


- 1 Department of Pathology and Laboratory Medicine, Children’s Hospital Los Angeles, Los Angeles, CA, USA
- 2 Center for Personalized Medicine, Children’s Hospital Los Angeles, Los Angeles, CA, USA
- 3 Department of Pathology, Keck School of Medicine of the University of Southern California, Los Angeles, CA, USA
Long non-coding RNAs (ncRNAs) have been shown to regulate important biological processes that support normal cellular functions. Aberrant regulation of these essential functions can promote tumor development. In this review, we underscore the importance of the regulatory role played by this distinct class of ncRNAs in cancer-associated pathways that govern mechanisms such as cell growth, invasion, and metastasis. We also highlight the possibility of using these unique RNAs as diagnostic and prognostic biomarkers in malignancies.
Introduction
Common knowledge until recently would suggest that only about 1% of the genome produces biologically meaningful RNA transcripts, specifically those that encode for proteins (Guttman et al., 2009). In the past decade, however, numerous papers have appeared that clearly document widespread transcription across most of the genome. However, the biological relevance of this transcription has been a matter of much debate, as witnessed by numerous original peer-reviewed articles, reviews, letters to the editor, and rebuttals. In particular, van Bakel and Hughes believe this pervasive transcription is simply transcriptional “noise,” while Mattick and Kapranov, among others, are of the strong belief these transcripts are functionally relevant (Kapranov, 2009; Mercer et al., 2009; van Bakel and Hughes, 2009). An in-depth review of published data in combination with unpublished observations from our work in this field has led to support the latter interpretation, based on a strong association of patterns of non-coding RNA (ncRNA) transcription with diagnosis and prognosis in cancer. We believe these data can only be interpreted as non-random, biologically meaningful patterns that point to a general functional role for ncRNA over much of the genome. Further, the character and pattern of transcription is itself of considerable interest, as there is no parallel among annotated, protein-encoding genes for many of the transcripts we have identified in several different types of cancer.
The findings reported here are consonant with several historical observations. First, as early as 1980, in a landmark publication, “The RNA World,” Watson, Crick, and others present compelling evidence that RNA was the first nucleic acid associated with life on this planet (Atkins et al., 2011). Several lines of evidence converge on the idea that DNA, and subsequently protein-encoding RNA, only appeared eons later. Prior to that time, RNA subserved the functions necessary for life currently associated with DNA (organismal memory) and protein (enzymatic cleavage, regulation of transcription, and many others). This “RNA First” hypothesis is widely accepted now, but the implications for genome-wide pervasive RNA transcription have only recently garnered attention. Most students today are still taught the central dogma of molecular biology, namely that DNA encodes RNA transcripts that are transcribed from the genome and translated into protein after cleavage and migration from the nucleus to the cytoplasm, where the ribosome utilizes the messenger RNA (mRNA) strand as a specific template to produce protein. With the vast amounts and variety of ncRNA transcripts found in every cell at all stages of development, this central dogma does not fully capture the role of RNA as a regulatory molecule independent of protein (Pauli et al., 2011; Suh and Blelloch, 2011).
This review provides evidence that these non-coding transcripts as a group are not only functional, but may well be some of the most basic and ancient functional RNAs of all. Certainly they are strikingly different than mRNA, in both structure and function, yet certain features are shared with mRNA, including, in some cases, “exons” and “introns,” poly-adenylation, and alternate splice variants. Conversely, unlike coding genes, ncRNAs occur in poorly conserved regions of the genome, in stark contrast to the highly conserved regions associated with coding genes.
Beyond these easily documented features, relatively little is known about the general structure, function, and transcriptional control of ncRNAs, and even less about their potential functions as a group or singly. Unlike coding genes, there is no easily documented protein product linked to the RNA sequence (Clamp et al., 2007). However, numerous individual examples have been documented, with very different structure and function. On the one hand, H19, a long recognized ncRNA, shows significant sequence conservation and is known to control IGF2 on the opposite paternal chromosome by epigenetic control mechanisms (Feil et al., 1994; Juan et al., 2000). On the other hand, XIST shows almost no sequence conservation between species yet consistently silences one X chromosome in females (Hendrich et al., 1993; Panning et al., 1997). In yet another well documented example, a ncRNA transcribed from the HOX locus, HOTAIR, has been shown to bind to the polycomb repressor complex 2 (PRC2) and affect expression of over 300 coding gene targets as part of a general reprogramming of breast cancer cells from a locally aggressive epithelial phenotype to an invasive and metastatic “mesenchymal” phenotype (Gupta et al., 2010). Clearly there are specific examples of functional ncRNA. There remains the larger question though of whether ncRNAs are predominantly functional, and if so, how this might be determined. In the following examples, we present evidence that ncRNAs are in fact strongly associated with cancer diagnosis and prognosis, functions that can hardly be ascribed to genome-wide random transcription.
Classes of ncRNA Transcription
Non-coding RNAs are an integral part of the mammalian transcriptome. Once described as “dark matter,” these underestimated molecules can play important functional and structural roles in the cell (Kapranov et al., 2010; Qureshi and Mehler, 2011). Based on size, these RNA can be grouped into three major classes: small ncRNAs, which include microRNA (miRNA), PIWI-interacting RNA (piRNA), endogenous short interfering RNA (siRNA), and other non-coding transcripts of less than 200 nucleotides (nt); long ncRNAs (lncRNA) that are greater than 200 nt and arise from intergenic regions or are organized around protein-coding regions; and very long ncRNA (vlncRNA) that can stretch through hundreds of kilobases, often across intergenic regions. While miRNAs post-transcriptionally regulate mRNA through the RNA-induced silencing complex, piRNA and siRNA are implicated in maintaining genomic integrity by silencing of transposable elements in cells. lncRNAs are involved in various levels of genomic regulation and related fundamental epigenetic processes: genomic imprinting, dosage compensation, and chromatin modifications. These can assist in subcellular transport, recruitment of transcription factors, and RNA processing and editing by forming ribonucleoprotein complexes.
