 Ismael A. Vergara1†
Ismael A. Vergara1† Nicholas Erho1†
Nicholas Erho1† Timothy J. Triche1* Mercedeh Ghadessi1
Timothy J. Triche1* Mercedeh Ghadessi1 Anamaria Crisan1
Anamaria Crisan1 Thomas Sierocinski1 Peter C. Black2
Thomas Sierocinski1 Peter C. Black2 Christine Buerki1
Christine Buerki1 Elai Davicioni1
Elai Davicioni1
- 1 GenomeDx Biosciences, Inc., Vancouver, BC, Canada
- 2 Department of Urologic Sciences, University of British Columbia, Vancouver, BC, Canada
Prostate cancer is the most diagnosed cancer among men in the United States. While the majority of patients who undergo surgery (prostatectomy) will essentially be cured, about 30–40% men remain at risk for disease progression and recurrence. Currently, patients are deemed at risk by evaluation of clinical factors, but these do not resolve whether adjuvant therapy will significantly attenuate or delay disease progression for a patient at risk. Numerous efforts using mRNA-based biomarkers have been described for this purpose, but none have successfully reached widespread clinical practice in helping to make an adjuvant therapy decision. Here, we assess the utility of non-coding RNAs as biomarkers for prostate cancer recurrence based on high-resolution oligonucleotide microarray analysis of surgical tissue specimens from normal adjacent prostate, primary tumors, and metastases. We identify differentially expressed non-coding RNAs that distinguish between the different prostate tissue types and show that these non-coding RNAs can predict clinical outcomes in primary tumors. Together, these results suggest that non-coding RNAs are emerging from the “dark matter” of the genome as a new source of biomarkers for characterizing disease recurrence and progression. While this study shows that non-coding RNA biomarkers can be highly informative, future studies will be needed to further characterize the specific roles of these non-coding RNA biomarkers in the development of aggressive disease.
Introduction
Prostate cancer is a major public health concern, with over 240,000 newly diagnosed men in the United States alone (Siegel et al., 2011). This clinically heterogeneous disease ranges from indolent forms of cancer with good long term prognosis to life-threatening disease associated with only a couple of months of survival (Rubin et al., 2011). After initial diagnosis, one of the most successful treatments with curative intent is radical prostatectomy, i.e., the complete removal of the prostate gland. It is, however, known that patients who present with aggressive clinical features after surgery, such as positive surgical margins (SM), extracapsular extension (ECE), and seminal vesicle invasion (SVI) likely will require further therapy in order to delay the onset of life-threatening metastasis (Bolla et al., 2005; Thompson et al., 2009; Wiegel et al., 2009). The efficient delivery of such therapies after prostatectomy is currently hampered by a lack of predictive tools to assess the risk of clinically significant recurrence and progression.
Biochemical recurrence (BCR), defined as a detectable prostate specific antigen (PSA) level above a certain threshold or as a rising PSA level after surgery, is a widely used surrogate for disease progression and prostate cancer specific mortality (PCSM). Still, BCR has been deemed an unreliable surrogate since, even though BCR always precedes metastatic progression and PCSM, not every patient with BCR will experience metastatic disease (Simmons et al., 2007). Given this, numerous efforts using mRNA-based biomarkers as a tool to assess the risk of recurrence and progression have been described, but none have successfully reached widespread clinical practice (Sorensen and Orntoft, 2010). Recently, the clinical utility of micro RNAs (or miRNAs) as potential biomarkers for disease diagnosis and prognosis has been assessed (Schaefer et al., 2010; Sevli et al., 2010; Catto et al., 2011; Martens-Uzunova et al., 2011). miRNAs have shown altered expression in prostate cancer and were found to be involved in the regulation of key pathways such as androgen signaling and apoptosis (Catto et al., 2011). In general, recent evidence showing that a much larger fraction of normal and cancer transcriptomes are composed of non-coding RNAs (or ncRNAs) than previously anticipated (Kapranov et al., 2010) has driven researchers towards exploring the utility of not only short ncRNAs but also long ncRNAs as biomarkers. For example, Chung et al. (2011) identified PRNCR1 (prostate cancer non-coding RNA 1) as a long intergenic ncRNA (or lincRNA) transcribed in the gene desert of the prostate cancer susceptibility locus 8q24. The same genomic region was found to be transcribed into PCAT-1, a lincRNA highly expressed in metastatic tissue specimens from prostate cancer patients (Prensner et al., 2011).
While there is increasing knowledge of the importance of ncRNAs in cancer, their clinical usefulness for diagnosis and prognosis is limited. To date, only one ncRNA is routinely used in the clinical setting in prostate cancer: prostate cancer antigen 3 (PCA3), a non-coding antisense transcript that is highly overexpressed in prostate cancer compared to benign tissue (Bussemakers et al., 1999). PCA3 is used in a urinary-based diagnostic test for patient screening in conjunction with PSA serum testing and other clinical information (Day et al., 2011).
In this study, we perform high-resolution oligonucleotide microarray analysis of a publicly available dataset (Taylor et al., 2010) from different types of normal and cancerous prostate tissue. We find, by analysis of the entire set of exonic and non-exonic features, differentially expressed ncRNAs that accurately discriminate clinical outcomes such as BCR and metastatic disease.
Materials and Methods
Microarray and Clinical Data
The publically available genomic and clinical data was generated as part of the Memorial Sloan–Kettering Cancer Center (MSKCC) Prostate Oncogenome Project, previously reported by (Taylor et al., 2010). The Human Exon arrays for 131 primary prostate cancer, 29 normal adjacent, and 19 metastatic tissue specimens were downloaded from GEO Omnibus at http://www.ncbi.nlm.nih.gov/geoseries GSE21034. The patient and specimen details for the primary and metastases tissues used in this study are summarized in Table 1. For the analysis of the clinical data, the following ECE statuses were summarized to be concordant with the pathological tumor stage: inv-capsule: ECE−, focal: ECE+, established: ECE+.
