 Pappu Srinivasan1*
Pappu Srinivasan1* Sivakumar Prasanth Kumar1
Sivakumar Prasanth Kumar1 Muthusamy Karthikeyan1 Jeyaram Jeyakanthan1 Yogesh T. Jasrai2 Himanshu A. Pandya2 Rakesh M. Rawal3 and Saumya K. Patel2
Muthusamy Karthikeyan1 Jeyaram Jeyakanthan1 Yogesh T. Jasrai2 Himanshu A. Pandya2 Rakesh M. Rawal3 and Saumya K. Patel2
- 1 Department of Bioinformatics, Alagappa University, Karaikudi, Tamil Nadu, India
- 2 Department of Botany, University School of Sciences, Gujarat University, Ahmedabad, Gujarat, India
- 3 Division of Medicinal Chemistry and Pharmacogenomics, Department of Cancer Biology, The Gujarat Cancer & Research Institute, Ahmedabad, Gujarat, India
Crimean–Congo hemorrhagic fever virus (CCHFV), the fatal human pathogen is transmitted to humans by tick bite, or exposure to infected blood or tissues of infected livestock. The CCHFV genome consists of three RNA segments namely, S, M, and L. The unusual large viral L protein has an ovarian tumor (OTU) protease domain located in the N terminus. It is likely that the protein may be autoproteolytically cleaved to generate the active virus L polymerase with additional functions. Identification of the epitope regions of the virus is important for the diagnosis, phylogeny studies, and drug discovery. Early diagnosis and treatment of CCHF infection is critical to the survival of patients and the control of the disease. In this study, we undertook different in silico approaches using molecular docking and immunoinformatics tools to predict epitopes which can be helpful for vaccine designing. Small molecule ligands against OTU domain and protein–protein interaction between a viral and a host protein have been studied using docking tools.
Introduction
Crimean–Congo hemorrhagic fever (CCHF) is a viral hemorrhagic fever of the Nairovirus group, primarily distinguished as zoonosis. Occurrence rate of sporadic cases and outbreaks of CCHF affecting humans have been reported. It has a history of endemic infections in South Africa, Pakistan, Kosovo, Turkey, United Arab Emirates and some parts of Asia. It is so named because it was first observed in Cremia of Congo in 1944 (WHO, 2011). It belongs to Bunyaviridae family of Nairovirus group. It spreads in animals through ticks or fleas and can infect human beings through the animal reservoir (Whitehouse, 2004). CCHF virus, abbreviated as CCHFV, recently noted in Gujarat, killing four people, the first outbreak reported in India. National Institute of Virology (NIV), India confirmed this outbreak and found to be due to hyalomma ticks (International Society for Infectious Diseases, 2011). The infected person normally develops febrile illness with headache, joint, lumbar, and abdominal pain with nausea, initially. After 3–5 days, hemorrhaging develops with skin discoloration and blood in saliva, urine, and vomit, leading to vascular collapse and death around 10 days after onset (WHO, 2011).
Crimean–Congo hemorrhagic fever virus has a tripartite genome consisting of large (L), medium (M), and small (S) negative-sense RNA segments that encode the viral RNA polymerase, the glycoproteins, and the nucleocapsid protein (NP), respectively (Clerx et al., 1981). Viral infection is detected by host cells via Toll-like receptor (TLR)-dependent and -independent mechanisms which results in ubiquitination and ISGylation of target proteins to induce the innate immune system. Ubiquitination involves a regulatory protein called Ubiquitin (Ub) which conjugates with target (viral) proteins and forms an Ub-tag which in turn, recognized by proteosome for destruction (Staheli et al., 2007). ISGylation engages an Ub-like molecule known as interferon-stimulated gene product 15 (ISG15), which covalently conjugates and induce innate response (Arguello and Hiscott, 2007). The viral L protein has an ovarian tumor (OTU) domain of cysteine protease family, located in N terminus, is dispensable for RNA-dependent RNA polymerase (RdRp) and deubiquitinating (DUB) activity. In order to invade host antiviral mechanisms (Figure 1), OTU domain conjugates with Ub and ISG15 to establish viral state (Bergeron et al., 2010). It was also reported that host system makes use of interferon (IFN) – induced defense mechanism against a number of viruses belonging to Bunyaviridae. MxA protein, a member of dynamin superfamily of large GTPases is exclusively induced by alpha and beta IFNs (Haller and Kochs, 2002). It has been found that MxA interacted with NP and the yield of progeny virus was reduced to 1,000-fold in CCHFV infected host cells. Hence, it is clear that human MxA protein has antiviral activity against CCFHV (Andersson et al., 2004). Current status for the treatment of CCHFV infections utilizes ribavirin to combat the infection (WHO, 2011). Treatment in infected individuals was shown to exhibit the in vivo genotoxicity and potential side effects caused by ribavirin (Tatar et al., 2005).
