


- 1 Center for RNA Molecular Biology, Case Western Reserve University School of Medicine, Cleveland, OH, USA
- 2 Department of Genetics, Case Western Reserve University School of Medicine, Cleveland, OH, USA
- 3 Department of Biochemistry, Case Western Reserve University School of Medicine, Cleveland, OH, USA
- 4 Case Comprehensive Cancer Center, Case Western Reserve University School of Medicine, Cleveland, OH, USA
The recent discovery of thousands of long non-coding (lnc)RNAs in the human genome has prompted investigation of the potential roles of these molecules in human biology and medicine. Indeed, it is now well documented that many lncRNAs are involved in key biological processes, including dosage compensation, genomic imprinting, chromatin regulation, alternative splicing of pre-mRNA, nuclear organization; and potentially many other biological processes, which are yet to be elucidated. Recently, a number of studies have also reported that lncRNAs are dysregulated in a number of human diseases, including several cancers and neurological disorders. Although many of these studies have fallen short of implicating lncRNAs as causative, they suggest potential roles that warrant further in depth investigations. In this review, we discuss the current state of knowledge regarding the roles of lncRNAs in cancer and neurological disorders, and suggest potential future directions in this rapidly emerging field.
Introduction
A number of studies over the past decade have now shown that mammalian genomes encode thousands of long non-coding RNAs (lncRNAs), which are mRNA-like transcripts that lack protein-coding capacity. It is now estimated that the human and mouse genomes produce almost as many lncRNAs as mRNAs (Carninci et al., 2005; Mattick and Makunin, 2006; Carninci and Hayashizaki, 2007; He et al., 2008; Guttman et al., 2009; Khalil et al., 2009; Cabili et al., 2011). More importantly, recent studies have demonstrated that lncRNAs are involved in key biological processes including dosage compensation, genomic imprinting, chromatin regulation, alternative splicing of pre-mRNA and nuclear organization. Furthermore, some lncRNAs have been shown to regulate the expression of hundreds of protein-coding genes by various mechanisms (Khalil et al., 2009; Gupta et al., 2010; Tsai et al., 2010; Guttman et al., 2011; Nagano and Fraser, 2011). For example, the lncRNA Xist, which is required for X chromosome inactivation (Xi) in mammalian females (Brockdorff et al., 1992; Brown et al., 1992), represses hundreds of genes during Xi by recruiting and tethering repressive chromatin-modifying complexes to the inactive X chromosome (Zhao et al., 2008; Jeon and Lee, 2011). Perhaps equally interesting is the tangled web of several lncRNAs that are also involved in this process by regulating the expression and action of Xist (Lee et al., 1999; Ogawa and Lee, 2003).
Another lncRNA, termed HOTAIR, was shown to regulate the expression of hundreds of genes genome-wide in trans (Rinn et al., 2007; Khalil et al., 2009; Gupta et al., 2010; Tsai et al., 2010). HOTAIR functions by binding to both the polycomb repressive complex 2 (PRC2) and the histone demethylase LSD1, and facilitate their co-recruitment to specific regions of the genome (Rinn et al., 2007; Khalil et al., 2009; Gupta et al., 2010; Tsai et al., 2010). Subsequently, PRC2 and LSD1 alter histone modifications to induce heterochromatin formation and gene repression (Kouzarides, 2007). Combined, these observations illustrate how a single lncRNA can regulate the expression of hundreds of protein-coding genes; and thus, the dysregulation of such a lncRNA could contribute to the dysregulation of hundreds of genes leading to abnormal effects on the cell and the organism (Hu et al., 2011; Wapinski and Chang, 2011). Indeed, a number of recent publications have reported that lncRNAs are dysregulated in a wide range of human diseases and disorders. In this review, we will focus on the potential roles that lncRNAs may play in cancer and neurological disorders, as these disorders are the most commonly reported to be associated with dysregulation of lncRNAs.
Potential Roles for lncRNAs in Cancer Etiology
Cancer is a devastating human disease that remains a challenge to treat or cure. This is due to the ability of cancerous cells to proliferate rapidly, resist apoptosis, evade the immune system, and metastasize to new regions within the body. Despite the identification of over 30 tumor suppressor genes and 100 oncogenes within the human genome, it is yet to be determined how mutations of these genes contribute to the initiation and metastasis of various cancer types (Hudson, 2011). Recent studies utilizing next generation sequencing technologies complicated this matter further when it was shown that many cancer patients did not harbor mutations in these protein-coding genes (Hudson, 2011). Intriguingly, several studies in the past few years have reported that a subset of lncRNAs is dysregulated in various cancers (Luo et al., 2006; Perez et al., 2008; Silva et al., 2010). Furthermore, some of these lncRNAs are strongly correlated with poor patient prognosis and/or metastasis, suggesting a potential role that should be further investigated. Currently, the detailed functions and mechanisms of these lncRNAs are not completely known, however, they appear to be involved in cellular proliferation and invasiveness, which may explain their potential roles in cancer etiology (Table 1).
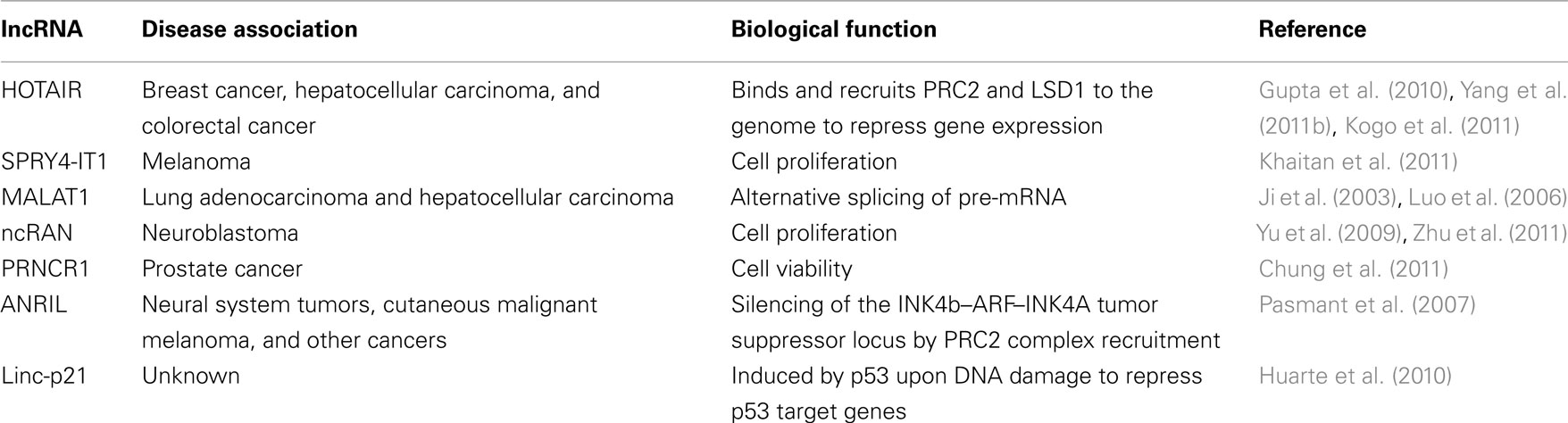
Table 1. Examples of IncRNAs that are dysregulated in cancer.
