 Brock C. Christensen1,2
Brock C. Christensen1,2 Carmen J. Marsit1,2*
Carmen J. Marsit1,2*- 1 Section on Biostatistics and Epidemiology, Department of Community and Family Medicine, Dartmouth Medical School, Hanover, NH, USA
- 2 Department of Pharmacology and Toxicology, Dartmouth Medical School, Hanover, NH, USA
This review considers the emerging relationships between environmental factors and epigenetic alterations and the application of genome-wide assessments to better define these relationships. First we will briefly cover epigenetic programming in development, one-carbon metabolism, and exposures that may disrupt normal developmental programming of epigenetic states. In addition, because a large portion of epigenetic research has focused on cancer, we discuss exposures associated with carcinogenesis including asbestos, alcohol, radiation, arsenic, and air pollution. Research on other exposures that may affect epigenetic states such as endocrine disruptors is also described, and we also review the evidence for epigenetic alterations associated with aging that may reflect cumulative effects of exposures. From this evidence, we posit potential mechanisms by which exposures modify epigenetic states, noting that understanding the true effect of environmental exposures on the human epigenome will require additional research with appropriate epidemiologic studies and application of novel technologies. With a more comprehensive understanding of the affects of exposures on the epigenome, including consideration of genetic background, the prediction of the toxic potential of new compounds may be more readily achieved, and may lead to the development of more personalized disease prevention and treatment strategies.
Introduction
Epigenetics can be broadly defined as mitotically and meiotically heritable changes in gene expression and gene expression potential that cannot be explained by changes in DNA sequence. Examples of epigenetic states that can be altered include methylation of cytosine residues on DNA, post-translational modification of histone tails, and microRNA (miRNA) expression. DNA methylation is the most widely studied epigenetic mark and refers to the catalytic addition of a methyl group to the fifth carbon position of a cytosine residue that is followed on the same strand by guanine, also known as a CpG dinucleotide. Though underrepresented in the genome, CpG dinucleotides occur in concentrations known as CpG-islands, and CpG-islands can be found in the promoter region of approximately half of all human genes. In pathologically normal cells promoter CpG-island regions are typically unmethylated. Aberrant methylation of a CpG-island can result in gene silencing, and this is a well-recognized mechanism by which epigenetic alterations can contribute to a disease phenotype.
Epigenetic regulatory mechanisms are stable mechanisms for the control of gene expression, though they also display some level of plasticity, likely in response to various cues from the environment. Thus, it is increasingly important to characterize the role for environmental exposures in epigenetic alterations. Much of the existing evidence implicating a role of environmental factors on the epigenome comes from studies of disease outcomes such as cancer and adverse reproductive/developmental events, although there is a growing literature in the role of the psychosocial environment’s impact on the epigenome as well. Initially, epigenetic alterations were identified in various human tumors and consequently, environmental exposures to toxicants known to have an etiologic role in cancer such as air pollutants, and metals have been studied and implicated in the modification of epigenetic marks in pathologic tissues.
To better understand the importance and function of such alterations in disease states, we might begin to consider how environmental factors may influence the epigenome in pathologically normal tissues, perhaps representing alterations necessary to initiate or predispose to disease phenotypes. To date, the majority of epigenetic research in human studies has focused on DNA methylation, though emerging work is beginning to address the affect of environmental exposures on histone modifications and miRNA expression. This review highlights the current evidence that epigenetic alterations are associated with environmental exposures, focusing on human studies, and considers through this research the potential mechanisms behind environmentally related epigenetic alterations.
Development and Epigenetic Programming
The establishment of somatic cell epigenetic patterns, in mammals, occurs early in fetal development. The re-establishment of appropriate epigenetic patterns is subsequent to genome-wide de-methylation following fertilization, required for allowing totipotency and pluripotency dynamics (Morgan et al., 2005). Thus, embryonic and fetal development represent a critical period during which nutrient availability as well as environmental stressors, including toxicant exposures, have great potential to affect the epigenetic reprogramming and patterning phenomena. Thus, these effects can not only have implications for proper development, but perhaps even life-long conditioning and health (Li, 2002; Gluckman et al., 2008). For instance, it is well-known that folate deficiency is associated with neural tube defects (NTD; Daly et al., 1995), animal studies have indicated the necessity of sufficient methyl group availability for proper neural tube development (Dunlevy et al., 2006), and hypomethylation of long interspersed nucleotide elements (LINE) and genomic DNA has been associated with increased risk of NTDs in humans (Wang et al., 2010). Beyond birth, though, epigenetic mechanisms may contribute to the developmental origins of health and disease hypothesis (Waterland and Michels, 2007), which links nutritional and environmental stimuli at critical developmental periods (pre- and post-natal) to susceptibility for various health outcomes throughout life, including metabolic and chronic diseases (Gluckman and Hanson, 2004).
The importance of folate, and other nutrients involved in one-carbon metabolism on the establishment and maintenance of DNA methylation and potentially other epigenetic mechanisms is one mode through which toxicants may have epigenome-wide effects. One-carbon metabolism is the network of biochemical reactions essential to both DNA synthesis and all cellular biomolecule (nucleic acids, proteins, lipids) methylation reactions that involve the transfer of one-carbon groups. Folate (vitamin B9), a central nutrient in this pathway, donates its methyl group for homocysteine remethylation to methionine. Subsequently, methionine is the methyl donor for all cellular methylation reactions, most notable for this discussion being DNA and histone methylation via S-adenosyl-methionine (SAM), and other B vitamins (B2, B6, B12) that act as enzymatic cofactors in the network. Collectively, B vitamins, homocysteine, and methionine are important contributors to the maintenance of DNA integrity and DNA methylation. The one-carbon pathway links nutrient availability with not only DNA methylation, but also with toxicant metabolism through glutathione and the glutathione transferase (GST) enzyme family (Figure 1). Therefore, interactions between one-carbon pathway participants may modify associations between exposures, pathway participants, and methylation alterations. In fact, a study of reduced GST enzyme activity in a mouse model of Alzheimer’s disease and neuronal health has shown that SAM can mediate the activity of GST enzymes (Tchantchou et al., 2008). This suggests that reactions dependent upon SAM are necessary for GST activity and that SAM may be a critical mediator of neuronal health (Tchantchou et al., 2008). Furthermore, it could be predicted that GST enzyme family genotypes (among other genotypes) will be shown to modify associations between certain exposures and DNA methylation alterations. Hence integrative studies that incorporate genetics (genotype and/or alterations) with measures of epigenetic alterations will allow a more comprehensive understanding of the relation among exposures, epigenetic alterations, and genetic states.
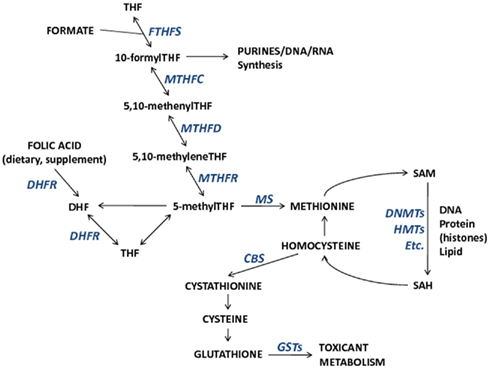
Figure 1. The relationships among metabolic components involved in one-carbon metabolism and important for DNA synthesis, DNA methylation, and toxicant metabolism. Enzymes that participate in metabolic reactions appear italicized in blue. Metabolic component abbreviations: tetrahydrofolate (THF), dihydrofolate (DHF), S-adenosyl-methionine (SAM), S-adenosyl-homocysteine (SAH).
The key role of one-carbon metabolism also brings to light the potential risks of high-dose nutrient supplementation practices. Folate supplementation is associated with a reduced risk of many forms of cancer, though once disease is present it is possible that a relative increase in one-carbon nutrient levels may speed disease progression or invasiveness dependent upon the epigenetic character of the disease state. Some evidence for this potential comes from our recent study of breast tumor methylation where increased folate intake (controlling for potential confounders) was associated with a methylation profile that was independently associated with increased tumor size (Christensen et al., 2010). Excessive folate supplementation may have potential adverse effects including masking of B12deficiency, disruption of zinc function, or interfering with one-carbon homeostasis and additional study is required to elucidate these potential effects (Hathcock, 1997; Sauer et al., 2009). In addition, more research is needed to ascertain best practices for assessing folate/methyl availability, perhaps comparing food-frequency-questionnaire data to homocysteine and/or SAM/SAH levels in a prospective manner.
