
- Departamento de Mejora Genética Animal, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Madrid, Spain
Livestock genetics is currently navigating through a genomic era promoted by advances in DNA technologies. For instance, massive amounts of genomic information (e.g., arrays that genotype more than 500K SNPs along the bovine genome) are incorporated into the prediction of genetic merit providing higher predictive accuracy (Van Raden et al., 2008). This increment has led to important changes in the animal breeding industry (Dekkers, 2004; Ibañez-Escriche and González-Recio, 2011). New advances continue, and efforts are currently placed in whole genome sequencing (e.g., in species such as cattle and swine) for its implementation future implementation to improve accuracy of genomic selection or mapping new QTL of interest (Meuwissen and Goddard, 2010).
There is, however, a promising field that has not yet been tackled in livestock breeding and genetics: epigenetics. The modern definition of epigenetics is the study of heritable changes in gene expression and other genomic functions without altering the underlying “DNA” sequence – hence the name “epi” – (derived from the Greek word meaning “over, above, outer” – combined with genetics; Richards, 2006). Epigenetic shows that not all genetic information is in the DNA sequence, but also in some modifications that occur along the epigenome, particularly DNA methylation (DNAm) in which a methyl group is added to the 5′ position of the cytosine pyrimidine ring (Figure 1). This variation in methylation patterns between individuals contributes to phenotypic variability even if these individuals are genotypically identical (Coolen et al., 2011). Epigenetic information can be thought as the grammar or orthography of the DNA alphabet because these DNAm events have been shown to be regulation mechanisms associated with gene silencing, expression, chromatin remodeling, or imprinting (Robertson, 2005; Khatib, 2012). This is an emerging field of research that is becoming increasingly important and has attracted much of the interest in genetic studies in cancer and other human diseases (Relton and Davey Smith, 2010; Rakyan et al., 2011).
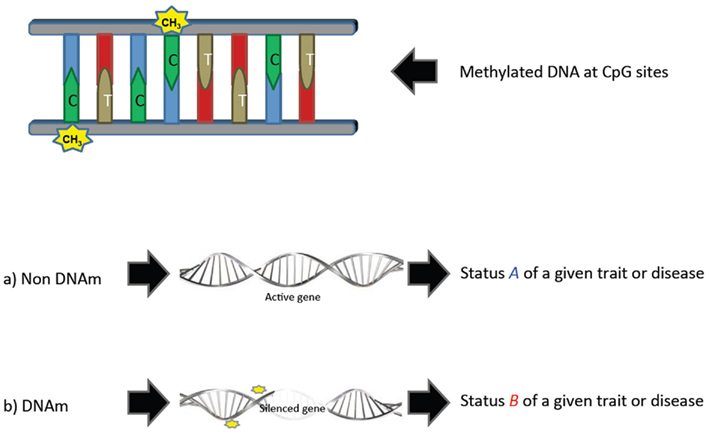
Figure 1. Simple representation of the effect of methylated DNA.
Epigenetics is attractive for animal breeding because it may help finding part of the missing causality and missing heritability of complex traits and diseases. DNAm patterns are modified along the life of an individual by environmental forces like diet, stress, drugs, or pollution among many others (Petronis, 2010). Therefore, some environments are more likely to increase certain methylation patterns, and these patterns would contribute to the phenotypic variation between individuals. Removing this noise from the phenotype decomposition equation (infinitesimal model) may help to estimate parameters more accurately.
Furthermore, the environment may affect the methylation pattern of up to three generations cohabiting under the same specific circumstances at a given time t during pregnancy: the productive female, the fetus, and the fetus’ germ cells (Figure 2). Hence, what happens to an animal during its lifetime may have consequences in future generations. Some examples can be found in humans: mothers who were pregnant during famine in The Netherlands in 1944, also known as the “Hunger Winter,” had children and grandchildren with a wide range of health problems (Heijmans et al., 2008). Nijland et al. (2008) recently showed a similar pattern in sheep: diet of pregnant ewes had some effect on the weight of their grand-daughters.
