
 Zané Lombard
Zané Lombard
- Division of Human Genetics, School of Pathology, Faculty of Health Sciences, National Health Laboratory Service and University of the Witwatersrand, Johannesburg, South Africa
Imprinted genes are expressed from one parental allele in a parent-of-origin manner. This monoallelic behavior is regulated by allele-specific DNA methylation that is confined to differentially methylated regions (DMRs). To date there are over 80 known human imprinted genes of which only three are known to have paternally methylated DMRs. In mice there exists an additional paternally methylated DMR associated with Rasgrf1. The Rasgrf1 gene forms part of the MAPK signaling pathway and plays a role in long-term memory formation and growth control. A RASGRF1-associated parent-of-origin specific DMR in humans and its methylation status in sperm DNA have not been explored. The primary aim of this study was to determine whether the human RASGRF1 gene contains a DMR and whether this DMR is paternally methylated and shows roughly 50% methylation in somatic tissue. Computational assessments were done to identify putative CTCF binding sites, CpG islands (CGIs) that could serve as potential RASGRF1 DMRs and tandem repeats within or adjacent to these CGIs. The methylation status of three putative CGIs was assessed using quantitative pyrosequencing technology. None of the putative CTCF binding sites was found to occur in the predicted CGIs. The three putative CGIs linked to RASGRF1 did not display allele-specific methylation. While one of the three CGIs was found to be hypomethylated in both blood DNA and sperm DNA, the other two were found to be hypermethylated. The CGIs evaluated in this study did not fit the criteria of being a allele-specific DMR. Unlike the mouse Rasgrf1 locus, the human RASGRF1-associated CpG rich regions do not exhibit differential methylation in a parent-of-origin manner.
Introduction
Differential marking at primary differentially methylated regions (DMRs) (also known as the imprinting control regions, ICRs) in a parent-of-origin manner is a key feature of all imprinted genes. Most ICRs overlap with CpG Island (CGIs) and the imprints at these regions are established during gametogenesis and are maintained throughout development (Morgan et al., 2005; Hutter et al., 2006; Lim and Ferguson-Smith, 2010). Although DNA methylation at the ICR is key for the reciprocal expression of these genes, differences in histone modifications at the ICR of the two parental chromosomes are also found to contribute to the reciprocal expression profiles (as reviewed in Kacem and Feil, 2009). The frequent presence of tandem repeats, simple repeats, and long interspersed nuclear elements (LINEs) in the vicinity of imprinted regions is another common feature of imprinted genes (Allen et al., 2003; Hutter et al., 2006). Tandem repeat elements tend to occur more frequently within or adjacent to CGIs associated with imprinted genes than they do with CGIs of biallelically expressed genes (Hutter et al., 2006). Transcription factors and chromatin insulators, Yin–Yang 1 (YY1) and CTCF, are other common features of imprinted genes and are often present within the ICR. They have prominent roles in the epigenetic modifications and transcriptional control of associated imprinted genes (Filippova et al., 1996; Kim et al., 2006). However, it is the ICR that is the key feature of imprinted genes due to its regulatory function. It is the methylation status of the ICR that has direct influence on gene expression at the locus (Yoon et al., 2002).
Rasgrf1 forms part of the MAPK signaling pathway and has been found to play a role in long-term memory consolidation and growth control (Brambilla et al., 1997; Itier et al., 1998). Rasgrf1, found on mouse chromosome 9 [NCBI Gene ID: 19417], is paternally imprinted and exclusively expressed from the paternal allele in the neonatal brain and liver (Dockery et al., 2009). A binary switch located 30 kbp upstream of the Rasgrf1 promoter is responsible for controlling the imprint status of the gene in mice. The first component of this binary switch is a repeat unit consisting of 40 copies of a 41-bp tandem repeat element which is responsible for establishing methylation of the second component of the switch, the DMR (Holmes et al., 2006). This DMR serves as a CCCTC-binding factor (CTCF) binding site on the unmethylated maternal allele. Binding of a CTCF protein blocks enhancer-to-promoter interactions resulting in the silencing of the maternal allele of Rasgrf1. On the paternal allele, the CTCF binding factor is unable to bind to the methylated DMR resulting in expression of Rasgrf1 from the paternal allele (Yoon et al., 2005).
The mouse DMR is a primary DMR established in the male germline (Yoon et al., 2002). It is located between Rasgrf1 and a brain-specific micro-RNA (Mir-184) that is imprinted and expressed from the paternal allele in the brain (Nomura et al., 2008). Both these genes have human homologs and are transcribed in opposite directions. The mouse and human genomes display conserved synteny of Rasgrf1 and Mir-184.
The human ortholog of Rasgrf1 is found on chromosome 15 [NCBI Gene ID: 5923] but its imprint status is unknown. A recent study by Yuen et al. (2011) set out to identify parent-of-origin specific methylation at many loci, including RASGRF1. While the results indicated some tissue-specific differential methylation ranging from 5 to 35% in trophoblast and mesenchyme tissue at this locus, there was no allele-specific or parent-of-origin specific differential methylation (Yuen et al., 2011). The aim of this study was to determine whether the human RASGRF1 gene is associated with a DMR and whether this DMR is paternally methylated. In order to assess the potential imprint status of the human RASGRF1 locus, a number of computational assessments were done to identify key imprinting features, while pyrosequencing analysis was used to assess the methylation status of various CGIs surrounding RASGRF1.
Methodology
Computational Assessment
Searching for sequence similarity
The BLAST-like alignment tool (BLAT) was used to identify sequence similarity using the mouse DMR [NCBI Reference Sequence: NT_039476.7] and the 40 copies of the 41-bp tandem repeat element unit as query sequences and comparing them against the human genome on Ensembl. The Ensembl database can be found at http://www.ensembl.org/index.html.
Identification of CTCF binding sites
Knowing that the mouse Rasgrf1 DMR is regulated by a CTCF binding protein (Yoon et al., 2005), a starting point for assessment was to identify all CTCF binding sites within human RASGRF1 and between RASGRF1 and the MIR-184. The in silico CTCF Binding site database (CTCFBSDB; http://insulatordb.uthsc.edu/storm.php) contains both experimentally identified and computationally predicted CTCF binding sites. The query sequence was uploaded in the CTCFBS prediction tool. This tool searches the query sequence for CTCFBS core motifs, which are represented by position weight matrices (PWM). A match between a query sequence and a PWM is represented by a score that corresponds to the log-odds of the observed sequence being generated by the motif versus being generated by the background. A large positive score is indicative of a good match for a potential CTCFBS.
