
 William M. Muir2
William M. Muir2
- 1 Avian Disease and Oncology Laboratory, Agricultural Research Service, United States Department of Agriculture, East Lansing, MI, USA
- 2 Department of Animal Sciences, Purdue University, West Lafayette, IN, USA
- 3 Genome Sequencing Center, Washington University in St. Louis, St. Louis, MO, USA
Marek’s disease (MD) is a commercially important neoplastic disease of chickens caused by Marek’s disease virus (MDV), a naturally occurring oncogenic alphaherpesvirus. Selecting for increased genetic resistance to MD is a control strategy that can augment vaccinal control measures. To identify high-confidence candidate MD resistance genes, we conducted a genome-wide screen for allele-specific expression (ASE) amongst F1 progeny of two inbred chicken lines that differ substantially in MD resistance. High throughput sequencing was initially used to profile transcriptomes from pools of uninfected and infected individuals at 4 days post-infection to identify any genes showing ASE in response to MDV infection. RNA sequencing identified 22,655 single nucleotide polymorphisms (SNPs) of which 5,360 in 3,773 genes exhibited significant allelic imbalance. Illumina GoldenGate assays were subsequently used to quantify regulatory variation controlled at the gene (cis) and elsewhere in the genome (trans) by examining differences in expression between F1 individuals and artificial F1 RNA pools over six time periods in 1,536 of the most significant SNPs identified by RNA sequencing. Allelic imbalance as a result of cis-regulatory changes was confirmed in 861 of the 1,233 GoldenGate assays successfully examined. Furthermore we have identified seven genes that display trans-regulation only in infected animals and ∼500 SNP that show a complex interaction between cis- and trans-regulatory changes. Our results indicate ASE analyses are a powerful approach to identify regulatory variation responsible for differences in transcript abundance in genes underlying complex traits. And the genes with SNPs exhibiting ASE provide a strong foundation to further investigate the causative polymorphisms and genetic mechanisms for MD resistance. Finally, the methods used here for identifying specific genes and SNPs have practical implications for applying marker-assisted selection to complex traits that are difficult to measure in agricultural species, when expression differences are expected to control a portion of the phenotypic variance.
Introduction
Marek’s disease virus (MDV, Gallid herpesvirus 2) is a naturally occurring oncogenic alphaherpesvirus of chickens that targets lymphoid tissues like the bursa of Fabricius, thymus, and particularly the spleen, which is an important lymphatic organ and repository of T and B cells. MDV infects both T and B cells, which may result in lymphoid tumors, nerve lesions, and immunosuppression. Clinical symptoms of infected birds include blindness, paralysis, and death in susceptible animals. MDV is highly contagious as virions shed from feathers and skin in the form of dander or dust persist in the environment and are readily spread horizontally by inhalation. Consequently, virtually every bird is exposed to MDV at a very young age. Immune transfer of maternal antibodies from hen to chick provides some protection from the virus in the first few days of life but the only effective control method, which prevents tumor formation, is vaccination. However, MD vaccines do not eliminate MDV infection, replication, or spread, thus, the virus is believed to have evolved for increased virulence in response to vaccination (Witter, 1997). The lack of new vaccines and the expectation that the virus will evolve to overcome existing vaccines means the identification of genes involved in natural immunity is of major interest to the poultry industry.
Genetic resistance to MD is complex and presumably controlled by a large number of genes. A number of loci have been identified that contribute to resistant phenotypes. One of the most well known is the major histocompatibility complex (MHC) or, as it is known in the chicken, the B complex. Alleles at this locus are well known for their involvement in genetic resistance to MD as a number of B haplotypes are known to be associated with resistance or susceptibility and also influence vaccine-mediated immunity (Bacon and Witter, 1994). However, the majority of disease resistance is expected to comprise non-MHC loci (Groot and Albers, 1992). A number of quantitative trait loci (QTL) associated with MD resistance have been reported outside of the MHC in various lines and breeds of chicken (Vallejo et al., 1998; Yonash et al., 1999; McElroy et al., 2005; Cheng et al., 2008; Heifetz et al., 2009). Nonetheless, like many other quantitative traits, only a very small number of genes have been identified that confer MD resistance (Liu et al., 2001b, 2003; Niikura et al., 2007; Praslickova et al., 2008), and from these, only a fraction of the phenotypic variance has been accounted for potentially due to small effect size, large type 1 error rates, and low LD or different phase between markers and quantitative trait nucleotides (QTN) (Goddard and Hayes, 2009; Manolio et al., 2009). Therefore, utilization of these regions in marker-assisted selection programs has been limited.
The importance of transcriptional regulation in controlling phenotypic variation within and between species is an old concept (King and Wilson, 1975). It is believed that polymorphisms within cis- or trans-elements that influence gene expression provide more opportunity for evolution and directional selection compared to mutations that alter protein coding regions in the genome (Wray, 2007). The fact that within an individual, all cells have the same DNA sequence and protein coding content yet vastly different forms and functions attests to this belief. Thus, regulation of gene expression within and amongst individuals may be important in the underlying fitness differences and the phenotypic variation associated with disease and other quantitative traits. Some of the first attempts to identify polymorphic cis- and trans-acting elements were accomplished by mapping expression quantitative trait loci (eQTL; Brem et al., 2002; Schadt et al., 2003). However, eQTL studies are subject to many of the limitations that traditional QTL studies face in that they suffer due to low mapping resolution and have difficulty detecting QTL of small effect. Measuring allele-specific expression (ASE) in a heterozygous individual offers an alternative and simpler approach to uncover cis-regulatory variation (Pastinen, 2010). Because levels of each allele are measured within the same RNA sample, changes in allele expression levels must be the result of differences within cis-acting regulatory elements that most probably influence transcription factor binding sites or mRNA stability as both alleles experience the same cellular environment. When expression differences are detected between samples that do not share a similar cellular environment, then both cis and trans-acting regulation can be measured. Therefore, comparing the ratio of expression levels in an F1 system to the expression levels in the parental strains or the difference in inter- vs. intra-variation in expression ratios should enable one to dissect the contribution of cis- from trans-regulation (Wittkopp et al., 2004).
Initially ASE was examined in subsets of candidate genes (Guo et al., 2004; Wittkopp et al., 2004). ASE has also been investigated using single nucleotide polymorphism (SNP) specific array-based platforms that increasingly have a genome-wide perspective (Bjornsson et al., 2008). However, with this approach, the sequence variations to differentiate the two alleles of each gene must be known a priori. The introduction of high throughput sequencing technologies and the ability to resequence entire transcriptomes has lead to a new approach to study ASE that can catalog sequence and transcriptional variation in tandem (Bradley et al., 2009; Heap et al., 2010). In addition platforms originally designed for SNP genotyping, have recently been adapted to quantify allele expression (Serre et al., 2008), which are thought to be less prone to genotyping errors and typically more cost–effective for screening a large number of individuals for a small subset of genes.
To this end, we have examined ASE in two highly inbred lines of chicken that are resistant (line 63) or susceptible (line 72) to MD. Both lines have been fixed for the same B haplotype over 50 generations ago, but differ greatly in their MD incidence following infection with MDV (Bacon et al., 2000), which also emphasizes the role that non-MHC genes play in genetic resistance to MD. The lack of gene flow between these lines, and direct inbreeding within lines, will result in a large number of fixed polymorphisms between the lines, which should have resulted in most polymorphisms being strictly heterozygous in the F1 progeny, a requirement of the ASE assay. Thus, we have undertaken an initial examination of ASE by re-sequencing the transcriptomes of infected and uninfected F1 animals to uncover genes with cis-acting regulatory elements that respond to MDV infection, and then examined in more detail 1,536 targeted SNP that show evidence of ASE using cost–effective and high throughput Illumina GoldenGate assays in an effort to assess the ability of ASE to uncover the genetic mechanisms responsible for MD resistance.