Several studies have now shown that the lncRNA HOTAIR is highly expressed in breast tumors, hepatocellular carcinoma, and colorectal cancer; and strongly correlates with poor patient prognosis and metastasis (Gupta et al., 2010; Kogo et al., 2011; Yang et al., 2011b). HOTAIR is expressed from the HOXC gene cluster and negatively regulates the expression of hundreds of genes genome-wide in trans (Rinn et al., 2007; Khalil et al., 2009; Gupta et al., 2010; Tsai et al., 2010; Chu et al., 2011). In vitro and in vivo studies suggest that HOTAIR is involved in metastasis by unknown mechanisms (Gupta et al., 2010). However, since HOTAIR is known to interact with several chromatin-modifying complexes, one can envision that the upregulation of HOTAIR, which is observed in several cancers, causes changes in the epigenomic landscape of cancer cells resulting in altered gene expression programs that promote an invasive phenotype (Figure 1A; Rinn et al., 2007; Khalil et al., 2009; Gupta et al., 2010; Tsai et al., 2010). Indeed, in vitro studies have shown that over-expression of HOTAIR in cell lines leads to the recruitment of the repressive complex PRC2 to over 800 additional loci genome-wide including tumor suppressor genes (Gupta et al., 2010). HOTAIR-mediated repression of these tumor suppressor genes could be responsible for the increased proliferation and invasiveness phenotype observed, but this hypothesis awaits further experimental evidence.
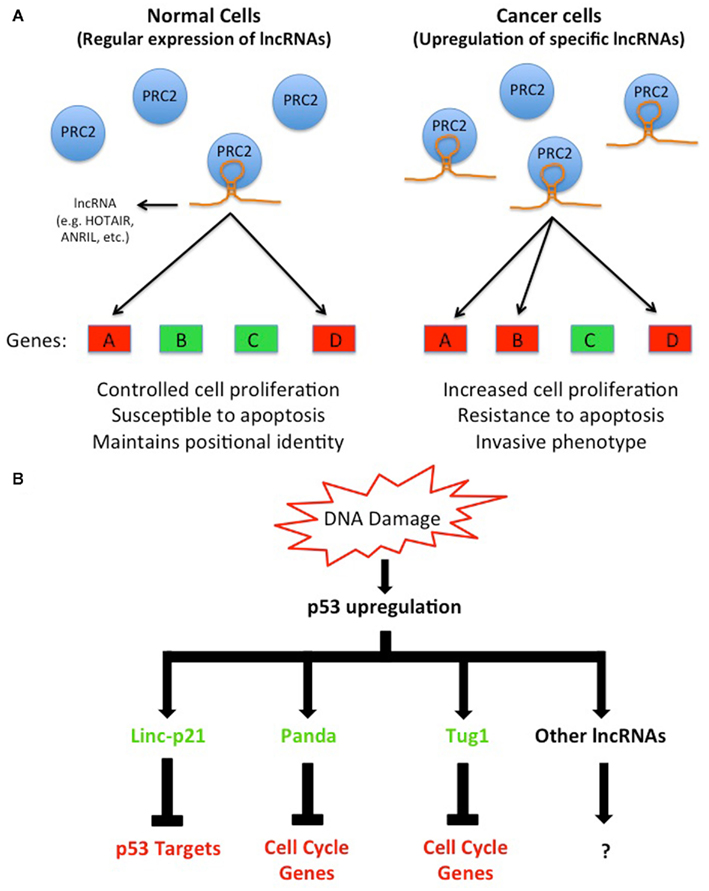
Figure 1. Dysregulation of lncRNAs in cancer cells alters gene expression by a variety of mechanisms. (A) A subset of lncRNAs (e.g., HOTAIR, ANRIL, and others) guide chromatin-modifying complexes such as PRC2 in cis or in trans to specific genes to modulate their expression. However, the upregulation of these lncRNAs, which is observed in several cancers, leads to non-specific guidance of chromatin-modifying complexes to the genome that alters normal gene expression programs. For example, the non-specific guidance of PRC2 by an upregulated lncRNA in cancer cells to one or more tumor suppressor genes leads to their repression; and consequently affects cells ability to regulate their normal proliferation pattern or induce apoptosis. Conversely, an upregulated lncRNA that leads an activating chromatin-modifying complex to oncogenes may also perturb normal cellular functions (not shown). (B) A group of lncRNAs is known to be induced directly by p53, which can be activated by various stimuli including DNA damage. These lncRNAs, once activated, modulate gene expression by a variety of mechanisms via their protein partners.
Similar to HOTAIR, the lncRNA ANRIL is also elevated in several cancers, and GWAS studies have linked ANRIL with acute lymphoblastic leukemia, glioma, basal cell carcinoma, nasopharyngeal carcinoma, breast cancer, and plexiform neurofibromas (Popov and Gil, 2010; Iacobucci et al., 2011; Pasmant et al., 2011a,b). ANRIL is an antisense lncRNA to several genes involved in cell cycle regulation: p15/INK4b, p16/INK4a, and p14/ARF (Pasmant et al., 2007). Studies of ANRIL have provided some insights into its mechanism of action. It is thought that ANRIL functions by directing the chromatin-modifying complexes PRC1 and PRC2 to chromatin, since the disruption of ANRIL interaction with PRC1 or knockdown of ANRIL abolishes the repression of INK4b/ARF/INK4a locus, which is a known target of PRC1 and PRC2 (Yap et al., 2010; Kotake et al., 2011). The elevated expression of ANRIL in cancer, and consequently the repression of the INK4b/ARF/INK4a locus, prevents these tumor suppressor genes from properly regulating the cell cycle. In essence, ANRIL and HOTAIR operate by similar mechanisms; that is by altering chromatin structure either locally or over long distances to repress the expression of tumor suppressor genes. Indeed, it is now well documented that the epigenomes of cancer cells are distinct from normal cells (Baylin and Jones, 2011); nevertheless, the molecular events that lead to these profound changes in cancer epigenomes are not completely understood. Environmental or genetics factors that cause lncRNAs to become dysregulated may turn out to be important players in such events. Further studies are needed to determine how the expression of lncRNAs, such as HOTAIR and ANRIL, become initially upregulated in cancer, and whether the elevated expression is sufficient to cause tumorigenesis and/or metastasis. Animal models might prove invaluable to get to the bottom of these key questions.
