

- 1 Department of Bioengineering, University of Illinois at Chicago, Chicago, IL, USA
- 2 Department of Pediatrics, University of Illinois at Chicago, Chicago, IL, USA
- 3 Institute of Human Genetics, University of Illinois at Chicago, Chicago, IL, USA
- 4 Cancer Center, University of Illinois at Chicago, Chicago, IL, USA
Variation in quantitative gene expression has been observed in natural populations and associated with various complex traits/phenotypes such as risks for common diseases and drug response. MicroRNAs (miRNAs), a family of small, non-coding RNA molecules, have been demonstrated to be an important class of gene regulators that mostly downregulate gene expression at the post-transcriptional level. A comprehensive and reliable catalogue of miRNAs and miRNA gene targets is critical to understanding the gene regulatory networks. Though experimental approaches have been used to identify many miRNAs and their gene targets, due to cost and efficiency, currently miRNA and target identification still largely relies on computational algorithms. We reviewed several widely used bioinformatic resources of miRNA sequences and gene targets that take advantage of the unique characteristics of miRNA–mRNA interactions, experimental validation, as well as the integration of sequence-based evidence and microarray expression data. Furthermore, given the importance of miRNAs in regulating gene expression, elucidating expression quantitative trait loci involved with miRSNPs or miR-polymorphisms will help improve our understanding of complex traits. We reviewed the available resources of miRNA genetic variation, and the current progress (e.g., the 1000 Genomes Project) in detailing the genetic variation in miRNA-related single nucleotide polymorphisms (SNPs). We also provided our perspectives of the potential impact of next-generation sequencing on the research of miRNAs, gene targets, and miRSNPs. These bioinformatic resources may help interpret experimental and association study results, thus enhancing our knowledge of the dynamic gene regulatory networks and the physiological pathways for complex traits/phenotypes. Prospectively, these bioinformatic resources of miRNAs will need to address the challenges raised by the application of next-generation sequencing in miRNA research.
Introduction
Alterations in gene expression, a quantitative phenotype, have been implicated in a variety of human diseases and traits, as well as response to therapeutic treatments (Zhang and Dolan, 2009). The variation of gene expression, therefore, could potentially explain the phenotypic variations (e.g., susceptibility to complex diseases, and drug response) among individuals and human populations. For example, differentially expressed genes enriched in ubiquitin proteasome and Parkinson’s disease pathways were found be associated with coffee consumption, a model for addictive behavior (Amin et al., 2011). In contrast, based on a cell-based pharmacogenomic discovery model using the HapMap Project (HapMap, 2003, 2005) lymphoblastoid cell line (LCL) samples, whole-genome gene expression profiling (i.e., transcriptome profiling; Zhang et al., 2008, 2009) has allowed identification of gene expression phenotypes associated with the cytoxicities to anticancer drugs, such as etoposide (Huang et al., 2007a), daunorubicin (Huang et al., 2008a), carboplatin (Huang et al., 2008b), cisplatin (Huang et al., 2007b), and Ara-C (cytarabine arabinoside; Hartford et al., 2009). In addition, significant variation in gene expression has been observed among individuals from the same human population, as well as among individuals from different ethnic background (Cheung et al., 2003a,b; Stranger et al., 2005, 2007a,b; Spielman et al., 2007; Storey et al., 2007; Duan et al., 2008; Zhang et al., 2008). For example, a substantial proportion of human genes (e.g., genes related to immune response) have been found to be differentially expressed between individuals of African and Northern/Western European ancestry (Zhang et al., 2008). Particularly, common genetic variants including single nucleotide polymorphisms (SNPs) and copy number variants (CNVs) have been identified to contribute to the variation in gene expression (e.g., through expression quantitative trait loci, eQTLs) both within and between human populations (Cheung et al., 2003a,b; Stranger et al., 2005, 2007a,b; Spielman et al., 2007; Storey et al., 2007; Duan et al., 2008; Zhang et al., 2008). Besides genetic factors, non-genetic factors such as environmental exposure, life style, diet may play at least as great a role as genetic divergence in modulating gene expression variation in humans (Idaghdour et al., 2008).
During the past decade, epigenetic mechanisms (e.g., DNA methylation, histone modifications) have begun to be appreciated as playing critical roles in regulating gene expression, and affecting complex traits including risks for common diseases. Recent process in biomedical research emphasizes the need to move beyond protein-coding genes and highlights the fact that continued investigation of non-coding RNAs (ncRNAs) will be necessary for a comprehensive understanding of human traits and diseases (Taft et al., 2010; Kaikkonen et al., 2011). Notably, microRNAs (miRNAs), a family of small, ncRNAs molecules, have been demonstrated to be an important class of gene regulators that mostly downregulate gene expression at the post-transcriptional level (Ambros, 2001; Bartel, 2004). Dysregulation of miRNAs and some other ncRNAs is being found to have relevance to tumorigenesis, neurological, cardiovascular, developmental, respiratory, and other diseases, as well as individual response to drugs (Zhang and Dolan, 2010; Esteller, 2011; Zhou et al., 2011). For example, dysregulation of miRNAs in brains has been observed after exposure to addictive drugs like cocaine (Eipper-Mains et al., 2011), while upregulation of some miRNAs have been identified in human alcoholics (Lewohl et al., 2011). In addition, pharmacogenomic studies have related miRNAs as an important mechanism for regulating genes responsible for drug response (Zhang and Dolan, 2010; Huang et al., 2011). A comprehensive understanding of miRNAs and other functional ncRNAs will be necessary for future integration of various “omics” data for elucidating the underpinning mechanisms of complex diseases, phenotypes, and traits including the fundamental problem of gene expression regulation.