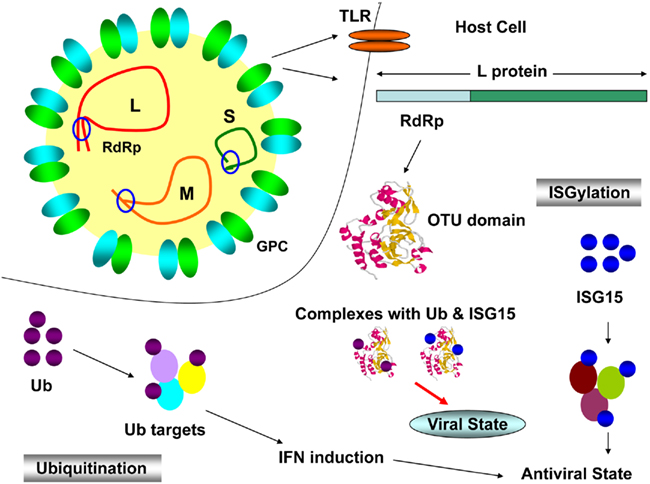
Figure 1. Crimean–Congo hemorrhagic fever virus infection cycle. L, M, and S, viral genomic segments; GPC, glycoprotein complex; TLR, Toll-like receptor; RdRp, RNA-dependent RNA polymerase; OTU domain, ovarian tumor domain; IFN, interferon; Ub, ubiquitin; ISG15, IFN simulated gene product 15.
In the present study, in silico drug designing and immunoinformatics strategies have been exploited using bioinformatics software and programs. Epitope-based immunoinformatics study was carried out for NP in order to predict informative epitopes which can be helpful for future vaccine designing. To examine the protein–protein interaction and the background of biochemical functionality between a viral and a host protein, protein–protein docking analysis of NP with human MxA protein was performed through structure-based approach. Finally, identification of effective ligand molecules to inhibit viral OTU domain were made using small molecule docking simulations.
Materials and Methods
Epitope-Based Immunoinformatics Study of NP
Protein sequence retrieval and epitope prediction parameters
We retrieved the protein sequence of NP of CCHFV (Accession No. AAX86921.1) from National Centre for Biotechnology Institute (NCBI) database (Benson et al., 2005). The locations of continuous epitopes have been correlated with various parameters (such as hydrophilicity, flexibility, accessibility, turns, exposed surface, polarity, and antigenic propensity of polypeptide chain). The epitope predictions are based on propensity scales for each of the 20 amino acids.
Assessment of solvent accessibility regions
The prediction for regions of surface accessibility is based on Emini’s surface accessibility scale. Accessibility profile is predicted using the formula 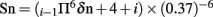 where Sn is the surface probability, δn is the fractional surface probability value, and i vary from 1 to 6. A hexapeptide sequence with Sn greater than 1.0 indicates an increased probability for being found on the surface (Emini et al., 1985).
where Sn is the surface probability, δn is the fractional surface probability value, and i vary from 1 to 6. A hexapeptide sequence with Sn greater than 1.0 indicates an increased probability for being found on the surface (Emini et al., 1985).
Epitope flexibility prediction
To strengthen the prediction accuracy, we used the Karplus and Schulz flexibility scale. This scale is derived from the mobility of protein segments based on the known temperature B factors of the alpha carbons of 31 proteins of known structure. The calculation is performed taking the center amino acid as the first amino acid of the six amino acid window length (threshold setting = 1.000; Karplus and Schulz, 1985).
Prediction of antigenicity
Peptides could be predicted in such a manner that it should possess the antigenic character and are likely to be antibody responsive. Antigenicity prediction was carried out using Kolaskar and Tongaonkar antigenicity scale. This prediction is based on a semi-empirical approach, developed on physicochemical properties of amino acid residues and their frequencies of occurrence in experimentally known segmental epitopes and has the efficiency to detect antigenic peptides with about 75% accuracy (threshold setting = 1.000; Kolaskar and Tongaonkar, 1990). The location of linear B-cell epitope was predicted using Bepipred linear epitope prediction which utilizes a combination of a hidden Markov model and a propensity scale method (threshold setting = 0.350; Larsen et al., 2006).
Hydrophobicity and hydrophilicity analysis
The amino acids making up the epitope are usually charged and hydrophilic in nature. Parker hydrophilicity scale was applied to evaluate the hydrophilicity. It is based on peptide retention time in high-performance liquid chromatography (HPLC) on a reversed-phase column. Using a window of seven residues, these experimental values are calibrated for each of the seven residues and the arithmetical mean of the seven residue value was assigned to the fourth, (i + 3), residue in the segment (threshold setting = 1.678; Parker et al., 1986).