Characteristics of ncRNA
Poly-Adenylated RNA versus Total RNA Transcription
Non-coding genomic DNA, some of which is genetically functional, has increased proportionally with genomic size and complexity (Taft et al., 2007). The human genome has more “dark matter” when compared to that of Drosophila. RNA polymerase II transcribes both protein-coding and non-coding transcripts. While transcription terminates at a poly-adenylation site for most protein-coding genes, there is substantial evidence that a fraction of ncRNAs do not necessarily end with a poly-A signal. This alternate termination of transcripts is sometimes associated with RNA-binding proteins Nrd1, Nab3, and Sen1 as is seen with non-poly-adenylated end of small nucleolar RNAs (snoRNAs) in S. cerevisiae (Creamer et al., 2011). Antisense asOct4-pg5 or the brain-associated BC200 are examples of functional lncRNA that are not poly-adenylated (Chen et al., 1997; Hawkins and Morris, 2010). ncRNA comprise approximately 30% of the total poly-A fraction, while they account for approximately 50–60% of total RNA with or devoid of ribosomal RNA, thus suggesting that a significant amount of these ncRNAs are non-poly-adenylated (Kapranov et al., 2010). The lack of poly-A tails has caused these transcripts to be underrepresented in cDNA libraries, SAGE, differential display, and microarrays which typically employ a 3′ poly-A labeling method.
Conserved versus not Conserved
While protein-coding genes are under high constraint, this is not the case with all ncRNAs. Recent studies have shown the emerging importance of lncRNAs as regulators of essential cellular functions that involve a great number of protein interactions (Guttman et al., 2009). Increased system complexity that enables highly skilled functions increases evolutionary pressure on regulators of this dynamic signaling network (Mattick, 2003). These RNAs are predicted to undergo more rapid evolution than pre-existing proteins or the de novo evolution of a unique set of signaling molecules (Ponjavic et al., 2007). Guttman et al. (2010) compared orthologous sequences of lncRNAs among 29 mammals and showed that their conservation is far greater than random genomic sequences or introns. On the other hand, Babak et al. (2005) found poor conservation between intergenic genomic transcripts and proposed that they may thus be non-functional. However, one of the non-coding elements of the genome, referred to as ultra-conserved elements (UCEs), is highly conserved. These regions span at least 200 base pairs in length and maintain 100% identity with no insertions or deletions between human, mouse, and rat genomes (Bejerano et al., 2004). Other than the exonic components, there are about 38.7% UCEs that are intergenic while another 42.6% are intronic (Mestdagh et al., 2010). The average distances observed among them (approximately 10 Mb) suggest that they are unlikely to function as exons of a gene. Some of these non-coding UCEs are transcribed (T-UCEs) and maintain evolutionary constraints.
Functional versus Non-Functional
Less than 1% of lncRNAs have been associated with a function. Their cell- and tissue-specific expression that changes in response to external factors such as stress and other environmental signals implies that their presence is dependent on the need of the cell. Many of these lncRNAs have binding sites for transcription factors Sp1, c-Myc, p53, and Creb, thus suggesting different levels of regulation (Cawley et al., 2004; Euskirchen et al., 2004). Their involvement varies from transcriptional to post-transcriptional regulation to translational control. There is evidence that some of these are essential for development (Rosenbluh et al., 2011; Han et al., 2012). For example, Mirg, a maternal ncRNA from the Dlk–Dio3 imprinted cluster, is expressed in different tissues at different time during murine embryonic development (Han et al., 2012).
General Patterns of ncRNA Expression in Normal Tissues and Cancer
The concept of a functional genome is being rewritten with the discovery of ncRNA. The abundance of these transcripts in cancer suggests their role in tumor pathogenesis. ncRNAs are abundant during embryogenesis (van Leeuwen and Mikkers, 2010; Pauli et al., 2011) and reactivation or non-suppression of some of these fetal lncRNAs may critically regulate pluripotency and uninhibited cellular growth, thus giving rise to adult or developmental cancers. For example, the H19 lncRNA is expressed during vertebrate embryogenesis but is downregulated after birth in most tissues except for cartilage and skeletal muscle (Lustig et al., 1994). However, loss of imprinting and overexpression of H19 in many cancers such as those of esophagus, liver, colon, and bladder cause it to function as an oncogene and promote tumor development (Hibi et al., 1996; Barsyte-Lovejoy et al., 2006; Matouk et al., 2007). Similarly, normal adult tissues express lncRNAs at various levels with lymph nodes and gall bladder reportedly having the most distinct lncRNAs (Gibb et al., 2011). Comparisons between normal and cancerous tissues revealed differential expression of at least 200 lncRNAs. The chromosome distribution of lncRNAs did not correlate with either protein-coding genes or miRNAs. Kapranov et al. (2010) also showed that in Ewing sarcoma, a childhood cancer, 43–63% of all non-ribosomal, non-mitochondrial RNAs by mass were non-exonic RNAs, and 24–37% of these were detected in intergenic regions. This study also suggested the presence of a vlncRNA of approximately 650 kb on chromosome 7 that was exclusively present in Ewing sarcoma and not in the leukemia cell line K562, normal brain, or liver. Similarly, another 300 kb intergenic region on chromosome 21 in the K562 cell line was not detected in Ewing sarcoma, suggesting that certain ncRNAs may be present in specific cancers.
Role of ncRNAs in Tumor Pathogenesis: Oncogenes or Tumor Suppressors
ncRNAs have been detected in cancer by various techniques including expression microarrays, tiling arrays, next generation sequencing, and methylation analysis (Cheung et al., 2010; Gupta et al., 2010; Sang et al., 2010; Trapnell et al., 2010). These approaches have led to the identification of several lncRNAs whose expression and epigenetic state are significantly associated with cancerous tissues.
Like protein-coding genes, ncRNAs may function as tumor oncogenes or tumor suppressors. Some T-UCEs are frequently located at fragile sites and cancer-associated genomic regions (CAGRs) such as minimal regions of amplification and of loss of heterozygosity, while others are known to act as oncogenes in cancer cells (Rossi et al., 2008). Functional analysis involving siRNAs identified uc.73A as a promoter of cell survival by evading cellular apoptosis in colorectal cancer (Calin et al., 2007). Enrichment analyses confirmed that UCEs are contained in genes involved in RNA processing and RNA binding (Licastro et al., 2010). They bear resemblance to enhancer-like sequences and are involved in transcription.