Toxicants and Epigenetic Alterations
Alcohol
Alcohol is known to interfere with folate absorption in the intestine and hepatic release of folate, and hence, supply to tissues (Hillman and Steinberg, 1982) and thus it too can be considered a toxicant with the potential for epigenomic alteration. More specifically, through interference with several steps of methionine metabolism alcohol can lead to activation of betaine homocysteine methyltransferase thereby activating a compensatory pathway for maintenance of SAM levels (Kharbanda, 2009). Continuous exposure though, can overload the compensatory pathway and result in liver injury. In addition to liver injury, an etiologic role for alcohol in several cancers is well established (International Agency for Research on Cancer, 1988). Alcohol consumption increases colon cancer risk (Giovannucci et al., 1995), though the association may be modified by dietary folate, and may be specific to LINE-1 hypomethylated tumors (Schernhammer et al., 2010). More specifically, subjects with high folate intake were less likely to develop LINE-1 hypomethylated colon cancers but subjects with high alcohol consumption had a significantly increased risk of developing LINE-1 hypomethylated colon cancers with no association for more highly methylated LINE-1 tumors (Schernhammer et al., 2010). In addition, in head and neck squamous cell carcinoma, where alcohol has a well-recognized etiologic role, alcohol consumption has been significantly associated with reduced LINE-1 methylation (Smith et al., 2007). Profiles of DNA methylation in HNSCC tumors are significantly and independently associated with alcohol intake (Marsit et al., 2009), suggesting that alcohol use may drive or select for the epigenetic alteration of specific gene regions. In multiple meta-analyses of prospective and case–control studies of breast cancer, a 10% excess risk for each alcoholic drink per day has been reported (Hamajima et al., 2002; Key et al., 2006). A study of breast tumor DNA methylation profiles demonstrated a significant, independent association (controlling for age, dietary folate, and other variables) between alcohol intake and tumor DNA methylation profile (based on 1413 CpG loci; Christensen et al., 2010). In addition to DNA methylation, ethanol exposure has been shown to selectively acetylate histone H3 at lysine 9 (H3K9) in primary culture of rat hepatocytes (Park et al., 2003) and in rats in vivo (Kim and Shukla, 2006), though studies in humans are necessary. Collectively, these studies suggest that a major carcinogenic mechanism of action of alcohol is interference with epigenetic regulation, in part through disruption of one-carbon metabolism.
Asbestos
Despite the known risks of exposure, asbestos continues to be mined and exported to developing nations where occupational exposures can be high. With approximately 80% of cases reporting a known exposure to asbestos (Robinson and Lake, 2005), asbestos exposure is the main risk factor for malignant pleural mesothelioma. Importantly, in contrast to tobacco smoke and radiation, asbestos is known to be a weak mutagen (Sugarbaker et al., 2008), and there have been several reports of tumor suppressor gene methylation in mesothelioma (Ohta et al., 1999; Hirao et al., 2002; Lee et al., 2004; He et al., 2005). In patients with mesothelioma, Tsou et al. (2007) described a significant association between self-reported asbestos exposure and methylation at the MT1A, and MT2A gene loci among 28 genes examined. A significant association has been reported between an increasing number of methylated cell cycle control genes and a quantitative asbestos burden measure (Christensen et al., 2008). This was followed by examination using an array-based approach, which demonstrated that quantitative measure of asbestos exposure was associated with over 100 discrete CpG loci, and that in almost all cases (94%) there was increased methylation associated with increased exposure (Christensen et al., 2009a). Further, overall methylation profiles for mesotheliomas were significantly associated with asbestos exposure burden (Christensen et al., 2009a).
Arsenic
Arsenic exposure has been associated with DNA methylation alterations in non-pathologic as well as tumor tissues and there is some data suggesting that changes in miRNA expression and histone tail modifications are also associated with exposure to arsenic. Although the mode (or modes) by which arsenic contributes to carcinogenesis is not completely clear, there is some speculation that arsenite-generated free radicals and arsenite-generated reactive oxygen species lead to genotoxic damage (Rossman, 2003). Despite continued speculation on the mechanisms by which arsenic can contribute to carcinogenesis, in vitro exposures to inorganic arsenic species have been shown to result in altered DNA methylation (Zhao et al., 1997; Zhong and Mass, 2001).
As more studies are conducted a complex picture of dose-dependent DNA methylation alterations with arsenic exposure is beginning to emerge. Chanda et al. (2006) have shown that arsenic exposure can result in CDKN2A hypermethylation in human blood DNA, but that a subgroup of cases had hypomethylation with high arsenic exposure. More recently, a group of some of the same authors has better quantified the relation showing that exposures to 250–500 μg/L of arsenic in drinking water results in global hypermethylation (3H methyl group uptake), but that >500 μg/L of arsenic results in global hypomethylation (Majumdar et al., 2010). In human bladder cancer, relatively low levels of inorganic arsenic exposure has been associated with methylation of RASSF1A and PRSS3 (but not CDKN2A; Marsit et al., 2006b). A recent study of a large number of human bladder tumors that examined hundreds of genes demonstrated a relationship in promoter methylation profiles with arsenic exposures, suggesting that more highly methylated tumors came from individuals with larger drinking water arsenic exposures, and that these tumors were more aggressive (Wilhelm-Benartzi et al., 2010). In mouse models of methyl- or folate-deficient diets, arsenic exposure through water supply led to hypomethylation in hepatic-derived DNA (Okoji et al., 2002), as well as to increases in chromosomal aberrations in blood lymphocytes (Mcdorman et al., 2002). In another study, Pilsner et al. (2007) showed increased DNA methylation to be associated with urinary and plasma arsenic and plasma folate, and that the association between arsenic and methylation was modified by folate in that it was restricted to individuals with high plasma folate. This same group has shown that folic acid supplementation lowers blood arsenic (Gamble et al., 2007), maternal and cord blood pairs have highly correlated arsenic levels (Hall et al., 2007), and folate deficiency, hyperhomocysteinemia, and leukocyte hypomethylation are associated with arsenic-induced skin lesions (Pilsner et al., 2009). Thus, perhaps a more plausible mechanism for arsenic-related carcinogenicity is via the depletion of S-adenosyl-methionine (SAM, the universal methyl donor) due to the metabolism of inorganic arsenic to its methylated forms resulting in altered DNA methylation.
Beyond interfering with one-carbon metabolism and affecting DNA methylation, treatment of human lymphoblastoid cells with sodium arsenite led to global increases in miRNA expression (Marsit et al., 2006a). The genotoxic effects of arsenic may also be mediated by altered chromatin and Ramirez et al. (2008) have shown in human hepatocarcinoma cells that treatment with sodium arsenite resulted in global increases in histone acetylation. Thus, it is reasonable to posit that increased expression of miRNAs associated with arsenic exposure may be due to increased histone acetylation. As with other toxicants, additional studies are necessary to further elucidate the associations between arsenic and all major forms of epigenetic alteration with particular attention being given to dose and modification by one-carbon metabolism pathway participants.
Other Metals (Nickel, Lead, Cadmium, Chromium)
There is a growing body of evidence for epigenetic alterations due to exposure to other metals in humans; several studies have suggested that exposures to metals such as nickel, lead, cadmium, and others may alter epigenetic marks. In vitro, nickel exposure has been associated with various histone modification and decreased histone H4 acetylation (Broday et al., 2000; Ke et al., 2006). Exposure of a rat cell line to cadmium resulted in a concentration-dependent reduction in DNA methyltransferase activity (Takiguchi et al., 2003).