Identification of evolutionary conserved regions among various species
The mouse DMR is located between Rasgrf1 and a brain-specific micro-RNA (Mir-184). The mouse and human genomes display conserved synteny of Rasgrf1 and Mir-184. Identifying regions of conservation between these two genes in multiple species could suggest the presence of regulatory elements. The start co-ordinate of Rasgrf1 (mouse chr9: 89,804,613 [NCBI37/mm9]; human chr15: 77,041,341 [NCBI36/hg18]) and the start co-ordinate of Mir-184 (mouse chr9: 89,697,098 [NCBI37/mm9]; human chr15: 77,289,185 [NCBI36/hg18]), in mice and in humans, was obtained from the UCSC Genome Browser and used in the evolutionary conserved region (ECR) browser to visualize conservation-profiles (Ovcharenko et al., 2004). These co-ordinates gave a defined region in which the mouse DMR lies. Hence, the mouse was used as a base genome first, followed by using the human (hg18) as a base genome. The ECRs were identified using a minimum identity of 70% per 100 bp sequence. The ECR browser can be found at http://ecrbrowser.dcode.org/whilst the UCSC Genome browser is accessible at http://genome.ucsc.edu/index.html.
Searching for CpG islands as potential sites for differential methylation
It is known that DMRs in mice have an average G + C content of 54%, suggesting that these regions are generally CpG rich (Kobayashi et al., 2006). Identifying CpG rich regions between the human RASGRF1 and MIR-184 would allow for identification of possible DMRs that would need to be validated in the laboratory.
A CGI Prediction tool, part of UCSC’s bioinformatic tools, was used to identify CGIs between RASGRF1 and MIR-184. All CGIs are scored quantitatively based on their CGI strength. High CGI strength is indicated by:
1. An absence of DNA methylation
2. Presence of promoter activity
3. Open chromatin structure.
The scores allow bona fide CGIs – which have high CGI strength – to be distinguished from CpG rich regions devoid of bona fide CGI characteristics (Bock et al., 2007). The best indicator of CGI strength and most predictive of bona fide CGI is the “combined epigenetic score,” which can assume one of three scores:
• 0.67 – high confidence for at least two of the three high CGI strengths
• 0.50 – equal chance of the CGI to be a bona fide island or a false positive
• 0.33 – high confidence for at least one of the three high CGI strengths (Bock et al., 2007).
The CGI Prediction tool can be found as a link in the eleventh paragraph of the “Custom Annotation Tracks” page. This page can be found by clicking on the “Custom Annotation Tracks” tab on the panel on the left on the UCSC Genome Browser homepage.
Search for tandem repeats within and adjacent to predicted CpG islands
The presence of tandem repeats within or adjacent to the CGIs predicted using the CGI Prediction Tool would allow for the identification of CGIs that could serve as potential ICRs.
The sequences of the CGIs, together with 1000 bp up- and downstream of the CGI regions, were used in the tandem repeat finder (TRF) program (version 4.03) to identify tandem repeats. The reason behind using 1000 bp up- and downstream was that tandem repeats associated with ICRs are either found within or adjacent to the ICRs (Thorvaldsen et al., 1998; Okamura et al., 2000; Takada et al., 2002). The repeats associated with Rasgrf1 in mice are found within the ICR (Brideau et al., 2010).
This program utilizes an algorithm to detect tandem repeats and does not require the need to specify the repeat pattern or repeat size. The repeats are modeled based on the percentage identity and frequency of indels between adjacent repeat copies in the sequence (Benson, 1999). Default parameters were used for this study. The TRF program can be found at http://tandem.bu.edu/trf/trf.html.
Molecular Assessment
Study participants
The methylation status of the putative DMRs was assessed using peripheral human blood (n = 10) and human sperm (n = 8) samples from unrelated participants, after obtaining written informed consent. Ethics approval for sample collection was obtained from the University of the Witwatersrand, Committee for Research on Human Subjects – Medical (M050706 and M090555, respectively).
DNA extraction and bisulfite modification
The blood samples were extracted using the salting-out procedure (Miller et al., 1988) while the sperm samples were extracted using the supplementary protocol “Purification of DNA from epithelial cells mixed with sperm cells” of the QIAamp® DNA Micro Kit (Qiagen, Valencia, CA, USA). The DNA samples (400 ng) were subjected to bisulfite modification using the EZ DNA Methylation Gold™ Kit (Zymo Research, Orange, CA, USA) in accordance with manufacturers’ instructions.
PCR amplification for pyrosequencing reactions
Two known imprinted loci were used as controls in this experiment. While SNRPN (a maternally imprinted gene), was used as a measure in sperm cell DNA for potential contamination with somatic cell DNA and expected to by unmethylated in sperm DNA, IG-DMR (a paternally imprinted gene) was expected to be fully methylated in sperm DNA.
All amplicons were generated in a 50-μl reaction containing 2 μl of bisulfite-treated DNA, 0.2 μM each of the forward and reverse PCR primers (primers are listed in Table 1) and 0.02 μM of the universal primer, 0.125 mM deoxynucleoside triphosphates (Bioline, London, UK), 2.0 mM MgCl2, 1× Buffer (Applied Biosystems, NJ, USA), 1 M Betaine (Sigma-Aldrich, Seelze, Germany), and 1 U of AmpliTaq Gold (Applied Biosystems, NJ, USA). For IG-DMR, a nested PCR was performed, first with the outer primer set and the same reaction mix as above. The second PCR was set up using 2 μl of round one PCR product, the inner primer set and the reaction mix as above.

Table 1. PCR primers for pyrosequencing PCR reactions.
PCR was performed in a GeneAmp® 2720 thermal cycler (Applied Biosystems, NJ, USA) with the following conditions for all reactions; 95°C for 5 min, followed by 50 cycles of 95°C for 15 s, Ta°C for 30 s [see Table 1 for annealing temperatures (Ta °C)], and 72°C for 15 s; with a final extension step of 72°C for 5 min; and a final hold at 15°C. The PCR amplifications for each sperm sample and each blood sample was performed in duplicate and triplicate, respectively.