Materials and Methods
Animals and mRNA Isolation
Highly inbred ADOL lines line 63 (MD resistant) or line 72 (MD susceptible) were reciprocally crossed to form F1 progeny where semen from the paternal male was pooled from either the MD resistant or susceptible line and used to inseminate females from the opposing line. The progeny from each reciprocal cross were split into uninfected and infected treatment groups each containing 12 animals.
Artificial F1 samples were used for comparison to the true F1s and their estimated cis-acting effects for infected and uninfected treatment groups. Fourteen replicate artificial F1s per infection status were created using a spectrophotometer to quantify concentrations of mRNA and to mix a single line 63 mRNA with a single line 72 mRNA. All mRNA was extracted from individuals and all individuals formed a treatment group. All infected animals were infected with a subcutaneous injection of MDV (2,000 pfu, JM strain) at 2 weeks of age. Birds from each treatment group were euthanized at 1, 4, 7, 11, 13, and 15 days post-infection (dpi). Animal management followed the ADOL Animal Care and Usage Committee policy.
All mRNA was extracted from splenic tissue and followed MIQE guidelines. Splenic tissue was recovered into RNAlater (Ambion; Austin, TX, USA), and frozen up to 2 weeks. Total RNA was extracted from splenic tissue, and mRNA was purified using the Stratagene Absolutely RNA Miniprep kit (Santa Clara, CA, USA). Transcriptome sequencing was undertaken on cDNA using the Promega Improm-II Reverse Transcription System (Sunnyvale, CA, USA). All RNA samples were analyzed using the Spectronic Instruments GENESYS 5 (Fisher Scientific; Pittsburgh, PA, USA) for absorbance and 260/280 ratios. Genomic DNA was also isolated from each F1 individual to control for dye and primer bias in the Illumina GoldenGate assay. Genomic DNA was isolated using QIA-amp DNA Blood mini-kits (Qiagen; Valencia, CA, USA) and concentrations examined using the Hoefer TKO 100 Fluorometer (Holliston, MA, USA) with Hoechst dye.
Next Generation Sequencing
A requirement of ASE is to identify at least one polymorphism within a transcript to allow tracking of both expressed alleles. Due to cost restrictions and the ability to simultaneously scan the genome for SNPs and get an initial indication of ASE, high throughput sequencing was used to profile the transcriptomes from two replicate pools for each reciprocal cross (6 × 7 and 7 × 6) at a single time point (4 dpi) from splenic mRNA isolated in six infected and six uninfected F1 individuals using ∼35 base single-end reads from the Illumina Genome Analyzer II (San Diego, CA, USA).
Reads were aligned to the chicken genome v3 (http://hgdownload.cse.ucsc.edu/downloads.html#chicken) with MAQ v0.50 (Li et al., 2008). FastQ files were initially parsed and all adaptor sequence was removed prior to mapping. Poor quality base calls with an Illumina quality score <40 were removed from the analysis to increase the chance of mapping reads to the reference genome. Alignments were parsed and mRNA transcripts were examined for the presence of SNP that differentiated transcripts in the two inbred lines. In an attempt to reduce sequencing and alignment errors, individual reads were only mapped back to the reference genome if they contained less than three mismatches per read. In addition, all SNP were filtered using standard options in MAQ and a Python wrapper that removed SNP represented by fewer than nine reads, had an allele frequency <0.2, were not bi-allelic or represented by ambiguous bases, or where there were more than 1 SNP in a flanking 40 base window. All mapped reads were BLASTed against a database combining RefSeq and all known chicken ESTs. All of the top unique hits were then parsed to give us an indication of how many genes were represented in the chicken spleen transcriptome.
Differential expression – RNAseq
As a first pass analysis, transcripts were considered suitable for further interrogation on the GoldenGate assay if they contained a SNP, an allele frequency >0.2, and showed a response to infection in at least one of the reciprocal crosses. Selection of each SNP was determined, by using a 2 × 2 factorial test examining each allele and infection status as classes in two biological replicates, SNPs with a significant effect for infection status or those with a significant interaction between allele and infection were selected for further investigation.
GoldenGate Assay
Alleles showing evidence of an allelic response to infection from the RNAseq pilot study were selected for a more comprehensive analysis using the Illumina GoldenGate assay (San Diego, CA, USA). ASE in each SNP was examined in 12 biological replicates from each reciprocal cross, over six time points in infected and uninfected individuals. Primer design was completed by retrieving 70 bases immediately up- and down-stream of the differentially expressed SNP using an automated computer program written in Python. In total, 1,536 SNPs were selected for further analysis on the GoldenGate platform based on evidence of ASE in response to MDV infection and a high primer design score as determined from the Illumina Assay Design Tool. The 1,536 SNPs originated from 1,297 transcripts. Of these, 239 SNPs were selected from 192 transcripts with >1 SNP on different exons. These SNPs were selected to act as a control and to examine evidence of alternative splicing.
Genomic DNA was extracted from 14 individuals from each reciprocal cross to help identify dye bias and, in the limited situations where the alternative genotype alleles were not fixed in the two parental lines, examine segregation of alleles within the F1. Scatter plots of the relative fluorescence within individuals for each A and B allele were completed using the genomic DNA samples. For all SNP the ratio of A to B alleles was expected to be 1:1 after accounting for any dye bias or technical variation. Any transcripts that were homozygous either for the A or B allele in any of the F1 samples were considered to be sequencing errors that managed to evade detection during RNAseq filtering and were removed. Thus, any SNP showing Mendelian errors in multiple individuals, i.e., two copies of either the A or B allele, were considered polymorphisms within a line that were still segregating, or sequencing errors and not fixed differences between lines and removed from the dataset.
Normalization and differential expression – goldenGate
When the GoldenGate assay was run on the Illumina Sentrix Array, an unexpected large dye bias (Cy3 vs. Cy5) was observed for all genes in samples ran in rows A and H of two 96 well plates, which coincided with treatments 1 dpi uninfected and 11 dpi infected individuals for both crosses. Therefore, raw intensities for alleles corresponding to each dye were normalized within samples. Normalized values for alleles of each dye was calculated within samples 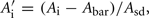 similarly
similarly 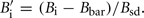 Where Ai and Bi are the raw intensities for each allele of SNP i, Abar, and Bbar are the mean intensities across all loci within a sample and Asd and Bsd are the SD across all loci within a sample for each dye. ASE was measured as
Where Ai and Bi are the raw intensities for each allele of SNP i, Abar, and Bbar are the mean intensities across all loci within a sample and Asd and Bsd are the SD across all loci within a sample for each dye. ASE was measured as 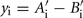 or the difference between adjusted A and B alleles, SNP found to significantly differ from 0 were categorized as showing ASE.
or the difference between adjusted A and B alleles, SNP found to significantly differ from 0 were categorized as showing ASE.