In a study among electric-furnace steel plant workers, experiencing metal-rich (lead, cadmium, chromium) particulate matter exposure has resulted in significant alterations of miRNA expression in peripheral blood leukocytes (Bollati et al., 2010). Patella lead levels, amongst participants in the Normative Aging Study were associated with reduced global DNA methylation (LINE-1 elements) though not Alu repeat regions (Wright et al., 2010), suggesting a role for lead exposure in epigenetic alterations. Interestingly, a sexually dimorphic global methylation (Luminometric methylation assay for CCGG) level decrease was associated with brain mercury levels in male polar bears (Pilsner et al., 2010).
The industrial applications of chromium include chrome plating and stainless steel welding and its exposure amongst those occupationally exposed as been related to lung cancers (Gibb et al., 2000). Beyond the recognized role for methylation induced silencing of CDKN2A in lung cancer that is related to tobacco smoking (Kim et al., 2001; Toyooka et al., 2003), Kondo et al. (2006) showed that chromate lung cancers were more likely to have methylated CDKN2A with increasing duration of occupational chromate exposure. An in vitro exposure of mouse hepatoma cells to chromium showed transcriptional repression of Cyp1a1 by local cross-linking of Hdac and Dnmt1 and altered histone marks (Schnekenburger et al., 2007). However, additional study of this diminishing exposure is necessary to validate potential chromium-induced epigenetic alterations.
Particulates and Air Pollution
Though few studies have addressed the potential for air pollution exposure to alter DNA methylation, environmental particulate exposure has been linked to increased risk of lung cancer and increased morbidity and mortality from cardiovascular and respiratory illnesses (Vineis and Husgafvel-Pursiainen, 2005). In rat lung cancer models of tumors induced by diesel exhaust, 59% were methylated at Cdkn2a, and 14% at Esr1, in those induced by carbon black 45% were methylated at Cdkn2a and 14% at Esr1, and in those induced by beryllium metal strikingly 80% were methylated at Cdkn2a and 50% at Esr1 (Belinsky et al., 2002), suggesting a role for epigenetic alteration in the mechanisms of these particulates’ carcinogenicity. A study using primary bronchial epithelial cells from non-smokers has shown dramatic alterations in miRNA expression following treatment with diesel exhaust particles (197 of 313 measured were up or down-regulated more than 1.5 fold), and suggested that the alterations were associated with inflammatory response pathways (Jardim et al., 2009). Significantly lower peripheral blood NOS2 promoter methylation was observed in steel plant workers with exposure to particulate matter of <10 μm (PM10; Tarantini et al., 2009). In addition, this same study reported significantly reduced methylation of LINE-1 and Alu repeat elements in blood DNA associated with long-term PM10 exposure (Tarantini et al., 2009). Consistent with this, a recent report examining the affect of ambient particulate pollutants on repeat element methylation in subjects from the Boston area Normative Aging Study found significantly decreased LINE-1 methylation following recent exposure to higher black carbon (Baccarelli et al., 2009).
Ren et al. (2010) found that interquartile range increases in PM2.5 and black carbon were associated with increases in plasma homocysteine (part of one-carbon metabolism), and glutathione (an antioxidant dependent upon homocysteine levels) transferase theta genotype. This same group of authors has also reported that increases in mean air pollution PM10 concentrations did not significantly alter fasting or post methionine-load total homocysteine in non-smokers, but was associated with significantly increased homocysteine levels in smokers, suggesting interactions between exposures may contribute to epigenetic alterations (Baccarelli et al., 2007). Another line of evidence for modification of exposure effects on epigenetics by genetics comes from Wilker et al. (2010) who have reported that increases in black carbon associated with increased blood pressure are modified by SNPs in miRNA processing machinery genes such as DICER and GEMIN genes.
Endocrine Disruptors
Exposures to endocrine disrupting chemicals such as the model toxicant diethylstilbestrol (DES) and the omnipresent bisphenol A (BPA) are of particular concern in the context of development. In utero exposure of mice to DES has been shown to result in the hypermethylation of the developmentally critical (specifically to uterine organogenesis) Hoxa10 gene (Bromer et al., 2009). Although not a genotoxic agent, epidemiologic evidence from individuals exposed to DES during the first 3 months in utero indicates an increase in vaginal clear cell carcinoma incidence and reproductive disorders as adults (Newbold, 2004). In addition, grandchildren of DES exposed women reported higher incidences of rare reproductive disorders; whether this reflects detection bias or possibly implicates a role for epigenetic transgenerational inheritance remains to be clarified (Newbold, 2004). Ruden et al. (2005) drawing similarities between DES and Hsp90 has proposed a mechanism through which DES establishes altered epigenetic marks capable of transgenerational inheritance, wherein DES plays a role in modifying H3K4 methylation by increasing the activity of the H3K4 methyltransferase SMYD3, thereby altering epigenetic control of various genes.
Animal studies have demonstrated that developmental exposure to BPA can alter epigenetic profiles. In agouti mice, Dolinoy et al. (2007) showed that in utero BPA exposure decreases CpG methylation and that methyl-donor supplementation negated BPA related hypomethylation. In human placental cell lines, BPA exposure has been shown to alter miRNA expression levels (Avissar-Whiting et al., 2010), and specifically, miR-146a was strongly induced by BPA treatment. This resulted in both slower proliferation rate and higher sensitivity to the DNA damaging agent bleomycin (Avissar-Whiting et al., 2010). BPA, like DES, has also been shown to alter the methylation status of the Hoxa10 gene in rodent in utero exposure models (Bromer et al., 2010), and breast cancer cell lines exposed to BPA showed altered DNA methylation of specific gene promoters (Weng et al., 2010).
Benzene
Benzene has been shown to be toxic to hematopoietic systems, is an established carcinogen (International Agency for Research on Cancer, 1999), and is known to be a cause of acute myeloid leukemia (AML) and myelodysplastic syndromes (Ye et al., 2008). Exposed to low-dose benzene, gasoline attendants and traffic officers have demonstrated significant leukocyte LINE-1 and Alu DNA hypomethylation as well as gene specific hypermethylation of CDKN2B and hypomethylation of MAGE1(Bollati et al., 2007). One preliminary study of benzene exposure using buffy coat DNA from six exposed workers and four unexposed workers have shown some evidence for relatively small changes in methylation (Zhang et al., 2010). Another preliminary study analyzed miRNA expression in seven exposed-control matched pairs and identified upregulated miR-154*, miR-487a, miR-493-3p, and miR-668 in exposed subjects (Zhang et al., 2010). Additional study of the potential epigenetic effects of benzene exposure is needed.
Radiation
Though normally thought to work through DNA damage in the form of large deletions and in some cases point mutation (Mcdonald et al., 1995; Little, 2000; Prise et al., 2001), radiation may also be pathogenic through epigenetic alteration. Silencing of Cdkn2a was detected in lung tumors of rats induced by exposure to 239plutonium (Swafford et al., 1997). In human lung adenocarcinoma, CDKN2A methylation occurred more often in workers compared to non-worker controls of the Russian MAYAK weapons-grade plutonium plant, and the prevalence of this methylation exhibited a dose–response with radiation internal exposure dose (Belinsky et al., 2004). Methylation of CDKN2A has also been linked to reactive oxygen species produced by radiation exposures (Romanenko et al., 2002), and murine models of radiation-induced lymphoma have also demonstrated hypermethylation of Cdkn2b (encoding p15ink4b (Malumbres et al., 1997, 1999; Cleary et al., 1999)).
Mechanisms of Toxicant Effects on Epigenomics
Beyond effects on the one-carbon metabolism pathway, one must consider other more directed mechanisms through which toxicants could lead to somatic changes in DNA methylation, which could be clonally propagated during pathologic processes. For example altered DNA methylation may be related to increased reactive oxygen species as a result of inflammatory response or as a result of exposure to an ROS producing toxicant. It has been reported that 5-hydroxymethylcytosine can be generated by oxidation of 5-methylcytosine (Masuda et al., 1975), and both 5-methylcytosine adjacent to 8-oxoguanine, and 5-hydroxymethylcytosine have been shown to inhibit binding of methyl-CpG binding protein 2, a critical epigenetic regulator that recruits cytosine methyltransferases and histone deacetylases (Valinluck et al., 2004). It is also known that 5-hydroxymethylcytosine is not recognized as 5-methylcytosine by the maintenance methyltransferase DNMT1, and hence, may lead to aberrant loss of methylation during cell replication (Valinluck and Sowers, 2007). Additional base alterations occur via neutrophil and eosinophil peroxidase-derived HOCl and HOBr which can react with DNA to form 5-chlorocytosine and 5-bromocytosine respectively (Henderson et al., 2003). These halogenated cytosines can be mistaken by DNMT1 as 5-methylcytosine during replication, thus providing a potential mechanism for inflammation-induced aberrant hypermethylation (Valinluck and Sowers, 2007).