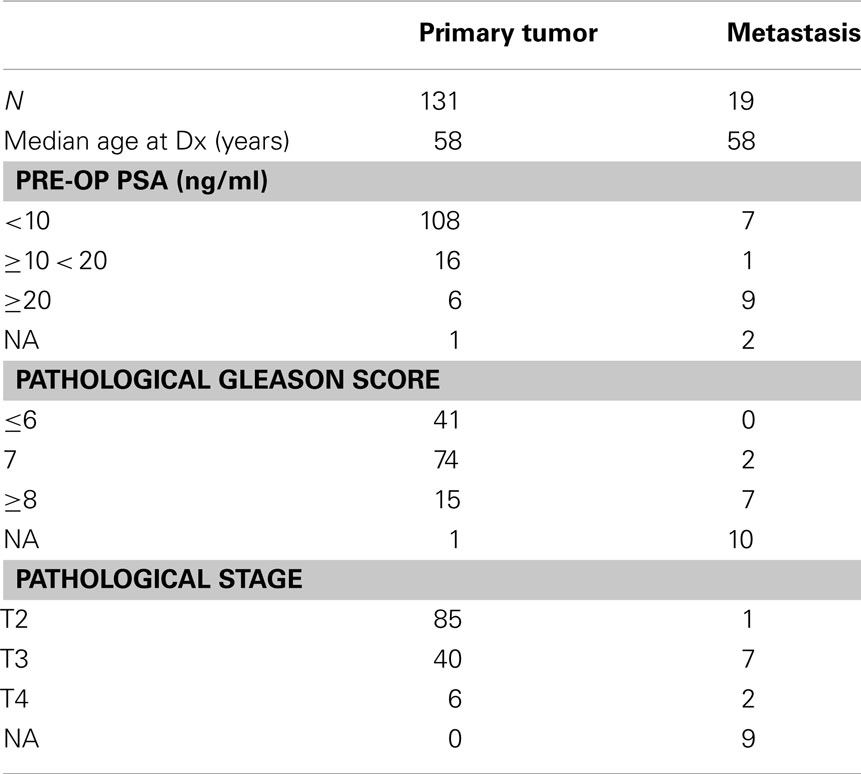
Table 1. Summary of the clinical characteristics of the dataset used in this study.
Microarray Pre-Processing
Normalization and summarization
The normalization and summarization of the 179 microarray samples (cell line samples were removed) were done with the frozen Robust Multiarray Average (fRMA) algorithm using custom frozen vectors (McCall et al., 2010). These custom vectors were created using the vector creation methods described in (McCall and Irizarry, 2011) including all MSKCC samples. Quantile normalization and robust weighted average methods were used for normalization and summarization, respectively, as implemented in fRMA.
Sample subsets
The normalized and summarized data was partitioned into three groups. The first group contains the matched samples from primary localized prostate cancer tumors and normal adjacent tissues (n = 58; used for the normal vs. primary comparison). The second group contains all the samples from metastatic tumors (n = 19) and all the localized prostate cancer tumors that were not matched with normal adjacent tissues (n = 102; used for the primary tumor vs. metastasis comparison). The third group corresponds to all samples from metastatic tumors (n = 19) and all the normal adjacent tissues (n = 29; used for the normal vs. metastasis comparison).
Feature selection
Probe sets (or PSRs) annotated as “unreliable” by the xmapcore package (Yates, 2010; defined as one or more probes that do not align uniquely to the genome) as well as those defined as class 2 and class 3 cross-hybridizing by Affymetrix annotation were excluded from further analysis. The remaining PSRs were subjected to univariate analysis to identify those associated to features differentially expressed between the labeled groups (primary tumor vs. metastatic, normal adjacent vs. primary tumor, and normal vs. metastatic). For this analysis, features were selected as differentially expressed if their Holm–Bonferroni adjusted (Holm, 1979) t-test p-value was significant (<0.05). The t-test was applied as implemented in the rowttests function of the genefilter package.1
The multiple testing correction was applied using the p.adjust function of the stats package in R.
This multiple testing correction was performed for the exonic (353k PSRs) and non-exonic (931k PSRs) sets independently due to differences in cardinality of the PSR sets. Data A1 in Appendix provides the detailed steps for the generation of differentially expressed features.
Feature evaluation and model building
Classical multidimensional scaling (MDS, Pearson’s distance) was used to evaluate the ability of the selected features to segregate primary tumor samples into clinically relevant clusters based on metastatic events and Gleason scores. MDS was applied as implemented in the cmdscale function of the stats package in R. The significance of the segregation in these two-dimensional MDS plots was assessed using permutational ANOVA as implemented within the vegan package in R2.
A custom implementation of the k-nearest-neighbor (KNN) model (k = 1, Pearson’s correlation distance metric) was trained on the normal and metastatic samples (n = 48) using only the features found to be differentially expressed between these two groups. Unmatched primary tumors were used as an independent set for validation.
Re-annotation of the human exon microarray probe sets
Affymetrix Human Exon 1.0 ST Arrays3 have about 1.4 million probe sets, with most probe sets containing four probes each. In order to properly assess the nature of the probe sets found differentially expressed in this study, we re-annotated them using the xmapcore R package4 (Yates, 2010) as follows: (i) exonic, if the probe set overlaps with the coding portion of a protein-coding exon or an untranslated region (UTR), and (ii) non-exonic if the probe set overlaps with an intron, an intergenic region, or a non-protein-coding transcript.
Annotation of non-coding transcripts was pursued using Ensembl Biomart available at http://www.ensembl.org
Statistical Analysis
Biochemical recurrence and metastatic disease progression end points are used as defined by the “BCR Event” and “Mets Event” columns of the supplementary material provided by (Taylor et al., 2010), respectively. Survival analysis for BCR was performed using the survfit function of the survival package5. Logistic regression for metastatic disease progression was performed using the lrm function of the rms package6.