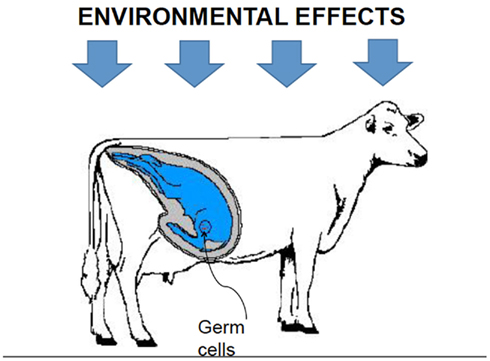
Figure 2. Environmental forces at time t, may affect the methylation patterns of three generations.
Interestingly, some authors have also related epigenetics with the missing heritability problem by demonstrating the existence of epigenetic variation and inheritance in plants and animals (Morgan et al., 1999; Henderson and Jacobsen, 2007; Migicovsky and Kovalchuk, 2011), although some controversy exists on this aspect. Epigenetic memory is supposed to be erased in mammals during the meiosis process, and only few cases of trans-generational epigenetic conservation is known in specific loci across the genomes (Jablonka and Raz, 2009). It is however well accepted that some genotypes are more susceptible to methylation than others (Coolen et al., 2011), but it is not clear what part of the genetic variation influences epigenetic variation. The causal relationship of genome–epigenome–phenotype is to be discovered, and the epigenome layer may confer additional knowledge on the relationship between genotype and phenotype. Thus, it will be important to detect the genotypes and practices associated to (un)favorable methylation affecting productive traits, functionality, and metabolic problems.
DNA Methylation and Sequencing Technology
Modification of histone proteins, non-coding RNA, and DNAm are known types of detectable epigenetic modifications. DNAm is so far the most studied epigenetic modification, and consists on the methylation of cytosines that are converted to 5-methylcytosines. They are more frequent on CpG sites (associated to promoter regions) along the genome, although CpH (H = A/C/T) methylation may also exist. The highly methylated regions tend to be less transcriptionally active, which may have some impact on the phenotypes of animals (Figure 1). DNAm at a single CpG or CpH position would be the equivalent of a polymorphism, and is called a methylation variable position. Detailed types of DNAm can be found in Rakyan et al. (2011) or Robinson et al. (2010). Methods to reveal the methylation status of individual CpG sites have been available since the early 1990s with the advent of bisulfite sequencing (Frommer et al., 1992). Recently, the identification of DNAm is possible using some state-of-the-art technologies based on second generation sequencing, which provide signals of DNAm on a genome-wide basis (Laird, 2010). These technologies allow obtaining different DNAm measurements with different levels of coverage and resolution. Bead-arrays exist for humans and mice that provide DNAm information of thousands of CpG sites throughout the genome (e.g., Human Infinium 450K Bead-array). The third generation technology for sequencing is expected to bring deeper and more accurate knowledge on the epi-genomic base modification, and may help to develop specific bead-arrays for its use in livestock.
Opportunities in Livestock
Selection for productive traits has led to amazing genetic gains in most livestock species during the last decades, and farms have intensified their production system with feeding practices based on concentrates from food plants rather than pasture based systems. Furthermore, current productive and reproductive practices demand high levels of metabolic efficiency and optimal energy partitioning. Without proper selection for genetic improvement of functional traits affected by these practices, a typical high producing animal is under higher stress, and increased risk for incidence of many metabolic diseases with negative effects on animal welfare. Based on studies in humans, one could assume all these environmental forces also affect the methylation patterns of the genome. For instance, animals with concentrate and uni-feed diet systems are expected to be differently methylated than animals in a less intensive system based on a pasture feeding systems. It will be important to detect what practices are associated to favorable methylation patterns that affect disease resistance and other economically important traits. Finding this missing causality would assist in rising animals under favorable circumstances and reduce unfavorable methylation patterns.
Breeding companies may also detect what genotypes are more susceptible to (un)favorable methylation patterns to select animals with a reduced susceptibility to unfavorable methylation patterns. Epigenome-wide association analyses are necessary to detect what methylation patterns negatively affect the traits of interest. Some correlations may exist between methylation patterns for different traits, which will also be a challenge for breeding strategies.
Farms could use epigenetic information to reduce disease incidence and the use of antibiotics in animal production. Personalized medicine using methylation on DNA is currently carried out on cancer research in humans (Peedicayil, 2008; Gomez and Ingelman-Sundberg, 2009), and seems to be a promising strategy for veterinary medicine as well. For instances, drugs may be created to modify the methylation pattern of a genomic region that is found to be associated to a given disease. These perspectives make epigenetics an interesting area of research at this time, as its potential application could lead to perform breeding and management of livestock in a more efficient and sustainable manner.