Recent studies have also identified lncRNAs that are induced in response to DNA damage in a p53-dependent manner, including linc-P21, PANDA, Tug1, and others (Guttman et al., 2009; Khalil et al., 2009; Huarte et al., 2010; Hung et al., 2011). Although these lncRNAs are all activated by p53, their mechanisms of action appear to be distinct (Figure 1B). For example, linc-P21 represses hundreds of genes in the p53 pathway by modulating hnRNPK localization to chromatin (Huarte et al., 2010), while PANDA interacts with the transcription factor NF-YA to reduce the expression of pro-apoptotic genes (Hung et al., 2011). Meanwhile, Tug1, which regulates cell cycle genes, functions via its interaction with the chromatin-modifying complex PRC2 and Pc2 (Khalil et al., 2009; Yang et al., 2011a).
The observations that p53 can induce the expression of specific lncRNAs are intriguing since they provide new insights into p53-mediated gene regulation. Although transcriptional activation by p53 is well studied, its ability to repress transcription remains enigmatic. The induction of specific lncRNAs by p53 that leads to gene repression could be part of the mechanism by which p53 represses its target genes. Thus, mutations in p53 that hinder its ability to activate these lncRNAs, or mutations in the p53 binding site in the promoter of one or more of these lncRNAs may contribute to cancer as they may cause p53 to lose its ability to repress downstream target genes. Ongoing whole genome sequencing of cancer genomes may uncover potential mutations in the promoters of lncRNAs that prevent p53 binding. Also, an in depth understanding of p53 induced lncRNAs may provide novel insights into cancer initiation and/or progression.
In addition to regulation of gene expression by lncRNAs at the transcriptional level, other lncRNAs regulate gene expression post-transcriptionally. Such lncRNAs, when dysregulated, can also lead to undesired biological consequences. For example, the lncRNA MALAT1 (Metastasis Associated Lung Adenocarcinoma Transcript 1), which is associated with high incidence of metastasis in lung tumors and poor patient prognosis (Ji et al., 2003), regulates alternative splicing of pre-mRNAs by modulating the phosphorylation of SR proteins (Tripathi et al., 2010). However, at this stage we can only speculate that the dysregulation of MALAT1 and consequently the altered ratios of alternative splicing products may contribute to cancer, for example, by increasing the production of protein isoforms that promote cell proliferation and invasiveness. This hypothesis is supported by studies that have reported altered ratios of spliced mRNAs in cancer cells and tissues (David and Manley, 2010).
The lncRNAs SPRY4-IT1, ncRAN, and PRNCR1 and others have also been shown to be dysregulated in various cancers; however, the mechanisms of these lncRNAs are not currently known (Table 1; Yu et al., 2009; Chung et al., 2011; Khaitan et al., 2011). By in vitro studies, all of the three aforementioned lncRNAs appear to affect cell viability or proliferation (Yu et al., 2009; Chung et al., 2011; Khaitan et al., 2011; Zhu et al., 2011). Importantly, depletion of the lncRNA ncRAN makes cancer cells more susceptible to treatments with chemotherapeutic agents in vitro (Zhu et al., 2011). Uncovering the molecular mechanisms of these lncRNAs will be invaluable in understanding why cancer cells become more susceptible to chemotherapy when a specific lncRNA levels are reduced. Also, future studies to validate these observations in vivo will be critical as in vitro studies have many caveats, and may not always reflect actual physiological conditions. Nevertheless, these observations, although preliminary, are potentially exciting since they suggest that combined chemotherapy and siRNAs against specific lncRNAs may prove to be a powerful approach in treating resistant tumors. Currently, the technology to target lncRNAs in vivo still requires years of research in animal models first, and then in clinical trials. If successful, such technology may provide new therapeutic strategies that increase the percentage of effective cancer treatments.
In summary, lncRNAs may prove to be potential drug targets for cancer therapies. These potential treatments can be combined with conventional methods to treat difficult cases or tumors that are highly resistant to conventional methods alone. Finally, elucidating the exact functions and mechanisms of lncRNAs that are dysregulated in cancer may provide fundamental new insights into cancer biology in general.
Potential Roles for lncRNAs in Neurological Disorders
Environmental and/or genetic factors can give rise to many types of neurological disorders that result in irregular brain function. Several mechanisms by which such factors lead to neurological disorders have been proposed including protein aggregation, oxidative stress, improper inflammatory response, and increased apoptosis (Double et al., 2010). A hallmark of neurological disorders is that they affect specific regions within the brain, while other regions remain functional (Double et al., 2010). Currently, a full understanding of neurological disorders and adequate therapies are both lacking. Previously, it was shown that hundreds of lncRNAs show specific localization to neuroanatomical regions, cell types, or subcellular compartments within the brain (Mercer et al., 2008) suggesting that a subset of these lncRNAs could contribute to neurological disorders when they become dysregulated. In this section, we highlight several studies that have reported dysregulation of lncRNAs in neurological disorders (Table 2), and attempt to provide some insights into their potential mechanisms of action.
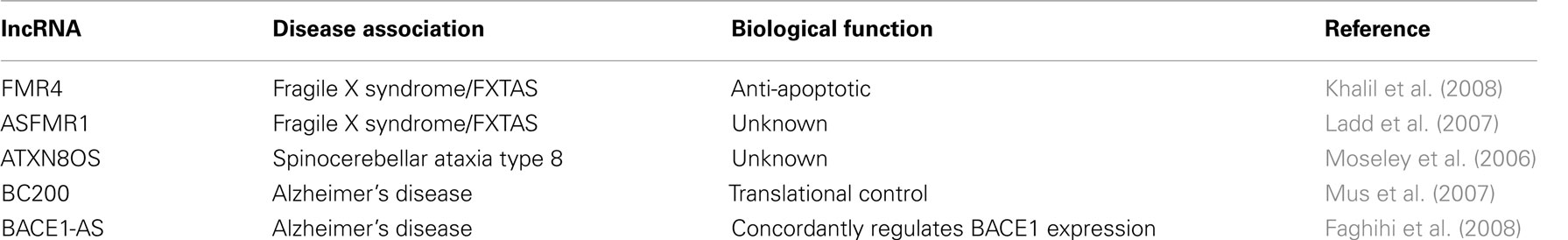
Table 2. Examples of IncRNAs that are dysregulated in neurological disorders.
Dysregulation of LncRNAs in Trinucleotide Repeat Neurological Disorders
Trinucleotide repeat disorders are a set of genetic disorders caused by the expansion of specific trinucleotide sequences (e.g., CAG, CGG, etc.) above a certain threshold leading to defects in the expression or functions of specific genes. Such disorders include fragile X syndrome (FXS), fragile X tremor ataxia syndrome (FXTAS), Huntington’s disease, spinocerebellar ataxias, and others. In addition to protein-coding genes, the expression, and/or function of several lncRNAs have been shown to be affected by trinucleotide repeats raising the possibility that these lncRNAs may also contribute to the disease phenotype.
