


- Department of Epidemiology, University of Alabama at Birmingham, Birmingham, AL, USA
When susceptibility to diseases is caused by cis-effects of multiple alleles at adjacent polymorphic sites, it may be difficult to assess with confidence the genetic phase and identify individuals carrying the risk haplotype. Experimental assessment of genetic phase is still challenging and most population studies use statistical approaches to infer haplotypes given the observed genotypes. While these statistical approaches are powerful and have been proven very useful in large scale genetic population studies, they may be prone to errors in studies with small sample size, especially in the presence of compound heterozygotes. Here, we describe a simple and novel approach using the popular PCR–RFLP based strategy to assess the genetic phase in compound heterozygotes. We apply this method to two extensively studied SNPs in two clustered immune-related genes: The −308 (G > A) and the +252 (A > G) SNPs of the tumor necrosis factor (TNF) alpha and the lymphotoxin alpha (LTA) genes, respectively. Using this method, we successfully determined the genetic phase of these two SNPs in known compound heterozygous individuals and in every sample tested. We show that the A allele of TNF −308 is carried on the same chromosome as the LTA +252(G) allele.
Introduction
SNPs are useful in mapping disease susceptibility genes. However, the SNPs identified from typical genetic mapping studies often are not the causal SNPs but may tag the true causal mutation if they are in linkage disequilibrium with each other (Ardlie et al., 2002; Gabriel et al., 2002; Ke et al., 2004). To increase heterozygosity, genetic information, and statistical power, haplotypes are usually reconstructed from observed genotypes to identify the individuals carrying the risk haplotypes. Haplotypes are also valuable for exploring cis-effects of specific combinations of intragenic polymorphisms, such as those variations located in gene promoters, or polymorphisms in closely linked genes where there may be interaction that affects gene expression (Levenstien et al., 2006; Fan et al., 2011).
Statistical and computational methods can provide highly accurate estimates of haplotypes reconstructed from conventional genotype data in most situations, but even the best method can have a large error rate of more than 35% for datasets where parental genotypes are unknown (Stephens and Donnelly, 2003; Marchini et al., 2006; Browning, 2008; Liu et al., 2008). Several experimental methods for haplotype and genetic phase determination have been reported, including cloning (Burgtorf et al., 2003; Kitzman et al., 2011), hybridization (Yan et al., 2000; Douglas et al., 2001; Boldt and Petzl-Erler, 2002), polony technology (Mitra et al., 2003), MALDI-TOF mass spectrometry (Tost et al., 2002), sequencing (Levy et al., 2007), and chromosome micro-dissection (Ma et al., 2010). However, these methods are labor-intensive and/or require specialized and expensive equipment. PCR-based methods, such as allele-specific PCR and isolation of single DNA molecules have been used in the past to determine haplotypes and genetic phase (Ruano et al., 1990; Michalatos-Beloin et al., 1996; Zhang et al., 2006), but experimental optimization is needed and/or is labor-intensive. All of the above methods may not be applicable in settings where cost is a premium and specialized equipment is not available or personnel have little or no training in labor-intensive methodologies.
We present here a simple application of the popular PCR–RFLP based strategy to determine the genetic phase at two adjacent SNPs in compound heterozygous individuals. Specifically, we looked at rs909253 (A > G) at position +252 of the lymphotoxin alpha (LTA) gene and about 3 kb downstream, rs1800629 (G > A), at position −308 of the tumor necrosis factor (TNF) alpha gene in the major histocompatibility complex. Using this method, we determined the phase of these two SNPs in nine individuals shown in a previous study to be heterozygous both at the LTA +252 and the TNF −308 positions (Aissani et al., 2009).