Given the critical roles of miRNAs in gene regulation and human diseases, it is imperative to have a comprehensive and reliable catalog of miRNA sequences (e.g., mature miRNAs, pre-, and pri-miRNAs) and miRNA gene targets. In addition to experimental approaches, computational algorithms have been developed for predicting miRNA sequences and their potential gene targets. To allow user-friendly queries, there are publicly available, web-based resources for miRNA sequences and gene targets. The current bioinformatic resources of miRNAs provide miRNA sequences and their gene targets from both experimental approaches and computational algorithms (in silico predictions). Furthermore, SNPs within the sequences of human miRNAs and their gene targets have been shown to have impact on various phenotypes and diseases such as cancer, blood pressure, and drug resistance (Mishra et al., 2007; Sethupathy et al., 2007; Landi et al., 2008). The polymorphisms in miRNA (miRSNPs) appear to have a differing effect on gene and protein expression, thus representing a novel type of genetic variation that may influence complex traits such as the risks of certain human diseases (Chen et al., 2008). With the advances in genotyping and sequencing technologies, genetic variation data on miRSNPs have become available for analyses in elucidating the mechanisms of miRNAs and complex phenotypes/traits. Therefore, we reviewed several publicly available miRNA-related (particularly, in humans) bioinformatic resources that accommodate miRNA sequences, miRNA gene targets, and miRSNPs (Table 1). Prospectively, these bioinformatic resources of miRNAs will need to address the challenges raised by the emerging applications of novel technologies (e.g., next-generation sequencing) in miRNA research.
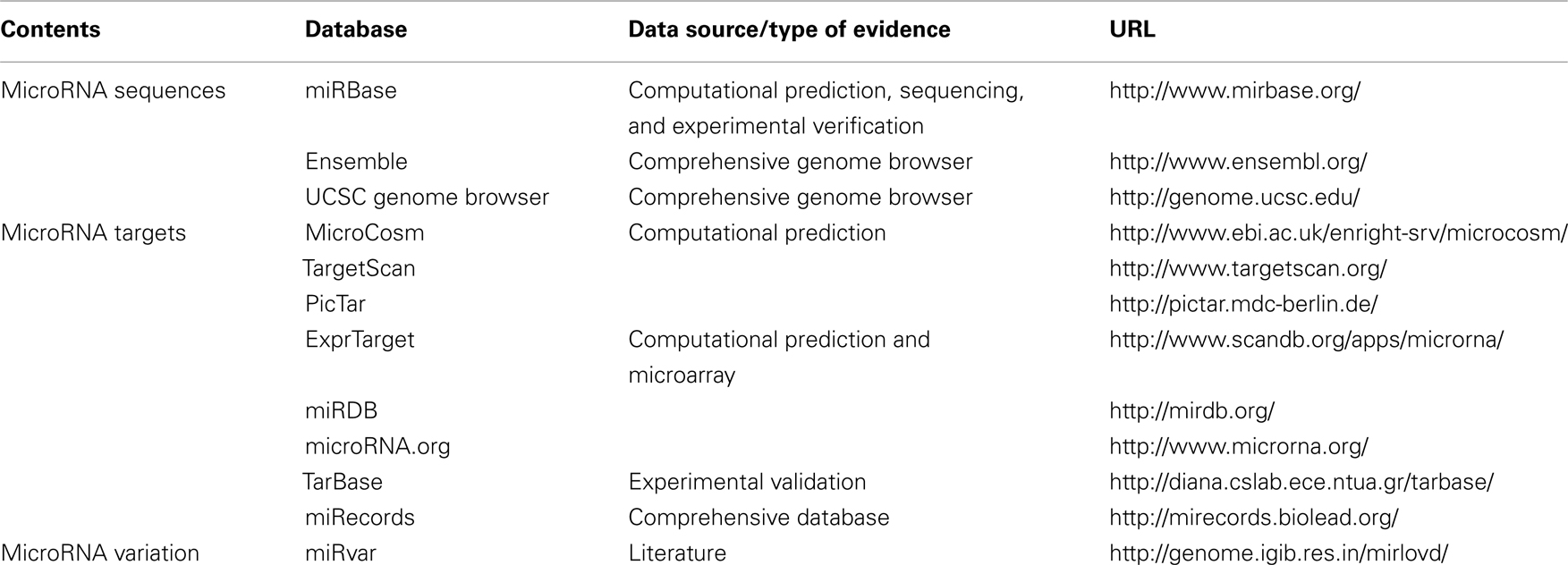
Table 1. Some widely used bioinformatic resources of microRNA sequences, gene targets and genetic variation.
miRBase – A Database of miRNA Sequences
Since its launch as the MicroRNA Registry (Griffiths-Jones, 2004), the miRBase database1 (Griffiths-Jones, 2004; Griffiths-Jones et al., 2006, 2008; Kozomara and Griffiths-Jones, 2011) has evolved into a central portal for miRNA sequences and other relevant information. The miRBase is a searchable database of published mature miRNA sequences, together with their predicted source hairpin precursors and annotations relating to their discovery, structure, and function (Griffiths-Jones et al., 2006). The miRBase contains miRNA sequence information from two fundamentally different sources: (1) experimentally verified mature miRNAs; and (2) miRNA sequences that are predicted homologs of miRNAs verified in a related organism (Griffiths-Jones et al., 2006). Each entry in the miRBase database represents a predicted hairpin portion of a miRNA transcript (termed “mir” in the database), with information on the location and sequence of the mature miRNA sequence (termed “miR”; Ambros et al., 2003).
The current miRBase Release 18 (November, 2011) contains 18,226 entries representing hairpin precursor miRNAs, expressing 21,643 mature miRNA products, in 168 species. Particularly, there are 1,527 human miRNA sequences provided bythe miRBase (accessed on December 1, 2011). The miRBase also provides links of miRNAs to potential gene targets predicted by MicroCosm, TargetScan (Lewis et al., 2003), and PicTar (Krek et al., 2005), which are described in the following section. Both hairpin and mature sequences are available for searching and browsing, and entries can also be retrieved by name (e.g., “let-7” or “miR-36”), keyword (e.g. “human”), references, and annotation. Queries can also be performed by genomic location, tissue expression (e.g., adenosquamous cell, embryonic stem cell for humans) and sequence. For searches by sequence, users can perform either BLASTN or SSEARCH to search against the intact precursor sequences or just the mature miRNAs. All sequence and annotation data served at the miRBase are also available for bulk download. In addition, novel miRNA sequences can be submitted to miRBase by researchers, therefore, the miRBase acts as a central depository of miRNA sequences. Besides the miRBase website, other major genome databases such as the Ensemble2 and UCSC Genome Browser3 (Kent et al., 2002) also provide access to miRNA sequences and annotations. For example, the Ensemble Genome Browser can be used to obtain miRNA sequences that have been predicted by BLASTN of genomic sequence slices against miRBase sequences (Guttman et al., 2009).