Protein-coding genes are known to be associated with antisense transcripts, and perturbation of these can alter protein expression that promotes cancer development (He et al., 2008). Antisense transcripts ANRIL and p21/CDKN1A-associated transcript repress tumor suppressor loci and promote cancer (Morris et al., 2008). Aberrant gene expression causes changes in chromatin structure leading to genomic instability that can give rise to uncontrollable growth and an invasive cellular phenotype. Therefore, proteins that control chromatin organization including polycomb repressor complexes, PRC1 and PRC2, and members of the trithorax family constitute key players in the molecular pathogenesis of cancer. Selective binding of lncRNAs, HOTAIR and ANRIL, with PRC1 and PRC2 to execute histone modifications at specific loci thus strongly supports the idea that lncRNAs may function as ideal regulators for epigenetic transcriptional repression (Gupta et al., 2010; Kotake et al., 2011). ANRIL and associated factors play critical roles in repression of the INK4b–ARF–INK4a locus that encodes for three critical tumor suppressors, p15INK4b, p14ARF (p19ARF in mice), and p16INK4a, which play central roles in cell-cycle inhibition, senescence, and stress-induced apoptosis (Pasmant et al., 2007; Yap et al., 2010; Kotake et al., 2011).
Long ncRNAs may also act as tumor suppressors. They may inhibit cell-cycle progression in response to DNA damage due to stress and environmental factors. lncRNA ncRNACCND1 is induced during DNA damage from the CCND1 promoter (Wang et al., 2008). This lncRNA recruits the TLS protein to the CCND1 promoter where it binds to histone acetyltransferases CBP/p300 and in turn inhibits CCND1 transcription thus affecting cell-cycle progression. Some lncRNAs may inhibit growth in cancer cells. MEG3, a lncRNA that is expressed in many normal tissues but not in human cancer cell lines, may function as a tumor suppressor as its ectopic expression in cancer cells suppressed their growth (Zhang et al., 2003).
Associations of lncRNAs with Cancer
Genome-wide association studies of cancer susceptibility have identified single nucleotide polymorphisms (SNPs) in some of the transcribed regions of the non-coding portions of the human genome (Manolio et al., 2008). T-UCEs differentially expressed in human cancers are located in CAGRs that are specifically associated with that type of cancer (Calin et al., 2007). These could be candidate players for cancer susceptibility. For example, differential expressions of uc.349A and uc.352 between normal and leukemic CD5-positive cells have been linked to susceptibility to familial chronic lymphocytic leukemia (Ng et al., 2007). Consistent with these findings, Yang et al. (2008) have reported that two SNPs in UCEs (rs9572903 and rs2056116) are associated with familial breast cancer risk. Recently, Pasmant et al. (2011) have also shown that modulation of ANRIL levels in patients with neurofibromatosis mediates susceptibility to plexiform neurofibromas. SNP rs2151280 located in ANRIL locus was statistically significantly associated with number of plexiform neurofibromas in these patients.
ncRNAs and Cancer Diagnosis
The differences in lncRNA profiling between normal and cancer cells may or may not be a mere secondary effect of cancerous transformations. Several lncRNAs can control transcriptional alteration, as seen with ANRIL and its interaction with PRC proteins that leads to repression of INK4b locus, a change observed in most cancers (Kotake et al., 2011). In other cases, altered expression of these RNAs may show a strong association with tumor progression, and thus can be used as classification markers for these malignancies. Most lncRNAs are expressed in various types of cancers; however, some have been associated with specific tumor types. A striking example is that of three lncRNAs in prostate cancer: PCGEM1, DD3, and PCNCR1 (Bussemakers et al., 1999; Petrovics et al., 2004; Chung et al., 2011). These lncRNAs either promote tumorigenicity or are associated with susceptibility to prostate adenocarcinoma. These unique lncRNAs could therefore potentially be used for prostate cancer diagnosis. The malignant cells have a unique spectrum of expressed UCEs when compared with the corresponding normal cells, suggesting that variations in T-UCE expression are involved in the malignant process. Moreover, distinct T-UCE signatures were differentially expressed in leukemias and carcinomas, and thus may offer a novel strategy for cancer diagnosis and prognosis (Calin et al., 2007).
Our experience with childhood tumors have led us to believe that ncRNAs play key roles in defining tumor subtypes (Bajaj et al., 2011). We have performed several exploratory analyses in pediatric tumors that provide evidence of unique non-coding intergenic regions that are characteristic of tumor types. One such preliminary analysis depicted in Figure 1 involved 40 unique primary tumors from patients with PAX–FKHR fusion-positive rhabdomyosarcoma (n = 10), fusion-negative rhabdomyosarcoma (n = 10), Ewing family of tumors (EFT, n = 5), osteosarcoma (n = 5), neuroblastoma (n = 5), and Wilms’ tumors (n = 5). An unsupervised nearest shrunken centroid model, a class prediction procedure that identifies transcripts that best characterize tumor subtypes, was used to analyze whole-transcriptome expression profiling data obtained from these tumors using Affymetrix Human Exon 1.0 ST microarrays. This procedure eliminates classifier transcripts from the prediction signature as the shrinkage parameter (Δ) increases, thereby creating highly class-specific profiles (Tibshirani et al., 2002). This revealed the presence of several classifier coding and non-coding transcripts, represented as probe set regions (PSRs) on the top histogram of Figure 1A, that were able to categorize tumors in the training (aqua line) and test (gold line) sets with 100 and 95% accuracy at Δ = 5.6, respectively. Examination of features contained in the centroid classes revealed the presence of a 250-kb stretch of non-coding transcript (locus marked by dashed black box in Figure 1C), a putative vlncRNA, which was unique to EFTs (tumor class 3 in Figure 1C). This tumor subgroup uniquely showed marked overexpression of this genomic stretch that does not code for any known proteins (aqua trace in Figure 1D); none of the other childhood tumors examined in this cohort appeared to express this vlncRNA at levels comparable to EFT. This demonstrates that the presence of such transcripts, if found on a larger scale with similar discriminatory power, may be extremely helpful in diagnosing such tumor types. In addition, it also suggests that such non-coding transcripts may play a role in the genesis and maintenance of these malignancies.