Results
Re-annotation and Categorization of Coding and Non-Coding Differentially Expressed Features
Previous transcriptome-wide assessments of differential expression using prostate tissues in the post-prostatectomy setting have been focused on protein-coding features (see Nakagawa et al., 2008 for a comparison of protein-coding gene-based panels). Recent evidence based on the characterization of transcriptomes from normal and cancerous tissues has shown that most of it is of non-coding nature (Kapranov et al., 2010). Human Exon Arrays provide a unique opportunity to explore the differential expression of non-coding parts of the genome, as 75% of their probe sets cover regions other than protein-coding sequences. In this study, we use the publicly available Human Exon Array data set from normal adjacent, localized primary tumors, and metastatic tissues generated as part of the MSKCC Prostate Oncogenome Project to explore the potential of non-coding regions in prostate cancer prognosis. Previous attempts on this dataset focused only on mRNA and gene-level analysis and concluded that expression analysis was inadequate for discrimination of outcome groups in primary tumors (Taylor et al., 2010). In order to assess the contribution of ncRNA probe sets in differential expression analysis between sample types, we re-assessed the annotation of all probe sets found to be differentially expressed according to their genomic location and categorized them into exonic and non-exonic (see Materials and Methods). Briefly, a probe set is classified as exonic if it falls in a region that encodes for a protein-coding transcript or an UTR; otherwise, it is annotated as non-exonic.
Based on the above categorization, we assessed the exonic and non-exonic sets for the presence of differentially expressed features for each possible pairwise comparison (i.e., primary vs. normal, normal vs. metastatic, and primary vs. metastatic). The majority of the differentially expressed features are labeled as exonic for a given pairwise comparison (81%, 81%, and 75% for normal-primary, primary-metastatic, and normal-metastatic comparisons, respectively; see Table S1 in Supplementary Material for the top 100 differentially expressed features for each pairwise comparison). For each category, the number of differentially expressed features is highest in normal vs. metastatic tissues, which is expected since the metastatic samples are a heterogeneous group that has likely undergone major genomic alterations through disease progression and through effects of therapy on the genome (Figure 1). Additional variation in expression may be due to contamination with metastatic site tissue as well as host cell-metastatic cell interactions for metastases that include distant lymph nodes (seven samples), bone (five samples), and brain (three samples). As expected, assessment of all gene loci with features found to be differentially expressed between normal and metastatic samples shows that those up-regulated in metastatic tissue compared to normal are enriched in cellular processes such as cell division, spindle check point, and cytokinesis, whereas those down-regulated are enriched in terms like cell adhesion, muscle contraction, neuron development, and urogenital system development (Table S2 in Supplementary Material).

Figure 1. Venn diagram of exonic (A) and non-exonic (B) features found differentially expressed in the following comparisons: normal vs. primary tumor tissue (N vs. P), primary tumor vs. metastatic tissue (P vs. M), and normal vs. metastatic tissue (N vs. M).
For each category of exonic and non-exonic features there is a significant number that are specific to each pairwise comparison. For example, 21% of the exonic features are specific to the differentiation between normal tissue and primary tumors and 10% are specific to the primary tumor vs. metastatic comparison. The same proportions are observed for the non-exonic category, suggesting that different genomic regions may play a role in the progression from normal tissue to primary tumor and from primary tumor to metastatic tumor.
Within the non-exonic category, the majority of the features are “intronic” for all pairwise comparisons (see Figures 2A–C). Also, a large proportion of features correspond to intergenic regions. Still, hundreds of features lie within non-coding transcripts, as reflected by the “NC Transcript” segment in Figure 2. These non-coding transcripts found to be differentially expressed in each pairwise comparison were categorized using the “Transcript Biotype” annotation of Ensembl. For all pairwise comparisons the “processed transcript”, “lincRNA”, “retained intron”, and “antisense” are the most prevalent (Figures 2D–F; see Table 2 for a definition of each transcript type). Even though “processed transcript” and “retained intron” categories are among the most frequent ones, they have a very broad definition.
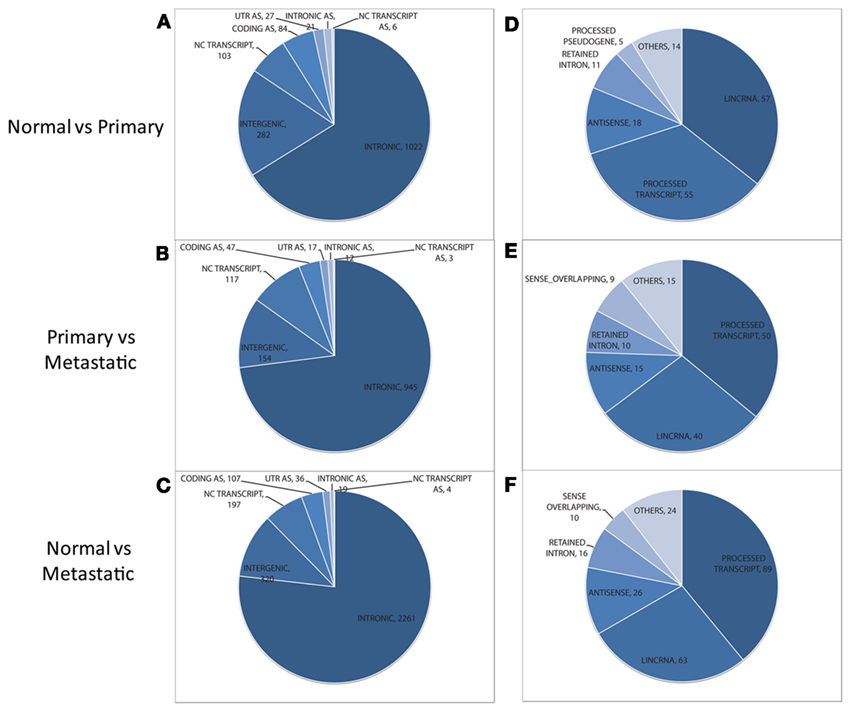
Figure 2. Distribution of non-exonic features (left) and overlapping annotated non-coding transcripts (right) found to be differentially expressed between normal and primary tumor (A,D), primary tumor and metastatic tissue (B,E), and normal vs. metastatic tissue (C,F). Features in the NC TRANSCRIPT slice of each pie chart (left) are assessed for their overlap with non-coding transcripts to generate the distribution of transcripts (shown at the right for each pairwise comparison). AS, antisense. UTR, untranslated region; lincRNA, long intergenic ncRNA.