Linear models yi = u + inf + dpi + cross + sex + cross:inf + cross:dpi + sex:inf + dpi:inf + cross:dpi:inf + e were used to sequentially test for significant differences between treatment groups and interactions between treatments, where yi = is as defined above, inf is the infection status, dpi is the days post-infection, cross = direction of the reciprocal cross, sex is the sex of the individual, and e is the error. The interactions cross:inf, sex:inf, dpi:inf, tested if allelic imbalance was induced by infection and was constant across cross, sex, or dpi. Specifically, if infection induced allelic imbalance, the dpi:inf interaction would be significant. The cross:dpi:inf interaction tested if an allelic imbalance was induced by infection and constant across direction of cross. To identify which SNP were reacting to infection, we analyzed all SNP individually using the model yi = u + inf + cross + dpi + dpi:inf + cross:dpi:inf + e. All p-values were outputted for infection and all interaction terms to determine those SNP that displayed a response to infection. Furthermore the regression coefficients for infection and all interactions were examined to determine the distribution and size of effect each marker had on infection. All p-values were corrected for multiple testing using the false discovery rate (FDR) method of Benjamini and Hochberg (1995).
To statistically test the relative contribution of cis- and trans-effects to ASE, we compared the expression differences between lines (AF1 or artificial F1) to the allelic differences within the F1 hybrid (TF1 or true F1) at the same loci. If cis-acting effects are completely responsible for the differences seen in ASE, the expression rates in the TF1 and the AF1 will be equal (t-test, h0: TF1 = AF1, p < 0.05) and when plotting means from each group, all SNP will fall along the 45° diagonal through the origin (i.e., X = Y). Alternatively, if trans-acting elements completely explain the difference between animals, both alleles will be equally expressed in the TF1 and all SNP will fall along the horizontal line with Y = 0. As defined by Wittkopp et al. (2004), a combination of cis- and trans-regulation will cause alleles to fall between the horizontal and diagonal lines, which can be dissected as either a cis-trans interaction where cis-effects explain a proportion of the variance between alleles and trans-effects explain the rest, and an antagonistic relationship, where opposing effects are identified between genetic backgrounds. Finally genes with no significant difference and do not cross the intercept show no cis- or trans-effects.
Gene Set Enrichment
Gene lists were populated from a Python program that designed primers for the GoldenGate assay from the RNAseq data that showed signs of allelic imbalance. Gene names were identified using a reciprocal best-hit algorithm (MacEachern et al., 2009). The Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.7 was used to examine whether genes with evidence of ASE where enriched in specific functions (Dennis et al., 2003; Huang et al., 2009). The DAVID set of web accessible programs was used to upload 1,042 genes with RefSeq mRNA or RNA IDs. Genes that returned an up-to-date annotation were then analyzed to identify enriched Gene Ontology annotations using the Fisher’s exact test and the standard options to ensure high annotation coverage. Likewise Enriched Pathways and DAVID annotations for chromosomal positions were examined for enrichment to identify pathways or genomic regions with a high proportion of genes showing signs of ASE in response to MDV challenge.
Results
Transcriptome Sequencing
Allele-specific expression assays require a SNP to monitor the abundance of each expressed allele. Next generation sequencing was selected to provide a cost–effective, unbiased, initial genome-wide survey of the expressed SNPs, and also give an indication of ASE between resistant and susceptible lines when challenged with MDV. To this end, the spleens from six infected and uninfected F1 individuals in each reciprocal cross (6 × 7 and 7 × 6) were harvested at a single time point (4 dpi) and the transcriptomes of two replicate pools were sequenced using a next generation sequencer (Illumina GA II). Both replicates from the four treatments yielded over 14 million reads per treatment group of which ∼75% were mapped to the reference genome resulting in more than 1.7 Gb of sequence (Table 1) at an average depth of 49× per gene. BLASTing all mapped reads against the most current version of the RefSeq database identified 12,696 genes, which is >65% of the genes represented in the RefSeq database. From the mapped reads over 670,000 polymorphisms were initially identified between the transcriptomes of lines 6 and 7. However, strict filtering of the raw SNP data reduced this number to 22,655 high quality SNPs. A χ2 test revealed that 5,360 SNPs in 3,773 genes exhibited some degree of allelic imbalance (p < 0.05 not corrected for multiple testing, Table S1 in Supplementary Material), which coincides with ∼30% of all the genes represented in our sample. With this procedure up to 5% of the genes identified will be false positives, but the next step with the GoldenGate Assay was designed as a more stringent test of ASE to eliminate potential false positives. Genes with at least one SNP displaying signs of allelic imbalance were further queried for an allele-specific response to infection as determined by a 2 × 2 factorial test, which examined the effect of disease status on ASE rates. In total, 1,089 SNPs showed ASE in response to MDV infection in at least one of the reciprocal crosses, with 803 SNP showing ASE in the 7 × 6 cross and 781 in the 6 × 7 cross.
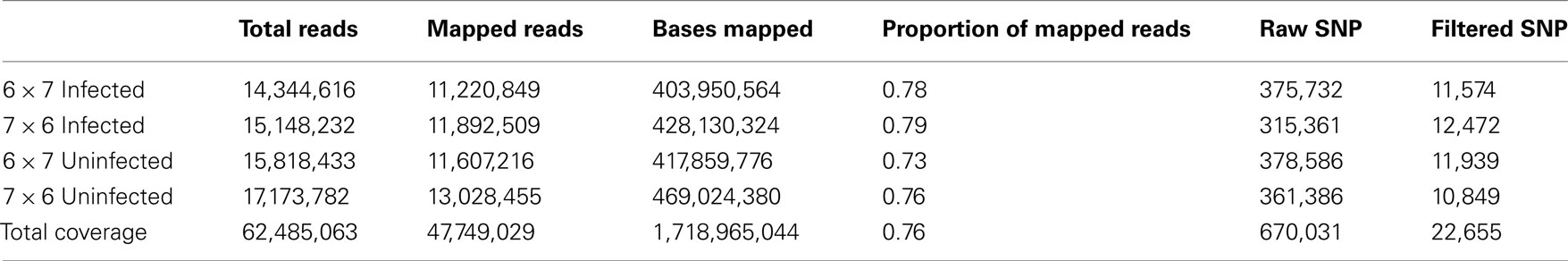
Table 1. Total reads from RNA sequence, the proportion of mapped sequences, and the number of raw and filtered SNPs from mapped reads.
Each of the 5,360 SNP were queried against the most current version of the dbSNP database (build 134) and 1,309 SNP had been described previously, but none had been previously associated with MD resistance or susceptibility. All SNP have been submitted to dbSNP and information on all SNP including the 1,536 SNP investigated with the GoldenGate assay are in Table S1 in Supplementary Material.
GoldenGate Assay
At the time of our experiment, the GoldenGate assay was a very cost–effective platform to examine ASE in a large number of individuals for up to 1,536 SNPs, therefore, 447 extra SNPs were included that were on transcripts with >1 SNP and those with strong evidence of ASE, but not necessarily a statistically significant response to infection (χ2 p > 0.05). Hence, 1,536 SNPs showing signs of ASE from our initial sequencing pilot study were selected for further investigation using the Illumina GoldenGate assay in 12 individuals from each treatment.
