 Jakelyne Lima1
Jakelyne Lima1 Louise Teixeira Cerdeira2
Louise Teixeira Cerdeira2 Erick Bol2 Maria Paula Cruz Schneider2 Artur Silva2
Erick Bol2 Maria Paula Cruz Schneider2 Artur Silva2 Vasco Azevedo3* and Antônio Jorge Gomes Abelém1
Vasco Azevedo3* and Antônio Jorge Gomes Abelém1
- 1 Institute of Exact and Natural Sciences, Federal University of Pará, Pará, Brazil
- 2 Institute of Biological Sciences, Federal University of Pará, Pará, Brazil
- 3 Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil
Improvements in genome sequencing techniques have resulted in generation of huge volumes of data. As a consequence of this progress, the genome assembly stage demands even more computational power, since the incoming sequence files contain large amounts of data. To speed up the process, it is often necessary to distribute the workload among a group of machines. However, this requires hardware and software solutions specially configured for this purpose. Grid computing try to simplify this process of aggregate resources, but do not always offer the best performance possible due to heterogeneity and decentralized management of its resources. Thus, it is necessary to develop software that takes into account these peculiarities. In order to achieve this purpose, we developed an algorithm aimed to optimize the functionality of de novo assembly software ABySS in order to optimize its operation in grids. We run ABySS with and without the algorithm we developed in the grid simulator SimGrid. Tests showed that our algorithm is viable, flexible, and scalable even on a heterogeneous environment, which improved the genome assembly time in computational grids without changing its quality.
Introduction
Genome studies radically changed when the first technique for sequencing DNA became available. Further improvements in sequencing techniques have allowed sequences to be produced on a large scale and inserted into a computer. Appropriate programs, capable of processing data, have been developed to facilitate access and make all of the information available throughout the process (Morozova and Marra, 2008).
After DNA sequencing is necessary to re-build the complete genome from the fragments obtained. This process is known as genome assembly. Genome assembly uses programs that read the sequences, in some cases find redundancies between them and merge the sequences, forming larger continuous consensus sequences. Multiple rounds of assembly are sometimes required for find the best genome assembly sets. In the end of assembly process is necessary ordering the contigs between them or by anchoring in a reference genome to obtain the genome scaffold (Fagin et al., 1992).
This entire process requires a large processing capacity, due to enormous amount of sequences generated by modern equipments. Even a simple organism requires many hours of processing and memory for its complete assembly.
The increasing availability of computers with massive computational power connected to high-speed networks has enabled the aggregation of geographically dispersed resources for the execution of large-scale applications. This aggregation of resources is called grid computing. Grid computing is a model that proposes the use of computing resources of several machines located in various places, even on separate continents, to solve problems that require massive computational power, such as data mining, weather forecasting, computational biology, and medical image processing (Foster, 2001; Foster et al., 2002; Berman et al., 2003).
Grids are currently being used as an alternative for clusters for achieving large-scale processing capacity. Grid computing differs from cluster computing because of the heterogeneity of their resources (which may consist of computers with different architectures, operating systems, and processing capabilities) and due to the dynamic character of the same. Cluster computing is defined as the sharing of resources working cooperatively and managed by a single global system synchronized and centralized. In grids, on other hand, each node has its own manager and resource allocation policy, which is not so visible (Foster et al., 2008).
Programs are composed of small pieces with specific responsibilities and clearly defined, called tasks. Each task has a set of attributes, among them the priority that should be assigned according to their importance. Each task is performed independently, but they need to interact with each other so that the system meets its objectives. In order for rules to be applied to a set of tasks that may use information from the various computers, it is necessary to adopt appropriate scheduling policies for each application. Applications of the type bag-of-tasks (BoT) facilitates scaling because they are composed of independent tasks, allowing the use of policies based on data from only a few systems. They do not require information on grid infrastructure, such as bandwidth, network topology, and network latency. Policies can be called static or dynamic, depending on how the schedule is set (Foster and Kesselman, 2004).
In spite of the BoT applications are simple, it is not easy job to make the scheduling in a heterogeneous and dynamic environment such as grid computing. The scheduling of independents tasks in grid is still difficult due to the use of resources that are shared and due restraint created by other applications, which are running simultaneously. To obtain a good performance in this type of situation requires the use of good information to make scheduling more efficient. In order to achieve this purpose, we developed a dynamic algorithm for BoT applications on grid environments because it has a better match and do not require detailed information (Cirne et al., 2007; Ghanem et al., 2010).