Resources of miRNA Gene Targets
Given the critical roles of miRNAs in regulating gene expression and cellular functions, a comprehensive and reliable catalog of miRNA gene targets will benefit the biomedical research community, for example, by enabling the construction of gene regulatory networks, as well as gene-set or pathway-based analyses. Though molecular biology approaches may be used to experimentally determine gene targets for miRNAs, the lack of high throughput techniques and the relatively high cost associated with experimental approaches limit their use in identifying all of miRNA gene targets. Therefore, to date, only some gene targets of a small number of the potential >1000 human miRNAs have been confirmed experimentally. Another major challenge with miRNA gene target identification is that an individual miRNA may regulate multiple mRNAs, and in contrast, an individual gene may also be regulated by multiple miRNAs, thus representing a complex network of miRNA–mRNA interactions. In lieu of experimental approaches, several computational approaches have been developed for genome-wide prediction of miRNA gene targets by taking advantage of the properties of miRNAs’ binding to gene targets. In animals, miRNAs typically bind to the 3′-UTRs (untranslated regions) of their target mRNAs to form miRNA–mRNA duplexes, leading to regulatory repression of translation (Lai et al., 2004), although the exact mechanism is not completely clear. Therefore, these approaches commonly depend on either miRNA–mRNA complementarity: e.g., miRanda (Enright et al., 2003; Griffiths-Jones et al., 2008), TargetScan (Lewis et al., 2003), or miRNA–mRNA duplex thermodynamics: e.g., PicTar (Krek et al., 2005). Notably, pair-wise comparisons of the miRanda scores (Enright et al., 2003; Griffiths-Jones et al., 2008), PicTar scores (Krek et al., 2005), and TargetScan scores (Lewis et al., 2003) were performed to evaluate the correlations between these computational algorithms (Gamazon et al., 2010). In general, these computational algorithms appeared to generate a significant number of overlapping miRNA gene targets between each pair of algorithms (Gamazon et al., 2010): 21,590 between miRanda (Enright et al., 2003; Griffiths-Jones et al., 2008) and TargetScan (Lewis et al., 2003), 2,465 between miRanda (Enright et al., 2003; Griffiths-Jones et al., 2008) and PicTar (Krek et al., 2005), as well as 8,707 between TargetScan (Lewis et al., 2003) and PicTar (Krek et al., 2005). Using TarBase (Papadopoulos et al., 2009), a database of experimentally supported miRNA targets, as gold standard, an receiver operating characteristic (ROC) analysis on each of the above-mentioned computational algorithms would seem to suggest that TargetScan (Lewis et al., 2003) may yield slightly better performance than the other computational methods (Gamazon et al., 2010). Recently, with the availability of whole-genome microarray data, approaches that aim to integrate both microarray data and sequence-based evidence have also been proposed to predict miRNA targets (e.g., ExprTarget; Gamazon et al., 2010). However, caution must be exercised in the interpretation of these results, given each prediction approach may be biased toward particular characteristics of miRNA–mRNA interactions, possibly a reason why some of these approaches may not provide consistent predictions. For example, since gene expression can be tissue-specific, results from integrative approaches utilizing both sequence-based evidence and microarray data, therefore, could be biased toward genes expressed in a particular tissue (e.g., the LCLs). Regarding bioinformatic resources, there are several widely used web-based databases that provide access to those computationally predicted miRNA gene targets, experimentally supported miRNA gene targets and miRNA gene targets derived from integrated approaches.
MicroCosm Targets
The MicroCosm Targets is a web resource (formerly the miRBase Targets)4 containing computationally predicted targets for miRNAs across many species including humans. The current Version 5 database contains detected relationships for 851 human miRNAs and ∼35,000 transcripts (accessed on December 1, 2011). The miRNA sequences were from the miRBase; Griffiths-Jones, 2004; Griffiths-Jones et al., 2006, 2008; Kozomara and Griffiths-Jones, 2011) and most genomic sequences from Ensembl (see text footnote 2). The miRanda algorithm (Enright et al., 2003; Griffiths-Jones et al., 2008) was used to identify potential binding sites for a given miRNA in genomic sequences. Particularly, the current Version 5 of the MicroCosm Targets database used dynamic programming alignment to identify highly complementary miRNA–mRNA pairs which are scored between 0 (i.e., no complementarity) and 100 (i.e., complete complementarity). The miRanda algorithm (Enright et al., 2003; Griffiths-Jones et al., 2008) uses a weighted scoring system and rewards complementarity at the 5′ end of miRNA. P-values are also calculated following statistical model proposed by Rehmsmeier et al. (2004). Since miRanda requires strict complementarity at the seed region (i.e., the 5′ end of miRNA) of miRNA–mRNA pairs, this algorithm does not allow alignments where more than one base in the seed region is not complementary to a target mRNA. Furthermore, gene targets selected by miRanda (Enright et al., 2003; Griffiths-Jones et al., 2008) were passed through the Vienna RNA folding routines (Hofacker, 2003; Gruber et al., 2008) for evaluating the property of thermodynamic stability. MicroCosm Targets supports queries for individual miRNAs, individual genes, gene ontology (GO; Ashburner et al., 2000) classes (e.g., cellular component, biological process) in a selected genome (e.g., the human genome), as well as batch queries. The MicroCosm Targets supports bulk download of the complete dataset.
TargetScan
TargetScan5 (Lewis et al., 2003) is a web resource that provided computationally predicted miRNA gene targets by searching for the presence of conserved 8 and 7 mer sites that match the seed region of each miRNA for a variety of species. TargetScanHuman (Release 6.0, November, 2011) is a sub-database for predicting human miRNA gene targets based on the TargetScan approach (Lewis et al., 2003). The current TargetScanHuman Release 6.0 contains ∼18,000 mRNAs, corresponding to ∼30,000 transcripts (accessed on December 1, 2011). As an option, TargetScanHuman also provides non-conserved sites and sites with mismatches in the seed region that are compensated by conserved 3′ pairing (i.e., conserved 3′-compensatory sites; Friedman et al., 2009). The TargetScan predictions are ranked based on the predicted efficacy of targeting (Grimson et al., 2007; Garcia et al., 2011) and by their probability of conserved targeting (i.e., PCT; Friedman et al., 2009), which reflects the Bayesian estimate of the probability that a site is conserved due to selective maintenance of miRNA targeting rather than by chance or any other reason not pertinent to miRNA targeting. TargetScan supports bulk download of the complete dataset.