Challenges and Difficulties for Livestock Breeding
As in most new fields or technologies, epigenetics also poses many difficulties for research and implementation in livestock that will need to be addressed. Probably, the main challenge is tracking epigenetic information that changes with each successive generation at the cellular level. Different from DNA, methylation is not constant during an individual lifetime, and environmental forces model methylation along time. Longitudinal studies create an interesting scenario to detect what feeding and management practices change methylation patterns. These studies will assist to acquire knowledge on the factors that influence methylation and help differentiate between causal and consequential epigenetic variation. Additional efforts on well-established management routines and data collection are encouraged in those cases. Dealing with these specific circumstances is challenging for geneticists and breeders.
Another interesting question to be answered is what proportion of phenotypic variance does DNAm account for, and how many generations will the current DNAm pattern have an effect on future generations of the lineage.
DNA methylation patterns are tissue specific, i.e., intestinal tract cells may be methylated at a given locus whereas germ cells may be not. Therefore, sample collection must be designed in accordance with the purpose of the research or field implementation. For instance, germ cells (e.g., semen, eggs) are of interest for breeding purposes, which would provide information on what methylated variable positions (if any) a given parent is transmitting to her/his progeny (imprinting) and will affect the latter during its lifetime (including fetal development). These trans-generational epigenetic variants will likely appear in all tissues. On the other hand, cells from specific organs or tissues may be of interest to study disease variation or etiology (e.g., intestinal tract cells may be of interest to study paratuberculosis, or mammary gland cells to study mastitis).
Most importantly, technologies to detect DNAm must gain in accuracy and efficiency to provide as much information as possible at affordable cost. The proposed capabilities of third generation sequencing instruments are the most promising for providing this sort of output. Experience and research are expected to bring insights to deal with these drawbacks.
Statistical Approaches Needed
Some approaches have been proposed to estimate the epigenetic contribution to covariance between relatives (Tal et al., 2010) and the epigenetic variation (Slatkin, 2009). The gain and loss rates, also called reset coefficients, refers to the probability of changing the epigenetic state during gametogenesis, so that the new generation has no memory of past epigenetic states. The complement of the reset coefficient is the coefficient of epigenetic transmissibility, and is important because it contributes to the epigenetic heritability estimate. However, these models are not suitable for a genome-wide scenario and incorporation of DNAm into association studies and breeding value predictions are still challenging. Statistical methods are needed to incorporate massive amounts of epigenetic information, and differentiate between missing heritability and missing causality. The inclusion of whole genome epigenetic information is expected to impair the course of dimensionality problem at DNAm effect estimation, because a several 1000 of methylated loci across the genome may be determined through bisulfite sequencing techniques. The sample size of animals with whole genome epigenetic information may initially be critical. Regularized regression (mainly in a Bayesian context), non-parametric approaches, or machine learning (González-Recio et al., 2008; de los Campos et al., 2009) are attractive alternatives for epigenome-wide association studies and epigenome-wide prediction. Epigenetics may add additional accuracy to current approaches by removing some noise from the data. Hence, the simultaneous incorporation of pedigree, SNP, “indels,” and DNAm poses new challenges for statistical methods for association studies and genome-wide prediction.
Important efforts are expected from the scientific community during the next years to build relevant theories and develop these or other statistical approaches to consider DNAm in inference and prediction analyses.
Concluding Remarks
There is still a long way to go to understand the relevance of DNAm on genome-wide prediction and elucidating the state of nature for complex traits. Epigenetics shows potential benefits and possibility of changes in the livestock industry, which is likely to evolve as information and experience accumulate, ultimately defining if epigenetic information is useful in animal breeding and genetics.
The study of epigenetic variation is an attractive challenge during the next decade. First efforts should focus on proving that DNAm marks contribute to variation of traits of interest in the livestock populations. Then, research efforts should focus on (1) the development of technology to detect DNAm on individuals in an affordable manner, (2) the development of statistical tools to accommodate genome-wide epigenetic information in the phenotype decomposition equation, and (3) an efficient implementation of epi-genomic selection in breeding and management programs.