The employment of an efficient algorithm for managing resources is crucial to reduce the time needed to finish tasks in a grid. Here, we propose a task scheduling algorithm that takes grid characteristics into account and can be implemented within ABySS, software used for parallelized de novo genome assembly, in order to optimize the performance of the genome assembly stage and consequently improve the efficiency of this process as a whole (Bittencourt and Madeira, 2006; Simpson et al., 2009).
Materials and Methods
The main goal of our experiments was to evaluate the performance of ABySS using the scheduling algorithm that we developed compared to the default scheduler bundled with ABySS. The ABySS is a software used for the de novo genome assembly and can be used in grids by using message passing interface (MPI) for the communication between nodes (Pacheco, 1996). Reads are distributed among the nodes to form a distributed graph, such that each node knows where the rest of the graph is. The proposed algorithm acts only at this stage of assembly of the distributed graph (Simpson et al., 2009).
These experiments allowed us to evaluate the influence of the heterogeneity of networks (different speeds), heterogeneity of the tasks (size variation), and the granularity of tasks (number of tasks per machine). We used genomic data available from the Institute of Biological Sciences, Federal University of Para, consisting in a set of 33 millions short reads with fixed length (25 bp) and 110× coverage of Corynebacterium pseudotuberculosis I19 genome. The data is available on http://sourceforge.net/projects/abyssgrid/files (Silva et al., 2011).
To perform the experiments, we used the toolkit SimGrid 3.6.1. This toolkit provides basic functions for simulation of distributed applications in grid computing environments. This is a structured set of functions implemented in C language for the simulation driven to events, using an extendend mackup language (XML) file as input to define the network topology and the characteristics of resources and responsibilities. In these simulations, the network transfer times are negligible, because the focus of this analysis to verify the processing efficiency of the CPUs using the proposed task scheduler (Casanova, 2001).
In order to run a simulation in SimGrid, it is necessary to perform the following steps (Legrand et al., 2003):
(a) Model the application defining the code of each agent.
(b) Model the physical platform defining the resources. This consists in to define hosts, links, and the network topology.
(c) Model the file of deployment of application, where is specified the location of the creation and allocation of agents.
(d) Run the simulation.
The experiments were conducted with six heterogeneous machines, i.e., with different operating systems, hardware, and processing power. The tasks used in the simulation also have different sizes. Simulations with ABySS were performed with and without our algorithm, for comparison purposes. Only the k-mer (length hash) parameter was changed to 17, such value has proved satisfactory.
The proposed scheduler does not change the mode of action of ABySS, just the way as the tasks are distributed among the nodes. Thus, the final assembly was not altered, because the main goal is not to improve the quality of the assembly using the ABySS, but minimize the processing time that this step demands.
Results
The scheduler developed in the C language provides ABySS with the capacity to distribute his tasks according to the rules above. All the files (the scheduler itself, the configuration files for SimGrid, the dataset used, and the detailed instructions) are available on http://sourceforge.net/p/abyssgrid.
Figure 1 shows the mean execution times of the scheduling algorithm. Each point summarizes six levels of heterogeneity of machines and tasks. The trend, which can be observed in Figure 1, is that in situations with a greater number of tasks per machine and less granularity, performance improves when there are many tasks. The scheduler dynamically manages to keep all processors busy most of the time. The situation changes only at the end of the execution due to load imbalance, which degrades performance. Another point to consider is the execution time of tasks, which is relatively long because of the large computational load in the case of genomic data.
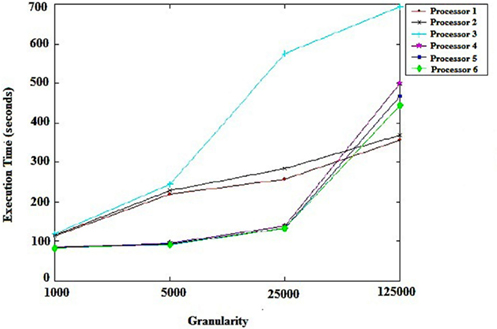
Figure 1. Schedule performance granularity.
Figure 2 shows the impact of the heterogeneity of the machines in the grid. Each point shows the levels of heterogeneity of tasks. We can conclude that the scheduler responds effectively in terms of processing time, even when machines are very heterogeneous. As shown in Figure 2, we can observe that processor 3 failed to perform well, because of the considerable heterogeneity of the machines. The possibility of a slow machine performing a large task is considerable, though it has only a small impact on the execution time of tasks, as was the case for processors 4 and 5.