To understand the distribution of polar and apolar residues along a protein sequence, hydrophobicity plot was studied using Kyte–Doolittle hydropathicity index (threshold setting ≥0 are hydrophobic) available at Expasy Protscale server (Gasteiger et al., 2005). Kyte–Doolittle is a widely applied scale for delineating hydrophobic character of a protein and useful in predicting membrane-spanning domains, potential antigenic sites, and regions that are likely exposed on the protein surface (Kyte and Doolittle, 1982).
HLA binding peptide prediction
Major histocompatibility complexes (MHC-I and MHC-II) display specificity for binding with their respective epitopes. In human, these MHC molecules are known by human leukocyte antigen (HLA) alleles. The HLA binding peptides were predicted (threshold setting = 3.000) using TmhcPred server. This prediction is based on the virtual and quantitative matrices based on 97 MHC alleles using position specific scoring matrices (PSSMs) and utilizes supervised learning method called support vector machine (SVM; Bhasin et al., 2003).
Note that the predictions based on the above mentioned scales viz. Emini, Karplus and Schulz, Kolaskar and Tongaonkar, Bepipred linear epitope prediction and Parker are different web-based tools centralized in a repository implemented by immune epitope database and analysis resource (IEDB) for the prediction and analysis of immune epitopes (Zhang et al., 2008).
Study on Protein–Protein Docking of Human MxA- Viral NP Proteins
Ab initio modeling of NP
An attempt was made to construct a homology model of NP using Swiss Model (Schwede et al., 2003). No suitable template was found by target-template alignment programs such as BLAST and HHSearch with the significant cutoffs for template identification (BLAST: e-value = 0.0001; HHSearch: e-value = 0.0001 and p-value = 50). Therefore, an ab initio model was constructed using Bhageerath server. Bhageerath, an energy based approach which narrow downs the search space of tertiary structures employing sequence and secondary structure information as building stage; and later filtered by atomic potential based biophysical methodology to present 10 plausible candidate structures (Jayaram et al., 2006).
Structure validation and energy minimization of modeled NP
Modeled NP structure was validated for structure correctness and stereochemistry using Ramachandran plot (Ramachandran et al., 1963) from RAMPAGE server (Lovell et al., 2003). The validated structure was subjected to energy minimization using NOMAD-Ref server (Lindahl et al., 2006). Normal mode analysis, deformation, and refinement, abbreviated as NOMAD-Ref, make use of elastic network model (ENM) as well as classical force fields to calculate normal modes for structural refinement and optimization.
Functional site identification of MxA protein
The protein structure of human MxA protein (Gao et al., 2010) was retrieved from Protein Databank (PDB ID: 3LJB; Bernstein et al., 1977). The MxA protein structure is comprised of two unique chains, functional site identification was carried out for a single chain using ConSurf server (Ashkenazy et al., 2010). This enables the identification of functionally important regions on the surface of a protein or domain of known three-dimensional protein structure and relied on the phylogenetic relations between its close sequence homologs.
Protein–protein docking
Protein–protein docking simulations were performed using global range molecular matching (GRAMM)-X protein–protein docking web server v.1.2.0 (Tovchigrechko and Vakser, 2006). In this process, optimal functional site of MxA protein deduced by ConSurf was implemented as potential interface region for docking simulations in order to avoid blind search and modeled NP was given as ligand input and best 10 returned docked conformations were analyzed. GRAMM, an empirical approach which smoothes the intermolecular energy function by changing the range of the atom–atom potentials, locates the area of the global minimum of intermolecular energy using a characteristic six-dimensional exhaustive search through the relative translations and rotations of the molecules. Moreover, this docking simulation was repeated using ClusPro protein–protein docking program (Kozakov et al., 2010). The program recruits PIPER, a FFT (Fast Fourier Transform) based rigid docking program in its first stage to generate 1,000 low energy docked conformations using pairwise interaction potentials. In the second stage, ClusPro clusters these conformations and retains 30 largest clusters having low energy. Later, the retained clusters are analyzed by SDU (Semi-Definite programming based Underestimation) program which predicts the stability of the clusters using medium-range optimization algorithm (resembling the funnel-like behavior of the free energy to attain local minima) and the stable clusters are then refined using Monte-Carlo simulation.
Study on Protein-Small Molecule Docking of Viral OTU Domain
Sequence retrieval and multiple sequence alignment to find active site
Active site of viral OTU domain was identified using multiple sequence alignment (MSA) with human Ub-specific protease otubains 1 and 2 (Accession No. AAO27702.1 and AAO27703.1). Protein sequences were retrieved from NCBI (Benson et al., 2005) and MSA was performed using EBI ClustalW program (Blosum weighting matrix, gap opening penalty = 10 and gap extension penalty = 0.1; Larkin et al., 2007). Further, active site pocket was structurally analyzed using CASTp program (Dundas et al., 2006).