Pyrosequencing reactions and data analysis
Pyrosequencing reactions were performed using the PyroMark® Gold Q96 Kit (Qiagen, Valencia, CA, USA). Single-stranded biotinylated PCR products were prepared for pyrosequencing by use of the Vacuum Prep Tool following the manufacturer’s instructions. The sequencing primers used for each pyrosequencing assay are listed in Table 2. The pyrosequencing assays were designed to include several internal controls for bisulfite treatment to evaluate the completion of the bisulfite conversion step. Pyrosequencing data were analyzed using the Pyro Q-CpG (Biotage, Uppsala, Sweden).
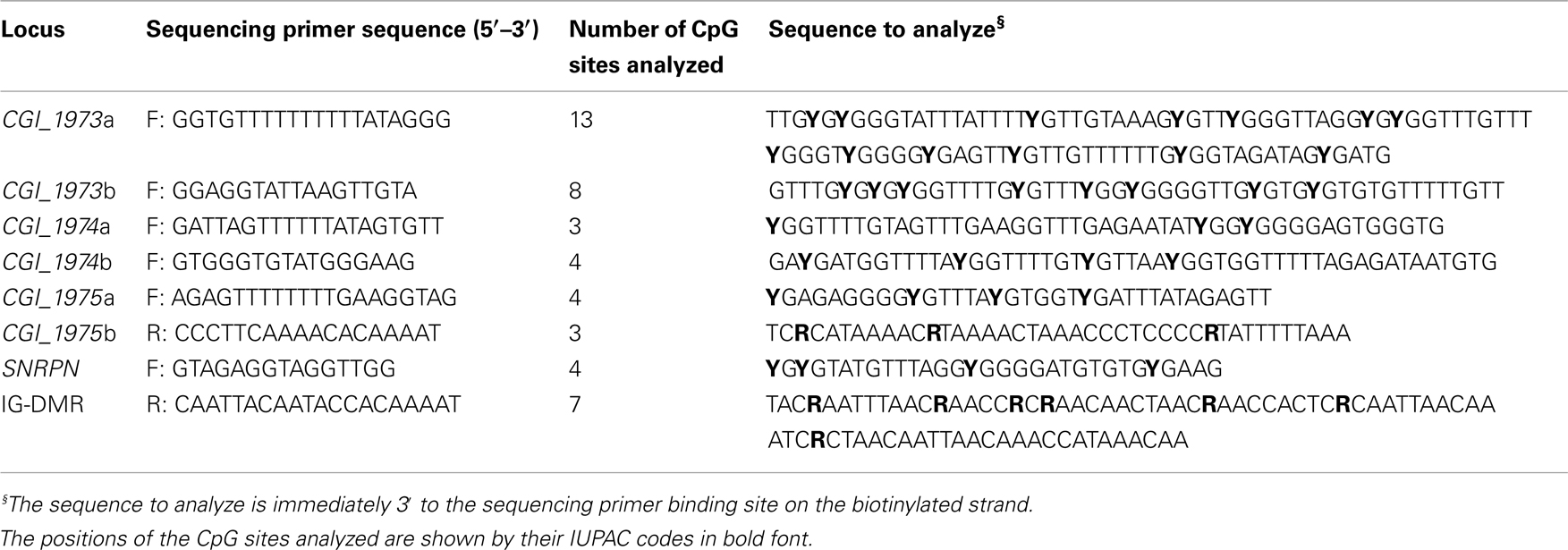
Table 2. Primers for pyrosequencing assays.
Results
Computational Assessment
Searching for sequence similarity
The BLAST algorithm was used to identify regions in the human genome that had sequence similarity to the mouse DMR and the 40 copies of the 41 bp repeat element. No sequence similarity was identified.
Identification of CTCF binding sites
REN_20, MIT_LM2, MIT_LM7, and MIT_LM23 are PWM motif sequences that correspond to CTCF binding sites (Kim et al., 2007b; Xie et al., 2007). The in silico CTCFBS prediction tool used these four motifs to search for putative CTCF binding sites in a region spanning 249841 bp (Ensembl co-ordinates chr 15: 79252289–79502130 [release 61, GRCh37.p2]), this region included RASGRF1 and 118915 bp upstream of the gene to the first base of MIR-184. Four putative CTCF binding sites were identified each with a PWN score greater than 3.0 (Table A1 in Appendix). A score greater than 3.0 indicates that the putative CTCF binding site matches more closely to the PWM motif compared with the random background sequence (Pan and Phan, 2008). The putative CTCF binding site sequences corresponding to REN_20 and MIT_LM7 were found within RASGRF1. MIT_LM2 and MIT_LM23 were both found in the intergenic region between MIR-184 and RASGRF1.
Identification of evolutionary conserved regions among various species
The mouse genome was selected as the base genome and the intergenic regions between Rasgrf1 and the Mir-184, within which the mouse DMR lies, was visualized for conservation-profiles. The mouse genome was compared to eight vertebrate species including Homo sapiens (humans), Pan troglodytes (chimp), Macaca mulatta (rhesus macaque), Xenopus tropicalis (frog), Gallus gallus (chicken), Fugu rubripes (pufferfish), and the Monodelphis domestica (opossum). A number of conserved intergenic regions were identified (not shown). We used the mouse DMR co-ordinates (chr9: 89774524–89774827) from UCSC to search for conservation between species in the ECR browser. This DMR was not found to be conserved in any of the eight vertebrate species. The human genome (hg18) was then used as a base genome and the region between RASGRF1 and MIR-184 was assessed to identify evolutionary conserved intergenic regions. The intergenic region at the promoter of RASGRF1 was conserved in five of eight species while the first coding exon was found to be conserved in all eight vertebrate species. The MIR-184 was conserved in five of the eight species.
Searching for CpG islands as potential sites for differential methylation
All the CGI tracks were viewed simultaneously on UCSC’s CGI prediction tool. We only focused on the panel containing combined epigenetic scores. The reason behind choosing this specific score is that it provides a precise estimate of CGI strength. The CGI strength indicates the CGI’s inherent tendency to exhibit an open and transcriptionally competent chromatin structure (Bock et al., 2007). Three CGIs were identified between the human RASGRF1 and MIR-184: CGI_1973 (chr15: 77168729–77171326) at the promoter of RASGRF1; CGI_1974 (chr15: 77258299–77258552) and CGI_1975 (chr15: 77269392–77269597) [NCBI36/hg18].
Identification of tandem repeats within or adjacent to CpG islands
The TRF program only detects repeats which occur in a head-to-tail manner (Hutter et al., 2006). Only one out of the three CGIs identified, using the CGI Prediction tool, had direct tandem repeats associated with it. There were a total of eight repeat elements found within and directly adjacent to CGI_1973 (Table A2 in Appendix). However, the TRF program identifies overlapping repeat elements therefore two elements were regarded as one if the smaller one overlapped with the larger one. The smaller repeat motif was chosen as a consensus sequence (Hutter et al., 2006).