From the 1,536 SNPs examined with the GoldenGate platform, we identified 69 (4%) to be segregating within one of the two lines, all of which were at a very low allele frequency (<0.05). In total 1,233 SNPs reliably passed QC filtering in that they were polymorphic but not segregating within a line, and their signals were above background levels. A moderate correlation was identified between SNP within the same transcript (r2 = 0.25). However, all SNP were selected from alternate exons and this may be evidence of alternative splicing in some genes. An analysis of variance for ASE in each of the remaining 1,233 SNP that passed QC filtering was completed for sex, reciprocal cross, and infection status across all dpi and we tested the interaction of each factor with infection. The results show that infection, dpi, and cross were all factors that significantly impact rates of expression. No significant interactions were detected for sex:infection and cross:infection, therefore, there is little evidence of parental imprinting in response to MDV infection. However, a significant interaction was detected for dpi:infection and dpi:cross:infection that indicates infection by MDV significantly impacts the expression levels in these genes (Table 2).
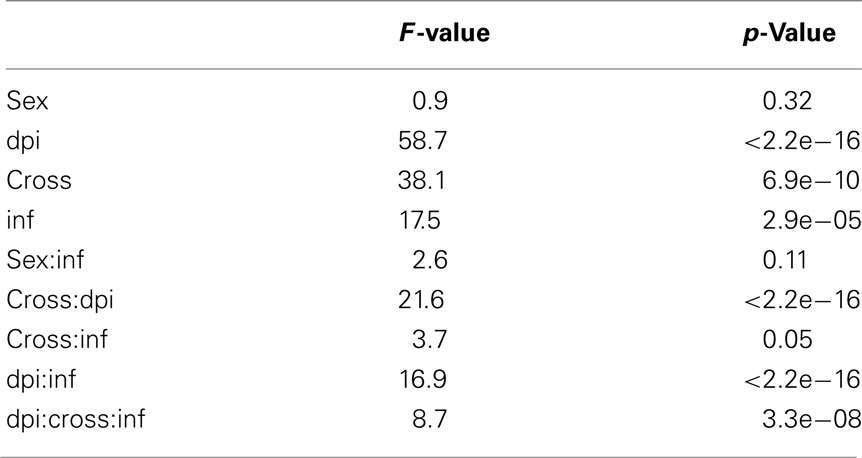
Table 2. Summary of p-values from ANOVA examining the covariates sex, cross, infection, and dpi and their interactions with infection status.
Linear models detected a number of SNP with significant ASE that were the result of cis-regulatory changes that responded to infection. We identified a number of SNP with a response to infection and those that respond to the two- and three-way interactions, dpi:infection, and cross:dpi:infection, respectively. In total, we identified 861 SNP with a cis-response to infection in at least one cross or 1 dpi. We found 581 SNP with evidence of an allele-specific response to infection, 65 of which were also significant for the interaction cross:dpi:infection and 345 were also significant for the dpi:infection interaction. These SNP responded to infection in both crosses and all dpi tested although a certain proportion changed the magnitude of response over time and to a smaller extent cross. We also identified 311 SNP with a significant interaction between dpi:infection with 50 SNP also significant for the cross:dpi:infection interaction indicating a cis-response to infection that varied over time or cross. We also distinguished 51 SNP with a significant three-way interaction between cross:dpi:infection, which highlights a relatively small but differential response to infection in each cross over time.
In Figure 1, we show the effects of infection on cis-regulation. If there were no cis-specific response to infection, we would expect a large number of genes with an effect of zero. However, we have found that the SNPs investigated here have a large distribution of effects, with a predominantly positive skew that appears to be driven by cis-regulatory changes, which may have some effect on resistance. Interaction terms had a much smaller distribution of effects and most SNP were centered on zero (results not shown).
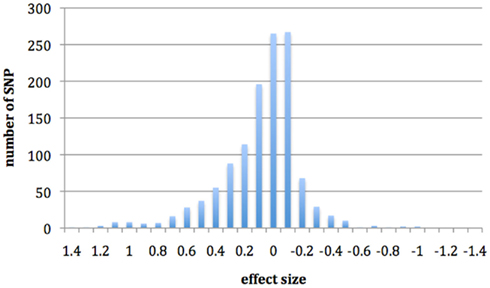
Figure 1. Histogram showing the distribution of SNP effects for the regression coefficient infection with evidence of cis-regulatory changes.
To further dissect the mechanisms controlling ASE, we compared expression between the TF1 and AF1 (t-test, h0: TF1 = AF1, p < 0.05) in infected and uninfected individuals. We plotted the relative expression of the true F1 against that of the artificial F1 for infected and uninfected individuals (Figure 2). Genes with a pure cis-effect would be expected to have a regression of 1, i.e., expression imbalance between genotypes of the parent lines (AF1) would correspond directly to allelic variation within the TF1, and lie on 45° diagonal through the origin, while those with a pure trans-effect would only show variation among the parent genotypes and no association with within locus variation and fall on the horizontal line. Alternatively SNP with a combination of cis- and trans-regulatory changes would lie somewhere in between. From the genes showing significant ASE, a large proportion showed a combination of cis- and trans-regulatory differences (337 SNPs or 27%, and 296 SNPs or 24% for uninfected and infected, respectively). SNPs displayed differences in cis-regulation that were smaller in the TF1 than in the AF1 (Figure 3). Changes to trans-regulatory regions were responsible for the remaining genes with significant ASE. A total of 253 (21%) and 239 (19%) SNPs in the uninfected and infected birds, respectively, had greater allelic expression differences in the AF1 when compared to the TF1 or the differences were in the opposite direction to the parental lines (Figure 4). Less than 1% of the genes examined (zero and seven for uninfected and infected, respectively) had a change in trans-regulation that explained all of the variance in ASE (Figure 5).
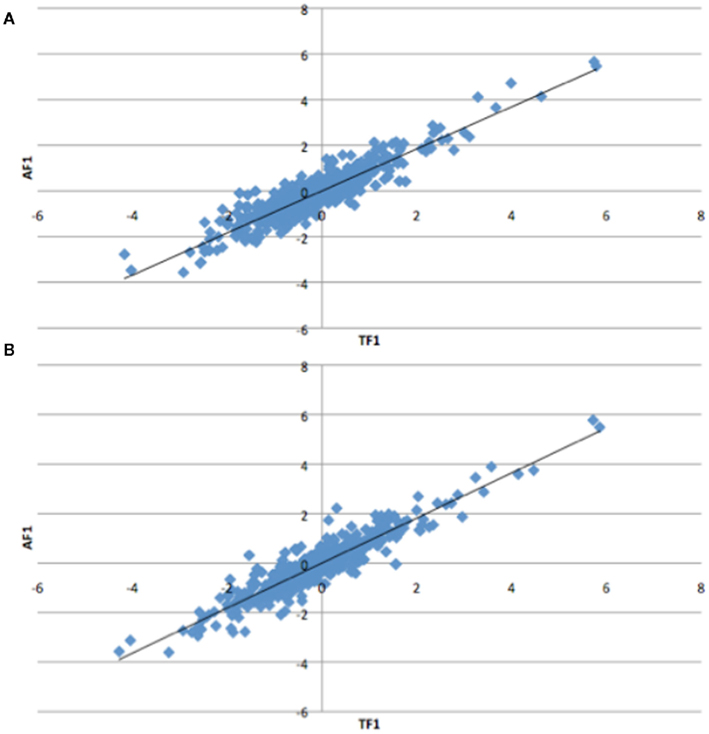
Figure 2. Plots of relative expression for each allele in the Artificial F1 (AF1) vs. True F1 (TF1) crosses. All crosses were generated from Marek’s disease resistant and susceptible lines of chicken. Expectations are pure cis-acting regulation will have a regression of one and lie along the 45° angle, while trans-acting regulation will fall off the 45° line for the (A) infected and (B) uninfected animals.