Table 2. Definitions of Ensembl “Transcript Biotype” annotations for non-coding transcripts found differentially expressed.
Previous studies have reported several long non-coding RNAs to play a role in prostate adenocarcinoma (Srikantan et al., 2000; Berteaux et al., 2004; Petrovics et al., 2004; Lin et al., 2007; Poliseno et al., 2010; Yap et al., 2010; Chung et al., 2011; Day et al., 2011). Close inspection of our data reveals that four of them (PCGEM1, PCA3, MALAT1, and PTENP1) have associated differentially expressed features in at least one pairwise comparison based on a 1.5 Median Fold Difference (MFD) threshold (Table 3). After adjusting the p-value for multiple testing however, only two ncRNA transcripts, PCA3 and MALAT1, remain significant (Table 3). In addition, we found three differentially expressed microRNA-encoding transcripts in primary tumor vs. metastatic (MIR143, MIR145, and MIR221) and two in normal vs. metastatic (MIR145 and MIR221) that have been previously reported as differentially expressed in prostate cancer (Porkka et al., 2007; Clape et al., 2009; Zaman et al., 2010).
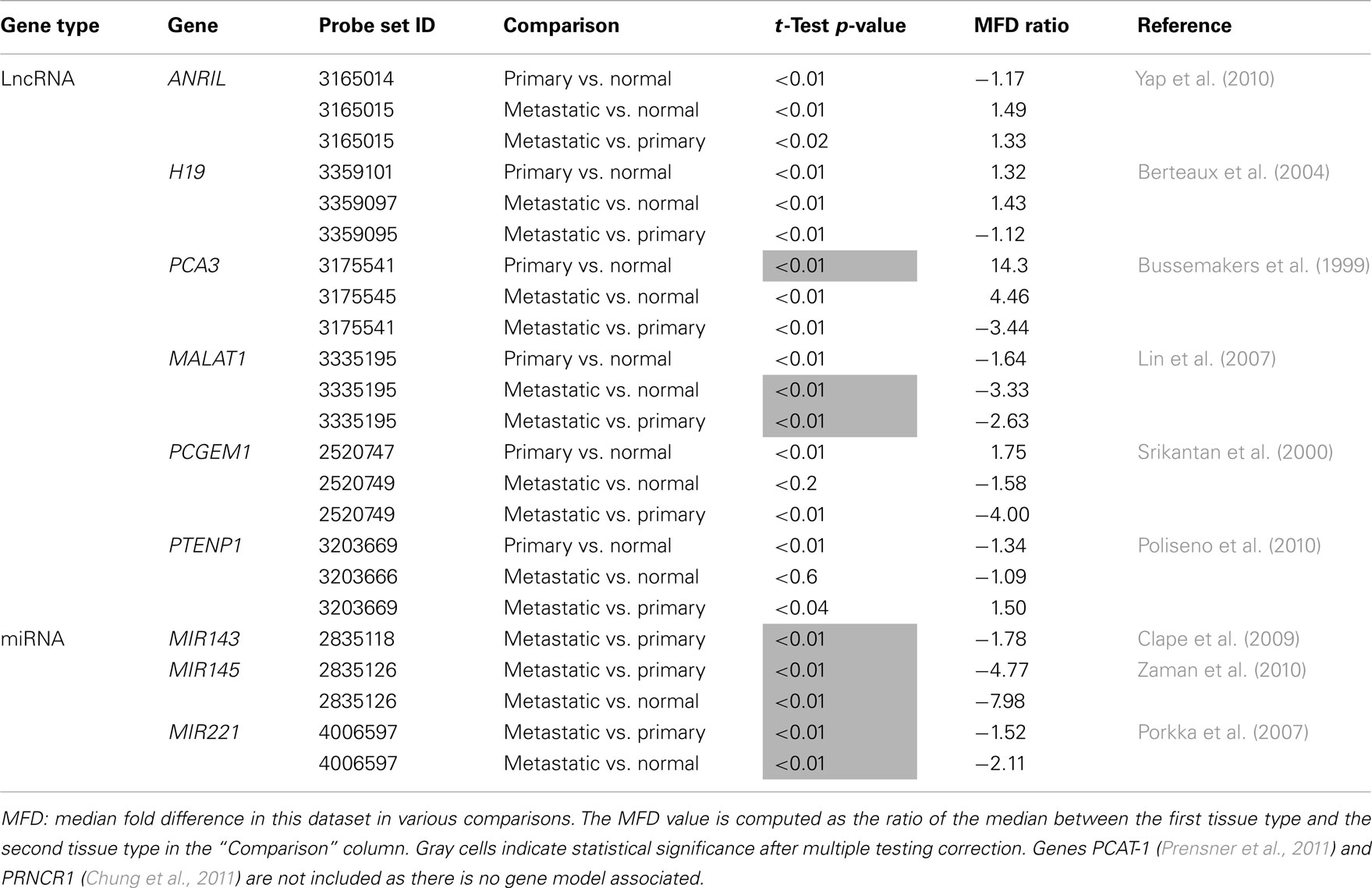
Table 3. Long non-coding RNAs previously reported as differentially expressed in prostate cancer.
Therefore, in addition to the handful of known ncRNAs, our analysis detected many other ncRNAs in regions that have yet to be explored in prostate cancer and that may play a role in the progression of the disease from normal glandular epithelium to distant metastases.