Molecular Assessment of the Putative CpG Islands Which Could Serve as Sites of Differential Methylation
Bisulfite-treated PCR products for each of the three CGIs and the two control loci were quantitatively assayed using pyrosequencing technology. Pyrosequencing was performed on PCR products from sperm (in duplicate on n = 8) and blood (in triplicate on n = 10) DNA samples, respectively. The C (methylated) and the T (unmethylated) alleles were quantified and subsequently expressed as a percentage. Figure 1 illustrates the methylation percentages, for RASGRF1 CGIs, in each tissue type for each region evaluated.
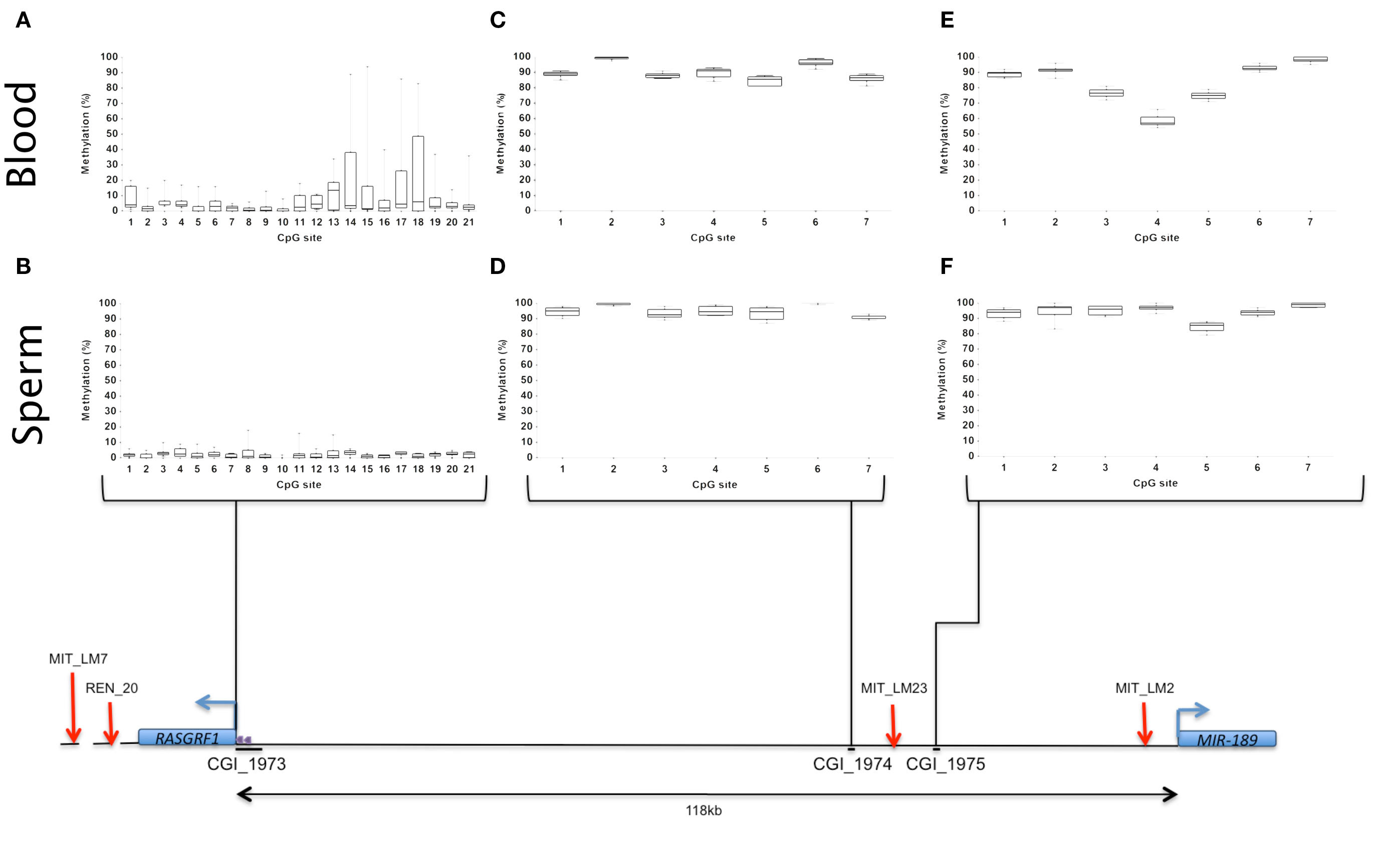
Figure 1. A schematic representation of RASGRF1 and MIR-184 and the positions of the putative CpG islands relative to the two genes. The box and whisker plots (A–F) represent the methylation percentages for individual CpG sites for the sperm (n = 8) and blood (n = 10) samples, respectively. The red arrows denote the CTCF binding sites while the purple arrows denote the tandem repeats associated with CGI_1973. The lines beneath the CGIs represent the approximate size of these regions relative to the other features in the region.
Due to the size of CGI_1973, found at the promoter of RASGRF1, a smaller region found to be rich in CG dinucleotides within this CGI was selected for further analysis. This smaller region was then split into two sub-regions CGI_1973a and CGI_1973b (bordering exon1 of RASGRF1) and each was assayed separately. The results were analyzed together. CGI_1974 and 1975 were difficult regions not easily amenable to pyrosequencing and therefore CGI_1974 was covered using two sequencing primers while CGI_1975 was amplified in two separate PCR reactions using two sets of PCR primers.
The entire CGI_1973 region was 2598 bp in length with a G + C content of 64%. However, only a region spanning 515 bp with a G + C content of 70% was investigated. CpG sites 1–13 were found to be hypomethylated in blood and sperm DNA. The methylation percentages for blood samples were very variable and displayed considerable inter-site and inter-individual variation at CpG sites 14–21 (Figure 1A). It should be kept in mind that these CpG sites border exon1 of the RASGRF1 gene. In comparison the methylation percentages of the sperm samples showed relative hypomethylation of this region with little or no inter-site and inter-individual variation (Figure 1B).
CGI_1974 was 212 bp in length with a G + C content of 56%. The mean methylation percentages of the blood and sperm samples showed relative hypermethylation (Figures 1C,D).