Likely critical to functional epigenetic alterations important in pathology is the characteristic ability of both mutagenic and non-mutagenic compounds to impart selective pressures on cellular clones. Some researchers believe that it is this effect, and not genotoxicity, that provides the carcinogenicity attributable to some exposures, by selecting pre-existing genetic or possibly epigenetic alterations (Thilly, 2003). If de novo methylation in the developing tumor occurred stochastically or due to the mechanisms described above, cells with a silenced tumor suppressor may be selected for growth or expansion due to exposure to specific carcinogens. In time these pressures will allow then for clonal expansion of the epigenetically altered cells into fields which now have an increased susceptibility for tumorigenesis (Slaughter et al., 1953; Wiencke and Kelsey, 2002).
Aging
The process of aging, and differences in the environment experienced over a lifetime has been hypothesized to influence clinically significant changes in methylation profiles. Aging also provides an interesting platform to consider additional mechanisms responsible for altered DNA methylation patterns. A study of monozygotic twins demonstrated that young twin-pairs seemed more epigenetically similar than older monozygotic twins, and suggests that such drift may be at least in part responsible for differences in disease susceptibility (Fraga et al., 2005). Motivated by studies of methylation in cancer, an early report from Issa et al. (1994) described an association between aging colonic mucosa and estrogen receptor methylation. This type of research has been extended more recently to show an overall trend of increased methylation associated with older age in normal human prostate and colon tissues in several genes (Shen et al., 2005; Kwabi-Addo et al., 2007). Although an increase in promoter methylation with aging is generally accepted, and is consistent with hypermethylation of tumor suppressor genes in cancer, a disease of aging, recent evidence from Bjornsson et al. (2008) suggests a more complex picture. These authors measured intra-individual global methylation changes over >10 years and found both increased and decreased methylation levels dependent on the individual, with over 50% of participants exhibiting >5% change in methylation in peripheral blood cell DNA over time.
A study of over 200 non-pathologic human tissues from 10 anatomic sites using the GoldenGate methylation array platform (1413 autosomal CpG loci) demonstrated consistent associations between methylation and age at previously reported candidate genes, and in conjunction with other reports, suggested that the relation between aging and promoter CpG methylation is complex (Christensen et al., 2009b). The direction and strength of correlation between age and methylation were largely dependent upon CpG-island status. Specifically, a propensity for CpG-island loci to gain methylation with age, and non-island CpGs to lose methylation with age was found. These results were consistent with (Issa, 2003; Shen et al., 2005; Kwabi-Addo et al., 2007), as well as with the findings of Tra et al. (2002) and Bjornsson et al. who showed bi-modal age-related methylation in normal tissues. Using restriction-landmark genome scanning of over 2000 CpG loci in T lymphocytes comparing newborns, middle age, and elderly people, reported that 29 loci had age-related methylation alterations, with 23 loci displaying increased methylation with age and six decreasing with age. In addition Teschendorff et al. (2010) have suggested that age-dependent methylation of polycomb-group target genes (genes suppressed in stem cells allowing differentiation) is a hallmark of cancer and is independent of gender, tissue type, or disease state, suggesting a mechanism for aging to predispose to carcinogenesis.
Additional Mechanistic Considerations
The observed pattern of age associated methylation in (Christensen et al., 2009b) was also irrespective of tissue type, suggesting a common mechanism or dysregulation to explain these alterations. Langevin et al. (2011) expanded these analyses to the larger, Illumina Infinium Methylation27 platform, assessing nearly 27,000 autosomal CpG loci in peripheral blood samples of healthy adults, and found again that the direction and magnitude of the association between methylation at CpG site and age was influenced by local sequence features. In addition, the study from Christensen et al. (2009b) that examined non-pathologic tissue methylation assessed different exposure types in different tissues. Though a detailed analysis that included genomic context was not published, these authors observed a consistent trend for exposure-related methylation alterations across tissue types. Assessment of pleural tissue methylation by asbestos exposure history, lung tissue by smoking history, and peripheral blood methylation by alcohol intake each indicated that exposure-associated methylation alterations were dependent on genomic context for CpGs analyzed. However, unlike age-related methylation alterations (increased methylation of CpG-island loci with age, and decreased methylation of non-CpG-island loci with age) exposure-related methylation alterations each had a contrasting pattern where CpG-island loci decreased with exposure, and non-CpG-island loci had increased methylation with exposure (unpublished). These reports highlight the importance of recognizing that contextual elements may differentially affect the likelihood and direction of exposure-related epigenetic alterations. Though these findings suggest that different mechanisms are responsible for age-related and exposure-related methylation alterations, and that some exposures may share common mechanisms for altering methylation, additional population-based research on appropriate target tissues is necessary to advance our understanding of any shared (within and across exposure types) or disparate mechanisms responsible for methylation alterations.
Beyond sequence context, one may also consider the chromatin environment to understand regions targeted for alterations of DNA methylation. Rakyan et al. (2010) showed that aging-associated hypermethylation was more likely to occur at promoters associated with bivalent chromatin domains. Additionally, distinct mechanisms may be responsible for altered repeat element (global) methylation and altered promoter methylation. Although traditional dogma indicates separate methyltransferase enzymes for maintenance (DNMT1) and de novo methylation (DNMT3A/B), a recent perspective from Jones and Liang (2009) proposes that maintenance methylation in CpG-islands may be a cooperative process. When considering the mechanisms of environmental exposure-related methylation alterations it is important to remain conscious of the fact that we do not have a complete understanding of de novo and maintenance methylation mechanisms. Reduced fidelity of maintenance methyltransferases with aging is one potential explanation for age-related decreases in methylation; while age-related increases in methylation could potentially reflect the accumulation of stochastic methylation events over time. Another mechanism that has been hypothesized is a spreading process, whereby CpG methylation from within the gene migrates into the promoter due to the loss of protective boundary elements which normally protect promoters from CpG-island methylation (Turker and Bestor, 1997; Sun and Elgin, 1999; Turker, 2002). Alternatively, for example in aging, a spreading process subsequent to a stochastic methylation alteration (in pathologically normal tissue) without functional consequences for gene expression, may allow the initiation of a spreading process that eventually confers an altered expression phenotype. Yet, these mechanisms do not fully explain the apparently sequence context specificity of these observations, thereby suggesting that the age-related alterations of methylation observed may be influence in part by targeted mechanisms that have yet to be fully explained. Using long-term culture of mesenchymal stem cells, Bork et al. (2010) show that for the majority of CpGs examined (using the Illumina Infinium Methylation27 platform), the methylation levels remain constant, and that only a specific set of CpGs undergo differential methylation with long-term culture, and that these sites were over-represented by homeobox genes. This same group (Koch et al., 2011) demonstrated similar findings in aging dermal fibroblasts, although often in directions opposite those observed in the MSCs. Such results suggest that aging related DNA methylation may be following a developmental program or resulting from the loss of some developmentally regulated maintenance.