PicTar
PicTar6 (Krek et al., 2005), a searchable website provides details (e.g., 3′-UTR alignments with computationally predicted sites, links to various public databases) of human miRNA targets that are not conserved but co-expressed (i.e., the miRNA and mRNA expressed in the same tissue; Chen and Rajewsky, 2006), as well as miRNA target predictions in vertebrates (Krek et al., 2005), Drosophila (Grun et al., 2005), and nematode species (Lall et al., 2006). PicTar (Krek et al., 2005) uses a probabilistic model to compute the likelihood that sequences are miRNA target sites when compared to the 3′-UTR background. In vivo experimental validation suggests a high degree of accuracy and sensitivity for the PicTar algorithm in flies (Stark et al., 2005). PicTar (Krek et al., 2005) supports queries and bulk download for more than 13,000 co-expressed human microRNA target predictions (Chen and Rajewsky, 2006) between 31 unique human miRNAs and ∼5,000 transcripts in multiple tissues (accessed on December 1, 2011).
TarBase
In contrast to those databases for computationally predicted miRNA gene targets, TarBase7 (Papadopoulos et al., 2009) v5.0 (accessed on December 1, 2011) houses a manually curated collection of more than 1,300 experimentally supported miRNA target interactions in a variety of species including human, mouse, and several other model organisms. TarBase (Papadopoulos et al., 2009) supports queries for individual miRNAs and genes in a selected organism. For humans, the current v5.0 database contains 1,094 miRNA–mRNA relationships (>100 miRNAs and ∼900 transcripts) that are supported by experimental evidence. For each miRNA–mRNA relationship, TarBase (Papadopoulos et al., 2009) provides the type of experimental support (e.g., microarray, pSILAC – pulsed stable isotope labeling by amino acids), detailed information on miRNA and mRNA target, as well as the scientific references.
ExprTarget
The efficacy of computational approaches to locate and rank potential genomic binding sites is supported by the relatively high degree of miRNA complementarity to experimentally determined binding sites (Maziere and Enright, 2007). In contrast, ExprTarget8 (Gamazon et al., 2010) provides a comprehensive catalog of miRNA targets supported by both sequence-based evidence and microarray gene expression association. Particularly, ExprTarget (Gamazon et al., 2010) combines miRNA (Huang et al., 2011) and exon array expression data (Zhang et al., 2009; mRNA-level) on 117 unrelated HapMap CEU (Caucasians from Utah, USA) and YRI (Yoruba people from Ibadan, Nigeria) LCL samples, thus providing an expression-corroborated catalog of miRNA targets (225 expressed miRNAs in LCLs and ∼9,000 transcripts) that were also predicted using the miRanda algorithm (Ambros, 2001; Enright et al., 2003; Griffiths-Jones et al., 2008). ExprTarget (Gamazon et al., 2010) will be expanded to accommodate miRNA target predictions in other human tissues such as liver.
miRDB
miRDB9 (Wang, 2008; Wang and El Naqa, 2008), is an online database for predicted miRNA targets in animals using an algorithm called MirTarget2, which was developed by analyzing thousands of genes impacted by miRNAs with an support vector machine (SVM) algorithm and a microarray training dataset. Particularly, by systematically analyzing public microarray data, statistically significant features that are important to gene target downregulation were identified (Wang and El Naqa, 2008). For human miRNAs, the current miRDB v4.0 (accessed on January 10, 2012) contains 1,919 miRNAs and more than >16,000 unique gene targets. About half of the predicted human miRNA target sites are not conserved in other organisms (Wang, 2008; Wang and El Naqa, 2008). MirTarget2 has been validated with independent experimental data for its improved performance on predicting miRNA downregulated gene targets (Wang and El Naqa, 2008). miRDB (Wang, 2008; Wang and El Naqa, 2008) supports queries for individual targets or miRNAs, as well as bulk download of the complete dataset.
microRNA.org
The microRNA.org website10 (John et al., 2004; Landgraf et al., 2007; Betel et al., 2008) is a comprehensive resource of miRNA target predictions, target downregulation scores, and experimentally observed expression patterns. Particularly, the microRNA.org target predictions are based on a development of the miRanda algorithm (Enright et al., 2003; Griffiths-Jones et al., 2008), which incorporates current biological knowledge on target rules and on the use of a compendium of mammalian miRNAs. The miRNA expression profiles were derived from a comprehensive sequencing project of mammalian tissues and cell lines of normal and disease origin (John et al., 2004; Landgraf et al., 2007; Betel et al., 2008). The current version of the microRNA.org database (released in August, 2010) contains predicted interactions between 249 human miRNAs and more than 19,000 human genes (accessed on December 1, 2011). The microRNA.org miRNA target predictions and expression data (John et al., 2004; Landgraf et al., 2007; Betel et al., 2008) are also available as tab-delimited files for bulk downloads.
miRecords
miRecords11 (Xiao et al., 2009) is a comprehensive resource for animal miRNA–mRNA interactions from some of the above-mentioned algorithms and experimental approaches. miRecords (Xiao et al., 2009) consists of two components: (1) Validated Targets component – a large, high-quality database of experimentally validated miRNA targets resulting from manual literature curation; and (2) The Predicted Targets component – an integration of predicted miRNA targets produced by some established miRNA target prediction programs: e.g., miRanda (Enright et al., 2003; Griffiths-Jones et al., 2008) and TargetScan (Lewis et al., 2003). miRecords (Xiao et al., 2009) allows queries of individual miRNAs in a selected organism. The search results provide detailed evidence for miRNA–mRNA interactions from a variety of algorithms and databases. The current Validated Targets component of miRecords (Xiao et al., 2009; released in November, 2010) hosts the interactions between 384 miRNAs and more than 1,000 target genes in nine animal species. miRecords (Xiao et al., 2009) supports bulk download of the complete list of validated gene targets collected in the database.