The CGI_1975 region was 231 bp in length with a G + C content of 51%. The mean methylation percentages of the blood and sperm samples showed relative hypermethylation. The blood samples showed an average methylation percentage of between 50 and 60% at CpG site 4 and the two sites (CpG 3 and 5) on either side displayed a mean methylation of between 70 and 90% (Figure 1E). The average methylation percentages of the sperm samples at these sites ranged between 90 and 100% except for site 5 where the methylation percentage was between 70 and 90% (Figure 1F).
The mean methylation of the SNRPN DMR was found to be negligible (2.43%) in sperm samples. The theoretical methylation value for DMRs in somatic tissue is 50% (Cooper and Constancia, 2010). However studies have shown that somatic DMRs display methylation levels between 35 and 65% (Woodfine et al., 2011). Therefore the low level of methylation at the SNRPN DMR within the sperm samples may suggest that the paternal allele may not be completely devoid of methylation but rather displays basal levels of methylation or that there could have been low levels of contamination with somatic cells. It was concluded that the sperm samples were not significantly contaminated with somatic cell DNA.
The IG-DMR locus was found to be hypermethylated in the sperm samples, as expected, displaying a mean methylation of 89.17%. These results confirmed that the quantitative methylation technique was working optimally.
Discussion
Given the role of imprinted genes in human development and health, it is important to identify key imprinted genes in the genome and to understand how they shape the phenotype. The primary aim of this study was to determine whether the human RASGRF1 gene has a DMR and whether this DMR is paternally methylated. Methylation analysis of three putative DMRs in the vicinity of this gene in sperm and blood samples indicate that RASGRF1-associated CpG rich regions do not exhibit differential methylation in a parent-of-origin manner, as is seen in its mouse counter-part.
These results demonstrate that the human genome does not contain any homologous sequences to the mouse Rasgrf1-associated DMR or the 40 copies of the 41 nucleotide tandem repeat element. Two of the most common features of any imprinted region are the presence of a DMR and adjacent to this, tandem repeats. It is often expected that DMRs and the tandem repeats associated with them are conserved between the mouse and human due to their key regulatory function in imprinted regions (Paulsen et al., 2005; Hutter et al., 2010). The lack of these two common features in the syntenic region between the RASGFRF1 gene and the MIR-184 suggest that the expression of RASGRF1 may not be regulated by an imprinted region in humans, as it is in the mouse.
The mouse Rasgrf1 and the human ortholog RASGRF1 display both organizational similarities and differences. The mouse gene contains 26 exons while the human gene possesses two extra exons. The sizes of exons and intron–exon junction sequences are highly conserved. The 5′ sequence upstream of exon 1 is not only highly conserved between the mouse and the human but is also highly conserved in other vertebrates. This is consistent with the presence of important regulatory elements in this region (de la Puente et al., 2002). However it should be noted that organizational similarity in the genomes of species does not necessarily relate to the conservation of imprinting between species (Okamura et al., 2000). As an example, the mouse and human IGF2R genes share conserved intronic CGIs with large direct repeats that are maternally expressed in the mouse but are biallelically expressed in humans (Kim et al., 2007a).
The CTCF binding protein is a ubiquitously expressed 11 zinc finger protein that plays a key role in chromatin insulation and is the only protein known to exhibit enhancer-blocking activity (Bao et al., 2008; Williams and Flavell, 2008). Two of the four putative CTCF binding sites corresponding to MIT_LM2 and MIT_LM2 were located in the intergenic region between MIR-184 and RASGRF1. The position of these binding sites is consistent with a potential role of CTCF as an insulator protein (Kim et al., 2007b). The other two putative binding sites corresponding to REN_20 and MIT_LM7 were found within RASGRF1. It is known that almost half of the CTCF binding sites within the human genome are located within genes and many of these segregate alternative promoters of a single gene (Kim et al., 2007b). The protocadherin gamma locus, the T-cell receptor locus and the immunoglobulin heavy chain locus all have intragenic CTCF binding sites that segregate transcriptional start sites that display differential activities across tissues (Kim et al., 2007b). RASGRF1 is known to have four transcripts therefore the identification of two putative CTCF binding sites within this gene may have a role in segregating the transcriptional start sites based on their activities across various tissues. Functional studies would need to be done to determine whether these putative CTCF binding sites actually bind the CTCF protein.
In some instances CTCF binding sites are found within DMRs. An example of such a CTCF binding site is that which is present at the mouse Rasgrf1 DMR (Yoon et al., 2005). Another example is the CTCF binding site present at the H19/Igf2 ICR which serves as a primary DMR established in the germline (Szabo et al., 2004). The hypomethylated maternal ICR serves as a binding site for the CTCF protein. Once the protein binds to this region it acts as an insulator preventing the interactions of the enhancers with the promoter of Igf2 (Szabo et al., 2004). In this study the putative CTCF binding sites were not found to occur in the predicted CGIs. This suggests that RASGRF1, if imprinted, would be unlikely to be regulated by a CTCF binding protein as is its mouse ortholog. However, if RASGRF1 is imprinted in humans there are other factors, besides the CTCF binding protein which could mediate imprinting. These factors could be cis- or trans-acting and could contain multiple elements that work in synergy to establish and maintain the imprint status (Ogawa et al., 2006).
The mouse and human genomes display conserved synteny of Rasgrf1 and Mir-184. In mice the Rasgrf1 DMR is found in the intergenic region. The ECR browser results demonstrate that the majority of the intergenic regions are conserved in the P. troglodytes, M. mulatta, Canis familiaris, Mus musculus and M. domestica. The conservation of these regions across multiple vertebrate species suggests that one or more of these regions could contain possible cis-regulatory elements such as enhancers or repressors crucial for gene regulation (Visel et al., 2007; Nakagawa et al., 2008).
The majority of the CGIs in the mouse and human genomes are hypomethylated and a small proportion of the hypermethylated CGIs are located at imprinted loci in the germline (Henckel and Arnaud, 2010). The hypermethylated CGIs of potential imprinted loci are expected to display monoallelic methylation (50% methylation) in somatic tissues (Strichman-Almashanu et al., 2002). For a region to be regarded as a DMR, it is proposed that the following criteria need to be fulfilled: the sequence should be rich in CpG dinucleotides with an average G + C content of 54% (Kobayashi et al., 2006) or above and display methylation levels between 35 and 65% in somatic tissue (Woodfine et al., 2011) over four consecutive CpG sites (Kobayashi et al., 2006).