Materials and Methods
DNA was extracted from peripheral blood mononuclear cells (Qiagen, Valencia, CA, USA) obtained from nine subjects who were double heterozygotes for the LTA +252 and TNF −308 SNPs (Aissani et al., 2009). Human subject approval was given by the Institutional Review Board at UAB and informed consent was obtained from all subjects. DNA quantitation was determined by a fluorometric method using the Qubit™ fluorometer (Invitrogen, Carlsbad, CA, USA). For the first PCR, 50 ng of DNA from each subject were used to amplify the 3.4-kb fragment separating the LTA and TNF SNPs. Due to the large size of the fragment to be amplified and its high GC content (75%), we used the Qiagen LongRange PCR kit. All primers were designed using Primer 3 (http://frodo.wi.mit.edu/primer3/). A final concentration of 0.4 μM of primers L–T forward (5′-GCTTCGTGCTTTGGACTACC-3′) and reverse (5′-GTCCTTTCCAGGGGAGAGAG-3′) were added to a 25-μl reaction containing 2.5 mM Mg++, 500 μM of each dNTP, 1× Q-solution, and two units of enzyme. DNA amplification was performed at an initial denaturation at 95°C for 3 min followed by 35 cycles of denaturation at 95°C/30 s, annealing at 62°C/30 s, and extension at 68°C for 3 min. After product specificity was tested and confirmed by agarose gel electrophoresis, the products were purified using the Qiagen PCR purification kit. Approximately 5 μl of 25 μl purified product were digested with two and one-half units of NcoI restriction endonuclease enzyme (NEB, Ipswich, MA, USA) in a total reaction of 25 μl for 1 h at 37°C. The digested and undigested samples were run in adjacent lanes along with a 1-kb size standard (Fermentas, Glen Burnie, MD, USA) on a 1.2% agarose gel. The samples were run first for 16 h at 20 V constant overnight and then 40 V for 7 h in order to obtain sufficient separation of the fragments for removal from the gel. The gel was stained with ethidium bromide for 30 min, followed by destaining in 1× TBE for 10 min. Under low UV light, the shorter 3.2 kb digested NcoI fragment containing the G allele at LTA +252 was identified and sliced from the gel.
The DNA fragment was eluted from the gel slice using the QIAquick Gel Extraction Kit. For the second PCR, TNF forward (5′-GCCCCTCCCAGTTCTAGTTC-3′) and reverse (5′-TTGGAAAGTTGGGGACACAC-3′) primers were designed to flank the TNF −308 site. We generated a 248-bp PCR fragment from 1 μl of gel purified product in a 20-μl reaction containing 0.5 μM each of primers and standard PCR reagents (200 μM dNTPs, 1.5 mM Mg++, and one unit of Amplitaq; Applied Biosystems, Foster City, CA, USA). After an initial denaturation at 95°C for 3 min, 35 cycles of denaturation at 95°C/30 s, annealing at 56°C/30 s, and extension at 72°C/30 s were performed, followed by a final extension of 72°C for 5 min. The PCR product was purified through silica columns (Qiagen, Valencia, CA, USA) to remove dNTPs and non-incorporated primers. To identify the allele at the TNF −308 site, the nucleotide sequence of the purified 248 bp fragment was determined by Big Dye terminator cycle sequencing on a 3730 Genomic Analyzer (Applied Biosystems, Forrest City, CA, USA) using the TNF reverse primer.
Results
Figure 1 shows the locations of the LTA +252 and TNF −308 SNPs in the initial 3.4 kb PCR product and in the resulting 3.2 kb NcoI digested fragment when the “G” allele is present at the LTA +252 site. Since each of the nine samples was heterozygous for the LTA +252 SNP (A > G), digestion of the PCR product with NcoI generated DNA fragments of 3.4 and of 3.2 kb of length carrying the “A” and the “G” alleles, respectively (Figure 2). Because allele “G” of LTA +252 can occur with either “A” or “G” allele of TNF −308, genetic phase was determined by sequencing the 3′ end of the 3.2-kb NcoI fragment that encompasses the TNF −308 site. Figure 3 shows the DNA sequences surrounding the TNF −308 site in the nine tested samples. As can be seen, all nine samples carry the TNF −308 “A” allele (displayed as the “T” allele on the reverse strand). Therefore, all nine subjects who had the “G” allele at the LTA +252 site possess the “A” allele at the TNF −308 site. Since all nine individuals are known compound heterozygotes (Aissani et al., 2009), the phase on the other chromosome must be LTA +252 (A) – TNF −308 (G).