Importance of Epigenomics in Environmental Health
We have highlighted what is a growing body of literature beginning to link epigenetic alterations to toxicant exposures from in vitro work, animal models, and human observational studies. Toxicologists and environmental health scientists, in addition to studying the effects of exposures at the genotoxic and transcriptional levels, must also consider a toxicants effect on the epigenome, a task which requires multi-disciplinary work from in vitro and model systems to epidemiologic studies of human disease. New technologies now allow for global analysis of epigenetic alterations and these may provide insight into the extent and patterns of alteration in normal and diseased tissue. Notably, the latest generation of CpG methylation arrays interrogates hundreds of thousands of CpG loci, includes all designable RefSeq genes, and is low-cost enough for application in population-level studies. Next generation sequencing approaches, as their cost decreases and throughput increases will also become increasingly used as tools for annotating the epigenome and its relation to exposures. Appropriate models must also be considered, and may include mammalian stem cells which could prove extremely useful in bettering the understanding of epigenetics in development and beyond. Finally, epidemiologist must begin to consider how to incorporate examinations of the epigenome in population-based studies, in order to define consistent methodologies and data collection that is crucial to understanding the true effect of environmental exposures on the human epigenome. Although examinations of epigenetic alterations related to exposures are emerging it is still important to note that the mechanisms by which toxicants and the environment modulate the epigenetic landscape of individual cells are yet to be elucidated and require further exploration. This work is urgently needed in order to better understand the biology of epigenetic alterations and the effects of toxic exposures on these disease-associated somatic epigenetic alterations. Better defined mechanisms will lead to better prediction of the toxic potential of new compounds introduced into the environment, and allow for more targeted and appropriate disease prevention strategies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by grants from the NIH, NIMH R01MH094609, and NIEHS/EPA P20ES018175.
References
Avissar-Whiting, M., Veiga, K. R., Uhl, K. M., Maccani, M. A., Gagne, L. A., Moen, E. L., and Marsit, C. J. (2010). Bisphenol A exposure leads to specific microRNA alterations in placental cells. Reprod. Toxicol. 29, 401–406.
Baccarelli, A., Wright, R. O., Bollati, V., Tarantini, L., Litonjua, A. A., Suh, H. H., Zanobetti, A., Sparrow, D., Vokonas, P. S., and Schwartz, J. (2009). Rapid DNA methylation changes after exposure to traffic particles. Am. J. Respir. Crit. Care Med. 179, 572–578.
Baccarelli, A., Zanobetti, A., Martinelli, I., Grillo, P., Hou, L., Lanzani, G., Mannucci, P. M., Bertazzi, P. A., and Schwartz, J. (2007). Air pollution, smoking, and plasma homocysteine. Environ. Health Perspect. 115, 176–181.
Belinsky, S. A., Klinge, D. M., Liechty, K. C., March, T. H., Kang, T., Gilliland, F. D., Sotnic, N., Adamova, G., Rusinova, G., and Telnov, V. (2004). Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma. Carcinogenesis 25, 1063–1067.
Belinsky, S. A., Snow, S. S., Nikula, K. J., Finch, G. L., Tellez, C. S., and Palmisano, W. A. (2002). Aberrant CpG island methylation of the p16(INK4a) and estrogen receptor genes in rat lung tumors induced by particulate carcinogens. Carcinogenesis 23, 335–339.
Bjornsson, H. T., Sigurdsson, M. I., Fallin, M. D., Irizarry, R. A., Aspelund, T., Cui, H., Yu, W., Rongione, M. A., Ekstrom, T. J., Harris, T. B., Launer, L. J., Eiriksdottir, G., Leppert, M. F., Sapienza, C., Gudnason, V., and Feinberg, A. P. (2008). Intra-individual change over time in DNA methylation with familial clustering. JAMA 299, 2877–2883.
Bollati, V., Baccarelli, A., Hou, L., Bonzini, M., Fustinoni, S., Cavallo, D., Byun, H. M., Jiang, J., Marinelli, B., Pesatori, A. C., Bertazzi, P. A., and Yang, A. S. (2007). Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 67, 876–880.
Bollati, V., Marinelli, B., Apostoli, P., Bonzini, M., Nordio, F., Hoxha, M., Pegoraro, V., Motta, V., Tarantini, L., Cantone, L., Schwartz, J., Bertazzi, P. A., and Baccarelli, A. (2010). Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ. Health Perspect. 118, 763–768.
Bork, S., Pfister, S., Witt, H., Horn, P., Korn, B., Ho, A. D., and Wagner, W. (2010). DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 9, 54–63.
Broday, L., Peng, W., Kuo, M. H., Salnikow, K., Zoroddu, M., and Costa, M. (2000). Nickel compounds are novel inhibitors of histone H4 acetylation. Cancer Res. 60, 238–241.
Bromer, J. G., Wu, J., Zhou, Y., and Taylor, H. S. (2009). Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: an epigenetic mechanism for altered developmental programming. Endocrinology 150, 3376–3382.
Bromer, J. G., Zhou, Y., Taylor, M. B., Doherty, L., and Taylor, H. S. (2010). Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 24, 2273–2280.
Chanda, S., Dasgupta, U. B., Guhamazumder, D., Gupta, M., Chaudhuri, U., Lahiri, S., Das, S., Ghosh, N., and Chatterjee, D. (2006). DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol. Sci. 89, 431–437.
Christensen, B. C., Godleski, J. J., Marsit, C. J., Houseman, E. A., Lopez-Fagundo, C. Y., Longacker, J. L., Bueno, R., Sugarbaker, D. J., Nelson, H. H., and Kelsey, K. T. (2008). Asbestos exposure predicts cell cycle control gene promoter methylation in pleural mesothelioma. Carcinogenesis 29, 1555–1559.
Christensen, B. C., Houseman, E. A., Godleski, J. J., Marsit, C. J., Longacker, J. L., Roelofs, C. R., Karagas, M. R., Wrensch, M. R., Yeh, R. F., Nelson, H. H., Wiemels, J. L., Zheng, S., Wiencke, J. K., Bueno, R., Sugarbaker, D. J., and Kelsey, K. T. (2009a). Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer Res. 69, 227–234.
Christensen, B. C., Houseman, E. A., Marsit, C. J., Zheng, S., Wrensch, M. R., Wiemels, J. L., Nelson, H. H., Karagas, M. R., Padbury, J. F., Bueno, R., Sugarbaker, D. J., Yeh, R. F., Wiencke, J. K., and Kelsey, K. T. (2009b). Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 5, e1000602. doi:10.1371/journal.pgen.1000602
Christensen, B. C., Kelsey, K. T., Zheng, S., Houseman, E. A., Marsit, C. J., Wrensch, M. R., Wiemels, J. L., Nelson, H. H., Karagas, M. R., Kushi, L. H., Kwan, M. L., and Wiencke, J. K. (2010). Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 6, e1001043. doi:10.1371/journal.pgen.1001043
Cleary, H. J., Boulton, E., and Plumb, M. (1999). Allelic loss and promoter hypermethylation of the p15INK4b gene features in mouse radiation-induced lymphoid – but not myeloid – leukaemias. Leukemia 13, 2049–2052.
Daly, L. E., Kirke, P. N., Molloy, A., Weir, D. G., and Scott, J. M. (1995). Folate levels and neural tube defects. Implications for prevention. JAMA 274, 1698–1702.
Dolinoy, D. C., Huang, D., and Jirtle, R. L. (2007). Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. U.S.A. 104, 13056–13061.
Dunlevy, L. P., Burren, K. A., Mills, K., Chitty, L. S., Copp, A. J., and Greene, N. D. (2006). Integrity of the methylation cycle is essential for mammalian neural tube closure. Birth Defects Res. Part A Clin. Mol. Teratol. 76, 544–552.
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., Heine-Suner, D., Cigudosa, J. C., Urioste, M., Benitez, J., Boix-Chornet, M., Sanchez-Aguilera, A., Ling, C., Carlsson, E., Poulsen, P., Vaag, A., Stephan, Z., Spector, T. D., Wu, Y. Z., Plass, C., and Esteller, M. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609.
Gamble, M. V., Liu, X., Slavkovich, V., Pilsner, J. R., Ilievski, V., Factor-Litvak, P., Levy, D., Alam, S., Islam, M., Parvez, F., Ahsan, H., and Graziano, J. H. (2007). Folic acid supplementation lowers blood arsenic. Am. J. Clin. Nutr. 86, 1202–1209.
Gibb, H. J., Lees, P. S., Pinsky, P. F., and Rooney, B. C. (2000). Lung cancer among workers in chromium chemical production. Am. J. Ind. Med. 38, 115–126.
Giovannucci, E., Rimm, E. B., Ascherio, A., Stampfer, M. J., Colditz, G. A., and Willett, W. C. (1995). Alcohol, low-methionine – low-folate diets, and risk of colon cancer in men. J. Natl. Cancer Inst. 87, 265–273.