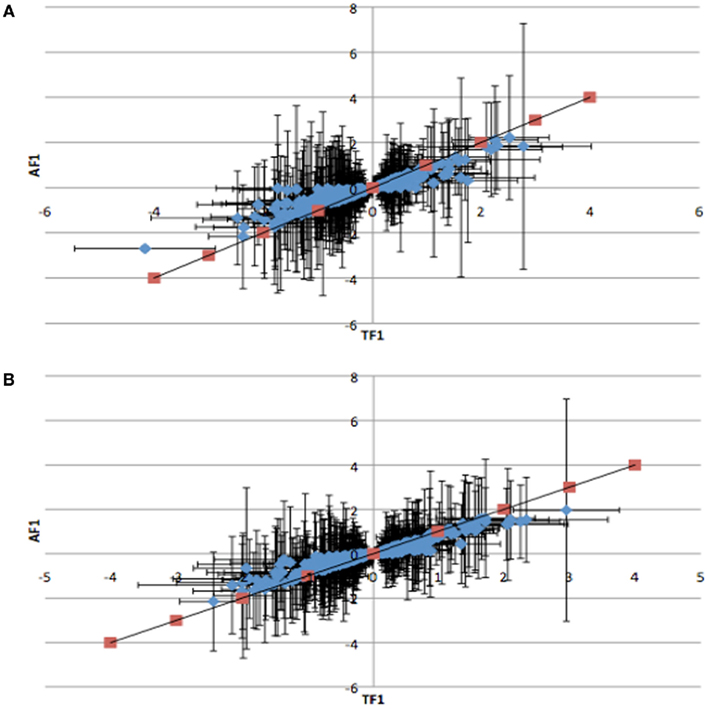
Figure 3. Plots of the relative expression with 95% confidence intervals of TF1 against AF1 cis-by-trans interactions for (A) infected and (B) uninfected animals. Red points indicate predicted response for cis-regulatory effect where TF1 = AF1. All cis-by-trans points show lower expression in AF1 than in TF1.
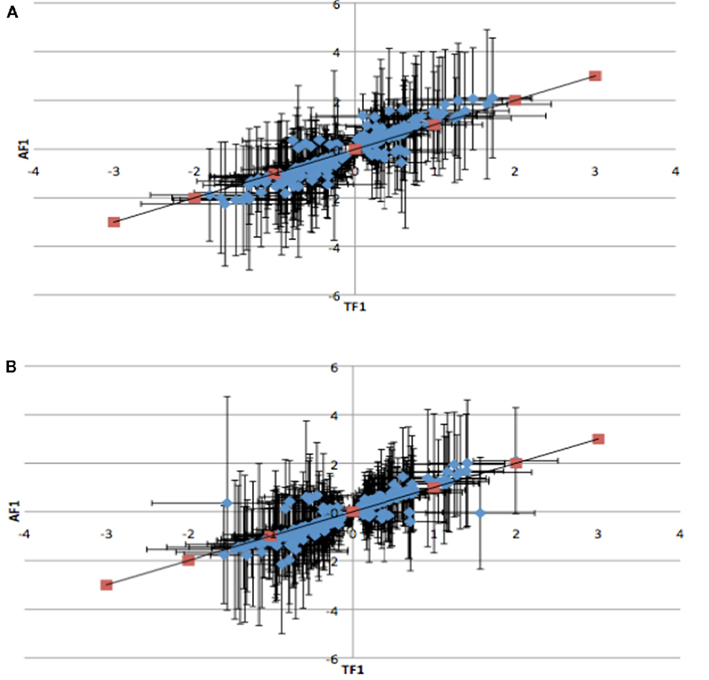
Figure 4. Plots of the relative expression with 95% confidence intervals of TF1 against AF1 with all antagonistic cis-by-trans interaction for infected (A) and uninfected (B) animals. Trans-regulatory changes cause AF1 to have expression higher or in the opposite direction of parental alleles in TF1.
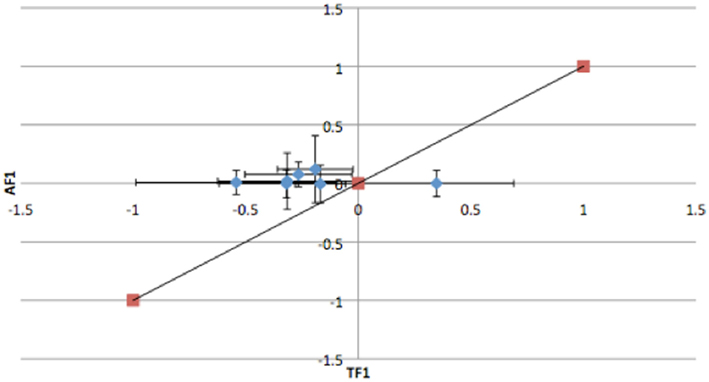
Figure 5. Plots of the relative expression with 95% confidence intervals of TF1 against AF1 with all trans-regulatory changes that explain variation in ASE in infected animals. All trans-effects lie on horizontal line and do not cross 45 line.
In an attempt to investigate the contribution of cis- and trans-regulation to ASE over time, data sets were examined within dpi and analyzed. There does not appear to be a linear trend for either the number of genes under cis- or trans-acting regulation (results not shown). However, we did witness a large number of SNPs with cis- and trans-effects early on following infection, which eventually decreases as time increases to 15 dpi for both cis- and trans-acting factors, which highlights the importance of examining a number of time periods when trying to make conclusions about the relative contribution of cis- vs. trans-regulation.
Gene Set Enrichment
The DAVID (Dennis et al., 2003; Huang et al., 2009) was used to examine all genes that show evidence of ASE to determine whether or not there was evidence of enrichment for particular biological processes, pathways, and chromosomal positions. We examined the main biological processes that were enriched and found genes with evidence of ASE that were enriched in the following processes: protein localization, protein transport, anti-apoptosis, adaptive immune response, leukocyte-mediated immunity, adaptive immune response, cytokine production, leukocyte-mediated immunity, and adaptive immune response. The pathways enriched were natural killer cell mediated cytotoxicity, apoptosis, and DNA replication (Table 3). Next we asked whether there were any chromosomes or genomic regions that may be enriched for genes that responds to MDV infection and found that chromosomes 1 and 17 were enriched for genes showing evidence of ASE and a near significant result for chromosome 3 (Table 3).