Previously, both Khalil et al. (2008) and Ladd et al. (2007) reported the discovery of two lncRNAs, FMR4, and ASFMR1 that are expressed from the fragile X mental retardation 1 gene (FMR1) locus; which is associated with FXS, FXTAS, and potentially autism (Penagarikano et al., 2007; Gallagher and Hallahan, 2011; Sokol et al., 2011). FXS is an X-linked neurological disorder and the leading cause of inherited mental retardation; and FXTAS is a late onset neurodegenerative disorder (Gallagher and Hallahan, 2011). Both disorders result from CGG trinucleotide repeat expansion in the 5′ UTR of FMR1 (Verkerk et al., 1991; Oostra and Willemsen, 2003; Godler et al., 2010; Gallagher and Hallahan, 2011). Subsequent studies to the discovery of the CGG expansion in the FMR1 locus demonstrated that there is high variability in the phenotype of FXS and FXTAS patients that could not be simply explained by FMR1 levels alone (Tassone et al., 2001; Gallagher and Hallahan, 2011). Thus, raising the possibility that dysregulation of FMR4 and ASFMR1 could be contributing to the variability in phenotype observed in these patients.
Both FMR4 and ASFMR1, similar to FMR1, are upregulated in FXTAS and are repressed in FXS patients (Ladd et al., 2007; Khalil et al., 2008). However, the underlying molecular mechanisms of FMR4 and ASFMR1 are yet to be determined. In vitro studies of FMR4 have shown that it has an anti-apoptotic function in human cells (Khalil et al., 2008). It is possible that FMR4 anti-apoptotic function protects neurons or their progenitors during development from apoptosis. This is consistent with a recent discovery from Harvey Lodish’s laboratory that the lncRNA ESlncRNA, which also has an anti-apoptotic function, protects progenitors of red blood cells from apoptosis (Hu et al., 2011). However, since FMR4 is only conserved in primates, it is difficult to assess its function in animal models such as mouse or Drosophila. One intriguing possibility is that FMR1, FMR4, and ASFMR1 work cooperatively in a RNA–protein network, and disruption of these delicate interactions may lead to undesired effects on brain function. Indeed, this may explain the wide range of variability in phenotype observed in FXS and FXTAS, as the magnitude of deviation from the physiological levels of FMR4 and/or ASFMR1 can lead to less or more severe levels of the phenotype (Penagarikano et al., 2007; Gallagher and Hallahan, 2011).
Long non-coding RNAs have also been reported to be dysregulated in spinocerebellar ataxia type 8 (SCA8), which is a neurodegenerative disease of the cerebellum that affects muscle and speech coordination (Koob et al., 1999; Mutsuddi et al., 2004). Within the SCA8 expansion region two genes were identified: the protein-coding gene ataxin 8 (ATXN8) with a CAG expansion that encodes a polyglutamine expansion tract protein, and ataxin 8 opposite strand (ATXN8OS), which is a lncRNA with a CUG repeat. Transgenic mice expressing both transcripts with (CTG)116 or with (CTG)11 repeat expansions demonstrated that the (CTG)116 but not (CTG)11 develop a progressive neurological phenotype (Moseley et al., 2006). Supporting a role for the lncRNA in the disease phenotype is a study in Drosophila where dysregulation of the lncRNA from the SCA8 region leads to neurodegeneration of the retina (Mutsuddi et al., 2004).
Currently, how a repeat expansion affects the function of a lncRNA is not completely understood; however, it is known that aberrant RNAs with a repeat expansion forms secondary structures that bind and sequester the splicing factor MBNL1 causing aberrant pre-mRNA splicing (Yuan et al., 2007). Indeed, a study in mice over-expressing ATXN8OS with the repeat expansion found that it also becomes associated with MBNL1 in neurons leading to aberrant splicing of GABT4 (GABA-A transporter 4) and loss of GABAergic inhibition in the granular cell layer, which is postulated to contribute to the observed phenotype (Daughters et al., 2009).
Collectively, these studies demonstrate that repeat expansions not only in protein-coding genes but also in lncRNAs may contribute to certain human neurological disorders. Some have referred to this phenomenon as RNA gain of function or RNA toxicity (Daughters et al., 2009). Studies in FXTAS patients have also found evidence that FMR1 mRNA with the repeat expansion remains sequestered in the nucleus, thus explaining the low levels of FMRP in these patients despite having higher levels of FMR1 mRNA than normal individuals (Tassone et al., 2004; Hagerman and Hagerman, 2007). It is quite conceivable that other lncRNAs with either a repeat expansion or mutations that change their protein-binding capacity could also lead to RNA gain of function that contributes to other human disorders with unknown mechanisms. The recent advances in RNA sequencing technologies may prove to be instrumental in discovering such cases by first identifying mutations in lncRNAs associated with human disease, and then experimentally testing the effects of such mutations on the function of lncRNAs.
Dysregulation of lncRNAs in Alzheimer’s Disease
Alzheimer’s disease (AD) is a form of dementia that gradually gets worse over time, affecting memory, cognition, and behavior (Goedert and Spillantini, 2006; Ballard et al., 2011). A common characteristic of AD is the formation of protein aggregates in neurons (Sokol et al., 2011). These aggregates, called amyloid plaques, are formed from the shift in processing of the amyloid-β precursor protein, APP, by the α-, β-, and γ-secretases (Minati et al., 2009; Ballard et al., 2011). This shift results in increased levels of the amyloid-β protein product, Aβ 1–42, which is the primary protein of the aggregates (Minati et al., 2009; Ballard et al., 2011). Normally, there is a balance between levels of Aβ 1–42 and Aβ 1–40 products, but this balance is disrupted in AD by unknown mechanisms, and is thought to cause amyloid plaques (Minati et al., 2009; Ballard et al., 2011). It is believed that the presence of these plaques is responsible for the atrophy and inflammation observed in the brains of Alzheimer’s patients. However, this conclusion has been debated (Sokol et al., 2011).
A number of studies have now shown that the expression of several lncRNAs is altered in AD patients. The primate-specific lncRNA BC200, which is expressed almost exclusively in neuronal cells and plays a role in regulating the translation of specific proteins, is dysregulated in AD (Tiedge et al., 1993; Muddashetty et al., 2002). Although the expression of BC200 steadily declines in the brain under normal aging conditions, it becomes upregulated in AD patients, and the severity of AD correlates with levels of BC200 (Mus et al., 2007). Functionally, BC200 has been implicated in regulating the translation of specific proteins in somatodendritic domains of neurons, and thus its upregulation in AD patients may lead to changes in protein production that are responsible, directly or indirectly, for plaque formation. Studies of BC1, the mouse functional homolog of BC200, have shown that BC1 knockout mice exhibit behavioral changes, demonstrating an important role for BC1 in brain function (Lewejohann et al., 2004). A deeper understanding of BC200 and BC1 regulation may provide new insights into AD pathogenesis.