Gluckman, P. D., and Hanson, M. A. (2004). Developmental origins of disease paradigm: a mechanistic and evolutionary perspective. Pediatr. Res. 56, 311–317.
Gluckman, P. D., Hanson, M. A., Cooper, C., and Thornburg, K. L. (2008). Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 359, 61–73.
Hall, M., Gamble, M., Slavkovich, V., Liu, X., Levy, D., Cheng, Z., Van Geen, A., Yunus, M., Rahman, M., Pilsner, J. R., and Graziano, J. (2007). Determinants of arsenic metabolism: blood arsenic metabolites, plasma folate, cobalamin, and homocysteine concentrations in maternal-newborn pairs. Environ. Health Perspect. 115, 1503–1509.
Hamajima, N., Hirose, K., Tajima, K., Rohan, T., Calle, E. E., Heath, C. W. Jr., Coates, R. J., Liff, J. M., Talamini, R., Chantarakul, N., Koetsawang, S., Rachawat, D., Morabia, A., Schuman, L., Stewart, W., Szklo, M., Bain, C., Schofield, F., Siskind, V., Band, P., Coldman, A. J., Gallagher, R. P., Hislop, T. G., Yang, P., Kolonel, L. M., Nomura, A. M., Hu, J., Johnson, K. C., Mao, Y., De Sanjose, S., Lee, N., Marchbanks, P., Ory, H. W., Peterson, H. B., Wilson, H. G., Wingo, P. A., Ebeling, K., Kunde, D., Nishan, P., Hopper, J. L., Colditz, G., Gajalanski, V., Martin, N., Pardthaisong, T., Silpisornkosol, S., Theetranont, C., Boosiri, B., Chutivongse, S., Jimakorn, P., Virutamasen, P., Wongsrichanalai, C., Ewertz, M., Adami, H. O., Bergkvist, L., Magnusson, C., Persson, I., Chang-Claude, J., Paul, C., Skegg, D. C., Spears, G. F., Boyle, P., Evstifeeva, T., Daling, J. R., Hutchinson, W. B., Malone, K., Noonan, E. A., Stanford, J. L., Thomas, D. B., Weiss, N. S., White, E., Andrieu, N., Bremond, A., Clavel, F., Gairard, B., Lansac, J., Piana, L., Renaud, R., Izquierdo, A., Viladiu, P., Cuevas, H. R., Ontiveros, P., Palet, A., Salazar, S. B., Aristizabel, N., Cuadros, A., Tryggvadottir, L., Tulinius, H., Bachelot, A., Le, M. G., Peto, J., Franceschi, S., Lubin, F., Modan, B., Ron, E., Wax, Y., Friedman, G. D., Hiatt, R. A., Levi, F., Bishop, T., Kosmelj, K., Primic-Zakelj, M., Ravnihar, B., Stare, J., Beeson, W. L., Fraser, G., Bullbrook, R. D., Cuzick, J., Duffy, S. W., Fentiman, I. S., Hayward, J. L., Wang, D. Y., McMichael, A. J., McPherson, K., Hanson, R. L., Leske, M. C., Mahoney, M. C., Nasca, P. C., Varma, A. O., Weinstein, A. L., Moller, T. R., Olsson, H., Ranstam, J., Goldbohm, R. A., van den Brandt, P. A., Apelo, R. A., Baens, J., de la Cruz, J. R., Javier, B., Lacaya, L. B., Ngelangel, C. A., La Vecchia, C., Negri, E., Marubini, E., Ferraroni, M., Gerber, M., Richardson, S., Segala, C., Gatei, D., Kenya, P., Kungu, A., Mati, J. G., Brinton, L. A., Hoover, R., Schairer, C., Spirtas, R., Lee, H. P., Rookus, M. A., van Leeuwen, F. E., Schoenberg, J. A., McCredie, M., Gammon, M. D., Clarke, E. A., Jones, L., Neil, A., Vessey, M., Yeates, D., Appleby, P., Banks, E., Beral, V., Bull, D., Crossley, B., Goodill, A., Green, J., Hermon, C., Key, T., Langston, N., Lewis, C., Reeves, G., Collins, R., Doll, R., Peto, R., Mabuchi, K., Preston, D., Hannaford, P., Kay, C., Rosero-Bixby, L., Gao, Y. T., Jin, F., Yuan, J. M., Wei, H. Y., Yun, T., Zhiheng, C., Berry, G., Cooper, B. J., Jelihovsky, T., MacLennan, R., Shearman, R., Wang, Q. S., Baines, C. J., Miller, A. B., Wall, C., Lund, E., Stalsberg, H., Shu, X. O., Zheng, W., Katsouyanni, K., Trichopoulou, A., Trichopoulos, D., Dabancens, A., Martinez, L., Molina, R., Salas, O., Alexander, F. E., Anderson, K., Folsom, A. R., Hulka, B. S., Bernstein, L., Enger, S., Haile, R. W., Paganini-Hill, A., Pike, M. C., Ross, R. K., Ursin, G., Yu, M. C., Longnecker, M. P., Newcomb, P., Bergkvist, L., Kalache, A., Farley, T. M., Holck, S., Meirik, O., and Collaborative Group on Hormonal Factors in Breast Cancer. (2002). Alcohol, tobacco and breast cancer – collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br. J. Cancer 87, 1234–1245.
He, B., Lee, A. Y., Dadfarmay, S., You, L., Xu, Z., Reguart, N., Mazieres, J., Mikami, I., Mccormick, F., and Jablons, D. M. (2005). Secreted frizzled-related protein 4 is silenced by hypermethylation and induces apoptosis in beta-catenin-deficient human mesothelioma cells. Cancer Res. 65, 743–748.
Henderson, J. P., Byun, J., Takeshita, J., and Heinecke, J. W. (2003). Phagocytes produce 5-chlorouracil and 5-bromouracil, two mutagenic products of myeloperoxidase, in human inflammatory tissue. J. Biol. Chem. 278, 23522–23528.
Hillman, R. S., and Steinberg, S. E. (1982). The effects of alcohol on folate metabolism. Annu. Rev. Med. 33, 345–354.
Hirao, T., Bueno, R., Chen, C. J., Gordon, G. J., Heilig, E., and Kelsey, K. T. (2002). Alterations of the p16(INK4) locus in human malignant mesothelial tumors. Carcinogenesis 23, 1127–1130.
Issa, J. P. (2003). Age-related epigenetic changes and the immune system. Clin. Immunol. 109, 103–108.
Issa, J. P., Ottaviano, Y. L., Celano, P., Hamilton, S. R., Davidson, N. E., and Baylin, S. B. (1994). Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat. Genet. 7, 536–540.
Jardim, M. J., Fry, R. C., Jaspers, I., Dailey, L., and Diaz-Sanchez, D. (2009). Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways. Environ. Health Perspect. 117, 1745–1751.
Jones, P. A., and Liang, G. (2009). Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 10, 805–811.
Ke, Q., Davidson, T., Chen, H., Kluz, T., and Costa, M. (2006). Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis 27, 1481–1488.
Key, J., Hodgson, S., Omar, R. Z., Jensen, T. K., Thompson, S. G., Boobis, A. R., Davies, D. S., and Elliott, P. (2006). Meta-analysis of studies of alcohol and breast cancer with consideration of the methodological issues. Cancer Causes Control 17, 759–770.
Kharbanda, K. K. (2009). Alcoholic liver disease and methionine metabolism. Semin. Liver Dis. 29, 155–165.
Kim, D. H., Nelson, H. H., Wiencke, J. K., Zheng, S., Christiani, D. C., Wain, J. C., Mark, E. J., and Kelsey, K. T. (2001). p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res. 61, 3419–3424.
Kim, J. S., and Shukla, S. D. (2006). Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohol Alcohol. 41, 126–132.