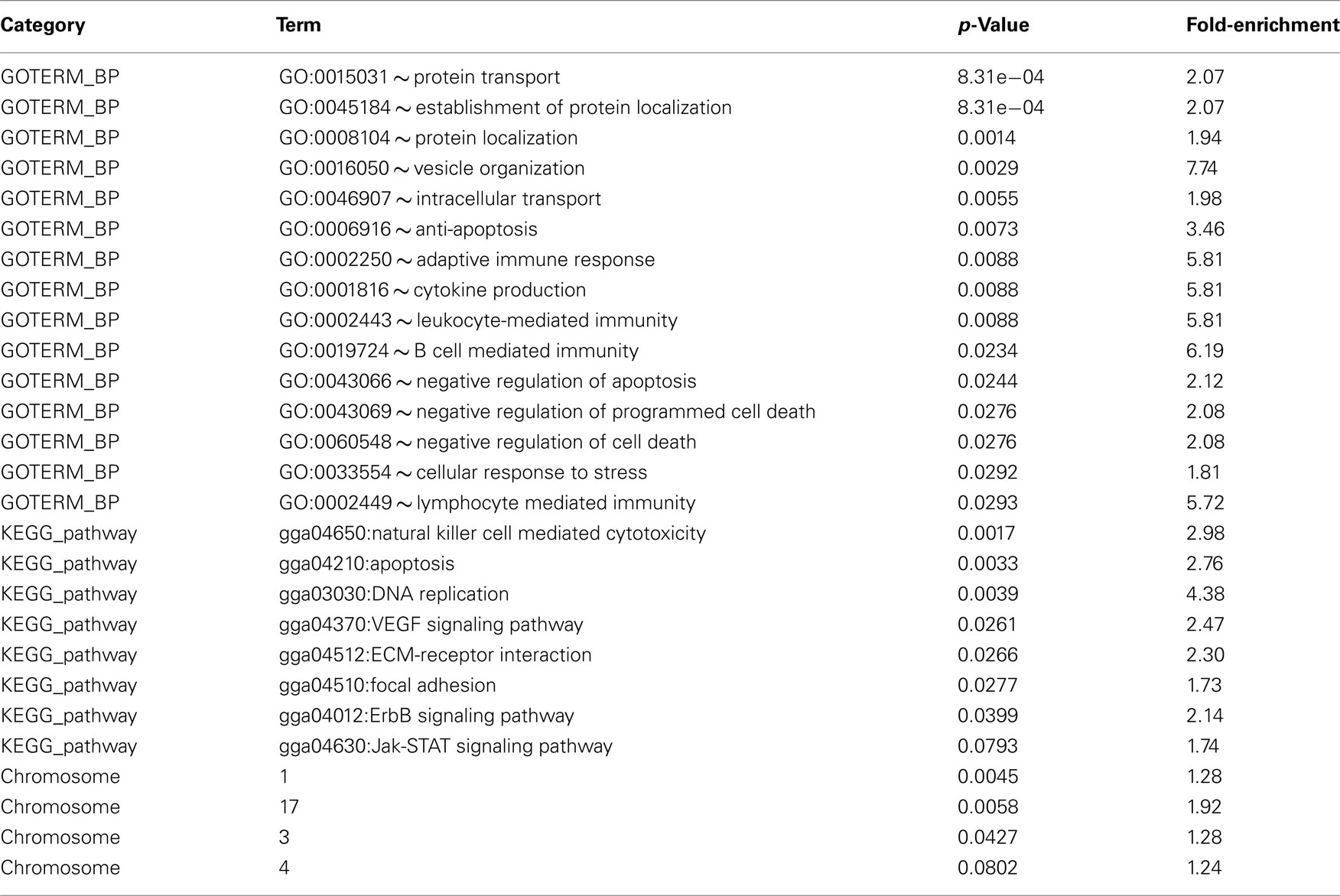
Table 3. Biological processes (BP), pathways, and chromosomes that are enriched with genes showing signs of an allele-specific response to infection.
Discussion
The majority of heritable traits that are of interest to scientists investigating model organisms, human diseases, and animal breeding are complex in nature and controlled by multiple loci. Most studies focusing on uncovering the genetic variation underlying these traits have only uncovered a small fraction of this variation (Goddard and Hayes, 2009; Manolio et al., 2009). The main hindrance in identifying more loci has been the use of stringent type-I error rates to limit false positives, the lack of statistical power to detect loci of small effect, and the lack of LD amongst common SNP markers that are typically used for association analysis, and potentially rare causative mutations (Goddard and Hayes, 2009). Here we present a method to identify potentially a large number of the genes underlying a complex trait that are controlled by transcriptional regulation. Increasing evidence points to transcriptional regulation as a major factor underlying the phenoytypes of complex traits and, therefore, a number of efforts have recently tried to uncover cis-regulatory polymorphisms and their contributions to complex phenotypes (Fay and Wittkopp, 2008; Montgomery et al., 2010).
Transcriptome Re-Sequencing
A transcriptome-wide scan of replicate pools for each reciprocal mating comprised of six infected and uninfected F1 individuals each created by mating resistant and susceptible inbred lines of chicken uncovered 22,655 high-confidence SNPs from a potential pool of over 670,000 polymorphisms. We developed an automated approach to remove false positive SNP calls in an effort to accurately track parental alleles and assess cis-regulatory variation within an individual. The 30-fold reduction in SNPs we uncovered highlights biases, such as sequencing errors and misalignments that can be introduced into RNA sequencing and potentially inflate expression differences with next generation sequencing technologies. Thus, the importance of screening transcriptome re-sequencing data is paramount to reduce false positives when examining ASE. Our automated approach based on sequence depth, sequence quality scores, and allele frequency revealed that 5,360 SNPs in 3,773 genes exhibited statistically significant allelic imbalance. Despite the high level of inbreeding, we have identified a number of polymorphisms that are still segregating within one or both lines. This finding was not surprising as inbred animals are rarely 100% inbred at all loci. Approximately 4% of the SNP we examined were segregating. The exact cause of SNPs that were found to be segregating within lines may be the result of new mutations, polymorphisms that remained segregating from the founder population, accidental breeding contamination, or potentially a faulty assay. Whatever their origin, the inclusion of sequence from the parent of origin would have helped identify segregating SNP and reduced the number of false positive GoldenGate assays developed for this study. While this number of positive SNPs found by RNAseq was high, it was less than the number we expected based on the RNAseq analysis. Of the 1,233 SNPs that passed quality control, based on the Type 1 error rate set, we expected 5%, or approximately 62 to be false positives, and 1,170 true positives. These results show that our RNAseq analysis was not as stringent a test of ASE as expected, perhaps due to the relatively low coverage. Nevertheless, the method was effective in finding a high proportion (>50%) of true ASE SNPs. As the price of sequencing continues to decrease, the level of heterozygosity in each of the resistant and susceptible lines would be interesting to examine in both the coding and non-coding regions of the genome. Also, lower sequencing costs help facilitate biological replicates as recommended by Auer and Doerge (2010), which would have provided higher confidence in identifying SNPs exhibiting ASE.
GoldenGate Assay
The Illumina GoldenGate assay is not free from error, but is generally thought to give data that is of high quality and reproducibility, and is currently a more economical option to examine large numbers of replicates for a targeted set of SNPs than next generation sequencing. Originally designed for SNP genotyping, it has been recently adapted to quantify allele expression (Serre et al., 2008). Thus, the GoldenGate platform was chosen as a cost–effective method to validate and extend the results from our RNAseq analysis. Our results demonstrate there is extensive ASE and that transcription is significantly impacted by cis- and a combination of cis- and trans-acting elements when animals are infected with MDV. We found a significant impact of each SNP on the degree of allelic imbalance, which would be expected if a large number of these genes and the extent to which they are regulated explain some of the genetic resistance to MD. We identified no significant effect for sex or any interaction with sex and infection suggesting there are no maternal effects impacting ASE. A significant cross effect was detected, thus, parental imprinting may play a role in ASE levels. However, no significant interaction between cross and infection suggests imprinting play little role in the differences observed for MDV resistance. The large number of cis-regulatory changes with an apparent response to infection was examined. A positive skew was identified for cis-regulatory changes that responded to infection suggesting that cis-variation has lead to over expression of the alleles in response to MDV infection.