The lncRNA BACE1-AS, which is transcribed antisense to the protein-coding gene BACE1, is also highly expressed in AD patients (Faghihi et al., 2008). BACE1 is a β-secretase that is responsible for APP cleavage into Aβ, and is the major Aβ producing β-secretase in the brain (Vassar et al., 2009). BACE1 expression is regulated post-transcriptionally by BACE1-AS both in vitro and in vivo (Faghihi et al., 2008). Importantly, levels of both the Aβ 1–40 and Aβ 1–42 become reduced upon BACE1-AS knockdown, without affecting APP levels (Faghihi et al., 2008). This suggests that BACE1-AS may regulate the levels of pathogenic Aβ by a shift in the processing ability of APP by BACE1. Collectively, these findings suggest that at least under some conditions, BACE1-AS upregulation may be responsible for the elevated levels of BACE1 seen in AD patients, and indicate a mechanism in which BACE1-AS upregulation serves to alter APP processing toward Aβ 1–42 (Faghihi et al., 2008). Further studies are needed to determine at which stage of AD the upregulation of BACE1-AS occurs, and if it is sufficient for AD pathogenesis.
Potential Mechanisms of lncRNAs and Implications for Human Disease
Although the full range of mechanisms of lncRNAs is yet to be elucidated, a number of studies have now provided some insights into these mechanisms. Here we will only highlight known mechanisms of lncRNAs as a full discussion of this topic has been published recently (Wang and Chang, 2011).
lncRNAs as Cellular “Navigation Systems”
For the past few years, much attention has been focused on the role of lncRNAs in regulating gene expression via their interactions with chromatin-modifying complexes. Previously, two genome-wide studies using RNA co-immunoprecipitation assays found numerous nuclear lncRNAs to be physically associated with chromatin-modifying complexes including PRC2, CoREST, SMCX, and others, which are responsible for regulating the expression of hundreds of genes (Khalil et al., 2009; Zhao et al., 2010). Moreover, these lncRNAs regulate similar genes to their protein-binding partners by unknown mechanisms (Khalil et al., 2009; Zhao et al., 2010; Guttman et al., 2011). Subsequent studies indicated that some of these lncRNAs may function by directing chromatin-modifying complexes to their genomic targets (Gupta et al., 2010; Tsai et al., 2010). In essence, some lncRNAs may turn out to be cellular “navigation systems” for proteins lacking direct DNA binding capacity.
The widespread interactions between lncRNAs and chromatin-modifying complexes suggest that disruption of these interactions could have profound effects on gene expression by misdirecting chromatin-modifying complexes to the genome. For example, a dysregulated lncRNA that directs a repressive chromatin-modifying complex inappropriately to a tumor suppressor gene, or away from an oncogene, may result in a similar outcome as a mutation in either one of these types of genes. Further studies are needed to uncover how lncRNAs recognize their protein-binding partners while excluding the thousands of other proteins in the cell. These studies may lead to the development of bioinformatic tools that can be used to predict lncRNA structures, and how some mutations in lncRNAs may lead to inappropriate lncRNA–protein interactions (Khalil and Rinn, 2011). Unfortunately, prediction of secondary structures of lncRNAs is an area that is still in its infancy, and may require tremendous efforts of research and innovation.
LncRNAs as Molecular Scaffolds
Some lncRNAs have been shown to play structural roles that are critical for organizing specific compartments within the cell. It has been known for over 20 years that Xist is required for the formation of the Barr body, which appears as a condensed chromatin usually at the periphery of nuclei (Brown et al., 1992; Chadwick and Willard, 2003). The lncRNAs MALAT1 and NEAT1 serve as molecular scaffolds for proteins in nuclear speckles and paraspeckles, respectively. Recently, HOTAIR was shown to also serve as a molecular scaffold for PRC2 and LSD1 at endogenous target genes (Tsai et al., 2010). It is very likely that many other lncRNAs serve as scaffolds similar to the lncRNAs mentioned above, as numerous lncRNAs are known to bind multiple protein complexes (Pandey et al., 2008; Khalil et al., 2009; Gupta et al., 2010; Tsai et al., 2010; Guttman et al., 2011). Nonetheless, lncRNAs recognition of their protein partners is a missing link in our understanding of this mechanism of action. Finally, mutations in lncRNAs, which function as molecular scaffolds for proteins, may result in disease state by altering the localization of proteins, or by altering the composition of nuclear and/or cytoplasmic compartments.
lncRNA as miRNA “Sponges”
Recently, lncRNAs have been implicated in regulating gene expression post-transcriptionally by binding to microRNAs (miRNAs) and preventing them from regulating their mRNA targets (Cesana et al., 2011). This finding is intriguing since it adds a new layer to post-transcriptional gene regulation by involving two distinct classes of non-coding RNAs. However, a number of key questions regarding this mechanism of action are yet to be elucidated. (1) What are the factors/proteins that regulate lncRNA interactions with miRNAs? (2) Do lncRNAs, which bind to miRNAs, become targeted for deadenylation and consequently degradation similar to mRNAs? (3) What are the dynamics of lncRNAs interactions with miRNAs? These key questions and others must be addressed to understand this novel mechanism of post-transcriptional gene regulation.
MicroRNAs have been previously implicated in a wide range of human diseases including cancer and neurological disorders (Bartel, 2004; Mattick and Makunin, 2005; Cho, 2007; Ikeda et al., 2007; Huang et al., 2008; Rosenfeld et al., 2008). However, the mechanisms by which miRNAs contribute to many of these diseases are not completely known. With a subset of lncRNAs potentially regulating the interactions of miRNAs to mRNAs, it is conceivable that dysregulation of such lncRNAs can lead to dramatic changes in mRNAs and/or protein levels reminiscent of disease state.
In summary, lncRNAs utilize various mechanisms (e.g., guidance of proteins to specific genomic loci, structural roles, and as molecular decoys, etc.) to carry out their cellular functions. In each case we have yet to fully elucidate the entire mechanism of action, nonetheless, we are beginning to get glimpses into the mysterious world of lncRNAs.
Concluding Remarks
It is now evident that lncRNAs play important roles in many key biological processes, although their mechanisms of action are yet to be fully elucidated. The abundance of information that remains to be learned about these fascinating macromolecules is vast, yet some key trends have emerged. In light of the importance of lncRNAs in regulating gene expression, it is not surprising that dysregulation of lncRNAs have been observed in disease states. While this review focused on the potential roles of lncRNAs in cancer and neurological disorders, it is likely that dysregulation of lncRNAs contributes to many other human diseases and disorders. Finally, many studies have focused simply on discovering lncRNAs that are dysregulated in human disease. However, the field must move toward functional and mechanistic studies of lncRNAs to fully appreciate the role of these novel molecules in human biology and medicine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Drs. Tim Nilsen and Jeff Coller; as well as Victoria Moran, Sarah Geisler, Mark Beno, and Colin Venner for their critical review of the article. Courtney Niland is a recipient of NIH training grant award (5T32GM008056-29).
References
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031.
Baylin, S. B., and Jones, P. A. (2011). A decade of exploring the cancer epigenome – biological and translational implications. Nat. Rev. Cancer 11, 726–734.
Brockdorff, N., Ashworth, A., Kay, G. F., McCabe, V. M., Norris, D. P., Cooper, P. J., Swift, S., and Rastan, S. (1992). The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71, 515–526.
Brown, C. J., Hendrich, B. D., Rupert, J. L., Lafreniere, R. G., Xing, Y., Lawrence, J., and Willard, H. F. (1992). The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71, 527–542.