CGI_1973, a putative CGI identified using the CGI Prediction tool, is located in the 5′ sequence upstream of exon 1, a region that harbors the promoter sequence for RASGRF1 and has been found to share high homology with the mouse sequence (de la Puente et al., 2002). The methylation assessment of this region in blood samples displayed a deviation from the expected 50% methylation for imprinted loci in somatic tissue, indicating that the selected CGI is not differentially methylated. This is due to the fact that CGIs associated with promoters of tissue-specific genes are generally unmethylated in both expressing and non-expressing tissues although exceptions are being identified (Bird, 2002; Song et al., 2005; Weber et al., 2007). It is however interesting to note that part of this CGI (CpG sites 14–21) displays great inter-individual and inter-site variation only in blood samples. The difference in methylation patterns between individuals in peripheral blood samples is unlikely to be attributed to technical errors. To test the reproducibility of the PCR and pyrosequencing results, we performed each PCR and pyrosequencing reaction in duplicate for sperm samples and in triplicate for blood samples. The similar methylation profiles observed between individuals in the sperm samples, for this region and other regions, demonstrates that our techniques were reproducible. The possible reason behind the high inter-individual variation observed in peripheral blood could be that CpG sites 14–21 in this specific region are not tightly regulated, making this region more responsive to the effects of various environmental factors including carcinogen exposures, inflammation, stress and diet which all have the ability to alter DNA methylation patterns (Jirtle and Skinner, 2007; Christensen et al., 2009; Murgatroyd et al., 2009).
The other two putative CGIs identified in this study, CGI_1974 and CGI_1975, lie in the intergenic region between RASGRF1 and the MIR-184. Both sample types were found to be hypermethylated in these regions and showed a lack of inter-individual variation. This suggests that this region is under tight regulation and may harbor transposable elements that are normally hypermethylated in the genome (Geiman and Robertson, 2002; Slotkin and Martienssen, 2007). The methylation profiles of the sperm samples for regions CGI_1974 and CGI_1975 displayed tight methylation patterns. These results are contrary to that found by Flanagan et al. (2006) who demonstrated the presence of significant inter-individual DNA methylation variation in human male germ cells. However, studies in mice and other organisms have shown that certain loci display partial epigenetic stability during meiosis which may be a possible explanation for our results (Flanagan et al., 2006). In somatic tissues DMRs are expected to exhibit 50% average methylation over several consecutive CpG sites (Cooper and Constancia, 2010). However the paternal allele, represented by the sperm samples, is found to be almost completely hypermethylated over seven CpG sites of the CGI_1975 region, with an average methylation between 90 and 100%, suggesting a possible paternal parent-of-origin effect. CpG site 4 in the blood samples assayed displayed an average methylation percentage between 50 and 60%. The surrounding CpG sites had significantly higher methylation, indicating that this is not a DMR for RASGRF1.
Tandem repeats are found to occur frequently within the human genome, however they tend to occur more frequently within or adjacent to CGIs associated with imprinted genes (Hutter et al., 2006). The tandem repeats identified using the TRF tool were found to be associated with only one of the CGIs identified, CGI_1973. This CGI is located at the promoter region of RASGRF1 and was found to be hypomethylated both in the peripheral blood and in the sperm samples. Owing to the fact that the CGI_1973 was unmethylated in both tissue types, it can be deduced that the tandem repeats associated with this promoter CGI is not involved in establishing methylation at this locus but rather plays a role in regulating gene expression by binding transcription factors and influencing splicing efficiency, RNA stability and RNA-to-protein interactions (Martin et al., 2005; Shah et al., 2010). The presence of repeats but the absence of differential methylation at CGI_1973 in peripheral blood suggests that this CGI does not serve as a DMR for RASGRF1.
One of the limitations of this study is that no expression data was available to confirm the imprint status of RASGRF1. Rasgrf1 is expressed in the neonatal brain and liver, in mice. Therefore, in order to assess allele-specific gene expression in humans, large numbers of neonatal human brain and liver samples would be needed to identify an individual heterozygous for a SNP within the coding region of the gene. The gene expression work was not within the scope of the current study but could be pursued in the future. In addition to the gene expression work, it would be useful to obtain DNA methylation profiles of other tissues including ovarian teratomas, brain tissue and liver tissue in the event that RASGRF1 displays tissue-specific differential methylation.
Conclusion
The CGIs evaluated in this study did not fit the criteria of being a DMR. The human RASGRF1-associated CpG rich regions do not exhibit differential methylation in a parent-of-origin manner. RASGRF1 does not have the same imprinting mechanism as the mouse Rasgrf1 locus and the human genome lacks the DMR, CTCF binding site and repeat sequences that control differential gene expression at this locus in mice.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the participants of this study and the contribution from the Cryobank, a private andrology laboratory and sperm bank in Parktown, Johannesburg, South Africa. We thank Shaun Aron for bioinformatics analyses. This study was funded by the University of the Witwatersrand FRC Medical Endownment Grant (001 410 8466101 PITA000) and NHLS Research Trust grants awarded to Michele Ramsay (94039) and Punita Pitamber (94146).
References
Allen, E., Horvath, S., Tong, F., Kraft, P., Spiteri, E., Riggs, A. D., and Marahrens, Y. (2003). High concentrations of long interspersed nuclear element sequence distinguish monoallelically expressed genes. Proc. Natl. Acad. Sci. U.S.A. 100, 9940–9945.
Bao, L., Zhou, M., and Cui, Y. (2008). CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res. 36, D83–D87.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580.
Bock, C., Walter, J., Paulsen, M., and Lengauer, T. (2007). CpG island mapping by epigenome prediction. PLoS Comput. Biol. 3, 1055–1070. doi:10.1371/journal.pcbi.0030110
Brambilla, R., Gnesutta, N., Minichiello, L., White, G., Roylance, A. J., Herron, C. E., and Ramsey, M. (1997). A role for the ras signaling pathway in synaptic transmission and long-term memory. Nature 390, 281–286.
Brideau, C. M., Kauppinen, K. P., Holmes, R., and Soloway, P. D. (2010). A non-coding RNA within the rasgrf1 locus in mouse is imprinted and regulated by its homologous chromosome in trans. PLoS ONE 5, 1–10. doi:10.1111/j.1601-183X.2011.00678.x
Christensen, B. C., Houseman, E. A., Marsit, C. J., Zheng, S., Wrensch, M. R., Wiemels, J. L., and Nelson, H. H. (2009). Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 5, 1–13. doi:10.1371/journal.pgen.1000602
Colella, S., Shen, L., Baggerly, K. A., Issa, J. P., and Krahe, R. (2003). Sensitive and quantitative universal pyrosequencing methylation analysis of CpG sites. BioTechniques 35, 146–150.