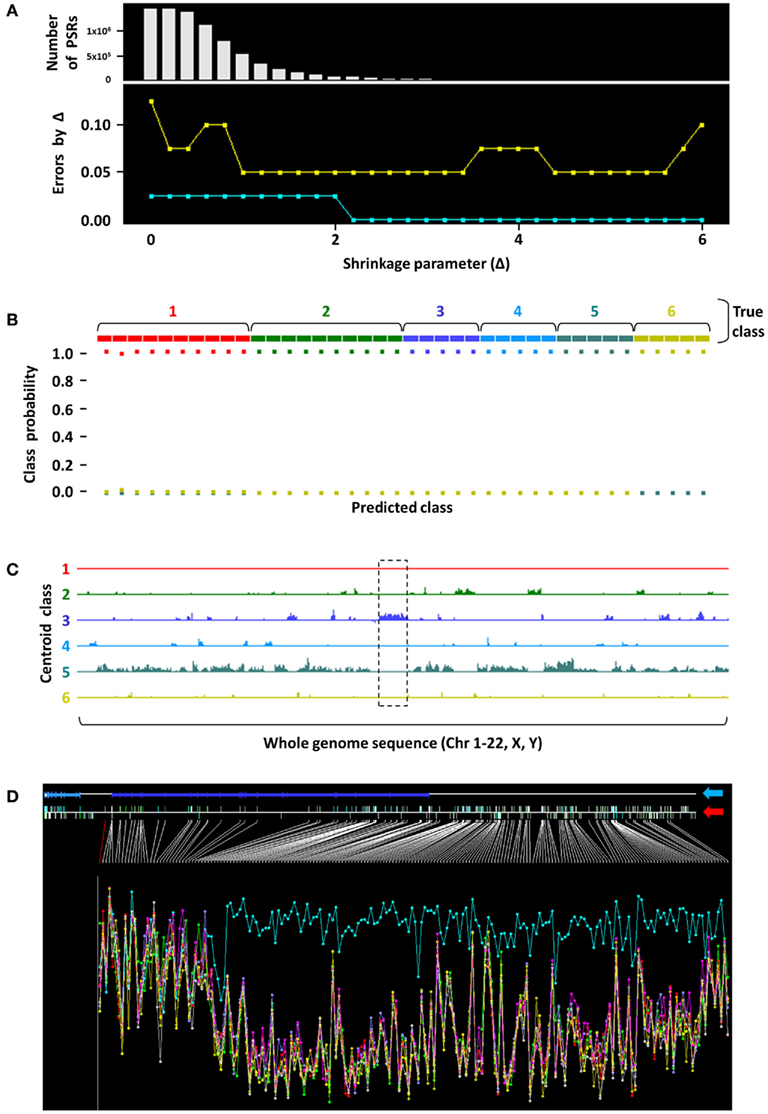
Figure 1. Nearest shrunken centroid analysis to identify a putative EFT-specific vlncRNA. (A) Nearest shrunken centroid modeling was performed on 40 unique primary childhood tumors. Shrinkage parameter (X-axis) Δ = 5.6 was selected as the threshold where the fewest number of PSRs (Y-axis, top panel) were required to categorize tumors in the training (aqua line) and test (gold line) sets with 0 and 5% error, respectively (Y-axis, bottom panel). (B) Classification performance of training set samples is shown, where probability of samples belonging to each color-coded tumor class (1, PAX–FKHR fusion-positive rhabdomyosarcoma; 2, fusion-negative rhabdomyosarcoma; 3, EFT; 4, osteosarcoma; 5, neuroblastoma; 6, Wilms’ tumors) was predicted with 100% accuracy at Δ = 5.6. Note that only squares of the like color are found at the 100% probability level in each true class. (C) Whole-genome plot of positions of the diagnostic PSRs (X-axis) that characterize the respective tumor groups versus their expression levels (Y-axis). A 250-kb stretch corresponding to a putative vlncRNA region (dashed black box) was observed as being uniquely overexpressed in EFT. (D) When zoomed in at this genomic segment (blue arrow points to the RefSeq annotation; red arrow indicates positions of PSRs across the region), evidence of significant overexpression of this transcript in EFTs (aqua trace) was clear compared to other childhood tumor types. Height of the Y-axis corresponds to the logarithm of PSR expression levels, and samples are aggregated into their respective tumor groups.
ncRNAs and Cancer Prognosis
Differential expressions of protein-coding genes and small ncRNAs between cancers have been used as a valuable tool to generate signatures that can reliably predict disease outcomes (Martens-Uzunova et al., 2012). A panel of 10 biomarkers that included 8 protein-coding genes and 2 miRNAs, miR-519d and miR-647, could significantly predict clinical recurrence in prostate cancer following radical prostatectomy (Long et al., 2011).
With recent growing evidence of similar expression patterns of lncRNAs in cancers, these transcripts may be profiled as prognostic candidates. A similar strategy may be adopted to develop lncRNA-dependent gene signatures that may predict disease outcomes and response to treatments. The lncRNA MALAT1 is upregulated in many solid tumors and is associated with cancer metastasis and recurrence. In hepatocellular carcinoma, MALAT1 levels corresponded to advanced disease stage and were inversely related to disease-free survival after liver transplantation (Lai et al., 2012). Similarly, an expression profile based on 28 T-UCEs in 14 patients with neuroblastoma was able to significantly distinguish between short-term and long-term survivors (Scaruffi et al., 2009).
Our group’s efforts in identifying non-coding transcripts that are associated with outcome have focused on childhood tumors. In one such analysis shown in Figure 2, we initially analyzed Affymetrix Human Exon 1.0 ST array-derived whole-transcriptome expression profiling data on primary EFT samples from 40 patients at surgical resection with long subsequent follow-up. Thirteen (32.5%) patients eventually metastasized (depicted in red in Figures 2A–D). An unsupervised nearest shrunken centroid model was used to identify coding and non-coding features that could categorize these tumors based on their probability of eventually metastasizing. At Δ = 1.2, several features were identified that could categorize the tumors into two groups based on risk of metastasis with 92.5 and 70% accuracies in the training and test sets, respectively (Figure 2A).