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., and Rinn, J. L. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927.
Carninci, P., and Hayashizaki, Y. (2007). Noncoding RNA transcription beyond annotated genes. Curr. Opin. Genet. Dev. 17, 139–144.
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., Oyama, R., Ravasi, T., Lenhard, B., Wells, C., Kodzius, R., Shimokawa, K., Bajic, V. B., Brenner, S. E., Batalov, S., Forrest, A. R., Zavolan, M., Davis, M. J., Wilming, L. G., Aidinis, V., Allen, J. E., Ambesi-Impiombato, A., Apweiler, R., Aturaliya, R. N., Bailey, T. L., Bansal, M., Baxter, L., Beisel, K. W., Bersano, T., Bono, H., Chalk, A. M., Chiu, K. P., Choudhary, V., Christoffels, A., Clutterbuck, D. R., Crowe, M. L., Dalla, E., Dalrymple, B. P., De Bono, B., Della Gatta, G., Di Bernardo, D., Down, T., Engstrom, P., Fagiolini, M., Faulkner, G., Fletcher, C. F., Fukushima, T., Furuno, M., Futaki, S., Gariboldi, M., Georgii-Hemming, P., Gingeras, T. R., Gojobori, T., Green, R. E., Gustincich, S., Harbers, M., Hayashi, Y., Hensch, T. K., Hirokawa, N., Hill, D., Huminiecki, L., Iacono, M., Ikeo, K., Iwama, A., Ishikawa, T., Jakt, M., Kanapin, A., Katoh, M., Kawasawa, Y., Kelso, J., Kitamura, H., Kitano, H., Kollias, G., Krishnan, S. P., Kruger, A., Kummerfeld, S. K., Kurochkin, I. V., Lareau, L. F., Lazarevic, D., Lipovich, L., Liu, J., Liuni, S., McWilliam, S., Madan Babu, M., Madera, M., Marchionni, L., Matsuda, H., Matsuzawa, S., Miki, H., Mignone, F., Miyake, S., Morris, K., Mottagui-Tabar, S., Mulder, N., Nakano, N., Nakauchi, H., Ng, P., Nilsson, R., Nishiguchi, S., Nishikawa, S., Nori, F., Ohara, O., Okazaki, Y., Orlando, V., Pang, K. C., Pavan, W. J., Pavesi, G., Pesole, G., Petrovsky, N., Piazza, S., Reed, J., Reid, J. F., Ring, B. Z., Ringwald, M., Rost, B., Ruan, Y., Salzberg, S. L., Sandelin, A., Schneider, C., Schönbach, C., Sekiguchi, K., Semple, C. A., Seno, S., Sessa, L., Sheng, Y., Shibata, Y., Shimada, H., Shimada, K., Silva, D., Sinclair, B., Sperling, S., Stupka, E., Sugiura, K., Sultana, R., Takenaka, Y., Taki, K., Tammoja, K., Tan, S. L., Tang, S., Taylor, M. S., Tegner, J., Teichmann, S. A., Ueda, H. R., van Nimwegen, E., Verardo, R., Wei, C. L., Yagi, K., Yamanishi, H., Zabarovsky, E., Zhu, S., Zimmer, A., Hide, W., Bult, C., Grimmond, S. M., Teasdale, R. D., Liu, E. T., Brusic, V., Quackenbush, J., Wahlestedt, C., Mattick, J. S., Hume, D. A., Kai, C., Sasaki, D., Tomaru, Y., Fukuda, S., Kanamori-Katayama, M., Suzuki, M., Aoki, J., Arakawa, T., Iida, J., Imamura, K., Itoh, M., Kato, T., Kawaji, H., Kawagashira, N., Kawashima, T., Kojima, M., Kondo, S., Konno, H., Nakano, K., Ninomiya, N., Nishio, T., Okada, M., Plessy, C., Shibata, K., Shiraki, T., Suzuki, S., Tagami, M., Waki, K., Watahiki, A., Okamura-Oho, Y., Suzuki, H., Kawai, J., Hayashizaki, Y., FANTOM Consortium, and RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group). (2005). The transcriptional landscape of the mammalian genome. Science 309, 1559–1563.
Cesana, M., Cacchiarelli, D., Legnini, I., Santini, T., Sthandier, O., Chinappi, M., Tramontano, A., and Bozzoni, I. (2011). A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147, 358–369.
Chadwick, B. P., and Willard, H. F. (2003). Barring gene expression after XIST: maintaining facultative heterochromatin on the inactive X. Semin. Cell Dev. Biol. 14, 359–367.
Chu, C., Qu, K., Zhong, F. L., Artandi, S. E., and Chang, H. Y. (2011). Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 44, 667–678.
Chung, S., Nakagawa, H., Uemura, M., Piao, L., Ashikawa, K., Hosono, N., Takata, R., Akamatsu, S., Kawaguchi, T., Morizono, T., Tsunoda, T., Daigo, Y., Matsuda, K., Kamatani, N., Nakamura, Y., and Kubo, M. (2011). Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 102, 245–252.
Daughters, R. S., Tuttle, D. L., Gao, W., Ikeda, Y., Moseley, M. L., Ebner, T. J., Swanson, M. S., and Ranum, L. P. (2009). RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 5, e1000600. doi:10.1371/journal.pgen.1000600
David, C. J., and Manley, J. L. (2010). Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 24, 2343–2364.
Double, K. L., Reyes, S., Werry, E. L., and Halliday, G. M. (2010). Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Prog. Neurobiol. 92, 316–329.
Faghihi, M. A., Modarresi, F., Khalil, A. M., Wood, D. E., Sahagan, B. G., Morgan, T. E., Finch, C. E., St Laurent, G. III, Kenny, P. J., and Wahlestedt, C. (2008). Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 14, 723–730.
Gallagher, A., and Hallahan, B. (2011). Fragile X-associated disorders: a clinical overview. J. Neurol. doi:10.1007/s00415-011-6161-3.
Godler, D. E., Tassone, F., Loesch, D. Z., Taylor, A. K., Gehling, F., Hagerman, R. J., Burgess, T., Ganesamoorthy, D., Hennerich, D., Gordon, L., Evans, A., Choo, K. H., and Slater, H. R. (2010). Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 19, 1618–1632.
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., Tsai, M. C., Hung, T., Argani, P., Rinn, J. L., Wang, Y., Brzoska, P., Kong, B., Li, R., West, R. B., Van De Vijver, M. J., Sukumar, S., and Chang, H. Y. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076.
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., Huarte, M., Zuk, O., Carey, B. W., Cassady, J. P., Cabili, M. N., Jaenisch, R., Mikkelsen, T. S., Jacks, T., Hacohen, N., Bernstein, B. E., Kellis, M., Regev, A., Rinn, J. L., and Lander, E. S. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227.