Koch, C. M., Suschek, C. V., Lin, Q., Bork, S., Goergens, M., Joussen, S., Pallua, N., Ho, A. D., Zenke, M., and Wagner, W. (2011). Specific age-associated DNA methylation changes in human dermal fibroblasts. PLoS ONE 6, e16679. doi:10.1371/journal.pone.0016679
Kondo, K., Takahashi, Y., Hirose, Y., Nagao, T., Tsuyuguchi, M., Hashimoto, M., Ochiai, A., Monden, Y., and Tangoku, A. (2006). The reduced expression and aberrant methylation of p16(INK4a) in chromate workers with lung cancer. Lung Cancer 53, 295–302.
Kwabi-Addo, B., Chung, W., Shen, L., Ittmann, M., Wheeler, T., Jelinek, J., and Issa, J. P. (2007). Age-related DNA methylation changes in normal human prostate tissues. Clin. Cancer Res. 13, 3796–3802.
Langevin, S. M., Houseman, E. A., Christensen, B. C., Wiencke, J. K., Nelson, H. H., Karagas, M. R., Marsit, C. J., and Kelsey, K. T. (2011). The influence of aging, environmental exposures and local sequence features on the variation of DNA methylation in blood. Epigenetics 6, 908–919.
Lee, A. Y., He, B., You, L., Dadfarmay, S., Xu, Z., Mazieres, J., Mikami, I., Mccormick, F., and Jablons, D. M. (2004). Expression of the secreted frizzled-related protein gene family is downregulated in human mesothelioma. Oncogene 23, 6672–6676.
Li, E. (2002). Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 3, 662–673.
Majumdar, S., Chanda, S., Ganguli, B., Mazumder, D. N., Lahiri, S., and Dasgupta, U. B. (2010). Arsenic exposure induces genomic hypermethylation. Environ. Toxicol. 25, 315–318.
Malumbres, M., Perez De Castro, I., Santos, J., Fernandez Piqueras, J., and Pellicer, A. (1999). Hypermethylation of the cell cycle inhibitor p15INK4b 3′-untranslated region interferes with its transcriptional regulation in primary lymphomas. Oncogene 18, 385–396.
Malumbres, M., Perez De Castro, I., Santos, J., Melendez, B., Mangues, R., Serrano, M., Pellicer, A., and Fernandez-Piqueras, J. (1997). Inactivation of the cyclin-dependent kinase inhibitor p15INK4b by deletion and de novo methylation with independence of p16INK4a alterations in murine primary T-cell lymphomas. Oncogene 14, 1361–1370.
Marsit, C. J., Christensen, B. C., Houseman, E. A., Karagas, M. R., Wrensch, M. R., Yeh, R. F., Nelson, H. H., Wiemels, J. L., Zheng, S., Posner, M. R., Mcclean, M. D., Wiencke, J. K., and Kelsey, K. T. (2009). Epigenetic profiling reveals etiologically distinct patterns of DNA methylation in head and neck squamous cell carcinoma. Carcinogenesis 30, 416–422.
Marsit, C. J., Eddy, K., and Kelsey, K. T. (2006a). MicroRNA responses to cellular stress. Cancer Res. 66, 10843–10848.
Marsit, C. J., Karagas, M. R., Danaee, H., Liu, M., Andrew, A., Schned, A., Nelson, H. H., and Kelsey, K. T. (2006b). Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 27, 112–116.
Masuda, T., Shinoara, H., and Kondo, M. (1975). Reactions of hydroxyl radicals with nucleic acid bases and the related compounds in gamma-irradiated aqueous solution. J. Radiat. Res. 16, 153–161.
Mcdonald, J. W., Taylor, J. A., Watson, M. A., Saccomanno, G., and Devereux, T. R. (1995). p53 and K-ras in radon-associated lung adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 4, 791–793.
Mcdorman, E. W., Collins, B. W., and Allen, J. W. (2002). Dietary folate deficiency enhances induction of micronuclei by arsenic in mice. Environ. Mol. Mutagen. 40, 71–77.
Morgan, H. D., Santos, F., Green, K., Dean, W., and Reik, W. (2005). Epigenetic reprogramming in mammals. Hum. Mol. Genet. 1, R47–R58.
Newbold, R. R. (2004). Lessons learned from perinatal exposure to diethylstilbestrol. Toxicol. Appl. Pharmacol. 199, 142–150.
Ohta, Y., Shridhar, V., Kalemkerian, G. P., Bright, R. K., Watanabe, Y., and Pass, H. I. (1999). Thrombospondin-1 expression and clinical implications in malignant pleural mesothelioma. Cancer 85, 2570–2576.
Okoji, R. S., Yu, R. C., Maronpot, R. R., and Froines, J. R. (2002). Sodium arsenite administration via drinking water increases genome-wide and Ha-ras DNA hypomethylation in methyl-deficient C57BL/6J mice. Carcinogenesis 23, 777–785.
Park, P. H., Miller, R., and Shukla, S. D. (2003). Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochem. Biophys. Res. Commun. 306, 501–504.
Pilsner, J. R., Lazarus, A. L., Nam, D. H., Letcher, R. J., Sonne, C., Dietz, R., and Basu, N. (2010). Mercury-associated DNA hypomethylation in polar bear brains via the LUminometric Methylation Assay: a sensitive method to study epigenetics in wildlife. Mol. Ecol. 19, 307–314.
Pilsner, J. R., Liu, X., Ahsan, H., Ilievski, V., Slavkovich, V., Levy, D., Factor-Litvak, P., Graziano, J. H., and Gamble, M. V. (2007). Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am. J. Clin. Nutr. 86, 1179–1186.
Pilsner, J. R., Liu, X., Ahsan, H., Ilievski, V., Slavkovich, V., Levy, D., Factor-Litvak, P., Graziano, J. H., and Gamble, M. V. (2009). Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ. Health Perspect. 117, 254–260.
Prise, K. M., Pinto, M., Newman, H. C., and Michael, B. D. (2001). A review of studies of ionizing radiation-induced double-strand break clustering. Radiat. Res. 156, 572–576.
Rakyan, V. K., Down, T. A., Maslau, S., Andrew, T., Yang, T. P., Beyan, H., Whittaker, P., Mccann, O. T., Finer, S., Valdes, A. M., Leslie, R. D., Deloukas, P., and Spector, T. D. (2010). Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 20, 434–439.
Ramirez, T., Brocher, J., Stopper, H., and Hock, R. (2008). Sodium arsenite modulates histone acetylation, histone deacetylase activity and HMGN protein dynamics in human cells. Chromosoma 117, 147–157.
Ren, C., Park, S. K., Vokonas, P. S., Sparrow, D., Wilker, E., Baccarelli, A., Suh, H. H., Tucker, K. L., Wright, R. O., and Schwartz, J. (2010). Air pollution and homocysteine: more evidence that oxidative stress-related genes modify effects of particulate air pollution. Epidemiology 21, 198–206.
Robinson, B. W., and Lake, R. A. (2005). Advances in malignant mesothelioma. N. Engl. J. Med. 353, 1591–1603.
Romanenko, A., Morell-Quadreny, L., Lopez-Guerrero, J. A., Pellin, A., Nepomnyaschy, V., Vozianov, A., and Llombart-Bosch, A. (2002). P16INK4A and p15INK4B gene alteration associated with oxidative stress in renal cell carcinomas after the chernobyl accident (pilot study). Diagn. Mol. Pathol. 11, 163–169.
Rossman, T. G. (2003). Mechanism of arsenic carcinogenesis: an integrated approach. Mutat. Res. 533, 37–65.
Ruden, D. M., Xiao, L., Garfinkel, M. D., and Lu, X. (2005). Hsp90 and environmental impacts on epigenetic states: a model for the trans-generational effects of diethylstibesterol on uterine development and cancer. Hum. Mol. Genet. 1, R149–R155.
Sauer, J., Mason, J. B., and Choi, S. W. (2009). Too much folate: a risk factor for cancer and cardiovascular disease? Curr. Opin. Clin. Nutr. Metab. Care 12, 30–36.
Schernhammer, E. S., Giovannucci, E., Kawasaki, T., Rosner, B., Fuchs, C. S., and Ogino, S. (2010). Dietary folate, alcohol and B vitamins in relation to LINE-1 hypomethylation in colon cancer. Gut 59, 794–799.
Schnekenburger, M., Talaska, G., and Puga, A. (2007). Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol. Cell. Biol. 27, 7089–7101.