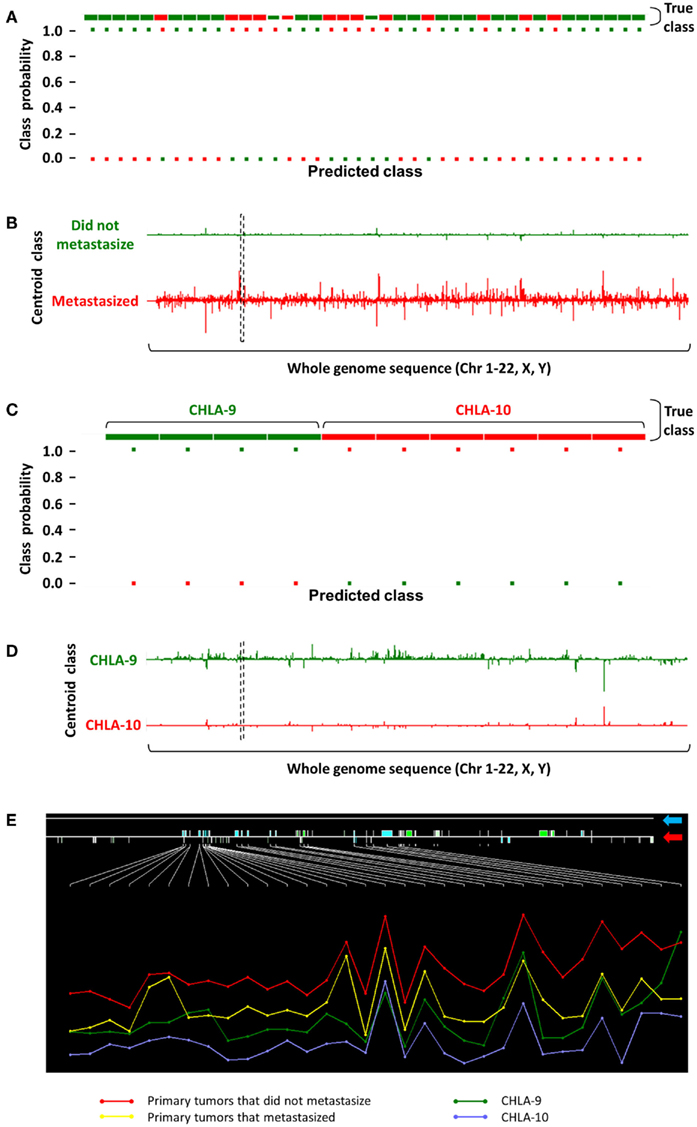
Figure 2. Identification of a non-coding transcript showing differential expression in EFTs with respect to metastasis. (A) Classification performance of a nearest shrunken centroid model is shown, where 40 primary EFTs were categorized based on their eventual metastatic fate (green, did not metastasize; red, eventually metastasized) in the training set with 92.5% accuracy at Δ = 1.2. (B) PSRs identified by this analysis that distinguish between non-metastasized versus metastasized groups are plotted over a whole-genome sequence, where height of the Y-axis over and under the baseline corresponds to their log fold change. (C) A similar nearest shrunken centroid analysis on CHLA-9 and CHLA-10 achieved 100% classification accuracy at Δ = 6.0. (D) Comparing the PSR profiles between both nearest shrunken centroid models resulted in the identification of a common 26 kb intergenic non-coding transcript [dashed black box in (B) and (D)]. (E) A zoomed in inspection of this genomic segment (blue arrow points to the RefSeq annotation; red arrow indicates positions of PSRs across the region) showed that the transcript was highly expressed in tumors that never metastasized, moderately expressed in tumors that eventually metastasized and CHLA-9, and showed low expression in CHLA-10. Height of the Y-axis corresponds to the logarithm of PSR expression levels, and samples are aggregated into their respective tumor groups.
To further investigate the biological implications of ncRNA features that could predict tumor metastasis, expression profiles on two EFT cell lines, CHLA-9 and CHLA-10, were analyzed using a similar nearest shrunken centroid model. At Δ = 6.0, the selected features were able to classify samples in the training and test sets with 100% accuracy (Figure 2C). When this set of classifier features was compared to those obtained from the analysis of the above EFT samples, a unique 26 kb intergenic non-coding transcript was identified on chromosome 2 (dashed black box in Figures 2B,D). The expression of this transcript was seemingly protective in nature – its expression was highest in primary tumors that did not metastasize, and lower in those primary tumors that eventually metastasized (Figure 2E). Following this trend, its expression was comparably lower in CHLA-9, a cell line generated from the primary tumor of an EFT patient, and lowest in CHLA-10, a cell line generated from a subsequent metastatic tumor in the same patient (Batra et al., 2004). Such observations provide credence to the argument that non-coding transcripts play crucial roles in the modulation of tumor behavior and can be used as markers in the primary malignancy to determine long-term prognosis.
ncRNAs: Bridging Normal Tissue Development and Oncogenesis
The data and studies presented here offer compelling evidence that transcription of ncRNAs in cancer is tightly linked to key biological processes, from differentiation to metastasis. The parallel with normal tissue differentiation during fetal development is striking and reminiscent of another well documented phenomenon in cancer: to reprise the expression of fetal antigens during oncogenesis. Given the documented higher levels of ncRNA transcription during normal tissue development, it should be no surprise that ncRNA levels in cancer are elevated compared to normal tissue development. Many parallels between oncogenesis and development are well known, such that oncogenesis is often viewed as a poorly executed mimicry of normal tissue development. Environmental influences may allow embryonic expression of lncRNAs in adult tissues that alter gene expression, thereby increasing cancer susceptibility. The chromatin-interacting ncRNA KCNQ1OT1 causes imprinting of CDKN1C gene in embryonic tissues (Lewis et al., 2004). CDKN1C gene expression is suppressed in breast cancers by estrogen through epigenetic mechanisms involving the highly expressed KCNQ1OT1 gene (Rodriguez et al., 2011). It is therefore not unreasonable to deduce that ncRNA expression is of fundamental importance, to the extent that ncRNA expression may well control coding RNA expression, using the latter to execute complex and fundamental programs responsible for organismal development. From a combined viewpoint, therefore, ncRNA is primary and coding RNA is secondary. The fact that a ncRNA gene like HOTAIR can orchestrate the expression of over 300 coding genes via complex formation with PRC2 and epigenetic regulation, leading to altered tumor cell differentiation and behavior, is entirely consistent with this concept. It will not be surprising, therefore, if a general pattern of ncRNA control of coding gene expression emerges from the many current studies on ncRNAs.