miRSNPs – Novel Players in Complex Traits
Genomic variations including germ line or somatic mutations may count for miRNA abnormal expression by altering their biogenesis and/or affect the ability of miRNAs to bind to their target mRNAs. For example, aberrant allele frequencies of the SNPs located in miRNA target sites were found to be potentially associated with human diseases (e.g., cancer; Yu et al., 2007; Landi et al., 2008). With the availability of comprehensive genetic variation data: e.g., the International HapMap Project (HapMap, 2003, 2005) and the 1000 Genomes Project (1000 Genomes Consortium, 2010) genotypic data, studies of miRSNPs or miR-polymorphisms have begun to shed novel light to the distribution and potential biological/clinical implication of these polymorphisms. For example, using the HapMap Phase 1/2 genotypic data (HapMap, 2003, 2005) on the CEU and YRI, a genome-scale search for the regulatory polymorphisms in the loci of pre-miRNAs and their gene targets demonstrated 187 SNPs in the pre-miRNAs, 497 consensus SNPs in the seed-matching 3-UTR of target genes, 385 CNVs harboring pre-miRNA precursors and 9 CNVs covering important miRNA processing genes (Duan et al., 2009), indicating potential roles of miR-polymorphisms in regulating miRNAs and their gene targets. Future studies that take advantage of more comprehensive genotypic data such as those from the 1000 Genomes Project (1000 Genomes Consortium, 2010) and other sequencing efforts will help generate a more comprehensive list of miR-polymorphisms, thus facilitating future association studies between the genetic polymorphisms in miRNA targets/pre-miRNAs and disease susceptibility or therapeutic outcome.
Since the research of miRSNPs is a relatively new field in miRNA research, especially relative to the research of miRNA gene targets, currently there are few bioinformatic tools or resources that specifically devote to miRSNPs. Efforts such as the miRvar database (Bhartiya et al., 2011) may prove valuable to the miRNA research community for integrating this important type of genetic variation in biomedical research.
miRvar – A Database for Genomic Variations in miRNAs
miRvar12 (Bhartiya et al., 2011) is a comprehensive database for genomic variations in miRNAs. This database contains curated genetic variations in miRNA loci in the human genome. Particularly, the miRNA genetic variation data is made available on the Leiden Open (source) Variation Database (LOVD) platform to provide ease of aggregation and analysis. For each genetic variant in a particular miRNA, miRvar (Bhartiya et al., 2011) provides detailed information and links to other publicly available resources such as miRBase (Griffiths-Jones, 2004; Griffiths-Jones et al., 2006, 2008; Kozomara and Griffiths-Jones, 2011), miRecords (Xiao et al., 2009), UCSC Genome Browser (Kent et al., 2002). In addition, miRvar (Bhartiya et al., 2011) is open for community curation efforts.
Conclusion
Since the discovery of miRNAs a decade ago, significant progress has been made in identifying both miRNAs and their gene targets. A number of bioinformatic resources of miRNAs have emerged to accommodate the needs for the miRNA research community. Given the unique properties of these small ncRNA molecules, in addition to experimental approaches, the identification of miRNAs and their gene targets have substantially relied upon computational and systems biology approaches. The current bioinformatic resources of miRNAs have greatly facilitated the research of miRNAs and their gene targets, as well as the integration of miRNAs into other “omics” (e.g., transcriptomics, proteomics, metabolomics) studies in elucidating the networks of complex phenotypes (e.g., gene expression, individual response to drugs) as well as the pathogenesis of human diseases. However, a big challenge facing these current bioinformatic resources is the integration of various resources, not only with the miRNA resources (e.g., the miRNA gene target prediction databases/approaches), but also with other relevant resources to facilitate the next stage of systems biomedicine research. For example, the integration of miRNA prediction databases with the PharmGKB13 (McDonagh et al., 2011), which provides the published genetic variants implicated in drug response, may help elucidate the role of the interactions between genetic and non-genetic factors (e.g., epigenetic factors including miRNAs) in determining individual drug response, thus facilitating the realization of personalized medical care.
The current knowledge of miRNA and their interactions with gene targets have been largely uncovered by experimental approaches, microarray-based studies, and computational algorithms. During the past several years, with the advancement of the next-generation sequencing technologies (e.g., the Illumina HiSeq Sequencing System, Roche 454 GS System; Mardis, 2008), it is now possible to perform RNA-sequencing to comprehensively catalog miRNAs in cells under various phenotypic contexts or disease statuses, thus enhancing our knowledge of the distribution, abundance and roles of miRNAs in real-time cellular environments. For example, deep sequencing platforms have revealed unexpected complexity in relation to miRNAs, including 5′ and 3′-end-length heterogeneity and RNA editing (Vaz et al., 2012). Notably, the majority of the evidence supporting miRNA annotations are now coming from deep sequencing experiments, as recorded in the miRbase (Kozomara and Griffiths-Jones, 2011). Though analysis tools such as miRDeep (Friedlander et al., 2008) have facilitated the current miRNA-targeted sequencing studies (Dhahbi et al., 2011), however, challenges remain to be addressed for properly analyzing these data, particularly the difficulties in aligning short reads from RNA-sequencing projects to profile miRNAs in various cellular environments, as well as distinguishing true miRNAs from fragments of other transcripts (Kozomara and Griffiths-Jones, 2011). The availability of these large-scale sequencing data, therefore, underscores the importance of data analysis tools and bioinformatic resources that enable users to efficiently analyze the unprecedented amounts of RNA-sequencing data. Notably, bioinformatic tools such as deepBase14 (Yang et al., 2010) have begun to emerge with the ambitious attempt to provide a universal platform for mapping, storage, retrieval, analysis, integration, annotation, mining and visualization of next-generation sequencing data on small and long ncRNAs including miRNAs. In addition, new research resources such as the Sanger Institute mirKO (Prosser et al., 2011), a library of mouse embryonic stem cell clones with deletions for the “majority” of known miRNAs may prove to be invaluable for the rigorous investigation of miRNA function. Furthermore, data from sequencing efforts such as the 1000 Genomes Project (1000 Genomes Consortium, 2010) may greatly improve our understanding of miRSNPs and their potential contribution to complex phenotypes/traits. Given the fast-evolving technologies in the general biomedical research, future development of bioinformatic resources of miRNAs would need to address significant challenges that remain to be met for better integration and utilization of these tremendous resources for the benefit of the miRNA research community.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported, in part, by a grant from the NHGRI/NIH (R21HG006367).