Assessment of Clinically Significant Prostate Cancer Risk Groups
Using MDS we observed that both exonic and non-exonic subsets of features present a statistically significant segregation of primary tumors from patients that progressed to metastatic disease (Figure A1 in Appendix), in contrast to the findings of Taylor et al. (2010). Similarly, we found the exonic and non-exonic subsets to discriminate high and low Gleason score samples (Figure A2 in Appendix). In order to assess the prognostic significance of differentially expressed exonic and non-exonic features, we trained a KNN classifier for each group using features from the comparison of normal and metastatic tissue samples (see Materials and Methods). Next, we used unmatched primary tumors (i.e., removing those tumors that had a matched normal in the training subset) as an independent validation set for the KNN classifiers. Each primary tumor in the validation set was classified by KNN as either more similar to normal or metastatic tissue. Subsequent Kaplan–Meier analysis of the classified primary tumor samples using BCR as end point showed that, as expected, primary tumors classified as belonging to the metastatic group had a higher rate of BCR (Figure 3). However, the KNN classifier trained on the exonic subset of features showed no statistically significant difference in BCR-free survival using a log-rank test (p < 0.08) whereas the difference was highly significant for the non-exonic KNN classifier (p < 0.00003).
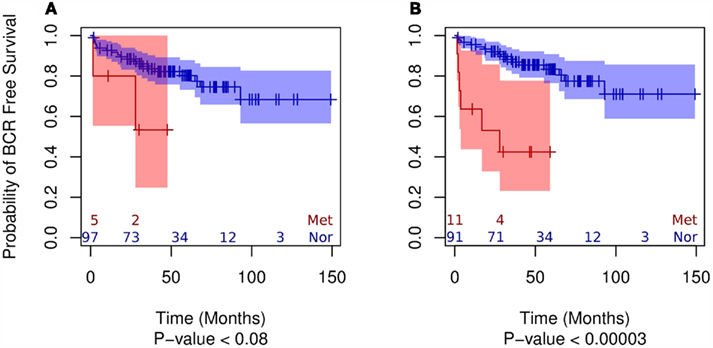
Figure 3. Kaplan–Meier plots of the two groups of primary tumor samples classified by KNN (“normal-like” vs. “metastatic-like”) using the BCR end point for exonic (A) and non-exonic (B) features.
Next, we used logistic regression analysis to determine the odds ratio of metastatic disease progression (i.e., castrate or non-castrate resistant clinical metastatic patients) for the exonic and non-exonic KNN classifiers. The univariable analysis shows that, while the exonic set is significant (OR = 8.57, p < 0.04), the non-exonic set had more than double the odds ratio (OR = 18.13, p < 0.0003). Multivariable logistic regression further revealed that, after adjusting for clinicopathological variables using the Kattan nomogram (Kattan et al., 1999), the non-exonic KNN classifier retains a significant odds ratio for predicting metastatic disease progression (OR = 11.7, p < 0.003) whereas the exonic KNN classifier does not (OR = 9.8, p < 0.07; Table 4). These results suggest that additional prognostic information can be obtained from analysis of non-exonic RNAs and that these may have the potential to be used as biomarkers along with individual clinical variables and nomograms to enhance the prediction of metastatic disease progression post-prostatectomy.

Table 4. Multivariable logistic regression analysis for prediction of the probability of metastatic disease progression.
Discussion
One of the key challenges in prostate cancer is clinical and molecular heterogeneity (Rubin et al., 2011); therefore this common disease provides an appealing opportunity for genomic-based personalized medicine to identify diagnostic, prognostic, or predictive biomarkers to assist in clinical decision making. There have been extensive efforts to identify biomarkers based on high-throughput molecular profiling such as protein-coding mRNA expression microarrays (Sorensen and Orntoft, 2010). While many different biomarkers signatures have been identified, none of them are actively being used in clinical practice. The major reason that no new biomarker signatures have widespread use in the clinic is because they fail to show meaningful improvement for prognostication over PSA testing or established pathological variables (e.g., Gleason).
In this study, we assessed the utility of ncRNAs, and particularly non-exonic ncRNAs as potential biomarkers to be used for patients who have undergone prostatectomy but are at risk for recurrent disease and hence further treatment would be considered. We identified thousands of exonic and non-exonic RNAs differentially expressed between different tissue specimens from the MSKCC Oncogenome Project. Of the non-exonic features, the majority fall within intronic regions. This further confirms the potential utility of intronic transcripts as biomarkers, given that previous studies have shown differential expression of these ncRNAs to correlate with Gleason score (Reis et al., 2004) and with tumor vs. benign prostate tissue types (Romanuik et al., 2009). In a more focused analysis of these feature subset groups (derived from comparison of normal adjacent to primary tumor and metastatic prostate cancer) three lines of evidence showed that the non-exonic feature subset contained substantial prognostic information as measured by its ability to discriminate two clinically relevant end points. First, we observed clustering of those primary tumor samples from patients that progressed to metastatic disease with true metastatic disease samples when using the non-exonic features. Second, Kaplan–Meier analysis showed that only the KNN classifier trained on the non-exonic feature set predicts risk groups (i.e., “normal-like” and “metastatic-like”) with statistically significant differences in BCR-free survival. Finally, multivariable analysis showed that only the non-exonic KNN classifier had a statistically significant odds ratio of 11.7 for predicting metastatic disease progression in primary tumors after adjustment for Kattan nomogram.
Based on these three main results, we conclude that non-exonic RNAs contain previously unrecognized prognostic information that may be relevant in the clinic for the prediction of cancer progression post-prostatectomy. This goes in hand with the increasing evidence of ncRNAs being involved in metastasis, their key role in the regulation of protein-coding genes (Gibb et al., 2011) and their significantly higher tissue-specific expression compared to protein-coding genes (Cabili et al., 2011).