Guttman, M., Donaghey, J., Carey, B. W., Garber, M., Grenier, J. K., Munson, G., Young, G., Lucas, A. B., Ach, R., Bruhn, L., Yang, X., Amit, I., Meissner, A., Regev, A., Rinn, J. L., Root, D. E., and Lander, E. S. (2011). lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300.
Hagerman, P. J., and Hagerman, R. J. (2007). Fragile X-associated tremor/ataxia syndrome-an older face of the fragile X gene. Nat. Clin. Pract. Neurol. 3, 107–112.
He, Y., Vogelstein, B., Velculescu, V. E., Papadopoulos, N., and Kinzler, K. W. (2008). The antisense transcriptomes of human cells. Science 322, 1855–1857.
Hu, W., Yuan, B., Flygare, J., and Lodish, H. F. (2011). Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes Dev. 25, 2573–2578.
Huang, Q., Gumireddy, K., Schrier, M., Le Sage, C., Nagel, R., Nair, S., Egan, D. A., Li, A., Huang, G., Klein-Szanto, A. J., Gimotty, P. A., Katsaros, D., Coukos, G., Zhang, L., Pure, E., and Agami, R. (2008). The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat. Cell Biol. 10, 202–210.
Huarte, M., Guttman, M., Feldser, D., Garber, M., Koziol, M. J., Kenzelmann-Broz, D., Khalil, A. M., Zuk, O., Amit, I., Rabani, M., Attardi, L. D., Regev, A., Lander, E. S., Jacks, T., and Rinn, J. L. (2010). A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419.
Hudson, T. J. (2011). Cancer genome variation in children, adolescents, and young adults. Cancer 117, 2262–2267.
Hung, T., Wang, Y., Lin, M. F., Koegel, A. K., Kotake, Y., Grant, G. D., Horlings, H. M., Shah, N., Umbricht, C., Wang, P., Kong, B., Langerod, A., Borresen-Dale, A. L., Kim, S. K., Van De Vijver, M., Sukumar, S., Whitfield, M. L., Kellis, M., Xiong, Y., Wong, D. J., and Chang, H. Y. (2011). Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 43, 621–629.
Iacobucci, I., Sazzini, M., Garagnani, P., Ferrari, A., Boattini, A., Lonetti, A., Papayannidis, C., Mantovani, V., Marasco, E., Ottaviani, E., Soverini, S., Girelli, D., Luiselli, D., Vignetti, M., Baccarani, M., and Martinelli, G. (2011). A polymorphism in the chromosome 9p21 ANRIL locus is associated to Philadelphia positive acute lymphoblastic leukemia. Leuk. Res. 35, 1052–1059.
Ikeda, S., Kong, S. W., Lu, J., Bisping, E., Zhang, H., Allen, P. D., Golub, T. R., Pieske, B., and Pu, W. T. (2007). Altered microRNA expression in human heart disease. Physiol. Genomics 31, 367–373.
Jeon, Y., and Lee, J. T. (2011). YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146, 119–133.
Ji, P., Diederichs, S., Wang, W., Boing, S., Metzger, R., Schneider, P. M., Tidow, N., Brandt, B., Buerger, H., Bulk, E., Thomas, M., Berdel, W. E., Serve, H., and Muller-Tidow, C. (2003). MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22, 8031–8041.
Khaitan, D., Dinger, M. E., Mazar, J., Crawford, J., Smith, M. A., Mattick, J. S., and Perera, R. J. (2011). The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 71, 3852–3862.
Khalil, A. M., Faghihi, M. A., Modarresi, F., Brothers, S. P., and Wahlestedt, C. (2008). A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS ONE 3, e1486. doi:10.1371/journal.pone.0001486
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., Thomas, K., Presser, A., Bernstein, B. E., Van Oudenaarden, A., Regev, A., Lander, E. S., and Rinn, J. L. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672.
Khalil, A. M., and Rinn, J. L. (2011). RNA-protein interactions in human health and disease. Semin. Cell Dev. Biol. 22, 359–365.
Kogo, R., Shimamura, T., Mimori, K., Kawahara, K., Imoto, S., Sudo, T., Tanaka, F., Shibata, K., Suzuki, A., Komune, S., Miyano, S., and Mori, M. (2011). Long non-coding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 71, 6320–6326.
Koob, M. D., Moseley, M. L., Schut, L. J., Benzow, K. A., Bird, T. D., Day, J. W., and Ranum, L. P. (1999). An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat. Genet. 21, 379–384.
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., and Xiong, Y. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962.
Ladd, P. D., Smith, L. E., Rabaia, N. A., Moore, J. M., Georges, S. A., Hansen, R. S., Hagerman, R. J., Tassone, F., Tapscott, S. J., and Filippova, G. N. (2007). An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 16, 3174–3187.
Lee, J. T., Davidow, L. S., and Warshawsky, D. (1999). Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 21, 400–404.
Lewejohann, L., Skryabin, B. V., Sachser, N., Prehn, C., Heiduschka, P., Thanos, S., Jordan, U., Dell’Omo, G., Vyssotski, A. L., Pleskacheva, M. G., Lipp, H. P., Tiedge, H., Brosius, J., and Prior, H. (2004). Role of a neuronal small non-messenger RNA: behavioural alterations in BC1 RNA-deleted mice. Behav. Brain Res. 154, 273–289.
Luo, J. H., Ren, B., Keryanov, S., Tseng, G. C., Rao, U. N., Monga, S. P., Strom, S., Demetris, A. J., Nalesnik, M., Yu, Y. P., Ranganathan, S., and Michalopoulos, G. K. (2006). Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 44, 1012–1024.
Mattick, J. S., and Makunin, I. V. (2005). Small regulatory RNAs in mammals. Hum. Mol. Genet. 14, R121–R132.
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F., and Mattick, J. S. (2008). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 105, 716–721.
Minati, L., Edginton, T., Bruzzone, M. G., and Giaccone, G. (2009). Current concepts in Alzheimer’s disease: a multidisciplinary review. Am. J. Alzheimers Dis. Other Demen. 24, 95–121.
Moseley, M. L., Zu, T., Ikeda, Y., Gao, W., Mosemiller, A. K., Daughters, R. S., Chen, G., Weatherspoon, M. R., Clark, H. B., Ebner, T. J., Day, J. W., and Ranum, L. P. (2006). Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 38, 758–769.
Muddashetty, R., Khanam, T., Kondrashov, A., Bundman, M., Iacoangeli, A., Kremerskothen, J., Duning, K., Barnekow, A., Huttenhofer, A., Tiedge, H., and Brosius, J. (2002). Poly(A)-binding protein is associated with neuronal BC1 and BC200 ribonucleoprotein particles. J. Mol. Biol. 321, 433–445.
Mus, E., Hof, P. R., and Tiedge, H. (2007). Dendritic BC200 RNA in aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 104, 10679–10684.
Mutsuddi, M., Marshall, C. M., Benzow, K. A., Koob, M. D., and Rebay, I. (2004). The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with Staufen in Drosophila. Curr. Biol. 14, 302–308.
Nagano, T., and Fraser, P. (2011). No-nonsense functions for long noncoding RNAs. Cell 145, 178–181.