References
Coolen, M. W., Statham, A. L., Qu, W., Campbell, M. J., Henders, A. K., Montgomery, G. W., Martin, N. G., and Clark, S. J. (2011). Impact of the genome on the epigenome is manifested in dna methylation patterns of imprinted regions in monozygotic and dizygotic twins. PLoS ONE 6, e25590. doi: 10.1371/journal.pone.0025590
de los Campos, G., Naya, H., Gianola, D., Crossa, J., Legarra, A., Manfredi, E., Weigel, K. A., and Cotes, J. M. (2009). Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics 182, 375–385.
Dekkers, J. (2004). Commercial application of marker- and gene-assisted selection in livestock: strategies and lessons. J. Anim. Sci. 82, E313–E328.
Frommer, M., McDonalD, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., Molloy, P. L., and Paul, C. L. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U.S.A. 89, 1827–1831.
Gomez, A., and Ingelman-Sundberg, M. (2009). Pharmacoepigenetics: its role in interindividual differences in drug response. Clin. Pharmacol. Ther. 2009 85, 426–430.
González-Recio, O., Gianola, D., Long, N., Weigel, K. A., Rosa, G. J. M., and Avendaño, S. (2008). Non-parametric methods for incorporating genomic information into genetic evaluations: an application to mortality in broilers. Genetics 178, 2305–2313.
Heijmans, B. T., Tobi, E. W., Stein, A. D., Putter, H., Blauw, G. J., Susser, E. S., Slagboom, P. E., and Lumey, L. H. (2008). Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 44.
Henderson, I. R., and Jacobsen, S. E. (2007). Epigenetic inheritance in plants. Nature 447, 418–424.
Ibañez-Escriche, N., and González-Recio, O. (2011). Review. Promises, pitfalls and challenges of genomic selection in breeding programs. Span. J. Agric. Res. 9, 404–413.
Jablonka, E., and Raz, G. (2009). Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 84, 131–175.
Laird, P. W. (2010). Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. 11, 191–203.
Meuwissen, T. H. E., and Goddard, M. (2010). Accurate prediction of genetic values for complex traits by whole-genome resequencing. Genetics 185, 623–631.
Migicovsky, Z., and Kovalchuk, I. (2011). Epigenetic memory in mammals. Front. Gene. 2:28. doi: 10.3389/fgene.2011.00028
Morgan, H. D., Sutherland, H. G., Martin, D. I., and Whitelaw, E. (1999). Epigenetic inheritance at the agouti locus in mouse. Nat. Genet. 23, 314–318.
Nijland, M. J., Ford, S. P., and Nathanielsz, P. W. (2008). Prenatal origins of adult disease. Curr. Opin. Obstet. Gynecol. 20, 132–138.
Petronis, A. (2010). Epigenetics as a unifying principle in the aetology of complex traits and diseases. Nature 465, 721–727.
Rakyan, V. K., Down, T. A., Balding, D. J., and Beck, S. (2011). Epigenome-wide association studies for common human diseases. Nat. Rev. 12, 529–541.
Relton, C. L., and Davey Smith, G. (2010). Epigeneticepidemiology of commoncomplexdisease: prospectsforprediction, prevention, and treatment. PLoS Med. 7, e1000356. doi: 10.1371/journal.pmed.1000356
Richards, E. J. (2006). Inherited epigenetic variation-revisiting soft inheritance. Nat. Rev. Genet. 7, 395–401.
Robinson, M. D., Statham, A. L., Speed, T. P., and Clark, S. J. (2010). Protocol matters: which methylome are you actually studying? Epigenetics 2, 587–598.
Slatkin, M. (2009). Epigenetic inheritance and the missing heritability problem. Genetics 182, 845–850.
Tal, O., Kisdi, K., and Jablonka, E. (2010). Epigenetic contribution to covariance between relatives. Genetics 184, 1037–1050.
Citation: González-Recio O (2012) Epigenetics: a new challenge in the post-genomic era of livestock. Front. Gene. 2:106. doi: 10.3389/fgene.2011.00106
Received: 01 October 2011;
Accepted: 21 December 2011;
Published online: 09 January 2012.
Copyright: © 2012 González-Recio. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence:Z29uemFsZXoub3NjYXJAaW5pYS5lcw==