Preparation of protein target structure and ligand molecules
The protein structure of viral OTU domain protease (James et al., 2011) was retrieved from PDB (ID: 3PT2). The dataset comprised of four ligands having known inhibitory activity chosen from literature, out of which one molecule is N-ethylmaleimide (NEM) and the other three were its analogs. All the molecules were computationally designed using Marvin Sketch 5.2 and subsequently, geometrically optimized using three-dimensional cleaning utility (ChemAxon LLC, 2008).
Protein-small molecule docking
Active site residues predicted from sequence alignment was specified for active site directed molecular docking. Ligand dataset under study were docked separately into the binding site of the receptor using PATCHDOCK (Duhovny et al., 2005). PATCHDOCK performs docking simulations in three phases viz. molecular shape representation, surface patch matching, and finally, filtering and scoring. Molecular shape representation step initially computes the molecular surface of given molecule, followed by detection of geometric patches using segmentation algorithm. Surface patch matching stage applies geometric hashing and pose-clustering techniques to match the filtered-hot spot residues containing patches. The candidate molecule is refined at last from patch penetration between ligand and receptor molecules and ranked by shape complementarity score in its final stage.
Results
NP Epitope Predictions
Epitope prediction for the NP showed regions spanning the sequence between positions 39 and 125, and scored well when all the scales were considered (Figure 2). Kolaskar and Tongaonkar antigenicity scale predicted a 24 length peptide in the positions 16–39. When all the scales were taken into consideration, this peptide did not show any significance in terms of surface accessibility, flexibility, and hydrophobicity/hydrophilicity. Similar setback occurred when a Bepipred linear epitope prediction showed an 11 length peptide in the positions of 8–18, which is in low significance region. Therefore, these two predicted epitopes were discarded. Peptides in the sequence positions 42–48, 68–77, 79–88, and 104–125 predicted using Kolaskar and Tongaonkar antigenicity scale displayed antigenicity. Further, Bepipred analysis (B-cell epitope prediction) revealed a continuous predicted epitope with 13 amino acid residues in the sequence positions 56–68 (Table 1). Beside, we found that these predicted epitope regions were observed in the surface of the modeled NP protein. Hence, the region spanning the sequence positions of 42–48, 56–68, 68–77, and 79–88 will be of greater importance for epitope-based vaccine design.
Figure 2. Epitope prediction of NP using (A) Emini surface accessibility prediction (threshold:1.000), (B) Karplus and Schulz flexibility prediction (threshold:1.000), (C) Kolaskar and Tongaonkar antigenicity prediction (threshold:1.000), (D) Parker hydrophilicity prediction (threshold:1.678), and (E) Kyte and Doolittle hydrophobicity prediction (threshold:0.000).
Table 1. Predicted epitopes of NP protein via Kolaskar and Tongaonkar antigenicity scale (S.no. 1–5) and Bepipred continuous epitope predictions (S.no. 6 and 7).
HLA Binding Peptides Predictions of NP and OTU Domain
Nucleocapsid protein and OTU domain sequences generated 141 and 179 nanomers, respectively. Promiscuous HLA binders are those peptides which bind most HLA alleles and its respective sub-alleles with increased affinity. Prediction of promiscuous HLA peptide binders displayed similarity as well as variation. Dissimilar pattern was found in NP analysis. For example, H-2Db and H-2Dd alleles expressed different peptides, 96-RVNANTAAL-104 and 71-VEVPKIEQL-79. Similar peptide prediction was observed in OTU domain sequence (PDB ID: 3PT2 chain A). For example, HLA-B7 and B8 alleles exhibited similar peptide 81-EARLVGLSL-89 (Table 2). Despite the use of scoring system of Tmhcpred server to rank its predictions, an in vitro and pharmacogenetic study will help to detect promiscuous HLA binders.