Ogawa, Y., and Lee, J. T. (2003). Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol. Cell 11, 731–743.
Oostra, B. A., and Willemsen, R. (2003). A fragile balance: FMR1 expression levels. Hum. Mol. Genet. 12, R249–R257.
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J., Nagano, T., Mancini-Dinardo, D., and Kanduri, C. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 32, 232–246.
Pasmant, E., Laurendeau, I., Heron, D., Vidaud, M., Vidaud, D., and Bieche, I. (2007). Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 67, 3963–3969.
Pasmant, E., Sabbagh, A., Masliah-Planchon, J., Ortonne, N., Laurendeau, I., Melin, L., Ferkal, S., Hernandez, L., Leroy, K., Valeyrie-Allanore, L., Parfait, B., Vidaud, D., Bieche, I., Lantieri, L., Wolkenstein, P., and Vidaud, M. (2011a). Role of noncoding RNA ANRIL in genesis of plexiform neurofibromas in neurofibromatosis type 1. J. Natl. Cancer Inst. 103, 1713–1722.
Pasmant, E., Sabbagh, A., Vidaud, M., and Bieche, I. (2011b). ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 25, 444–448.
Penagarikano, O., Mulle, J. G., and Warren, S. T. (2007). The pathophysiology of fragile X syndrome. Annu. Rev. Genomics Hum. Genet. 8, 109–129.
Perez, D. S., Hoage, T. R., Pritchett, J. R., Ducharme-Smith, A. L., Halling, M. L., Ganapathiraju, S. C., Streng, P. S., and Smith, D. I. (2008). Long, abundantly expressed non-coding transcripts are altered in cancer. Hum. Mol. Genet. 17, 642–655.
Popov, N., and Gil, J. (2010). Epigenetic regulation of the INK4b-ARF-INK4a locus: in sickness and in health. Epigenetics 5, 685–690.
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., Goodnough, L. H., Helms, J. A., Farnham, P. J., Segal, E., and Chang, H. Y. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323.
Rosenfeld, N., Aharonov, R., Meiri, E., Rosenwald, S., Spector, Y., Zepeniuk, M., Benjamin, H., Shabes, N., Tabak, S., Levy, A., Lebanony, D., Goren, Y., Silberschein, E., Targan, N., Ben-Ari, A., Gilad, S., Sion-Vardy, N., Tobar, A., Feinmesser, M., Kharenko, O., Nativ, O., Nass, D., Perelman, M., Yosepovich, A., Shalmon, B., Polak-Charcon, S., Fridman, E., Avniel, A., Bentwich, I., Bentwich, Z., Cohen, D., Chajut, A., and Barshack, I. (2008). MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol. 26, 462–469.
Silva, J. M., Perez, D. S., Pritchett, J. R., Halling, M. L., Tang, H., and Smith, D. I. (2010). Identification of long stress-induced non-coding transcripts that have altered expression in cancer. Genomics 95, 355–362.
Sokol, D. K., Maloney, B., Long, J. M., Ray, B., and Lahiri, D. K. (2011). Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology 76, 1344–1352.
Tassone, F., Hagerman, R. J., Taylor, A. K., and Hagerman, P. J. (2001). A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J. Med. Genet. 38, 453–456.
Tassone, F., Iwahashi, C., and Hagerman, P. J. (2004). FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol. 1, 103–105.
Tiedge, H., Chen, W., and Brosius, J. (1993). Primary structure, neural-specific expression, and dendritic location of human BC200 RNA. J. Neurosci. 13, 2382–2390.
Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T., Freier, S. M., Bennett, C. F., Sharma, A., Bubulya, P. A., Blencowe, B. J., Prasanth, S. G., and Prasanth, K. V. (2010). The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 39, 925–938.
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., Shi, Y., Segal, E., and Chang, H. Y. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693.
Vassar, R., Kovacs, D. M., Yan, R., and Wong, P. C. (2009). The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 29, 12787–12794.
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., Reiner, O., Richards, S., Victoria, M. F., Zhang, F. P., Eussen, B. E., van Ommen, G.-J. B., Blonden, L. A. J., Riggins, G. J., Chastain, J. L., Kunst, C. B., Galjaard, H., Caskey, C. T., Nelson, D. L., Oostra, B. A., and Warren, S. T. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914.
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914.
Wapinski, O., and Chang, H. Y. (2011). Long noncoding RNAs and human disease. Trends Cell Biol. 21, 354–361.
Yang, L., Lin, C., Liu, W., Zhang, J., Ohgi, K. A., Grinstein, J. D., Dorrestein, P. C., and Rosenfeld, M. G. (2011a). ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 147, 773–788.
Yang, Z., Zhou, L., Wu, L. M., Lai, M. C., Xie, H. Y., Zhang, F., and Zheng, S. S. (2011b). Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 18, 1243–1250.
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., Gil, J., Walsh, M. J., and Zhou, M. M. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674.
Yu, M., Ohira, M., Li, Y., Niizuma, H., Oo, M. L., Zhu, Y., Ozaki, T., Isogai, E., Nakamura, Y., Koda, T., Oba, S., Yu, B., and Nakagawara, A. (2009). High expression of ncRAN, a novel non-coding RNA mapped to chromosome 17q25.1, is associated with poor prognosis in neuroblastoma. Int. J. Oncol. 34, 931–938.
Yuan, Y., Compton, S. A., Sobczak, K., Stenberg, M. G., Thornton, C. A., Griffith, J. D., and Swanson, M. S. (2007). Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 35, 5474–5486.
Zhao, J., Ohsumi, T. K., Kung, J. T., Ogawa, Y., Grau, D. J., Sarma, K., Song, J. J., Kingston, R. E., Borowsky, M., and Lee, J. T. (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 40, 939–953.
Zhao, J., Sun, B. K., Erwin, J. A., Song, J. J., and Lee, J. T. (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756.
Keywords: long non-coding RNAs, large non-coding RNAs, lincRNAs, cancer, neurological disorders, neurodegeneration, human disease
Citation: Niland CN, Merry CR and Khalil AM (2012) Emerging roles for long non-coding RNAs in cancer and neurological disorders. Front. Gene. 3:25. doi: 10.3389/fgene.2012.00025
Received: 25 November 2011;
Paper pending published: 20 December 2011;
Accepted: 08 February 2012;
Published online: 27 February 2012.
Edited by:
Peng Jin, Emory University School of Medicine, USAReviewed by:
Alexander Pertsemlidis, UT Southwestern Medical Center, USADavid Corey, UT Southwestern, USA
Xuekun Li, Emory University, USA
Copyright: © 2012 Niland, Merry and Khalil. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ahmad M. Khalil, Center of RNA Molecular Biology and Departments of Genetics and Biochemistry, Khalil Lab, Wood Research Building W-106, 10900 Euclid Avenue, SOM_Wood W106, 44114, OH, Cleveland, USA. e-mail:ZHIuYWhtYWQua2hhbGlsQGdtYWlsLmNvbQ==