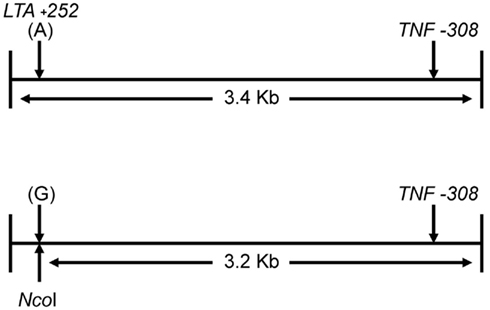
Figure 1. Location of LTA +252 and TNF −308 SNPs in PCR product and NcoI fragment.
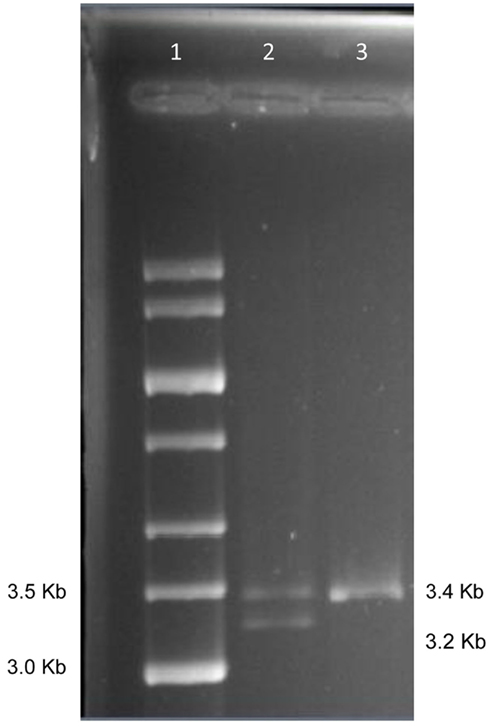
Figure 2. Gel separation of digested and undigested NcoI fragments. Lane 1: 1 kb size std.; Lane 2: NcoI digest of PCR from subject 1; Lane 3: first PCR product from subject 1.
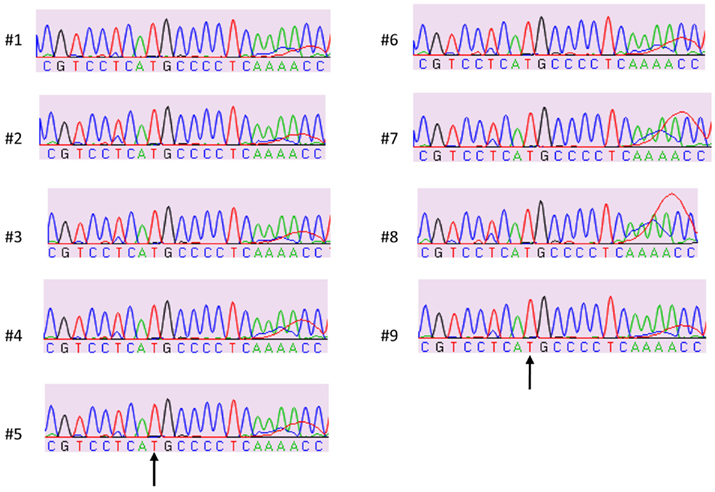
Figure 3. Sample sequences of TNF −308 SNP on NcoI digested fragment (3′ strand). “T” nucleotide on the 3′ strand represents the TNF −308 “A” allele on 5′ strand.
Discussion
For population studies where often hundreds or thousands of individuals are sampled, phase determination can usually be statistically inferred, i.e., probabilistically. Family data can also assist in determining phase. However, if only a few subjects are genotyped, determining phase usually must rely on laborious methods such as purification of single molecules to isolate the alleles. Allele-specific PCR is relatively simple but is limited by the demand of allele-specific oligos which may not always be compatible with efficient amplification. The PCR–RFLP technique we present here is a simple, cost–effective way to determine phase when compound heterozygotes are present, especially in studies of small sample size or when family data are not available. This method does not require expensive equipment and can be performed with the basic tools found in most laboratories with only moderate technical experience. There are a couple of factors that can affect or even restrict the use of this technique. If one of the SNPs is not a restriction site, allele-specific PCR could be utilized as an alternative. However, our method is more flexible than allele-specific PCR as any heterozygous site upstream of the SNP sites of interest could be used to generate fragments with different electrophoretic mobilities, although sequencing across both SNPs on the shorter fragment would be necessary. Here, there was no need to sequence through the LTA +252 site because the allele was determined by RFLP. Size is an obvious limitation of the technique we present here since a template of ∼40 kb approaches the upper limit for successful amplification. Finally, some initial optimization of PCR and electrophoretic conditions, such as the presence of difficult sequences (e.g., GC content) and similarly sized cut and uncut fragments, may be necessary.