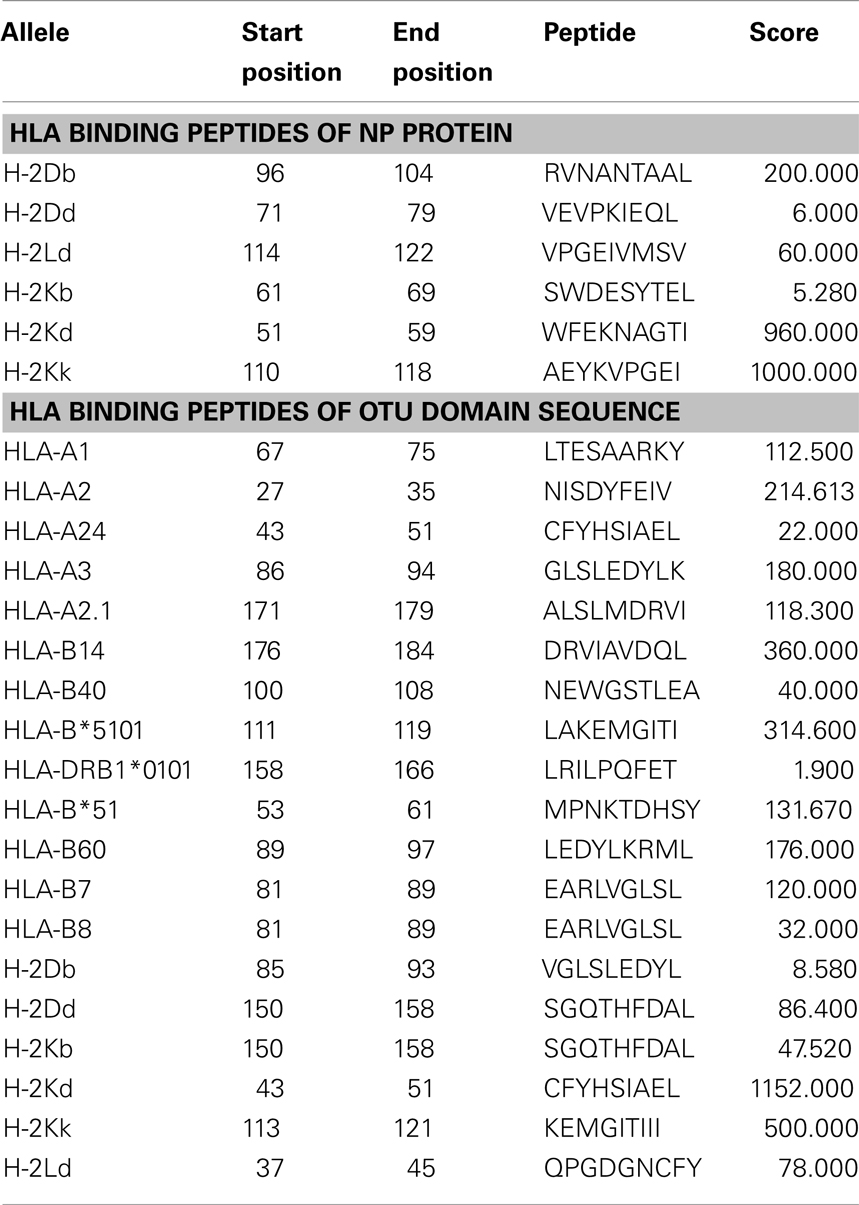
Table 2. Predicted HLA binding peptides of selected alleles (only highly scored peptides were reported here).
Protein–Protein Docking Study of MxA–NP Proteins
Among the 10 plausible candidate structures generated, a best ab initio model for the NP was selected using Ramachandran plot. Selected model exhibited 98% amino acids in allowed regions with the exception of two outliers (Figure 3A). Subsequently, the selected model displayed the structure with energy minimized to −5760.383 KJ/mol (Figure 3B).
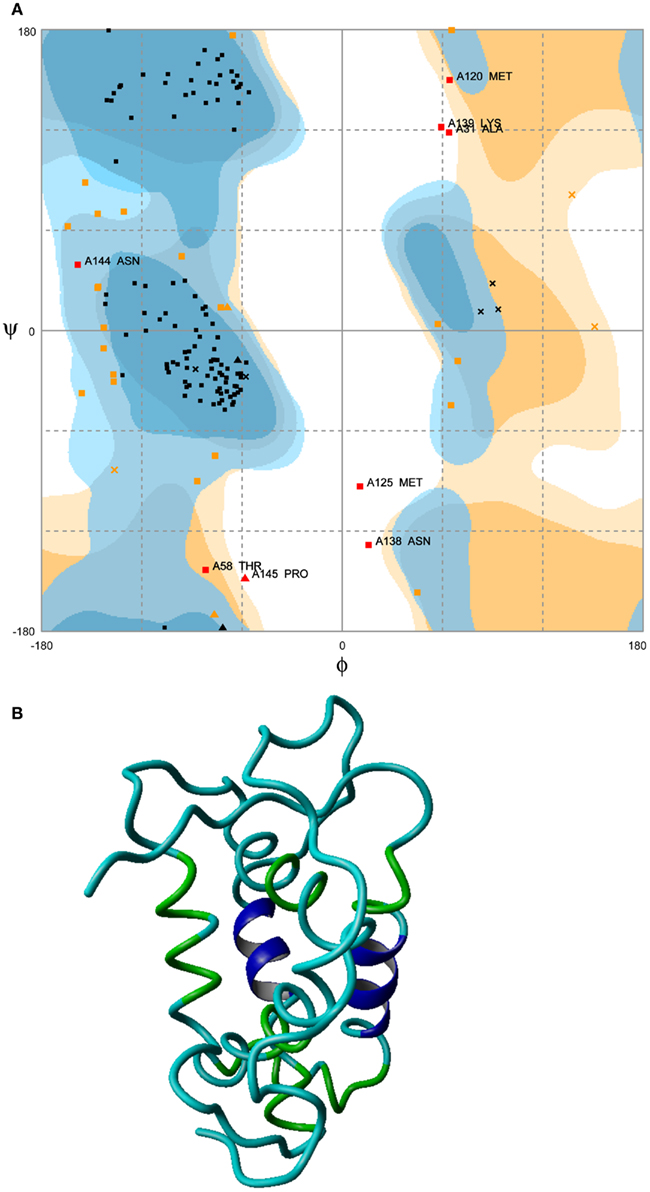
Figure 3. Energy minimized structure of NP. (A) Ramachandran plot favoring 98% in allowed regions with the exception of two outliers (boxed regions) and (B) ab initio model.