Perhaps the reason that previous efforts to develop new biomarker-based predictors of outcome in prostate cancer have not translated into the clinic is the focus on mRNA and proteins, therefore largely ignoring the wealth of information contained within the non-coding transcriptome. As more high-resolution data sets of the prostate cancer transcriptome become available (e.g., by new technologies such as RNA-Seq; Prensner et al., 2011) and as expression profiles of specific ncRNA transcripts are further validated, the results presented here can be further tested. While the clinical utility of these results require further validation on larger numbers of patients, they do show the potential of prognostic information encoded within ncRNAs, a part of the genome largely ignored in the immediate post-human genome project era.
These results add to the growing body of literature showing that the “dark matter” of the genome has potential to shed light on tumor biology, characterize aggressive cancer and improve in the prognosis and prediction of disease progression.
Conflict of Interest Statement
Ismael A. Vergara, Anamaria Crisan, Thomas Sierocinski, Christine Buerki, Nicholas Erho, Mercedeh Ghadessi, Timothy J. Triche, and Elai Davicioni are employes of GenomeDx Biosciences, Inc.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Non-Coding_RNA/10.3389/fgene.2012.00023/abstract
Table S1. Top 100 differentially expressed exonic and non-exonic features for each pairwise comparison. The features were ranked according to their adjusted p-value.
Table S2. Gene ontology and pathway enrichment analysis for all, up-regulated and down-regulated features.
Acknowledgments
We would like to thank Zaid Haddad and Benedikt Zimmermann for valuable input regarding this project. This research was supported in part by the National Research Council of Canada Industrial Research Assistance Program.
Footnotes
- ^http://www.bioconductor.org/packages/2.3/bioc/html/genefilter.html
- ^http://cran.r-project.org/web/packages/vegan/index.html
- ^www.affymetrix.com/
- ^http://www.bioconductor.org/packages/2.6/bioc/html/xmapcore.html
- ^http://cran.r-project.org/web/packages/survival/index.html
- ^http://biostat.mc.vanderbilt.edu/twiki/bin/view/Main/RmS
References
Berteaux, N., Lottin, S., Adriaenssens, E., Van Coppenolle, F., Leroy, X., Coll, J., Dugimont, T., and Curgy, J. J. (2004). Hormonal regulation of H19 gene expression in prostate epithelial cells. J. Endocrinol. 183, 69–78.
Bolla, M., Van Poppel, H., Collette, L., Van Cangh, P., Vekemans, K., Da Pozzo, L., De Reijke, T. M., Verbaeys, A., Bosset, J. F., Van Velthoven, R., Marechal, J. M., Scalliet, P., Haustermans, K., and Pierart, M. (2005). Postoperative radiotherapy after radical prostatectomy: a randomised controlled trial (EORTC trial 22911). Lancet 366, 572–578.
Bussemakers, M. J., Van Bokhoven, A., Verhaegh, G. W., Smit, F. P., Karthaus, H. F., Schalken, J. A., Debruyne, F. M., Ru, N., and Isaacs, W. B. (1999). DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 59, 5975–5979.
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., and Rinn, J. L. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927.
Catto, J. W., Alcaraz, A., Bjartell, A. S., De Vere White, R., Evans, C. P., Fussel, S., Hamdy, F. C., Kallioniemi, O., Mengual, L., Schlomm, T., and Visakorpi, T. (2011). MicroRNA in prostate, bladder, and kidney cancer: a systematic review. Eur. Urol. 59, 671–681.
Chung, S., Nakagawa, H., Uemura, M., Piao, L., Ashikawa, K., Hosono, N., Takata, R., Akamatsu, S., Kawaguchi, T., Morizono, T., Tsunoda, T., Daigo, Y., Matsuda, K., Kamatani, N., Nakamura, Y., and Kubo, M. (2011). Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 102, 245–252.
Clape, C., Fritz, V., Henriquet, C., Apparailly, F., Fernandez, P. L., Iborra, F., Avances, C., Villalba, M., Culine, S., and Fajas, L. (2009). miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS ONE 4, e7542. doi:10.1371/journal.pone.0007542
Day, J. R., Jost, M., Reynolds, M. A., Groskopf, J., and Rittenhouse, H. (2011). PCA3: from basic molecular science to the clinical lab. Cancer Lett. 301, 1–6.
Gibb, E. A., Brown, C. J., and Lam, W. L. (2011). The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 10, 38.
Holm, S. (1979). A simple sequentially rejective multiple test procedure. Scand. J. Statist. 6, 65–70.
Kapranov, P., St Laurent, G., Raz, T., Ozsolak, F., Reynolds, C. P., Sorensen, P. H., Reaman, G., Milos, P., Arceci, R. J., Thompson, J. F., and Triche, T. J. (2010). The majority of total nuclear-encoded non-ribosomal RNA in a human cell is “dark matter” un-annotated RNA. BMC Biol. 8, 149. doi:10.1186/1741-7007-8-149
Kattan, M. W., Wheeler, T. M., and Scardino, P. T. (1999). Postoperative nomogram for disease recurrence after radical prostatectomy for prostate cancer. J. Clin. Oncol. 17, 1499–1507.
Lin, R., Maeda, S., Liu, C., Karin, M., and Edgington, T. S. (2007). A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 26, 851–858.
Martens-Uzunova, E. S., Jalava, S. E., Dits, N. F., Van Leenders, G. J., Moller, S., Trapman, J., Bangma, C. H., Litman, T., Visakorpi, T., and Jenster, G. (2011). Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene. doi: 10.1038/onc.2011.304. [Epub ahead of print]..
McCall, M. N., Bolstad, B. M., and Irizarry, R. A. (2010). Frozen robust multiarray analysis (fRMA). Biostatistics 11, 242–253.