Shen, L., Kondo, Y., Rosner, G. L., Xiao, L., Hernandez, N. S., Vilaythong, J., Houlihan, P. S., Krouse, R. S., Prasad, A. R., Einspahr, J. G., Buckmeier, J., Alberts, D. S., Hamilton, S. R., and Issa, J. P. (2005). MGMT promoter methylation and field defect in sporadic colorectal cancer. J. Natl. Cancer Inst. 97, 1330–1338.
Slaughter, D. P., Southwick, H. W., and Smejkal, W. (1953). Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 6, 963–968.
Smith, I. M., Mydlarz, W. K., Mithani, S. K., and Califano, J. A. (2007). DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. Int. J. Cancer 121, 1724–1728.
Sugarbaker, D. J., Richards, W. G., Gordon, G. J., Dong, L., De Rienzo, A., Maulik, G., Glickman, J. N., Chirieac, L. R., Hartman, M. L., Taillon, B. E., Du, L., Bouffard, P., Kingsmore, S. F., Miller, N. A., Farmer, A. D., Jensen, R. V., Gullans, S. R., and Bueno, R. (2008). Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc. Natl. Acad. Sci. U.S.A. 105, 3521–3526.
Swafford, D. S., Middleton, S. K., Palmisano, W. A., Nikula, K. J., Tesfaigzi, J., Baylin, S. B., Herman, J. G., and Belinsky, S. A. (1997). Frequent aberrant methylation of p16INK4a in primary rat lung tumors. Mol. Cell. Biol. 17, 1366–1374.
Takiguchi, M., Achanzar, W. E., Qu, W., Li, G., and Waalkes, M. P. (2003). Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp. Cell Res. 286, 355–365.
Tarantini, L., Bonzini, M., Apostoli, P., Pegoraro, V., Bollati, V., Marinelli, B., Cantone, L., Rizzo, G., Hou, L., Schwartz, J., Bertazzi, P. A., and Baccarelli, A. (2009). Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ. Health Perspect. 117, 217–222.
Tchantchou, F., Graves, M., Falcone, D., and Shea, T. B. (2008). S-adenosylmethionine mediates glutathione efficacy by increasing glutathione S-transferase activity: implications for S-adenosyl methionine as a neuroprotective dietary supplement. J. Alzheimers Dis. 14, 323–328.
Teschendorff, A. E., Menon, U., Gentry-Maharaj, A., Ramus, S. J., Weisenberger, D. J., Shen, H., Campan, M., Noushmehr, H., Bell, C. G., Maxwell, A. P., Savage, D. A., Mueller-Holzner, E., Marth, C., Kocjan, G., Gayther, S. A., Jones, A., Beck, S., Wagner, W., Laird, P. W., Jacobs, I. J., and Widschwendter, M. (2010). Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–446.
Thilly, W. G. (2003). Have environmental mutagens caused oncomutations in people? Nat. Genet. 34, 255–259.
Toyooka, S., Maruyama, R., Toyooka, K. O., Mclerran, D., Feng, Z., Fukuyama, Y., Virmani, A. K., Zochbauer-Muller, S., Tsukuda, K., Sugio, K., Shimizu, N., Shimizu, K., Lee, H., Chen, C. Y., Fong, K. M., Gilcrease, M., Roth, J. A., Minna, J. D., and Gazdar, A. F. (2003). Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int. J. Cancer 103, 153–160.
Tra, J., Kondo, T., Lu, Q., Kuick, R., Hanash, S., and Richardson, B. (2002). Infrequent occurrence of age-dependent changes in CpG island methylation as detected by restriction landmark genome scanning. Mech. Ageing Dev. 123, 1487–1503.
Tsou, J. A., Galler, J. S., Wali, A., Ye, W., Siegmund, K. D., Groshen, S., Laird, P. W., Turla, S., Koss, M. N., Pass, H. I., and Laird-Offringa, I. A. (2007). DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer 58, 220–230.
Turker, M. S. (2002). Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 21, 5388–5393.
Turker, M. S., and Bestor, T. H. (1997). Formation of methylation patterns in the mammalian genome. Mutat. Res. 386, 119–130.
Valinluck, V., and Sowers, L. C. (2007). Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 67, 946–950.
Valinluck, V., Tsai, H. H., Rogstad, D. K., Burdzy, A., Bird, A., and Sowers, L. C. (2004). Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 32, 4100–4108.
Vineis, P., and Husgafvel-Pursiainen, K. (2005). Air pollution and cancer: biomarker studies in human populations. Carcinogenesis 26, 1846–1855.
Wang, L., Wang, F., Guan, J., Le, J., Wu, L., Zou, J., Zhao, H., Pei, L., Zheng, X., and Zhang, T. (2010). Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects. Am. J. Clin. Nutr. 91, 1359–1367.
Waterland, R. A., and Michels, K. B. (2007). Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 27, 363–388.
Weng, Y. I., Hsu, P. Y., Liyanarachchi, S., Liu, J., Deatherage, D. E., Huang, Y. W., Zuo, T., Rodriguez, B., Lin, C. H., Cheng, A. L., and Huang, T. H. (2010). Epigenetic influences of low-dose bisphenol A in primary human breast epithelial cells. Toxicol. Appl. Pharmacol. 248, 111–121.
Wiencke, J. K., and Kelsey, K. T. (2002). Teen smoking, field cancerization, and a “critical period” hypothesis for lung cancer susceptibility. Environ. Health Perspect. 110, 555–558.
Wilhelm-Benartzi, C. S., Koestler, D. C., Houseman, E. A., Christensen, B. C., Wiencke, J. K., Schned, A. R., Karagas, M. R., Kelsey, K. T., and Marsit, C. J. (2010). DNA methylation profiles delineate etiologic heterogeneity and clinically important subgroups of bladder cancer. Carcinogenesis 31, 1972–1976.
Wilker, E. H., Baccarelli, A., Suh, H., Vokonas, P., Wright, R. O., and Schwartz, J. (2010). Black carbon exposures, blood pressure, and interactions with single nucleotide polymorphisms in MicroRNA processing genes. Environ. Health Perspect. 118, 943–948.
Wright, R. O., Schwartz, J., Wright, R. J., Bollati, V., Tarantini, L., Park, S. K., Hu, H., Sparrow, D., Vokonas, P., and Baccarelli, A. (2010). Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ. Health Perspect. 118, 790–795.
Ye, Y., Wang, K. K., Gu, J., Yang, H., Lin, J., Ajani, J. A., and Wu, X. (2008). Genetic variations in microRNA-related genes are novel susceptibility loci for esophageal cancer risk. Cancer Prev. Res. (Phila) 1, 460–469.
Zhang, L., Mchale, C. M., Rothman, N., Li, G., Ji, Z., Vermeulen, R., Hubbard, A. E., Ren, X., Shen, M., Rappaport, S. M., North, M., Skibola, C. F., Yin, S., Vulpe, C., Chanock, S. J., Smith, M. T., and Lan, Q. (2010). Systems biology of human benzene exposure. Chem. Biol. Interact. 184, 86–93.
Zhao, C. Q., Young, M. R., Diwan, B. A., Coogan, T. P., and Waalkes, M. P. (1997). Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U.S.A. 94, 10907–10912.
Keywords: exposures, asbestos, aging, arsenic, endocrine disruptors, metals, DNA methylation, microRNA
Citation: Christensen BC and Marsit CJ (2011) Epigenomics in environmental health. Front. Gene. 2:84. doi: 10.3389/fgene.2011.00084
Received: 20 October 2011;
Accepted: 04 November 2011;
Published online: 22 November 2011.
Edited by:
David Mittelman, Virginia Tech, USAReviewed by:
Natalie C. Fonville, Virginia Tech, USAJonathan Daniel Wren, Oklahoma Medical Research Foundation, USA
Copyright: © 2011 Christensen and Marsit. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Carmen J. Marsit, Department of Pharmacology and Toxicology, Dartmouth Medical School, 7650 Remsen, Room 520, Hanover, NH 03755, USA. e-mail:Y2FybWVuLmoubWFyc2l0QGRhcnRtb3V0aC5lZHU=