We present here a simple, cost–effective method for determining genetic phase between SNPs in compound heterozygotes. We were able to use this PCR–RFLP procedure to determine phase for two SNPs that are 3 kb apart in nine such individuals. Specifically, using this procedure, we show that allele A of TNF −308 is in complete disequilibrium with allele G of LTA +252. This procedure would be very helpful for diseases or conditions where the clinical manifestation and/or outcome are associated with haplotypes. Except for sequencing, which can be outsourced to relatively inexpensive sequencing services, this procedure can be performed in most labs with only moderate technical experience and with no expensive equipment or high up-front cost. This technique could eventually be applied in clinical settings where disease susceptibility or variability in expression is an urgent need. Although there are certain situations that can limit the practical use of this approach, the simple design, and relatively inexpensive cost makes it a viable alternative to current methods.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by P30-AI045008 Penn Center for AIDS Research subcontract (Brahim Aissani). The sampled individuals are from the Multicenter AIDS Cohort Study (MACS) with centers (Principal Investigators) at The Johns Hopkins University Bloomberg School of Public Health (Joseph B. Margolick, Lisa Jacobson), Howard Brown Health Center, and Northwestern University Medical School (John Phair), University of California, Los Angeles (Roger Detels), and University of Pittsburgh (Charles Rinaldo). Website located at http://www.statepi.jhsph.edu/macs/macs.html.
References
Aissani, B., Ogwaro, K. M., Shrestha, S., Tang, J., Breen, E. C., Wong, H. L., Jacobson, L. P., Rabkin, C. S., Ambinder, R. F., Martinez-Maza, O., and Kaslow, R. A. (2009). The major histocompatibility complex conserved extended haplotype 8.1 in AIDS-related non-Hodgkin lymphoma. J. Acquir. Immune Defic. Syndr. 52, 170–179.
Ardlie, K. G., Kruglyak, L., and Seielstad, M. (2002). Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 3, 299–309.
Boldt, A. B., and Petzl-Erler, M. L. (2002). A new strategy for mannose-binding lectin gene haplotyping. Hum. Mutat. 19, 296–306.
Browning, S. R. (2008). Missing data imputation and haplotype phase inference for genome-wide association studies. Hum. Genet. 124, 439–450.
Burgtorf, C., Kepper, P., Hoehe, M., Schmitt, C., Reinhardt, R., Lehrach, H., and Sauer, S. (2003). Clone-based systematic haplotyping (CSH): a procedure for physical haplotyping of whole genomes. Genome Res. 13, 2717–2724.
Douglas, J. A., Boehnke, M., Gillanders, E., Trent, J. M., and Gruber, S. B. (2001). Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nat. Genet. 28, 361–364.
Fan, H. C., Wang, J., Potanina, A., and Quake, S. R. (2011). Whole-genome molecular haplotyping of single cells. Nat. Biotechnol. 29, 51–57.
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., Higgins, J., DeFelice, M., Lochner, A., Faggart, M., Liu-Cordero, S. N., Rotimi, C., Adeyemo, A., Cooper, R., Ward, R., Lander, E. S., Daly, M. J., and Altshuler, D. (2002). The structure of haplotype blocks in the human genome. Science 296, 2225–2229.
Ke, X., Hunt, S., Tapper, W., Lawrence, R., Stavrides, G., Ghori, J., Whittaker, P., Collins, A., Morris, A. P., Bentley, D., Cardon, L. R., and Deloukas, P. (2004). The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum. Mol. Genet. 13, 577–588.