Cooper, W. N., and Constancia, M. (2010). How genome-wide approaches can be used to unravel the remaining secrets of the imprintome. Brief. Funct. Genomics 9, 315–328.
de la Puente, A., Hall, J., Wu, Y. Z., Leone, G., Peters, J., Yoon, B. J., Soloway, P., and Plass, C. (2002). Structural characterization of Rasgrf1 and a novel linked imprinted locus. Gene 291, 287–297.
Dockery, L., Gerfen, J., Harview, C., Rahn-Lee, C., Horton, R., Park, Y., and Davis, T. L. (2009). Differential methylation persists at the mouse Rasgrf1 DMR in tissues displaying monoallelic and biallelic expression. Epigenetics 4, 241–247.
Filippova, G. N., Fagerlie, S., Klenova, E. M., Myers, C., Dehner, Y., Goodwin, G., Neiman, P. E., Collins, S. J., and Lobanenkov, V. V. (1996). An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 16, 2802–2813.
Flanagan, J. M., Popendikyte, V., Pozdniakovaite, N., Sobolev, M., Assadzadeh, A., Schumacher, A., and Zangeneh, M. (2006). Intra- and interindividual epigenetic variation in human germ cells. Am. J. Hum. Genet. 79, 67–84.
Geiman, T. M., and Robertson, K. D. (2002). Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J. Cell. Biochem. 87, 117–125.
Henckel, A., and Arnaud, P. (2010). Genome-wide identification of new imprinted genes. Brief. Funct. Genomics 9, 304–314.
Holmes, R., Chang, Y., and Soloway, P. D. (2006). Timing and sequence requirements defined for embryonic maintenance of imprinted DNA methylation at Rasgrf1. Mol. Cell. Biol. 26, 9564–9570.
Hutter, B., Bieg, M., Helms, V., and Paulsen, M. (2010). Imprinted genes show unique patterns of sequence conservation. BMC Genomics 11, 649. doi:10.1186/1471-2164-11-649
Hutter, B., Helms, V., and Paulsen, M. (2006). Tandem repeats in the CpG islands of imprinted genes. Genomics 88, 323–332.
Itier, J. M., Tremp, G. L., Leonard, J. F., Multon, M. C., Ret, G., Schweighoffer, F., Tocque, B., Bluet-Pajot, M. T., Cormier, V., and Dautry, F. (1998). Imprinted gene in postnatal growth role. Nature 393, 125–126.
Jirtle, R. L., and Skinner, M. K. (2007). Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 8, 253–262.
Kacem, S., and Feil, R. (2009). Chromatin mechanisms in genomic imprinting. Mamm. Genome 20, 544–556.
Kim, J. D., Hinz, A. K., Bergmann, A., Huang, J. M., Ovcharenko, I., Stubbs, L., and Kim, J. (2006). Identification of clustered YY1 binding sites in imprinting control regions. Genome Res. 16, 901–911.
Kim, K. P., Thurston, A., Mummery, C., Ward-van Oostwaard, D., Priddle, H., Allegrucci, C., Denning, C., and Young, L. (2007a). Gene-specific vulnerability to imprinting variability in human embryonic stem cell lines. Genome Res. 17, 1731–1742.
Kim, T. H., Abdullaev, Z. K., Smith, A. D., Ching, K. A., Loukinov, D. I., Green, R. D., Zhang, M. Q., Lobanenkov, V. V., and Ren, B. (2007b). Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128, 1231–1245.
Kobayashi, H., Suda, C., Abe, T., Kohara, Y., Ikemura, T., and Sasaki, H. (2006). Bisulfite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet. Genome Res. 113, 130–137.
Lim, A. L., and Ferguson-Smith, A. C. (2010). Genomic imprinting effects in a compromised in utero environment: implications for a healthy pregnancy. Semin. Cell Dev. Biol. 21, 201–208.
Martin, P., Makepeace, K., Hill, S. A., Hood, D. W., and Moxon, E. R. (2005). Microsatellite instability regulates transcription factor binding and gene expression. Proc. Natl. Acad. Sci. U.S.A. 102, 3800–3804.
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215.
Morgan, H. D., Santos, F., Green, K., Dean, W., and Reik, W. (2005). Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14, R47–R58.
Murgatroyd, C., Patchev, A. V., Wu, Y., Micale, V., Bockmuhl, Y., Fischer, D., Holsboer, F., Wotjak, C. T., Almeida, O. F., and Spengler, D. (2009). Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 12, 1559–1566.
Nakagawa, H., Tategu, M., Yamauchi, R., Sasaki, K., Sekimachi, S., and Yoshida, K. (2008). Transcriptional regulation of an evolutionary conserved intergenic region of CDT2-INTS7. PLoS ONE 3, e1484. doi:10.1371/journal.pone.0001484
Nomura, T., Kimura, M., Horii, T., Morita, S., Soejima, H., Kudo, S., and Hatada, I. (2008). MeCP2-dependent repression of an imprinted miR-184 released by depolarization. Hum. Mol. Genet. 17, 1192–1199.
Ogawa, H., Wu, Q., Komiyama, J., Obata, Y., and Kono, T. (2006). Disruption of parental-specific expression of imprinted genes in uniparental fetuses. FEBS Lett. 580, 5377–5384.
Okamura, K., Hagiwara-Takeuchi, Y., Li, T., Vu, T. H., Hirai, M., Hattori, M., Sakaki, Y., Hoffman, A. R., and Ito, T. (2000). Comparative genome analysis of the mouse imprinted gene impact and its non-imprinted human homolog IMPACT: toward the structural basis for species-specific imprinting. Genome Res. 10, 1878–1889.
Ovcharenko, I., Nobrega, M. A., Loots, G. G., and Stubbs, L. (2004). ECR browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 32, W280–W286.
Paulsen, M., Khare, T., Burgard, C., Tierling, S., and Walter, J. (2005). Evolution of the Beckwith-Wiedemann syndrome region in vertebrates. Genome Res. 15, 146–153.
Shah, S. N., Hile, S. E., and Eckert, K. A. (2010). Defective mismatch repair, microsatellite mutation bias, and variability in clinical cancer phenotypes. Cancer Res. 70, 431–435.
Slotkin, R. K., and Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285.