Beyond simple primary–secondary control mechanisms, it also appears that ncRNA itself is likely tightly regulated in an interactive network (Sumazin et al., 2011). This model of self-regulating RNA networks is intuitively attractive, as it allows for a degree of subtle control via multiple interacting regulatory networks that is essential to account for the development of higher organisms such as humans. The observation that ncRNA expression levels are highest in developing brain is consonant with this concept. The challenge going forward will be to unravel and understand these complex interactions. The reward will almost certainly be a far more sophisticated understanding of how biology works, and by extension, how it is perturbed in cancer.
Conclusion
This review provides some evidence of the multifaceted roles of lncRNAs in cancer. It underscores the importance of the functional existence of these transcripts that are proving to be much more than “transcriptional noise.” Understanding their biological relevance in normal development may provide an insight into their perturbed functions in cancer. This will allow use of these enigmatic molecules as diagnostic or predictive biomarkers. They may be further developed into cancer-specific RNA targets to improve treatment sensitivity for various malignancies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Atkins, J. F., Gesteland, R. F., and Cech, T. R. (2011). RNA Worlds: From Life’s Origins to Diversity in Gene Regulation. Woodbury, NY: Cold Spring Harbor Laboratory Press.
Babak, T., Blencowe, B. J., and Hughes, T. R. (2005). A systematic search for new mammalian noncoding RNAs indicates little conserved intergenic transcription. BMC Genomics 6, 104. doi:10.1186/1471-2164-6-104
Bajaj, S. V., Wai, D. H., Buckley, J. D., Kapranov, P., Lawlor, E. R., and Triche, T. J. (2011). A large non-coding RNA that is characteristic of Ewing sarcoma family of tumors. Paper Presented at 102nd Annual Meeting of the American Association for Cancer Research, Orlando, FL: American Association for Cancer Research.
Barsyte-Lovejoy, D., Lau, S. K., Boutros, P. C., Khosravi, F., Jurisica, I., Andrulis, I. L., Tsao, M. S., and Penn, L. Z. (2006). The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 66, 5330–5337.
Batra, S., Reynolds, C. P., and Maurer, B. J. (2004). Fenretinide cytotoxicity for Ewing’s sarcoma and primitive neuroectodermal tumor cell lines is decreased by hypoxia and synergistically enhanced by ceramide modulators. Cancer Res. 64, 5415–5424.
Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W. J., Mattick, J. S., and Haussler, D. (2004). Ultraconserved elements in the human genome. Science 304, 1321–1325.
Bussemakers, M. J., van Bokhoven, A., Verhaegh, G. W., Smit, F. P., Karthaus, H. F., Schalken, J. A., Debruyne, F. M., Ru, N., and Isaacs, W. B. (1999). DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 59, 5975–5979.
Calin, G. A., Liu, C. G., Ferracin, M., Hyslop, T., Spizzo, R., Sevignani, C., Fabbri, M., Cimmino, A., Lee, E. J., Wojcik, S. E., Shimizu, M., Tili, E., Rossi, S., Taccioli, C., Pichiorri, F., Liu, X., Zupo, S., Herlea, V., Gramantieri, L., Lanza, G., Alder, H., Rassenti, L., Volinia, S., Schmittgen, T. D., Kipps, T. J., Negrini, M., and Croce, C. M. (2007). Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12, 215–229.
Cawley, S., Bekiranov, S., Ng, H. H., Kapranov, P., Sekinger, E. A., Kampa, D., Piccolboni, A., Sementchenko, V., Cheng, J., Williams, A. J., Wheeler, R., Wong, B., Drenkow, J., Yamanaka, M., Patel, S., Brubaker, S., Tammana, H., Helt, G., Struhl, K., and Gingeras, T. R. (2004). Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116, 499–509.
Chen, W., Bocker, W., Brosius, J., and Tiedge, H. (1997). Expression of neural BC200 RNA in human tumours. J. Pathol. 183, 345–351.
Cheung, H. H., Lee, T. L., Davis, A. J., Taft, D. H., Rennert, O. M., and Chan, W. Y. (2010). Genome-wide DNA methylation profiling reveals novel epigenetically regulated genes and non-coding RNAs in human testicular cancer. Br. J. Cancer 102, 419–427.
Chung, S., Nakagawa, H., Uemura, M., Piao, L., Ashikawa, K., Hosono, N., Takata, R., Akamatsu, S., Kawaguchi, T., Morizono, T., Tsunoda, T., Daigo, Y., Matsuda, K., Kamatani, N., Nakamura, Y., and Kubo, M. (2011). Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 102, 245–252.
Clamp, M., Fry, B., Kamal, M., Xie, X., Cuff, J., Lin, M. F., Kellis, M., Lindblad-Toh, K., and Lander, E. S. (2007). Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. U.S.A. 104, 19428–19433.
Creamer, T. J., Darby, M. M., Jamonnak, N., Schaughency, P., Hao, H., Wheelan, S. J., and Corden, J. L. (2011). Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet. 7, e1002329. doi:10.1371/journal.pgen.1002329
Euskirchen, G., Royce, T. E., Bertone, P., Martone, R., Rinn, J. L., Nelson, F. K., Sayward, F., Luscombe, N. M., Miller, P., Gerstein, M., Weissman, S., and Snyder, M. (2004). CREB binds to multiple loci on human chromosome 22. Mol. Cell. Biol. 24, 3804–3814.
Feil, R., Walter, J., Allen, N. D., and Reik, W. (1994). Developmental control of allelic methylation in the imprinted mouse Igf2 and H19 genes. Development 120, 2933–2943.