In order to evade blind docking of MxA protein, functional site identification was carried out to understand the region of protein interaction sites (Figure 4A). It is well known that MxA protein is a homodimer of unique chains. Dimer interface site was predicted to be non-interacting site since it is required for its dimerization. The regions exposed to solvent were predicted to be entirely accessible for protein–protein interaction. Further, MSA of MxA protein with its close sequence homology revealed similar results. Hence, regions excluding dimer interface was utilized as potential receptor-interaction site for protein–protein docking.
Figure 4. (A) Predicted MxA protein functional site (only one chain shown). Conserved regions >90% (dark purple) and 80–89% (pink color) in MSA alignment with its close sequence homologs (B) MxA protein (surface with electrostatic potential) as docked with NP (surface with B factor).
Protein–protein docking simulations with MxA protein as receptor and modeled NP as ligand molecule was also performed. Among the best 10 docked conformations, a best docked conformation (Figure 4B) was selected in which the docked regions between two proteins exhibited significant surface complementarity. To examine the possibility of any penetrations between two molecules, the selected conformation was manually studied using standard three-dimensional protein structure visualizer.
Further, this docking simulation was repeated using ClusPro protein–protein docking program to confirm the interactions of MxA–NP protein. Rigid body dock PIPER program yielded a weighted score of −1066.40 Kcal/mol whereas ClusPro provided a largest cluster (first cluster as per rank) in which 154 docked conformations were found with cluster center scored −923.80 Kcal/mol. The variation observed in the energy value of lowest energy docked conformer with its cluster centered conformer was −142.60 Kcal/mol. The comparative analysis of the docked conformations generated from GRAMM-X and ClusPro showed that the interacting site was found to be the MxA protein’s dimer interface and in good correspondence with the predictions of ConSurf server. Such studies on expression level of human MxA protein in CCHFV infected human subjects will help us to demonstrate that a purified MxA protein usually a product of recombinant genetic engineering will be a best candidate for CCHFV vaccinomics, if the MxA protein expression in infected human cells is expected to below.
Protein-Small Molecule Docking of Viral OTU Domain
Active site prediction of OTU domain was achieved using sequence and structure-based approaches. MSA of OTU domain with human otubains 1 and 2 revealed that a conserved pattern, DGNCFY was preserved in these cysteine proteases. The MSA analysis (Figure 5A) showed that the active site consisted of Asp40 and Cys43. Further, structure-based active site prediction was made to find whether the conserved pattern containing the two active site residues, Asp40 and Cys43, is exposed or buried. It was observed that conserved motif is exposed and solvent accessible (Figure 5B).
Figure 5. Conserved pattern DGNCFY (boxed) around active site residues, (A) Asp40 and Cys43 (B) Spacefill model of OTU domain showing conserved pattern (shown as patch).
Small inhibitor molecules targeting the active site residues will be of greater importance for designing specific inhibitors for viral OTU domain. Since OTU domain belongs to cysteine protease family and has DUB activity, the literature survey could not reveal any DUB small molecule inhibitors. Hence, search was carried on for cysteine protease small molecule inhibitors having proven inhibitory activity and the potential to modify active site cysteine residue (Singh and Liu, 2000). N-ethylmaleimide (NEM) and its analogs, N-Phenyl maleimide (NPM), N,N′-(1,2-phenylene) dimaleimide (NNPD) and N-(1-pyrenyl) maleimide (NPYM) was used as ligand dataset (Figure 6). Conserved motif was employed as active site residues (residues: 37–42) for active site directed docking procedure using PATCHDOCK. Among the 200 docked conformations generated for each procedure, best conformation was selected using the shape complementarity score and area of docking translations (Table 3; Figure 6). Due to non-specificity of potent cysteine proteases having the ability to harm all the cellular proteins exposing cysteine residues on surface, there arises a requirement of potent cysteine protease inhibitors which should target the complete conserved motif. On the other hand, fragment based and de novo drug design of DUB inhibitors will prove to be a best drug candidate to treat against this deadliest CCHFV infection.