Song, F., Smith, J. F., Kimura, M. T., Morrow, A. D., Matsuyama, T., Nagase, H., and Held, W. A. (2005). Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc. Natl. Acad. Sci. U.S.A. 102, 3336–3341.
Strichman-Almashanu, L. Z., Lee, R. S., Onyango, P. O., Perlman, E., Flam, F., Frieman, M. B., and Feinberg, A. P. (2002). A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 12, 543–554..
Szabo, P. E., Tang, S. H., Silva, F. J., Tsark, W. M., and Mann, J. R. (2004). Role of CTCF binding sites in the Igf2/H19 imprinting control region. Mol. Cell. Biol. 24, 4791–4800.
Takada, S., Paulsen, M., Tevendale, M., Tsai, C. E., Kelsey, G., Cattanach, B. M., and Ferguson-Smith, A. C. (2002). Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: implications for imprinting control from comparison with Igf2-H19. Hum. Mol. Genet. 11, 77–86.
Thorvaldsen, J. L., Duran, K. L., and Bartolomei, M. S. (1998). Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 12, 3693–3702.
Visel, A., Bristow, J., and Pennacchio, L. A. (2007). Enhancer identification through comparative genomics. Semin. Cell Dev. Biol. 18, 140–152.
Weber, M., Hellmann, I., Stadler, M. B., Ramos, L., Paabo, S., Rebhan, M., and Schubeler, D. (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–466.
Williams, A., and Flavell, R. A. (2008). The role of CTCF in regulating nuclear organization. J. Exp. Med. 205, 747–750.
Woodfine, K., Huddleston, J. E., and Murrell, A. (2011). Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenetics Chromatin 4, 1.
Xie, X., Mikkelsen, T. S., Gnirke, A., Lindblad-Toh, K., Kellis, M., and Lander, E. S. (2007). Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc. Natl. Acad. Sci. U.S.A. 104, 7145–7150.
Yoon, B. J., Herman, H., Hu, B., Park, Y. J., Lindroth, A., Bell, A., and West, A. G. (2005). Rasgrf1 imprinting is regulated by a CTCF-dependent methylation-sensitive enhancer blocker. Mol. Cell. Biol. 25, 11184–11190.
Yoon, B. J., Herman, H., Sikora, A., Smith, L. T., Plass, C., and Soloway, P. D. (2002). Regulation of DNA methylation of Rasgrf1. Nat. Genet. 30, 92–96.
Yuen, R. K., Jiang, R., Peñaherrera, M. S., McFadden, D. E., and Robinson, W. P. (2011). Genome-wide mapping of imprinted differentially methylated regions by DNA methylation profiling of human placentas from triploidies. Epigenetics Chromatin 4, 10.
Appendix

Table A1. In silico predicted CTCF binding site motif sequences within RASGRF1 and between RASGRF1 and MIR-184.
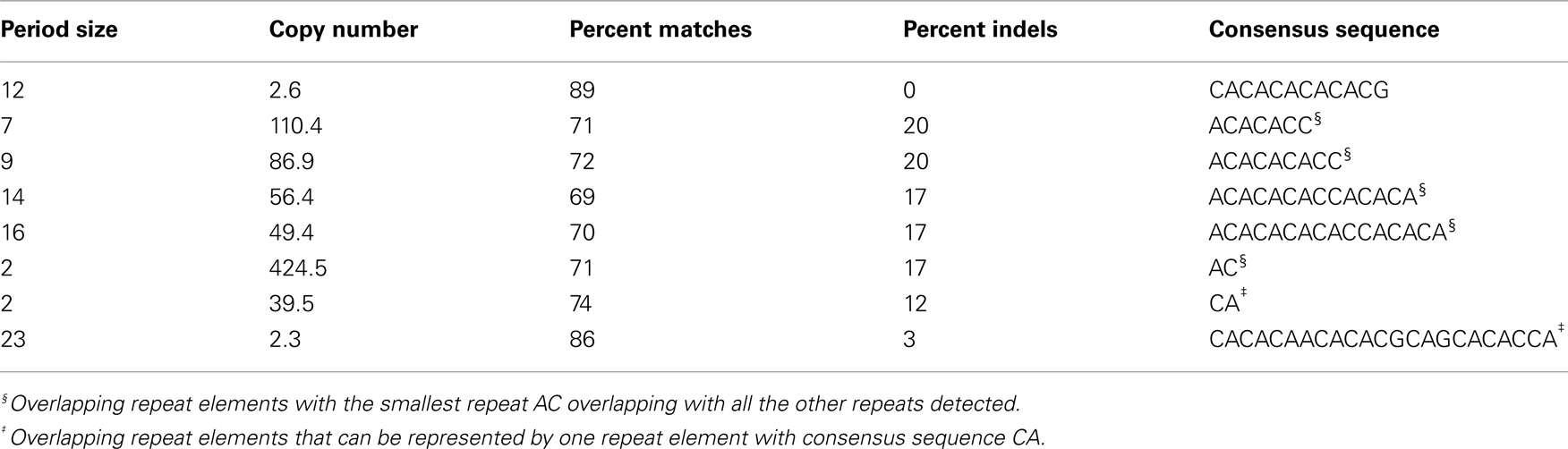
Table A2. Tandem repeats detected within and adjacent to CGI_1973.
Keywords: genomic imprinting, DNA methylation, CpG island, DMR, RASGRF1, Mir-184
Citation: Pitamber PN, Lombard Z and Ramsay M (2012) No evidence for a parent-of-origin specific differentially methylated region linked to RASGRF1. Front. Gene. 3:41. doi: 10.3389/fgene.2012.00041
Received: 14 September 2011; Accepted: 02 March 2012;
Published online: 28 March 2012.
Edited by:
Richard Saffery, Murdoch Childrens Research Institute, AustraliaReviewed by:
Igor Pogribny, National Center for Toxicological Research, USAJafar Sharif, RIKEN, Japan
David Jim Martino, Murdoch Childrens Research Institute, Australia
Boris Novakovic, Murdoch Childrens Research Institute, Australia
Copyright: © 2012 Pitamber, Lombard and Ramsay. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Punita Navnitlal Pitamber, Division of Human Genetics, National Health Laboratory Service, Corner Hospital and De Korte Street, Johannesburg 2000, South Africa. e-mail:cHVuaXRhLnBpdGFtYmVyQG5obHMuYWMuemE=;cHVuaXRhLnBpdGFtYmVyQGdtYWlsLmNvbQ==