Gibb, E. A., Vucic, E. A., Enfield, K. S., Stewart, G. L., Lonergan, K. M., Kennett, J. Y., Becker-Santos, D. D., MacAulay, C. E., Lam, S., Brown, C. J., and Lam, W. L. (2011). Human cancer long non-coding RNA transcriptomes. PLoS ONE 6, e25915. doi:10.1371/journal.pone.0025915
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., Tsai, M. C., Hung, T., Argani, P., Rinn, J. L., Wang, Y., Brzoska, P., Kong, B., Li, R., West, R. B., van de Vijver, M. J., Sukumar, S., and Chang, H. Y. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076.
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., Huarte, M., Zuk, O., Carey, B. W., Cassady, J. P., Cabili, M. N., Jaenisch, R., Mikkelsen, T. S., Jacks, T., Hacohen, N., Bernstein, B. E., Kellis, M., Regev, A., Rinn, J. L., and Lander, E. S. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227.
Guttman, M., Garber, M., Levin, J. Z., Donaghey, J., Robinson, J., Adiconis, X., Fan, L., Koziol, M. J., Gnirke, A., Nusbaum, C., Rinn, J. L., Lander, E. S., and Regev, A. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 28, 503–510.
Han, Z., He, H., Zhang, F., Huang, Z., Liu, Z., Jiang, H., and Wu, Q. (2012). Spatiotemporal expression pattern of Mirg, an imprinted non-coding gene, during mouse embryogenesis. J. Mol. Histol. 43, 1–8.
Hawkins, P. G., and Morris, K. V. (2010). Transcriptional regulation of Oct4 by a long non-coding RNA antisense to Oct4-pseudogene 5. Transcription 1, 165–175.
He, Y., Vogelstein, B., Velculescu, V. E., Papadopoulos, N., and Kinzler, K. W. (2008). The antisense transcriptomes of human cells. Science 322, 1855–1857.
Hendrich, B. D., Brown, C. J., and Willard, H. F. (1993). Evolutionary conservation of possible functional domains of the human and murine XIST genes. Hum. Mol. Genet. 2, 663–672.
Hibi, K., Nakamura, H., Hirai, A., Fujikake, Y., Kasai, Y., Akiyama, S., Ito, K., and Takagi, H. (1996). Loss of H19 imprinting in esophageal cancer. Cancer Res. 56, 480–482.
Juan, V., Crain, C., and Wilson, C. (2000). Evidence for evolutionarily conserved secondary structure in the H19 tumor suppressor RNA. Nucleic Acids Res. 28, 1221–1227.
Kapranov, P. (2009). Studying chromosome-wide transcriptional networks: new insights into disease? Genome Med. 1, 50.
Kapranov, P., St Laurent, G., Raz, T., Ozsolak, F., Reynolds, C. P., Sorensen, P. H., Reaman, G., Milos, P., Arceci, R. J., Thompson, J. F., and Triche, T. J. (2010). The majority of total nuclear-encoded non-ribosomal RNA in a human cell is ‘dark matter’ un-annotated RNA. BMC Biol. 8, 149. doi:10.1186/1741-7007-9-86
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., and Xiong, Y. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene 30, 1956–1962.
Lai, M. C., Yang, Z., Zhou, L., Zhu, Q. Q., Xie, H. Y., Zhang, F., Wu, L. M., Chen, L. M., and Zheng, S. S. (2012). Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med. Oncol. (in press).
Lewis, A., Mitsuya, K., Umlauf, D., Smith, P., Dean, W., Walter, J., Higgins, M., Feil, R., and Reik, W. (2004). Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat. Genet. 36, 1291–1295.
Licastro, D., Gennarino, V. A., Petrera, F., Sanges, R., Banfi, S., and Stupka, E. (2010). Promiscuity of enhancer, coding and non-coding transcription functions in ultraconserved elements. BMC Genomics 11, 151. doi:10.1186/1471-2164-11-151
Long, Q., Johnson, B. A., Osunkoya, A. O., Lai, Y. H., Zhou, W., Abramovitz, M., Xia, M., Bouzyk, M. B., Nam, R. K., Sugar, L., Stanimirovic, A., Williams, D. J., Leyland-Jones, B. R., Seth, A. K., Petros, J. A., and Moreno, C. S. (2011). Protein-coding and microRNA biomarkers of recurrence of prostate cancer following radical prostatectomy. Am. J. Pathol. 179, 46–54.
Lustig, O., Ariel, I., Ilan, J., Lev-Lehman, E., De-Groot, N., and Hochberg, A. (1994). Expression of the imprinted gene H19 in the human fetus. Mol. Reprod. Dev. 38, 239–246.
Manolio, T. A., Brooks, L. D., and Collins, F. S. (2008). A HapMap harvest of insights into the genetics of common disease. J. Clin. Invest. 118, 1590–1605.
Martens-Uzunova, E. S., Jalava, S. E., Dits, N. F., van Leenders, G. J., Moller, S., Trapman, J., Bangma, C. H., Litman, T., Visakorpi, T., and Jenster, G. (2012). Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene (in press).
Matouk, I. J., DeGroot, N., Mezan, S., Ayesh, S., Abu-lail, R., Hochberg, A., and Galun, E. (2007). The H19 non-coding RNA is essential for human tumor growth. PLoS ONE 2, e845. doi:10.1371/journal.pone.0000845
Mattick, J. S. (2003). Challenging the dogma: the hidden layer of non-protein-coding RNAs in complex organisms. Bioessays 25, 930–939.
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159.
Mestdagh, P., Fredlund, E., Pattyn, F., Rihani, A., Van Maerken, T., Vermeulen, J., Kumps, C., Menten, B., De Preter, K., Schramm, A., Schulte, J., Noguera, R., Schleiermacher, G., Janoueix-Lerosey, I., Laureys, G., Powel, R., Nittner, D., Marine, J. C., Ringnér, M., Speleman, F., and Vandesompele, J. (2010). An integrative genomics screen uncovers ncRNA T-UCR functions in neuroblastoma tumours. Oncogene 29, 3583–3592.