Comparisons between the average expression in both reciprocal crosses and an artificial RNA pool as outlined in Wittkopp et al. (2004) show that trans-acting elements play a minor role in regulating expression changes in infected and uninfected individuals. The small number of loci with purely trans-regulatory changes we identified confirmed previous studies that suggest cis-acting elements are dominant (Brem et al., 2002; Schadt et al., 2003; Osada et al., 2006; Genissel et al., 2008; Pastinen, 2010). However, we did confirm that a large proportion of cis-regulatory changes show an overlapping change in trans-regulation (Wittkopp et al., 2004). In particular, we were able to dissect cis-by-trans interactions into those where the expression of the F1 hybrid was lower than the expression in the parental lines, and those where the expression in the F1 was greater or even in a different direction to the parent or origin, which may highlight some difference in genetic backgrounds for cis-by-trans-regulation. The observed antagonist interactions are consistent with our previous results that demonstrated two-way epistatic interactions between genetic markers that account for MDV viremia levels (Cheng et al., 2007). Overall the strong bias of a response of cis-regulation to infection may highlight the ability of cis-variation to evolve by drift and directional selection to a greater extent than trans, which could be under more selective constraint. However, this should be confirmed in additional outbred populations.
In order to extract further biological insight, genes exhibiting ASE were analyzed for biological enrichment. We identified a number of pathways and processes that are involved in apoptosis and immunity, which may play a role in susceptibility or resistance to MDV infection. We have reported a number of generic pathways that are involved with protein localization that may have some role in MDV resistance or susceptibility. However, the most interesting pathways that show moderate to highly significant p-values (p < 0.01) were involved in leukocyte-mediated immunity, anti-apoptosis, cytokine production, and adaptive immune response. These pathways appear to confirm previous findings that show different responses to infection for cytokine production and peripheral leukocyte expression and cytotoxicity between resistant and susceptible chicken lines (Liu et al., 2001a; Garcia-Camacho et al., 2003; Sarson et al., 2008). We also report evidence of localization of some of these genes on certain chromosomes in the genome, implying joint regulation of some biological functions, which has also been found previously in other studies examining cis- and trans-acting regulation (Zhang and Borevitz, 2009; Emerson et al., 2010); chromosome 16, which contains the MHC, did not come up as being enriched as the two lines examined are fixed for the same B haplotype. The pathways and genomic regions that we identified to be enriched for genes with evidence of an allele-specific response to MDV infection may be useful to investigate further when a genome-wide study is not necessary or affordable. With further completion and annotation of the chicken genome, a more complete understanding of the pathways and regions of the chicken genome that is important for resistance and susceptibility may become clear.
Conclusion
The results presented here show ASE is an effective approach to identify genes with cis-regulatory elements that respond to MDV infection, therefore, this study should provide a strong start to identifying a large proportion of the genes with an allele-specific response to infection, which may underlie at least some of the genetic resistance to MD resistance, a complex trait. It is hoped that many of the genes sampled in this study contribute to genetic resistance to MD, which can be directly monitored in resource populations using the same SNPs. Future work is planned to integrate results from genomic DNA sequences of the two parental lines to identify possible causative mutations. The described method, although demonstrated in inbred chicken lines, is applicable to all traits in any diploid species, and should prove to be a facile method to identify those genes whose alleles can be traced, and where differences in expression are responsible for controlling at least some proportion of the variance in a complex trait. Finally, our results suggest that ASE screens are simple and powerful approaches to identify genetic elements and specific alleles for genetic resistance to MD and other complex traits, especially those that involve two-state situations in disease challenge experiments where parental alleles can be tracked by SNPs or other polymorphisms. The use of a segregating marker can also be applied to determine whether the expression differences are linked to measurable phenotypic changes (e.g., disease incidence) and, if so, it might be used in genomic selection programs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by National Research Initiative Competitive Grant no. 2007-04201 from the USDA National Institute of Food and Agriculture. We also appreciate Cobb-Vantress, Inc., for funding and material support, and Laurie Molitor for excellent technical and lab assistance. Thank you to Ruth-Marie Putnam Smith whose unwavering support and confidence contributed to the completion of this study. You are dearly missed.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Livestock_Genomics/10.3389/fgene.2011.00113/abstract
Table S1. Information on the SNP identified with allelic imbalance from RNAseq including position (galGal3), flanking sequence, p-value for ASE, gene description, rsID and those selected for GoldenGate assay. *RSID are for pre-existing SNP, SSID coincide with new submissions.
References
Auer, P. L., and Doerge, R. W. (2010). Statistical design and analysis of RNA sequencing data. Genetics 185, 405–416.
Bacon, L. D., Hunt, H. D., and Cheng, H. H. (2000). A review of the development of chicken lines to resolve genes determining resistance to diseases. Poult. Sci. 79, 1082–1093.
Bacon, L. D., and Witter, R. L. (1994). B haplotype influence on the relative efficacy of Marek’s disease vaccines in commercial chickens. Poult. Sci. 73, 481–487.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Bjornsson, H. T., Albert, T. J., Ladd-Acosta, C. M., Green, R. D., Rongione, M. A., Middle, C. M., Irizarry, R. A., Broman, K. W., and Feinberg, A. P. (2008). SNP-specific array-based allele-specific expression analysis. Genome Res. 18, 771–779.
Bradley, J., Main, B. J., Bickel, R. D, McIntyre, L. M., Graze, R. M., Calabrese, P. P., and Nuzhdin, S. V. (2009). Allele-specific expression assays using Solexa. BMC Genomics 10, 422. doi: 10.1186/1471-2164-10-422
Brem, R. B., Yvert, G., Clinton, R., and Kruglyak, L. (2002). Genetic dissection of transcriptional regulation in budding yeast. Science 296, 752–755.
Cheng, H., Niikura, M., Kim, T., Mao, W., MacLea, K. S., Hunt, H., Dodgson, J., Burnside, J., Morgan, R., Ouyang, M., Lamont, S., Dekkers, J., Fulton, J., Soller, M., and Muir, W. (2008). Using integrative genomics to elucidate genetic resistance to Marek’s disease in chickens. Dev. Biol. 132, 365–372.
Cheng, H. H., Zhang, Y., and Muir, W. M. (2007). Evidence for widespread epistatic interactions influencing Marek’s disease virus viremia levels in chicken. Cytogenet. Genome Res. 117, 313–318.
Dennis, G. Jr., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C., and Lempicki, R. A. (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, 3.
Emerson, J. J., Hsieh, L.-C., Sung, H-.M., Wang, T.-Y., Huang, C.-J., Lu, H. H.-S., Lu, M.-Y. J., Wu, S.-H., and Li, W.-H. (2010). Natural selection on cis and trans regulation in yeasts. Genome Res. 20, 826–836.
Fay, J. C., and Wittkopp, P. J. (2008). Evaluating the role of natural selection in the evolution of gene regulation. Heredity 100, 191–199.
Garcia-Camacho, L., Schat, K. A., Brooks, R. Jr., and Bonous, D. I. (2003). Early cell-mediated immune responses to Marek’s disease virus in two chicken lines with defined major histocompatibility complex antigens. Vet. Immunol. Immunopathol. 95, 145–153.
Genissel, A., McIntyre, L. M., Wayne, M. L., and Nuzhdin, S. V. (2008). Cis and trans regulatory effects contribute to natural variation in transcriptome of Drosophila melanogaster. Mol. Biol. Evol. 25, 101–110.
Goddard, M. E., and Hayes, B. J. (2009). Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10, 381–391.