Footnotes
- ^http://www.mirbase.org/
- ^http://www.ensembl.org/
- ^http://genome.ucsc.edu/
- ^http://www.ebi.ac.uk/enright-srv/microcosm/
- ^http://www.targetscan.org/
- ^http://pictar.mdc-berlin.de/
- ^http://diana.cslab.ece.ntua.gr/tarbase/
- ^http://www.scandb.org/apps/microrna/
- ^http://mirdb.org/
- ^http://www.microrna.org/
- ^http://mirecords.biolead.org/
- ^http://genome.igib.res.in/mirlovd/
- ^http://www.pharmgkb.org/
- ^http://deepbase.sysu.edu.cn/
References
1000 Genomes Consortium. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073.
Ambros, V., Bartel, B., Bartel, D. P., Burge, C. B., Carrington, J. C., Chen, X., Dreyfuss, G., Eddy, S. R., Griffiths-Jones, S., Marshall, M., Matzke, M., Ruvkun, G., and Tuschl, T. (2003). A uniform system for microRNA annotation. RNA 9, 277–279.
Amin, N., Byrne, E., Johnson, J., Chenevix-Trench, G., Walter, S., Nolte, I. M., Vink, J. M., Rawal, R., Mangino, M., Teumer, A., Keers, J. C., Verwoert, G., Baumeister, S., Biffar, R., Petersmann, A., Dahmen, N., Doering, A., Isaacs, A., Broer, L., Wray, N. R., Montgomery, G. W., Levy, D., Psaty, B. M., Gudnason, V., Chakravarti, A., Sulem, P., Gudbjartsson, D. F., Kiemeney, L. A., Thorsteinsdottir, U., Stefansson, K., Van Rooij, F. J., Aulchenko, Y. S., Hottenga, J. J., Rivadeneira, F. R., Hofman, A., Uitterlinden, A. G., Hammond, C. J., Shin, S. Y., Ikram, A., Witteman, J. C., Janssens, A. C., Snieder, H., Tiemeier, H., Wolfenbuttel, B. H., Oostra, B. A., Heath, A. C., Wichmann, E., Spector, T. D., Grabe, H. J., Boomsma, D. I., Martin, N. G., and Van Duijn, C. M. (2011). Genome-wide association analysis of coffee drinking suggests association with CYP1A1/CYP1A2 and NRCAM. Mol. Psychiatry. doi: 10.1038/mp.2011.101. [Epub ahead of print].
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., and Sherlock, G. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29.
Betel, D., Wilson, M., Gabow, A., Marks, D. S., and Sander, C. (2008). The microRNA.org resource: targets and expression. Nucleic Acids Res. 36, D149–D153.
Bhartiya, D., Laddha, S. V., Mukhopadhyay, A., and Scaria, V. (2011). miRvar: a comprehensive database for genomic variations in microRNAs. Hum. Mutat. 32, E2226–E2245.
Chen, K., and Rajewsky, N. (2006). Natural selection on human microRNA binding sites inferred from SNP data. Nat. Genet. 38, 1452–1456.
Chen, K., Song, F., Calin, G. A., Wei, Q., Hao, X., and Zhang, W. (2008). Polymorphisms in microRNA targets: a gold mine for molecular epidemiology. Carcinogenesis 29, 1306–1311.
Cheung, V. G., Conlin, L. K., Weber, T. M., Arcaro, M., Jen, K. Y., Morley, M., and Spielman, R. S. (2003a). Natural variation in human gene expression assessed in lymphoblastoid cells. Nat. Genet. 33, 422–425.
Cheung, V. G., Jen, K. Y., Weber, T., Morley, M., Devlin, J. L., Ewens, K. G., and Spielman, R. S. (2003b). Genetics of quantitative variation in human gene expression. Cold Spring Harb. Symp. Quant. Biol. 68, 403–407.
Dhahbi, J. M., Atamna, H., Boffelli, D., Magis, W., Spindler, S. R., and Martin, D. I. (2011). Deep sequencing reveals novel microRNAs and regulation of microRNA expression during cell senescence. PLoS ONE 6, e20509. doi:10.1371/journal.pone.0020509
Duan, S., Huang, R. S., Zhang, W., Bleibel, W. K., Roe, C. A., Clark, T. A., Chen, T. X., Schweitzer, A. C., Blume, J. E., Cox, N. J., and Dolan, M. E. (2008). Genetic architecture of transcript-level variation in humans. Am. J. Hum. Genet. 82, 1101–1113.
Duan, S., Mi, S., Zhang, W., and Dolan, M. E. (2009). Comprehensive analysis of the impact of SNPs and CNVs on human microRNAs and their regulatory genes. RNA Biol. 6, 412–425.
Eipper-Mains, J. E., Kiraly, D. D., Palakodeti, D., Mains, R. E., Eipper, B. A., and Graveley, B. R. (2011). microRNA-Seq reveals cocaine-regulated expression of striatal microRNAs. RNA 17, 1529–1543.
Enright, A. J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D. S. (2003). MicroRNA targets in Drosophila. Genome Biol. 5, R1.
Friedlander, M. R., Chen, W., Adamidi, C., Maaskola, J., Einspanier, R., Knespel, S., and Rajewsky, N. (2008). Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 26, 407–415.
Friedman, R. C., Farh, K. K., Burge, C. B., and Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105.
Gamazon, E. R., Im, H. K., Duan, S., Lussier, Y. A., Cox, N. J., Dolan, M. E., and Zhang, W. (2010). Exprtarget: an integrative approach to predicting human microRNA targets. PLoS ONE 5, e13534. doi:10.1371/journal.pone.0013534
Garcia, D. M., Baek, D., Shin, C., Bell, G. W., Grimson, A., and Bartel, D. P. (2011). Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 18, 1139–1146.
Griffiths-Jones, S., Grocock, R. J., Van Dongen, S., Bateman, A., and Enright, A. J. (2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144.