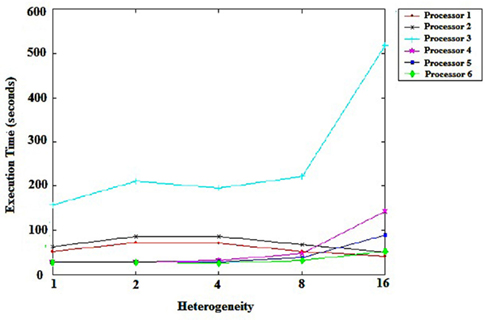
Figure 2. Machine heterogeneity.
Figure 3 shows the impact of heterogeneity on the performance of the tasks scheduling algorithm. Each dot shows the tasks that were performed. We can conclude that the heterogeneity of the tasks did not have a significant impact on the performance of the task scheduler that we proposed, demonstrating its efficiency in this context.
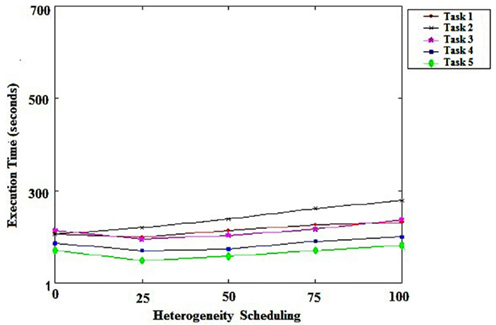
Figure 3. Schedule heterogeneity.
Figure 4 show that using ABySS without our algorithm spent more time spent to execute all the tasks in the same environment. This is due to the fact that the ABySS uses, to distribute its tasks, the first in, first out (FIFO) algorithm, which does not take into account the differences between the available resources in the environment or its load.
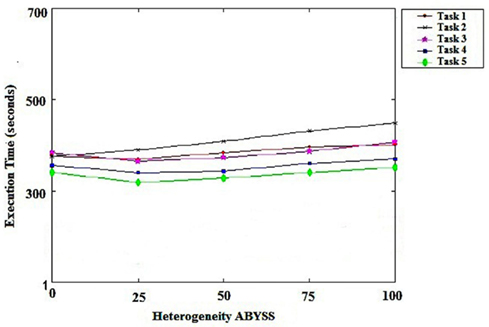
Figure 4. Heterogeneity of ABySS.
Discussion
Grid computing has greatly advanced because of the demand for scientific applications that require processing, storage, and manipulation of large amounts of information. Fields that require such processing power include molecular biology, astronomy, and earthquake prediction.
Genome sequencing techniques are also advancing and generating very large amounts of data that must be processed using resources with great computational power. Computational grids can be used to address this demand for execution of parallel or distributed applications. Though computers have become much more powerful, the genome assembly stage continues to confront limitations because of the type and volume of data generated by next-generation sequencers. Increasing amounts of data exponentially rise the processing time, which can overwhelm the assembly process, so that it becomes necessary to implement techniques that will minimize these limitations.
Future work will be based on improvements in the scheduler so the distribution of tasks is made by first checking if the processors available have enough computing power to speed up processing. These improvements will be checked by testing genome assembly in real computational grids.
Based on the experiments, the obtained results were considered satisfactory, since the assembly can be completed, and when such results are compared to the assembly without the use of the scheduler developed, they proved to be faster. The fact of the machines has used different configurations, and the tasks have different sizes, such fact did not affect significantly the completion and assembly performance. The simulations also showed that the scheduling algorithm submitted to testing can be considered scalable in its entirety, since it reached a minimum efficiency, and for any tested task, there was a minimum number of processors available or imminently available to be perform the processing.
Conclusion
We proposed a scheduling algorithm for computer grids, using the de novo assembly software ABySS. The algorithm proved being very effective in tests with heterogeneous tasks, and there was no impact on processor performance. However, when granularity increased, we identified a small imbalance, causing decreased processor performance. The runtime of the tasks did not affect processor performance in the tests that we run. When the tests were performed with a heterogeneous group of machines, the scheduler responded effectively to the processing challenge. Even when there was deterioration in performance due to a slow machine that was allocated a large task, other processes ended up compensating for the slight loss of time in the task. The task scheduler optimally performed the tests conducted on heterogeneous machines. Bioinformatics researchers can use this tool to optimize genome assembly processes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Rede Paraense de Genômica e Proteômica (genoma.ufpa.br), Conselho Nacional de Desenvolvimento Científico (www.cnpq.br), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (www.capes.gov.br), and Grupo de Estudos em Redes de Computadores e Comunicação Multimídia (gercom.ufpa.br).
