hodgson, J. G., Eden, A., Jackson-Grusby, L., Dausman, J., Gray, J. W., Leonhardt, H., and Jaenisch, R. (2003). Induction of tumors in mice by genomic hypomethylation. Science 300, 489–492.
Gifford, G., Paul, J., Vasey, P. A., Kaye, S. B., and Brown, R. (2004). The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin. Cancer Res. 10, 4420–4426.
Guil, S., and Esteller, M. (2009). DNA methylomes, histone codes and miRNAs: tying it all together. Int. J. Biochem. Cell Biol. 41, 87–95.
Hassan, Y. I., and Zempleni, J. (2006). Epigenetic regulation of chromatin structure and gene function by biotin. J. Nutr. 136, 1763–1765.
Herceg, Z. (2007). Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis 22, 91–103.
Ibanez De Caceres, I., Battagli, C., Esteller, M., Herman, J. G., Dulaimi, E., Edelson, M. I., Bergman, C., Ehya, H., Eisenberg, B. L., and Cairns, P. (2004). Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 64, 6476–6481.
Iorio, M. V., Piovan, C., and Croce, C. M. (2010). Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim. Biophys. Acta 1799, 694–701.
Iorio, M. V., Visone, R., Di Leva, G., Donati, V., Petrocca, F., Casalini, P., Taccioli, C., Volinia, S., Liu, C.-G., Alder, H., Calin, G. A., Ménard, S., and Croce, C. M. (2007). MicroRNA signatures in human ovarian cancer. Cancer Res. 67, 8699–8707.
Karpf, A. R., and Jones, D. A. (2002). Reactivating the expression of methylation silenced genes in human cancer. Oncogene 21, 5496–5503.
Kwon, M. J., Kim, S. S., Choi, Y. L., Jung, H. S., Balch, C., Kim, S. H., Song, Y. S., Marquez, V. E., Nephew, K. P., and Shin, Y. K. (2010). Derepression of CLDN3 and CLDN4 during ovarian tumorigenesis is associated with loss of repressive histone modifications. Carcinogenesis 31, 974–983.
Laird, P. W. (2003). The power and the promise of DNA methylation markers. Nat. Rev. Cancer 3, 253–266.
Lehmann, U., Celikkaya, G., Hasemeier, B., Länger, F., and Kreipe, H. (2002). Promoter hypermethylation of the death-associated protein kinase gene in breast cancer is associated with the invasive lobular subtype. Cancer Res. 62, 6634–6638.
Leskelä, S., Leandro-García, L. J., Mendiola, M., Barriuso, J., Inglada-Pérez, L., Muñoz, I., Martínez-Delgado, B., Redondo, A., De Santiago, J., Robledo, M., Hardisson, D., and Rodríguez-Antona, C. (2011). The miR-200 family controls beta-tubulin III expression and is associated with paclitaxel-based treatment response and progression-free survival in ovarian cancer patients. Endocr. Relat. Cancer 18, 85–95.
Li, M., Balch, C., Montgomery, J. S., Jeong, M., Chung, J. H., Yan, P., Huang, T. H., Kim, S., and Nephew, K. P. (2009). Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med. Genomics 2, 34. doi: 10.1186/1755-8794-2-34
Liang, D., Meyer, L., Chang, D. W., Lin, J., Pu, X., Ye, Y., Gu, J., Wu, X., and Lu, K. (2010). Genetic variants in MicroRNA biosynthesis pathways and binding sites modify ovarian cancer risk, survival, and treatment response. Cancer Res. 70, 9765–9776.
Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B. L., Mak, R. H., Ferrando, A. A., Downing, J. R., Jacks, T., Horvitz, H. R., and Golub, T. R. (2005). MicroRNA expression profiles classify human cancers. Nature 435, 834–838.
Lu, T. Y., Kao, C. F., Lin, C. T., Huang, D. Y., Chiu, C. Y., Huang, Y. S., and Wu, H. C. (2009). DNA methylation and histone modification regulate silencing of OPG during tumor progression. J. Cell. Biochem. 108, 315–325.
Lujambio, A., Portela, A., Liz, J., Melo, S. A., Rossi, S., Spizzo, R., Croce, C. M., Calin, G. A., and Esteller, M. (2010). CpG island hypermethylation-associated silencing of non-coding RNAs transcribed from ultraconserved regions in human cancer. Oncogene 29, 6390–6401.
Lyko, F., and Brown, R. (2005). DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst. 97, 1498–1506.
Magdinier, F., Ribieras, S., Lenoir, G. M., Frappart, L., and Dante, R. (1998). Down-regulation of BRCA1 in human sporadic breast cancer; analysis of DNA methylation patterns of the putative promoter region. Oncogene 17, 3169–3176.
Makarla, P. B., Saboorian, M. H., Ashfaq, R., Toyooka, K. O., Toyooka, S., Minna, J. D., Gazdar, A. F., and Schorge, J. O. (2005). Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin. Cancer Res. 11, 5365–5369.
Marson, C. M. (2009). Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med. Chem. 9, 661–692.
Matsumura, N., Huang, Z., Mori, S., Baba, T., Fujii, S., Konishi, I., Iversen, E. S., Berchuck, A., and Murphy, S. K. (2011). Epigenetic suppression of the TGF-beta pathway revealed by transcriptome profiling in ovarian cancer. Genome Res. 21, 74–82.
Meeran, S. M., Patel, S. N., Chan, T. H., and Tollefsbol, T. O. (2011). A Novel prodrug of epigallocatechin-3-gallate: differential epigenetic hTERT repression in human breast cancer cells. Cancer Prev. Res. (Phila). 4, 1243–1254.