Griffiths-Jones, S., Saini, H. K., Van Dongen, S., and Enright, A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158.
Grimson, A., Farh, K. K., Johnston, W. K., Garrett-Engele, P., Lim, L. P., and Bartel, D. P. (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105.
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neubock, R., and Hofacker, I. L. (2008). The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74.
Grun, D., Wang, Y. L., Langenberger, D., Gunsalus, K. C., and Rajewsky, N. (2005). microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput. Biol. 1, e13. doi:10.1371/journal.pcbi.0010013
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., Huarte, M., Zuk, O., Carey, B. W., Cassady, J. P., Cabili, M. N., Jaenisch, R., Mikkelsen, T. S., Jacks, T., Hacohen, N., Bernstein, B. E., Kellis, M., Regev, A., Rinn, J. L., and Lander, E. S. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227.
Hartford, C. M., Duan, S., Delaney, S. M., Mi, S., Kistner, E. O., Lamba, J. K., Huang, R. S., and Dolan, M. E. (2009). Population-specific genetic variants important in susceptibility to cytarabine arabinoside cytotoxicity. Blood 113, 2145–2153.
Huang, R. S., Duan, S., Bleibel, W. K., Kistner, E. O., Zhang, W., Clark, T. A., Chen, T. X., Schweitzer, A. C., Blume, J. E., Cox, N. J., and Dolan, M. E. (2007a). A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 104, 9758–9763.
Huang, R. S., Duan, S., Shukla, S. J., Kistner, E. O., Clark, T. A., Chen, T. X., Schweitzer, A. C., Blume, J. E., and Dolan, M. E. (2007b). Identification of genetic variants contributing to cisplatin-induced cytotoxicity by use of a genomewide approach. Am. J. Hum. Genet. 81, 427–437.
Huang, R. S., Duan, S., Kistner, E. O., Bleibel, W. K., Delaney, S. M., Fackenthal, D. L., Das, S., and Dolan, M. E. (2008a). Genetic variants contributing to daunorubicin-induced cytotoxicity. Cancer Res. 68, 3161–3168.
Huang, R. S., Duan, S., Kistner, E. O., Hartford, C. M., and Dolan, M. E. (2008b). Genetic variants associated with carboplatin-induced cytotoxicity in cell lines derived from Africans. Mol. Cancer Ther. 7, 3038–3046.
Huang, R. S., Gamazon, E. R., Ziliak, D., Wen, Y., Im, H. K., Zhang, W., Wing, C., Duan, S., Bleibel, W. K., Cox, N. J., and Dolan, M. E. (2011). Population differences in microRNA expression and biological implications. RNA Biol. 8, 692–701.
Idaghdour, Y., Storey, J. D., Jadallah, S. J., and Gibson, G. (2008). A genome-wide gene expression signature of environmental geography in leukocytes of Moroccan Amazighs. PLoS Genet. 4, e1000052. doi:10.1371/journal.pgen.1000052
John, B., Enright, A. J., Aravin, A., Tuschl, T., Sander, C., and Marks, D. S. (2004). Human microRNA targets. PLoS Biol. 2, e363. doi:10.1371/journal.pbio.0020363
Kaikkonen, M. U., Lam, M. T., and Glass, C. K. (2011). Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 90, 430–440.
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., and Haussler, D. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006.
Kozomara, A., and Griffiths-Jones, S. (2011). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, D152–D157.
Krek, A., Grun, D., Poy, M. N., Wolf, R., Rosenberg, L., Epstein, E. J., Macmenamin, P., Da Piedade, I., Gunsalus, K. C., Stoffel, M., and Rajewsky, N. (2005). Combinatorial microRNA target predictions. Nat. Genet. 37, 495–500.
Lai, E. C., Wiel, C., and Rubin, G. M. (2004). Complementary miRNA pairs suggest a regulatory role for miRNA:miRNA duplexes. RNA 10, 171–175.
Lall, S., Grun, D., Krek, A., Chen, K., Wang, Y. L., Dewey, C. N., Sood, P., Colombo, T., Bray, N., Macmenamin, P., Kao, H. L., Gunsalus, K. C., Pachter, L., Piano, F., and Rajewsky, N. (2006). A genome-wide map of conserved microRNA targets in C. elegans. Curr. Biol. 16, 460–471.
Landgraf, P., Rusu, M., Sheridan, R., Sewer, A., Iovino, N., Aravin, A., Pfeffer, S., Rice, A., Kamphorst, A. O., Landthaler, M., Lin, C., Socci, N. D., Hermida, L., Fulci, V., Chiaretti, S., Foa, R., Schliwka, J., Fuchs, U., Novosel, A., Muller, R. U., Schermer, B., Bissels, U., Inman, J., Phan, Q., Chien, M., Weir, D. B., Choksi, R., De Vita, G., Frezzetti, D., Trompeter, H. I., Hornung, V., Teng, G., Hartmann, G., Palkovits, M., Di Lauro, R., Wernet, P., Macino, G., Rogler, C. E., Nagle, J. W., Ju, J., Papavasiliou, F. N., Benzing, T., Lichter, P., Tam, W., Brownstein, M. J., Bosio, A., Borkhardt, A., Russo, J. J., Sander, C., Zavolan, M., and Tuschl, T. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414.
Landi, D., Gemignani, F., Barale, R., and Landi, S. (2008). A catalog of polymorphisms falling in microRNA-binding regions of cancer genes. DNA Cell Biol. 27, 35–43.
Lewis, B. P., Shih, I. H., Jones-Rhoades, M. W., Bartel, D. P., and Burge, C. B. (2003). Prediction of mammalian microRNA targets. Cell 115, 787–798.
Lewohl, J. M., Nunez, Y. O., Dodd, P. R., Tiwari, G. R., Harris, R. A., and Mayfield, R. D. (2011). Up-regulation of MicroRNAs in brain of human alcoholics. Alcohol. Clin. Exp. Res. 35, 1928–1937.
Mardis, E. R. (2008). Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402.
Maziere, P., and Enright, A. J. (2007). Prediction of microRNA targets. Drug Discov. Today 12, 452–458.