References
Berman, F., Fox, G., and Hey, A. J. G. (2003). Grid Computing: Making the Global Infrastructure a Reality, 1st Edn. New York: John Wiley & Sons.
Bittencourt, L. F., and Madeira, E. R. M. (2006). “A dynamic approach for scheduling dependent tasks on the Xavantes grid middleware,” in Proceedings of the 4th International Workshop on Middleware for Grid Computing (MCG 2006) (Melbourne: ACM Press).
Casanova, H. (2001). “Simgrid: a toolkit for the simulation of application scheduling,” in Proceedings First IEEEACM International Symposium on Cluster Computing and the Grid, Brisbane, 430–437.
Cirne, W., Brasileiro, F. V., Silva, D. P. D., Góes, L. F. W., and Voorsluys, W. (2007). On the efficacy, efficiency and emergent behavior of task replication in large distributed systems. J. Parallel Comput. 33, 213–234.
Fagin, B., Watt, J. G., and Gross, R. (1992). A special-purpose processor for gene sequence analysis. Comput. Appl. Biosci. 9, 221–226.
Foster, I. (2001). The anatomy of the grid: enabling scalable virtual organizations. Int. J. High Perform. Comput. Appl. 15, 200–222.
Foster, I., and Kesselman, C. (2004). Grid 2: Blueprint for a New Computing Infrastructure, 2nd Edn. San Francisco: Morgan Kaufmann.
Foster, I., Kesselman, C., Nick, J. M., and Tuecke, S. (2002). Grid services for distributed system integration. Computer 35, 37–46.
Foster, I., Zhao, Y., Raicu, I., and Lu, S. (2008). “Cloud computing and grid computing 360-degree compared,” in 2008 Grid Computing Environments Workshop, Austin, Vol. 5, 1–10.
Ghanem, A. M. A., Saleh, A. I., and Ali, H. A. (2010). “High performance adaptive framework for scheduling Grid Workflow applications,” in International Conference on Computer Engineering and Systems (ICCES), Cairo, 52–57.
Legrand, A., Marchal, L., and Casanova, H. (2003). “Scheduling distributed applications: the SimGrid simulation framework,” in Proceedings of Third IEEEACM International Symposium on Cluster Computing and the Grid (CCGrid 2003), Washington, 138–145.
Morozova, O., and Marra, M. A. (2008). Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264.
Silva, A., Schneider, M. P., Cerdeira, L., Barbosa, M. S., Ramos, R. T., Carneiro, A. R., Santos, R., Lima, M., D’Afonseca, V., Almeida, S. S., Santos, A. R., Soares, S. C., Pinto, A. C., Ali, A., Dorella, F. A., Rocha, F., de Abreu, V. A., Trost, E., Tauch, A., Shpigel, N., Miyoshi, A., and Azevedo, V. (2011). Complete genome sequence of Corynebacterium pseudotuberculosis I19, a Strain Isolated from a Cow in Israel with bovine mastitis. J. Bacteriol. 193, 323–324.
Keywords: computational grids, task scheduling, genome assembly, NGS
Citation: Lima J, Cerdeira LT, Bol E, Schneider MPC, Silva A, Azevedo V and Abelém AJG (2012) A scheduling algorithm for computational grids that minimizes centralized processing in genome assembly of next-generation sequencing data. Front. Gene. 3:38. doi: 10.3389/fgene.2012.00038
Received: 16 November 2011; Accepted: 27 February 2012;
Published online: 19 March 2012.
Edited by:
Raya Khanin, Memorial Sloan-Kettering Cancer Center, USAReviewed by:
Mario Inostroza-Ponta, Universidad de Santiago de Chile, ChileJan Aerts, Leuven University, Belgium
Copyright: © 2012 Lima, Cerdeira, Bol, Schneider, Silva, Azevedo and Abelém. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Vasco Azevedo, Institute of Biological Sciences, Federal University of Minas Gerais, Av. Antônio Carlos, 6627 – Pampulha, CEP 31270-901, Belo Horizonte, Minas Gerais, Brazil. e-mail:dmFzY29hcmlzdG9uQGdtYWlsLmNvbQ==