Kitzman, J. O., Mackenzie, A. P., Adey, A., Hiatt, J. B., Patwardhan, R. P., Sudmant, P. H., Ng, S. B., Alkan, C., Qiu, R., Eichler, E. E., and Shendure, J. (2011). Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat. Biotechnol. 29, 59–63.
Levenstien, M. A., Ott, J., and Gordon, D. (2006). Are molecular haplotypes worth the time and expense? A cost-effective method for applying molecular haplotypes. PLoS Genet. 2, e127. doi:10.1371/journal.pgen.0020127
Levy, S., Sutton, G., Ng, P. C., Feuk, L., Halpern, A. L., Walenz, B. P., Axelrod, N., Huang, J., Kirkness, E. F., Denisov, G., Lin, Y., MacDonald, J. R., Pang, A. W., Shago, M., Stockwell, T. B., Tsiamouri, A., Bafna, V., Bansal, V., Kravitz, S. A., Busam, D. A., Beeson, K. Y., McIntosh, T. C., Remington, K. A., Abril, J. F., Gill, J., Borman, J., Rogers, Y. H., Frazier, M. E., Scherer, S. W., Strausberg, R. L., and Venter, J. C. (2007). The diploid genome sequence of an individual human. PLoS Biol. 5, e254. doi:10.1371/journal.pbio.0050254
Ma, L., Xiao, Y., Huang, H., Wang, Q., Rao, W., Feng, Y., Zhang, K., and Song, Q. (2010). Direct determination of molecular haplotypes by chromosome microdissection. Nat. Methods 7, 299–301.
Marchini, J., Cutler, D., Patterson, N., Stephens, M., Eskin, E., Halperin, E., Lin, S., Qin, Z. S., Munro, H. M., Abecasis, G. R., and Donnelly, P. (2006). A comparison of phasing algorithms for trios and unrelated individuals. Am. J. Hum. Genet. 78, 437–450.
Michalatos-Beloin, S., Tishkoff, S. A., Bentley, K. L., Kidd, K. K., and Ruano, G. (1996). Molecular haplotyping of genetic markers 10 kb apart by allele-specific long-range PCR. Nucleic Acids Res. 24, 4841–4843.
Mitra, R. D., Butty, V. L., Shendure, J., Williams, B. R., Housman, D. E., and Church, G. M. (2003). Digital genotyping and haplotyping with polymerase colonies. Proc. Natl. Acad. Sci. U.S.A. 100, 5926–5931.
Ruano, G., Kidd, K. K., and Stephens, J. C. (1990). Haplotype of multiple polymorphisms resolved by enzymatic amplification of single DNA molecules. Proc. Natl. Acad. Sci. U.S.A. 87, 6296–6300.
Stephens, M., and Donnelly, P. (2003). A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 73, 1162–1169.
Tost, J., Brandt, O., Boussicault, F., Derbala, D., Caloustian, C., Lechner, D., and Gut, I. G. (2002). Molecular haplotyping at high throughput. Nucleic Acids Res. 30, e96.
Yan, H., Papadopoulos, N., Marra, G., Perrera, C., Jiricny, J., Boland, C. R., Lynch, H. T., Chadwick, R. B., de la Chapelle, A., Berg, K., Eshleman, J. R., Yuan, W., Markowitz, S., Laken, S. J., Lengauer, C., Kinzler, K. W., and Vogelstein, B. (2000). Conversion of diploidy to haploidy. Nature 403, 723–724.
Keywords: haplotype, PCR–RFLP, phase
Citation: Perry RT, Dwivedi H and Aissani B (2011) A simple PCR–RFLP method for genetic phase determination in compound heterozygotes. Front. Gene. 2:108. doi: 10.3389/fgene.2011.00108
Received: 25 October 2011;
Paper pending published: 15 November 2011;
Accepted: 22 December 2011;
Published online: 13 January 2012.
Edited by:
Manuel A. Palacios, General Electric Global Research, USACopyright: © 2012 Perry, Dwivedi and Aissani. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Brahim Aissani, Department of Epidemiology, University of Alabama at Birmingham School of Public Health, Ryals Building 217J, Birmingham, AL 35294-0022, USA. e-mail:YmFpc3NhbmlAdWFiLmVkdQ==