McDonagh, E. M., Whirl-Carrillo, M., Garten, Y., Altman, R. B., and Klein, T. E. (2011). From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark. Med. 5, 795–806.
Mishra, P. J., Humeniuk, R., Mishra, P. J., Longo-Sorbello, G. S., Banerjee, D., and Bertino, J. R. (2007). A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc. Natl. Acad. Sci. U.S.A. 104, 13513–13518.
Papadopoulos, G. L., Reczko, M., Simossis, V. A., Sethupathy, P., and Hatzigeorgiou, A. G. (2009). The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res. 37, D155–D158.
Prosser, H. M., Koike-Yusa, H., Cooper, J. D., Law, F. C., and Bradley, A. (2011). A resource of vectors and ES cells for targeted deletion of microRNAs in mice. Nat. Biotechnol. 29, 840–845.
Rehmsmeier, M., Steffen, P., Hochsmann, M., and Giegerich, R. (2004). Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–1517.
Sethupathy, P., Borel, C., Gagnebin, M., Grant, G. R., Deutsch, S., Elton, T. S., Hatzigeorgiou, A. G., and Antonarakis, S. E. (2007). Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3′ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes. Am. J. Hum. Genet. 81, 405–413.
Spielman, R. S., Bastone, L. A., Burdick, J. T., Morley, M., Ewens, W. J., and Cheung, V. G. (2007). Common genetic variants account for differences in gene expression among ethnic groups. Nat. Genet. 39, 226–231.
Stark, A., Brennecke, J., Bushati, N., Russell, R. B., and Cohen, S. M. (2005). Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123, 1133–1146.
Storey, J. D., Madeoy, J., Strout, J. L., Wurfel, M., Ronald, J., and Akey, J. M. (2007). Gene-expression variation within and among human populations. Am. J. Hum. Genet. 80, 502–509.
Stranger, B. E., Forrest, M. S., Clark, A. G., Minichiello, M. J., Deutsch, S., Lyle, R., Hunt, S., Kahl, B., Antonarakis, S. E., Tavare, S., Deloukas, P., and Dermitzakis, E. T. (2005). Genome-wide associations of gene expression variation in humans. PLoS Genet. 1, e78. doi:10.1371/journal.pgen.0010078
Stranger, B. E., Forrest, M. S., Dunning, M., Ingle, C. E., Beazley, C., Thorne, N., Redon, R., Bird, C. P., De Grassi, A., Lee, C., Tyler-Smith, C., Carter, N., Scherer, S. W., Tavare, S., Deloukas, P., Hurles, M. E., and Dermitzakis, E. T. (2007a). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853.
Stranger, B. E., Nica, A. C., Forrest, M. S., Dimas, A., Bird, C. P., Beazley, C., Ingle, C. E., Dunning, M., Flicek, P., Koller, D., Montgomery, S., Tavare, S., Deloukas, P., and Dermitzakis, E. T. (2007b). Population genomics of human gene expression. Nat. Genet. 39, 1217–1224.
Taft, R. J., Pang, K. C., Mercer, T. R., Dinger, M., and Mattick, J. S. (2010). Non-coding RNAs: regulators of disease. J. Pathol. 220, 126–139.
Vaz, C., Ahmad, H. M., Sharma, P., Gupta, R., Kumar, L., Kulshreshtha, R., and Bhattacharya, A. (2012). Analysis of microRNA transcriptome by deep sequencing of small RNA libraries of peripheral blood. BMC Genomics 11, 288. doi:10.1186/1471-2164-11-288
Wang, X. (2008). miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA 14, 1012–1017.
Wang, X., and El Naqa, I. M. (2008). Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics 24, 325–332.
Xiao, F., Zuo, Z., Cai, G., Kang, S., Gao, X., and Li, T. (2009). miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 37, D105–D110.
Yang, J. H., Shao, P., Zhou, H., Chen, Y. Q., and Qu, L. H. (2010). deepBase: a database for deeply annotating and mining deep sequencing data. Nucleic Acids Res. 38, D123–D130.
Yu, Z., Li, Z., Jolicoeur, N., Zhang, L., Fortin, Y., Wang, E., Wu, M., and Shen, S. H. (2007). Aberrant allele frequencies of the SNPs located in microRNA target sites are potentially associated with human cancers. Nucleic Acids Res. 35, 4535–4541.
Zhang, W., and Dolan, M. E. (2009). Use of cell lines in the investigation of pharmacogenetic loci. Curr. Pharm. Des. 15, 3782–3795.
Zhang, W., and Dolan, M. E. (2010). The emerging role of microRNAs in drug responses. Curr. Opin. Mol. Ther. 12, 695–702.
Zhang, W., Duan, S., Bleibel, W. K., Wisel, S. A., Huang, R. S., Wu, X., He, L., Clark, T. A., Chen, T. X., Schweitzer, A. C., Blume, J. E., Dolan, M. E., and Cox, N. J. (2009). Identification of common genetic variants that account for transcript isoform variation between human populations. Hum. Genet. 125, 81–93.
Zhang, W., Duan, S., Kistner, E. O., Bleibel, W. K., Huang, R. S., Clark, T. A., Chen, T. X., Schweitzer, A. C., Blume, J. E., Cox, N. J., and Dolan, M. E. (2008). Evaluation of genetic variation contributing to differences in gene expression between populations. Am. J. Hum. Genet. 82, 631–640.
Keywords: microRNA, gene expression, gene regulation, genetic variation, single nucleotide polymorphism, RNA-sequencing
Citation: Mu W and Zhang W (2012) Bioinformatic resources of microRNA sequences, gene targets, and genetic variation. Front. Gene. 3:31. doi: 10.3389/fgene.2012.00031
Received: 15 January 2012; Paper pending published: 02 February 2012;
Accepted: 20 February 2012; Published online: 05 March 2012.
Edited by:
Jonathan Pollock, National Institute on Drug Abuse, USAReviewed by:
David Langenberger, University Leipzig, GermanyElana J. Fertig, Johns Hopkins School of Medicine, USA
Copyright: © 2012 Mu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Wei Zhang, Department of Pediatrics, University of Illinois at Chicago, 840 South Wood Street, 1200 CSB (MC856), Chicago, IL 60612, USA. e-mail:d2VpemhhbjFAdWljLmVkdQ==