Figure 6. Two-dimensional structure of ligands and respective docked conformations with OTU domain (A) N-ethylmaleimide (NEM), (B) N-phenyl maleimide (NPM), (C) N,N ′-(1,2-phenylene) dimaleimide (NNPD), and (D) N-(1-pyrenyl) maleimide (NPYM).

Table 3. Docking results of OTU domain with NEM and its analogs.
Discussion
The present study was focused on in silico epitope-based immunoinformatics and molecular docking against the CCHFV deadliest virus. Despite the hindrances to initiate research on CCHFV as it requires biosafety level-4 (BSL-4), the high pathogenicity to establish endemic and the non-availability of effective treatments poses a great threat to human community. These predicted epitopes may play a highly informative and crucial role in antidote production against CCHFV. Common antigenic peptides are also important for synthetic peptide vaccine production or antidote production. Similarly, small molecule based drug designing can also be initiated simultaneously. Highly specific NEM analogs and/or DUB inhibitors designed using fragment based or de novo approaches will prove to be the best drug candidates to establish effective treatment against this infection. The real consequences will arise if this pathogenic form takes the form of a biological warfare as similar to Anthrax. We hope that our current findings would help the pharmacologists as well as scientific research bodies to instigate research on this very recent outbreak.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Alagappa University is gratefully acknowledged for providing the infrastructure facilities for carrying out this research work. The authors want to thank the anonymous reviewers for their critical comments.
References
Andersson, I., Bladh, L., Jazi, M. M., Magnusson, K. E., Lundkvist, A., Haller, O., and Mirazimi, A. (2004). Human MxA protein inhibits the replication of crimean-congo hemorrhagic fever virus. J. Virol. 78, 4323–4329.
Arguello, M. D., and Hiscott, J. (2007). Ub surprised: viral ovarian tumor domain proteases remove ubiquitin and ISG15 conjugates. Cell Host Microbe 2, 367–369.
Ashkenazy, H., Erez, E., Martz, E., Pupko, T., and Ben-Tal, N. (2010). ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, 529–533.
Benson, D. A., Mizrachi, I. K., Lipman, D. J., Ostell, J., and Wheeler, D. L. (2005). GenBank. Nucleic Acids Res. 33, 34–38.
Bergeron, E., Albarino, C. G., Khristova, M. L., and Nichol, S. T. (2010). Crimean-Congo hemorrhagic fever virus-encoded ovarian tumor protease activity is dispensable for virus RNA polymerase function. J. Virol. 84, 216–226.
Bernstein, F. C., Koetzle, T. F., Williams, G. J., Meyer, E. E., Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T., and Tasumi, M. (1977). The protein data bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112, 535–542.
Bhasin, M., Singh, H., and Raghava, G. P. S. (2003). MHCBN: a comprehensive database of MHC binding and non-binding peptides. Bioinformatics 19, 665–666.
ChemAxon LLC. (2008). “Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions,” in Marvin 5.2.1. Available at: www.chemaxon.com
Clerx, J. P., Casals, J., and Bishop, D. H. (1981). Structural characteristics of nairoviruses (genus Nairovirus, Bunyaviridae). J. Gen. Virol. 55, 165–178.
Duhovny, S. D., Inbar, Y., Nussinov, R., and Wolfson, H. J. (2005). PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, 363–367.
Dundas, J., Ouyang, Z., Tseng, J., Binkowski, A., Turpaz, Y., and Liang, J. (2006). CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, 116–118.
Emini, E. A., Hughes, J. V., Perlow, D. S., and Boger, J. (1985). Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 55, 836–839.
Gao, S., von der Malsburg, A., Paeschke, S., Behlke, J., Haller, O., Kochs, G., and Daumke, O. (2010). Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465, 502–506.
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., and Bairoch, A. (2005). “Protein identification and analysis tools on the ExPASy server,” in The Proteomics Protocols Handbook, ed. J. M. Walker (Totowa, NJ: Humana Press), 571–607.
Haller, O., and Kochs, G. (2002). Interferon-induced Mx proteins: dynamin-like GTPases with antiviral activity. Traffic 3, 710–717.