Morris, K. V., Santoso, S., Turner, A. M., Pastori, C., and Hawkins, P. G. (2008). Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 4, e1000258. doi:10.1371/journal.pgen.1000258
Ng, D., Toure, O., Wei, M. H., Arthur, D. C., Abbasi, F., Fontaine, L., Marti, G. E., Fraumeni, J. F. Jr., Goldin, L. R., Caporaso, N., and Toro, J. R. (2007). Identification of a novel chromosome region, 13q21.33-q22.2, for susceptibility genes in familial chronic lymphocytic leukemia. Blood 109, 916–925.
Panning, B., Dausman, J., and Jaenisch, R. (1997). X chromosome inactivation is mediated by Xist RNA stabilization. Cell 90, 907–916.
Pasmant, E., Laurendeau, I., Heron, D., Vidaud, M., Vidaud, D., and Bieche, I. (2007). Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 67, 3963–3969.
Pasmant, E., Sabbagh, A., Masliah-Planchon, J., Ortonne, N., Laurendeau, I., Melin, L., Ferkal, S., Hernandez, L., Leroy, K., Valeyrie-Allanore, L., Parfait, B., Vidaud, D., Bièche, I., Lantieri, L., Wolkenstein, P., Vidaud, M., and NF France Network. (2011). Role of noncoding RNA ANRIL in genesis of plexiform neurofibromas in neurofibromatosis type 1. J. Natl. Cancer Inst. 103, 1713–1722.
Pauli, A., Rinn, J. L., and Schier, A. F. (2011). Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 12, 136–149.
Petrovics, G., Zhang, W., Makarem, M., Street, J. P., Connelly, R., Sun, L., Sesterhenn, I. A., Srikantan, V., Moul, J. W., and Srivastava, S. (2004). Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene 23, 605–611.
Ponjavic, J., Ponting, C. P., and Lunter, G. (2007). Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 17, 556–565.
Qureshi, I. A., and Mehler, M. F. (2011). Non-coding RNA networks underlying cognitive disorders across the lifespan. Trends. Mol. Med. 17, 337–346.
Rodriguez, B. A., Weng, Y. I., Liu, T. M., Zuo, T., Hsu, P. Y., Lin, C. H., Cheng, A. L., Cui, H., Yan, P. S., and Huang, T. H. (2011). Estrogen-mediated epigenetic repression of the imprinted gene cyclin-dependent kinase inhibitor 1C in breast cancer cells. Carcinogenesis 32, 812–821.
Rosenbluh, J., Nijhawan, D., Chen, Z., Wong, K. K., Masutomi, K., and Hahn, W. C. (2011). RMRP is a non-coding RNA essential for early murine development. PLoS ONE 6, e26270. doi:10.1371/journal.pone.0026270
Rossi, S., Sevignani, C., Nnadi, S. C., Siracusa, L. D., and Calin, G. A. (2008). Cancer-associated genomic regions (CAGRs) and noncoding RNAs: bioinformatics and therapeutic implications. Mamm. Genome 19, 526–540.
Sang, X., Zhao, H., Lu, X., Mao, Y., Miao, R., Yang, H., Yang, Y., Huang, J., and Zhong, S. (2010). Prediction and identification of tumor-specific noncoding RNAs from human UniGene. Med. Oncol. 27, 894–898.
Scaruffi, P., Stigliani, S., Moretti, S., Coco, S., De Vecchi, C., Valdora, F., Garaventa, A., Bonassi, S., and Tonini, G. P. (2009). Transcribed-ultra conserved region expression is associated with outcome in high-risk neuroblastoma. BMC Cancer 9, 441. doi:10.1186/1471-2407-9-441
Suh, N., and Blelloch, R. (2011). Small RNAs in early mammalian development: from gametes to gastrulation. Development 138, 1653–1661.
Sumazin, P., Yang, X., Chiu, H. S., Chung, W. J., Iyer, A., Llobet-Navas, D., Rajbhandari, P., Bansal, M., Guarnieri, P., Silva, J., and Califano, A. (2011). An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 147, 370–381.
Taft, R. J., Pheasant, M., and Mattick, J. S. (2007). The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays 29, 288–299.
Tibshirani, R., Hastie, T., Narasimhan, B., and Chu, G. (2002). Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. U.S.A. 99, 6567–6572.
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J., and Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515.
van Bakel, H., and Hughes, T. R. (2009). Establishing legitimacy and function in the new transcriptome. Brief. Funct. Genomic. Proteomic. 8, 424–436.
van Leeuwen, S., and Mikkers, H. (2010). Long non-coding RNAs: guardians of development. Differentiation 80, 175–183.
Wang, X., Arai, S., Song, X., Reichart, D., Du, K., Pascual, G., Tempst, P., Rosenfeld, M. G., Glass, C. K., and Kurokawa, R. (2008). Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130.
Yang, R., Frank, B., Hemminki, K., Bartram, C. R., Wappenschmidt, B., Sutter, C., Kiechle, M., Bugert, P., Schmutzler, R. K., Arnold, N., Weber, B. H., Niederacher, D., Meindl, A., and Burwinkel, B. (2008). SNPs in ultraconserved elements and familial breast cancer risk. Carcinogenesis 29, 351–355.
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., Gil, J., Walsh, M. J., and Zhou, M. M. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674.
Keywords: non-coding RNA, tumor, diagnosis, prognosis, development
Citation: Mitra SA, Mitra AP and Triche TJ (2012) A central role for long non-coding RNA in cancer. Front. Gene. 3:17. doi: 10.3389/fgene.2012.00017
Received: 02 December 2011;
Paper pending published: 14 December 2011;
Accepted: 28 January 2012;
Published online: 15 February 2012.
Edited by:
Philipp Kapranov, St. Laurent Institute, USAReviewed by:
Robert Arceci, Johns Hopkins, USATim McCaffrey, The George Washington University Hospital, USA
Copyright: © 2012 Mitra, Mitra and Triche. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Timothy J. Triche, Department of Pathology and Laboratory Medicine, Children’s Hospital Los Angeles, 4650 Sunset Blvd., SRT 1016, MS 133, Los Angeles, CA 90027, USA. e-mail:dHJpY2hlQHVzYy5lZHU=