McCall, M. N., and Irizarry, R. A. (2011). Thawing Frozen Robust multi-array analysis (fRMA). BMC Bioinformatics 12, 369. doi:10.1186/1471-2105-12-369
Nakagawa, T., Kollmeyer, T. M., Morlan, B. W., Anderson, S. K., Bergstralh, E. J., Davis, B. J., Asmann, Y. W., Klee, G. G., Ballman, K. V., and Jenkins, R. B. (2008). A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS ONE 3, e2318. doi: 10.1371/journal.pone.0002318
Petrovics, G., Zhang, W., Makarem, M., Street, J. P., Connelly, R., Sun, L., Sesterhenn, I. A., Srikantan, V., Moul, J. W., and Srivastava, S. (2004). Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene 23, 605–611.
Poliseno, L., Salmena, L., Zhang, J., Carver, B., Haveman, W. J., and Pandolfi, P. P. (2010). A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465, 1033–1038.
Porkka, K. P., Pfeiffer, M. J., Waltering, K. K., Vessella, R. L., Tammela, T. L., and Visakorpi, T. (2007). MicroRNA expression profiling in prostate cancer. Cancer Res. 67, 6130–6135.
Prensner, J. R., Iyer, M. K., Balbin, O. A., Dhanasekaran, S. M., Cao, Q., Brenner, J. C., Laxman, B., Asangani, I. A., Grasso, C. S., Kominsky, H. D., Cao, X., Jing, X., Wang, X., Siddiqui, J., Wei, J. T., Robinson, D., Iyer, H. K., Palanisamy, N., Maher, C. A., and Chinnaiyan, A. M. (2011). Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 29, 742–749.
Reis, E. M., Nakaya, H. I., Louro, R., Canavez, F. C., Flatschart, A. V., Almeida, G. T., Egidio, C. M., Paquola, A. C., Machado, A. A., Festa, F., Yamamoto, D., Alvarenga, R., Da Silva, C. C., Brito, G. C., Simon, S. D., Moreira-Filho, C. A., Leite, K. R., Camara-Lopes, L. H., Campos, F. S., Gimba, E., Vignal, G. M., El-Dorry, H., Sogayar, M. C., Barcinski, M. A., Da Silva, A. M., and Verjovski-Almeida, S. (2004). Antisense intronic non-coding RNA levels correlate to the degree of tumor differentiation in prostate cancer. Oncogene 23, 6684–6692.
Romanuik, T. L., Ueda, T., Le, N., Haile, S., Yong, T. M., Thomson, T., Vessella, R. L., and Sadar, M. D. (2009). Novel biomarkers for prostate cancer including noncoding transcripts. Am. J. Pathol. 175, 2264–2276.
Rubin, M. A., Maher, C. A., and Chinnaiyan, A. M. (2011). Common gene rearrangements in prostate cancer. J. Clin. Oncol. 29, 3659–3668.
Schaefer, A., Jung, M., Mollenkopf, H. J., Wagner, I., Stephan, C., Jentzmik, F., Miller, K., Lein, M., Kristiansen, G., and Jung, K. (2010). Diagnostic and prognostic implications of microRNA profiling in prostate carcinoma. Int. J. Cancer 126, 1166–1176.
Sevli, S., Uzumcu, A., Solak, M., Ittmann, M., and Ozen, M. (2010). The function of microRNAs, small but potent molecules, in human prostate cancer. Prostate Cancer Prostatic Dis. 13, 208–217.
Siegel, R., Ward, E., Brawley, O., and Jemal, A. (2011). Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 61, 212–236.
Simmons, M. N., Stephenson, A. J., and Klein, E. A. (2007). Natural history of biochemical recurrence after radical prostatectomy: risk assessment for secondary therapy. Eur. Urol. 51, 1175–1184.
Sorensen, K. D., and Orntoft, T. F. (2010). Discovery of prostate cancer biomarkers by microarray gene expression profiling. Expert Rev. Mol. Diagn. 10, 49–64.
Srikantan, V., Zou, Z., Petrovics, G., Xu, L., Augustus, M., Davis, L., Livezey, J. R., Connell, T., Sesterhenn, I. A., Yoshino, K., Buzard, G. S., Mostofi, F. K., Mcleod, D. G., Moul, J. W., and Srivastava, S. (2000). PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 97, 12216–12221.
Taylor, B. S., Schultz, N., Hieronymus, H., Gopalan, A., Xiao, Y., Carver, B. S., Arora, V. K., Kaushik, P., Cerami, E., Reva, B., Antipin, Y., Mitsiades, N., Landers, T., Dolgalev, I., Major, J. E., Wilson, M., Socci, N. D., Lash, A. E., Heguy, A., Eastham, J. A., Scher, H. I., Reuter, V. E., Scardino, P. T., Sander, C., Sawyers, C. L., and Gerald, W. L. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22.
Thompson, I. M., Tangen, C. M., Paradelo, J., Lucia, M. S., Miller, G., Troyer, D., Messing, E., Forman, J., Chin, J., Swanson, G., Canby-Hagino, E., and Crawford, E. D. (2009). Adjuvant radiotherapy for pathological T3N0M0 prostate cancer significantly reduces risk of metastases and improves survival: long-term followup of a randomized clinical trial. J. Urol. 181, 956–962.
Wiegel, T., Bottke, D., Steiner, U., Siegmann, A., Golz, R., Storkel, S., Willich, N., Semjonow, A., Souchon, R., Stockle, M., Rube, C., Weissbach, L., Althaus, P., Rebmann, U., Kalble, T., Feldmann, H. J., Wirth, M., Hinke, A., Hinkelbein, W., and Miller, K. (2009). Phase III postoperative adjuvant radiotherapy after radical prostatectomy compared with radical prostatectomy alone in pT3 prostate cancer with postoperative undetectable prostate-specific antigen: ARO 96-02/AUO AP 09/95. J. Clin. Oncol. 27, 2924–2930.
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., Gil, J., Walsh, M. J., and Zhou, M. M. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674.