International Society for Infectious Diseases. (2011). Crimean-Congo Hemorrhagic Fever – India. Communications, Archive No. 20110123.0285. Available at: http://www.promedmail.org/pls/apex/f?p=2400:1001:229078591000888
James, T. W., Frias-Staheli, N., Bacik, J. P., Macleod, L. J. M., Khajehpour, M., Garcia-Sastre, A., and Mark, B. L. (2011). Structural basis for the removal of ubiquitin and interferon-stimulated gene 15 by a viral ovarian tumor domain-containing protease. Proc. Natl. Acad. Sci. U.S.A. 108, 2222–2227.
Jayaram, B., Bhushan, K., Shenoy, S. R., Narang, P., Bose, S., Agrawal, P., Sahu, D., and Pandey, V. (2006). Bhageerath: an energy based web enabled computer software suite for limiting the search space of tertiary structures of small globular proteins. Nucleic Acids Res. 34, 6195–6204.
Karplus, P. A., and Schulz, G. E. (1985). Prediction of chain flexibility in proteins - a tool for the selection of peptide antigens. Naturwissenschaften 72, 212–213.
Kolaskar, A. S., and Tongaonkar, P. C. (1990). A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 276, 172–174.
Kozakov, D., Hall, D. R., Beglov, D., Brenke, R., Comeau, S. R., Shen, Y., Li, K., Zheng, J., Vakili, P., Paschalidis, I. C., and Vajda, S. (2010). Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13-19. Proteins 78, 3124–3130.
Kyte, J., and Doolittle, R. (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948.
Larsen, J. E. P., Lund, O., and Nielsen, M. (2006). Improved method for predicting linear B-cell epitopes. Immunome Res. 2, 2–8.
Lindahl, E., Azuara, C., Koehl, P., and Delarue, M. (2006). NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 34, 52–56.
Lovell, S. C., Davis, I. W., Arendall, W. B., de Bakker, P. I. W., Word, J. M., Prisant, M. G., Richardson, J. S., and Richardson, D. C. (2003). Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50, 437–450.
Parker, J. M., Guo, D., and Hodges, R. S. (1986). New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 25, 5425–5432.
Ramachandran, G. N., Ramakrishnan, C., and Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99.
Schwede, T., Kopp, J., Guex, N., and Peitsch, M. C. (2003). Swiss-model: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385.
Singh, T. K., and Liu, L. (2000). Modification of cysteine residues by N-ethylmaleimide inhibits annexin II tetramer mediated liposome aggregation. Arch. Biochem. Biophys. 381, 235–240.
Staheli, N. F., Giannakopoulos, N. V., Kikkert, M., Taylor, S. L., Bridgen, A., Paragas, J. J., Richt, J. A., Rowland, R. R., Schmaljohn, C. S., Lenschow, D. J., Snijder, E. J., Sastre, A. G., and Virgin, H. W. (2007). Ovarian tumor (OTU)-domain containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2, 404–416.
Tatar, A., Ozkurt, A., and Kiki, I. (2005). Genotoxix effect of ribavirin in patients with Crimean-Congo hemorrhagic fever. Jpn. J. Infect. Dis. 58, 313–315.
Tovchigrechko, A., and Vakser, I. A. (2006). GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34, 310–314.
WHO. (2011). World Health Organization (WHO) Fact Sheet of Crimean-Congo Haemorrhagic Fever. Available at: http://www.who.int/mediacentre/factsheets/fs208/en/
Zhang, Q., Wang, P., Kim, Y., Haste-Andersen, P., Beaver, J., Bourne, P. E., Bui, H. H., Buus, S., Frankild, S., Greenbaum, J., Lund, O., Lundegaard, C., Nielsen, M., Ponomarenko, J., Sette, A., Zhu, Z., and Peters, B. (2008). Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 36, 513–518.
Keywords: CCHFV, OTU domain, polymerase, immunoinformatics, molecular docking
Citation: Srinivasan P, Kumar SP, Karthikeyan M, Jeyakanthan J, Jasrai YT, Pandya HA, Rawal RM and Patel SK (2011) Epitope-based immunoinformatics and molecular docking studies of nucleocapsid protein and ovarian tumor domain of Crimean–Congo hemorrhagic fever virus. Front. Gene. 2:72. doi: 10.3389/fgene.2011.00072
Received: 21 July 2011;
Accepted: 29 September 2011;
Published online: 02 November 2011.
Edited by:
John Hancock, Medical Research Council, UKReviewed by:
Yu Xue, Huazhong University of Science and Technology, ChinaJinn-Moon Yang, National Chiao Tung University, Taiwan
Copyright: © 2011 Srinivasan, Kumar, Karthikeyan, Jeyakanthan, Jasrai, Pandya, Rawal and Patel. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Pappu Srinivasan, Department of Bioinformatics, Alagappa University, Karaikudi-630003, Tamil Nadu, India. e-mail:c3JpLmJpb2luZm9ybWF0aWNzQGdtYWlsLmNvbQ==