Mezzanzanica, D., Canevari, S., Cecco, L. D., and Bagnoli, M. (2011). miRNA control of apoptotic programs: focus on ovarian cancer. Expert Rev. Mol. Diagn. 11, 277–286.
Mittal, A., Pate, M. S., Wylie, R. C., Tollefsbol, T. O., and Katiyar, S. K. (2004). EGCG down-regulates telomerase in human breast carcinoma MCF-7 cells, leading to suppression of cell viability and induction of apoptosis. Int. J. Oncol. 24, 703–710.
Moore, R. G., Maclaughlan, S., and Bast, R. C. (2010). Current state of biomarker development for clinical application in epithelial ovarian cancer. Gynecol. Oncol. 116, 240–245.
Mottet, D., and Castronovo, V. (2008). Histone deacetylases: target enzymes for cancer therapy. Clin. Exp. Metastasis 25, 183–189.
Oliver, S. S., and Denu, J. M. (2011). Dynamic interplay between histone H3 modifications and protein interpreters: emerging evidence for a “histone language.” Chembiochem 12, 299–307.
Richon, V. M., Sandhoff, T. W., Rifkind, R. A., and Marks, P. A. (2000). Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. U.S.A. 97, 10014–10019.
Schöndorf, T., Ebert, M. P., Hoffmann, J., Becker, M., Moser, N., Pur, S., Göhring, U. J., and Weisshaar, M. P. (2004). Hypermethylation of the PTEN gene in ovarian cancer cell lines. Cancer Lett. 207, 215–220.
Secrist, J. P., Zhou, X., and Richon, V. M. (2003). HDAC inhibitors for the treatment of cancer. Curr. Opin. Investig. Drugs 4, 1422–1427.
Steele, N., Finn, P., Brown, R., and Plumb, J. A. (2009). Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 100, 758–763.
Strathdee, G., Mackean, M. J., Illand, M., and Brown, R. (1999). A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene 18, 2335–2341.
Takahara, P. M., Rosenzweig, A. C., Frederick, C. A., and Lippard, S. J. (1995). Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature 377, 649–652.
Takai, N., and Narahara, H. (2010). Histone deacetylase inhibitor therapy in epithelial ovarian cancer. J. Oncol. 2010, 458431.
Terasawa, K., Sagae, S., Toyota, M., Tsukada, K., Ogi, K., Satoh, A., Mita, H., Imai, K., Tokino, T., and Kudo, R. (2004). Epigenetic inactivation of TMS1/ASC in ovarian cancer. Clin. Cancer Res. 10, 2000–2006.
Valls, E., Sánchez-Molina, S., and Martínez-Balbás, M. A. (2005). Role of histone modifications in marking and activating genes through mitosis. J. Biol. Chem. 280, 42592–42600.
Veeck, J., and Esteller, M. (2010). Breast cancer epigenetics: from DNA methylation to microRNAs. J. Mammary Gland Biol. Neoplasia 15, 5–17.
Walsh, C. P., Chaillet, J. R., and Bestor, T. H. (1998). Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20, 116–117.
Widschwendter, M., Jiang, G., Woods, C., Müller, H. M., Fiegl, H., Goebel, G., Marth, C., Müller-Holzner, E., Zeimet, A. G., Laird, P. W., and Ehrlich, M. (2004). DNA hypomethylation and ovarian cancer biology. Cancer Res. 64, 4472–4480.
Wilcox, C. B., Baysal, B. E., Gallion, H. H., Strange, M. A., and Deloia, J. A. (2005). High-resolution methylation analysis of the BRCA1 promoter in ovarian tumors. Cancer Genet. Cytogenet. 159, 114–122.
Yang, H., Kong, W., He, L., Zhao, J.-J., O’Donnell, J. D., Wang, J., Wenham, R. M., Coppola, D., Kruk, P. A., Nicosia, S. V., and Cheng, J. Q. (2008a). MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 68, 425–433.
Yang, H., Kong, W., He, L., Zhao, J. J., O’Donnell, J. D., Wang, J., Wenham, R. M., Coppola, D., Kruk, P. A., Nicosia, S. V., and Cheng, J. Q. (2008b). MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 68, 425–433.
Yang, N., Kaur, S., Volinia, S., Greshock, J., Lassus, H., Hasegawa, K., Liang, S., Leminen, A., Deng, S., Smith, L., Johnstone, C. N., Chen, X.-M., Liu, C.-G., Huang, Q., Katsaros, D., Calin, G. A., Weber, B. L., Bützow, R., Croce, C. M., Coukos, G., and Zhang, L. (2008c). MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res. 68, 10307–10314.
Yarbro, J. W., Page, D. L., Fielding, L. P., Partridge, E. E., and Murphy, G. P. (1999). American joint committee on cancer prognostic factors consensus conference. Cancer 86, 2436–2446.
Keywords: ovarian cancer, epigenomics, chemoprevention, biomarkers
Citation: Chen H, Hardy TM and Tollefsbol TO (2011) Epigenomics of ovarian cancer and its chemoprevention. Front. Gene. 2:67. doi: 10.3389/fgene.2011.00067
Received: 30 July 2011; Accepted: 13 September 2011;
Published online: 04 October 2011.
Edited by:
Jeff Schwartz, Griffith University, AustraliaReviewed by:
Tina Bianco-Miotto, University of Adelaide, AustraliaAlbert S. Mellick, Griffith University, Australia
Eugen Bogdan Petcu, Griffith University School of Medicine, Australia
Copyright: © 2011 Chen, Hardy and Tollefsbol. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Trygve O. Tollefsbol, Department of Biology, University of Alabama at Birmingham, CH175, 1300 University Boulevard, Birmingham, AL 35294-1170, USA. e-mail:dHJ5Z3ZlQHVhYi5lZHU=