Yates, T. (2010). Xmapcore: Core Access to the Xmap Database. Available at: http://xmap.picr.man.ac.uk
Zaman, M. S., Chen, Y., Deng, G., Shahryari, V., Suh, S. O., Saini, S., Majid, S., Liu, J., Khatri, G., Tanaka, Y., and Dahiya, R. (2010). The functional significance of microRNA-145 in prostate cancer. Br. J. Cancer 103, 256–264.
Appendix
Steps for the detection of differentially expressed features
1) Download raw CEL files from http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE21034
2) Pre-process all exon arrays using the fRMA algorithm (McCall et al., 2010; McCall and Irizarry, 2011) with custom fRMA vectors created from the files obtained in step 1.fRMA can be obtained from http://www.bioconductor.org/packages/release/bioc/html/frma.html
3) Exclude all probe selection regions (PSRs) annotated as “unreliable” by the xmapcore package (Yates, 2010; one or more probes do not align uniquely to the genome) as well as those not defined as class 1 cross-hybridizing by Affymetrix annotation (http://www.affymetrix.com).
4) Classify each PSR as “exonic” if they overlap with protein-coding regions or UTRs according to the xmapcore package annotation, and as “non-exonic” if they do not (this can be achieved with the “coding.probe sets” and “utr.probe sets” functions).
Then, the following steps need to be pursued separately for each pairwise comparison: (i) normal vs. primary, (ii) primary vs. metastatic, and (iii) normal vs. metastatic. For the normal vs. primary comparison, only matched samples were used and for the primary vs. metastatic comparison the matched samples were excluded.
5) Calculate the background expression level by taking the median of the Affymetrix defined anti-genomic PSRs (http://www.affymetrix.com/Auth/support/downloads/library_files/HuEx-1_0-st-v2.r2.zip; file HuEx-1_0-st-v2.r2.antigenomic.bgp). For each PSR, calculate the median expression level for each group. Filter PSRs where the median expression levels for both groups are below the background expression level.
6) Apply the rowttests function of the genefilter R package available at http://www.bioconductor.org/packages/2.3/bioc/html/genefilter.html in order to perform a t-test on each PSR.
7) Adjust the obtained p-values using the p.adjust function of the stats package in R for each group of PSRs (exonic and non-exonic) separately. Select the Holm–Bonferroni method for this purpose (Holm, 1979).
8) Filter out those PSRs that have an adjusted p-value higher than 0.05.
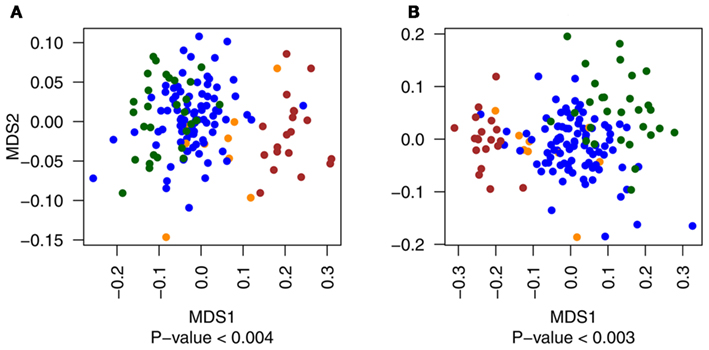
Figure A1. Multidimensional scaling plots of the distribution of primary tumor samples with (yellow) and without (blue) metastatic events compared to metastatic (red) and normal (green) tissues for exonic (A) and non-exonic (B) features. Metastatic and normal data points are included in the figure for illustrative purposes only.
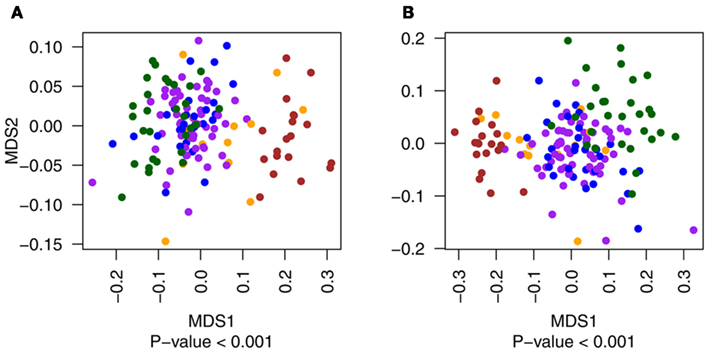
Figure A2. Multidimensional scaling plots of the distribution of primary tumor samples with Gleason score of 6 (blue), 7 (purple), 8 and 9 (both in yellow) compared to metastatic (red) and normal (green) tissues for exonic (A) and non-exonic (B) features. Metastatic and normal data points are included in the figure for illustrative purposes only.
Keywords: prostate cancer, prognosis, microarrays, clinical progression, non-coding RNA
Citation: Vergara IA, Erho N, Triche TJ, Ghadessi M, Crisan A, Sierocinski T, Black PC, Buerki C and Davicioni E (2012) Genomic “dark matter” in prostate cancer: exploring the clinical utility of ncRNA as biomarkers. Front. Gene. 3:23. doi: 10.3389/fgene.2012.00023
Received: 14 December 2011;
Paper pending published: 27 December 2011;
Accepted: 04 February 2012;
Published online: 22 February 2012.
Edited by:
Philipp Kapranov, St. Laurent Institute, USAReviewed by:
David Ting, Massachusetts General Hospital Cancer Center, USARobert Arceci, Johns Hopkins, USA
Eduardo M. Reis, Universidade de Sao Paulo, Brazil
Copyright: © 2012 Vergara, Erho, Triche, Ghadessi, Crisan, Sierocinski, Black, Buerki and Davicioni. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Timothy J. Triche, GenomeDx Biosciences, Inc., 201-1595 West 3rd Avenue, Vancouver, BC, Canada V6J 1J8. e-mail:dGltQGdlbm9tZWR4LmNvbQ==
†Ismael A. Vergara and Nicholas Erho have contributed equally to this work.