Groot, A. J. C., and Albers, G. A. A. (1992). “The effect of MHC on resistance to Marek’s disease by White Leghorn crosses,” in Proceedings of the World’s Poultry Congress, Amsterdam, 185–188.
Guo, M., Rupe, M. A., Zinselmeier, C., Habben, J., Bowen, B. A., and Smith, O. S. (2004). Allelic variation of gene expression in maize hybrids. Plant Cell 16, 1707–1716.
Heap, G. A., Yang, J. H. M., Downe, K., Healy, B. C., Hunt, K. A., Bockett, N., Franke, L., Dubois, P. C., Mein, C. A., Dobson, R. J., Albert, T. J., Rodesch, M. J., Clayton, D. G., Todd, J. A., van Heel, D. A., and Pagnol, V. (2010). Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing. Hum. Mol. Genet. 19, 122–134.
Heifetz, E. M., Fulton, J. E., O’Sullivan, N. P, Arthur, J. A., Cheng, H., Wang, J., Soller, M., and Dekkers, J. C. M. (2009). Mapping QTL affecting resistance to Marek’s disease in an F6 advanced intercross population of commercial layer chickens. BMC Genomics 10, 20. doi:10.1186/1471-2164-10-20
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57.
King, M. C., and Wilson, A. C. (1975). Evolution at two levels in humans and chimpanzees. Science 188, 107–116.
Li, H., Ruan, J., and Durbin, R. (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858.
Liu, H.-C., Cheng, H. H., Softer, L., and Burnside, J. (2001a). A strategy to identify positional candidate genes conferring Marek’s disease resistant by integrating DNA microarrays and genetic mapping. Anim. Genet. 32, 351–359.
Liu, H.-C., Kung, H.-J., Fulton, J. E., Morgan, R. W., and Cheng, H. H. (2001b). Growth hormone interacts with the Marek’s disease virus SORF2 protein and is associated with disease resistance in chicken. Proc. Natl. Acad. Sci. U.S.A. 98, 9203–9208.
Liu, H.-C., Niikura, M., Fulton, J., and Cheng, H. H. (2003). Identification of chicken stem lymphocyte antigen 6 complex, locus E (LY6E, alias SCA2) as a putative Marek’s disease resistance gene via a virus-host protein interaction screen. Cytogenet. Genome Res. 102, 304–308.
MacEachern, S., McEwan, J., and Goddard, M. (2009). Phylogenetic reconstruction and the identification of ancient polymorphism in the Bovini tribe (Bovidae, Bovinae). BMC Genomics 10, 177. doi:10.1186/1471-2164-10-177
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., McCarthy, M. I., Ramos, E. M., Cardon, L. R., Chakravarti, A., Cho, J. H., Guttmacher, A. E., Kong, A., Kruglyak, L., Mardis, E., Rotimi, C. N., Slatkin, M., Valle, D., Whittemore, A. S., Boehnke, M., Clark, A. G., Eichler, E. E., Gibson, G., Haines, J. L., Mackay, T. F. C., McCarroll, S. A., and Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753.
McElroy, J. P., Dekkers, J. C. M, Fulton, J. E., O’Sullivan, N. P., Soller, M., Lipkin, E., Zhang, W., Koehler, K. J., Lamont, S. J., and Cheng, H. H. (2005). Microsatellite markers associated with resistance to Marek’s disease in commercial layer chickens. Poult. Sci. 84, 1678–1688.
Montgomery, S. B, Sammeth, M., Gutierrez-Arcelus, M., Lach, R. P., Ingle, C., Nisbett, J., Guigo, R., and Dermitzakis, E. T. (2010). Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464, 773–777.
Niikura, M., Kim, T., Hunt, H. D., Burnside, J., Morgan, R. W., Dodgson, J. B., and Cheng, H. H. (2007). Marek’s disease virus up-regulates major histocompatibility complex class II cell surface expression in infected cells. Virology 359, 212–219.
Osada, M., Kohn, M. H., and Wu, C.-I. (2006). Genomic inference of cis- regulatory nucleotide polymorphism underlying gene expression differences between Drosophila melanogaster mating races. Mol. Biol. Evol. 23, 1585–1591.
Pastinen, T. (2010). Genome-wide allele-specific analysis: insights into regulatory variation. Nat. Rev. Genet. 11, 533–538.
Praslickova, D., Sharif, S., Sarson, A., Abdul-Careem, M. F., Zadworny, D., Kulenkamp, A., Ansah, G., and Kuhnlein, U. (2008). Association of a marker in the vitamin D receptor gene with Marek’s disease resistance in poultry. Poult. Sci. 87, 1112–1119.
Sarson, A. J., Parvizi, P., Lepp, D., Quinton, M., and Sharif, S. (2008). Transcriptional analysis of host responses to Marek’s disease virus infection in genetically resistant and susceptible chickens. Anim. Genet. 32, 232–240.
Schadt, E. E., Monks, S. A., Drake, T. A., Lusis, A. J., Che, N., Colinayo, V., Ruff, T. G., Milligan, S. B., Lamb, J. R., Cavet, G., Linsley, P. S., Mao, M., Stoughton, R. B., and Friend, S. H. (2003). Genetics of gene expression surveyed in maize, mouse and man. Nature 422, 269–270.
Serre, D., Gurd, S., Ge, B., Sladek, R., Sinnett, D., Harmsen, E., Bibikova, M., Chudin, E., Barker, D. L., Dickinson, T., Fan, J. B., and Hudson, T. J. (2008). Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoS Genet. 4, e1000006. doi:10.1371/journal.pgen.1000006
Vallejo, R. L., Bacon, L. D., Liu, H.-C., Witter, R. L., Groenen, M. A. M., Hillel, J., and Cheng, H. H. (1998). Genetic mapping of quantitative trait loci affecting susceptibility to Marek’s disease virus induced tumors in F2 intercross chickens. Genetics 148, 349–360.
Witter, R. L. (1997). Increased virulence of Marek’s disease field isolates. Avian Dis. 41, 149–163.
Wittkopp, P. J., Haerum, B. L., and Clark, A. G. (2004). Evolutionary changes in cis and trans gene regulation. Nature 430, 85–88.
Wray, G. A. (2007). The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8, 206–216.
Yonash, N., Bacon, L. D., Witter, R. L., and Cheng, H. H. (1999). Higher resolution mapping and identification of quantitative trait loci (QTL) affecting susceptibility to Marek’s disease. Anim. Genet. 30, 126–135.
Keywords: genetic resistance, allelic imbalance, Marek’s disease, RNA sequencing
Citation: MacEachern S, Muir WM, Crosby SD and Cheng HH (2011) Genome-wide identification and quantification of cis- and trans-regulated genes responding to Marek’s disease virus infection via analysis of allele-specific expression. Front. Gene. 2:113. doi: 10.3389/fgene.2011.00113
Received: 12 October 2011;
Accepted: 29 December 2011;
Published online: 13 January 2012.
Edited by:
Peng Xu, Chinese Academy of Fishery Sciences, ChinaReviewed by:
Yniv Palti, United States Department of Agriculture, USAMichael Bailey, Texas A&M University, USA
Eric Peatman, Auburn University, USA
Copyright: © 2012 MacEachern, Muir, Crosby and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sean MacEachern, Cobb-Vantress Inc., P.O. Box 1030, 4703 US HWY 412 East, Siloam Springs, AR 72761, USA. e-mail:c2Vhbi5tYWNlYWNoZXJuQGNvYmItdmFudHJlc3MuY29t