 Kerstin Pietsch1,2*
Kerstin Pietsch1,2* Nadine Saul1,2
Nadine Saul1,2 Suresh C. Swain2†
Suresh C. Swain2† Ralph Menzel1,2 Christian E. W. Steinberg1
Ralph Menzel1,2 Christian E. W. Steinberg1 Stephen R. Stürzenbaum2*
Stephen R. Stürzenbaum2*- 1 Laboratory of Freshwater and Stress Ecology, Department of Biology, Humboldt-Universität zu Berlin, Berlin, Germany
- 2 Analytical and Environmental Science Division, School of Biomedical Sciences, King’s College London, London, UK
Recent research has highlighted that the polyphenols Quercetin and Tannic acid are capable of extending the lifespan of Caenorhabditis elegans. To gain a deep understanding of the underlying molecular genetics, we analyzed the global transcriptional patterns of nematodes exposed to three concentrations of Quercetin or Tannic acid, respectively. By means of an intricate meta-analysis it was possible to compare the transcriptomes of polyphenol exposure to recently published datasets derived from (i) longevity mutants or (ii) infection. This detailed comparative in silico analysis facilitated the identification of compound specific and overlapping transcriptional profiles and allowed the prediction of putative mechanistic models of Quercetin and Tannic acid mediated longevity. Lifespan extension due to Quercetin was predominantly driven by the metabolome, TGF-beta signaling, Insulin-like signaling, and the p38 MAPK pathway and Tannic acid’s impact involved, in part, the amino acid metabolism and was modulated by the TGF-beta and the p38 MAPK pathways. DAF-12, which integrates TGF-beta and Insulin-like downstream signaling, and genetic players of the p38 MAPK pathway therefore seem to be crucial regulators for both polyphenols. Taken together, this study underlines how meta-analyses can provide an insight of molecular events that go beyond the traditional categorization into gene ontology-terms and Kyoto encyclopedia of genes and genomes-pathways. It also supports the call to expand the generation of comparative and integrative databases, an effort that is currently still in its infancy.
Introduction
Although numerous molecules of herbal origin have been hailed as powerful decelerators of aging, few studies have focused on the precise physiological and genetic mechanisms that drive this process. The nematode Caenorhabditis elegans is ideally suited for biogerontological research, not only because of its short life cycle (Gami and Wolkow, 2006; Gill, 2006; Kaletta and Hengartner, 2006) but also due to the considerable conservation of basic cellular and molecular principles (The C. elegans Sequencing Consortium, 1998).
Of the many bioactive polyphenols (PPs) previously shown to extend the lifespan of C. elegans we selected two well characterized compounds, namely Quercetin (Q; work in C. elegans: Kampkötter et al., 2007a,b, 2008; Saul et al., 2008; Pietsch et al., 2009, 2011; Surco-Laos et al., 2011; Grünz et al., 2012; for a detailed review about general beneficial effects of Q, tested in vivo and ex vivo, be referred to Boots et al., 2008) and Tannic acid (TA; work in C. elegans: Saul et al., 2010, 2011; Lublin et al., 2011; for a detailed review be referred to Koleckar et al., 2008). Both PPs are characterized by inverted J-shaped concentration–response curves, typical for a hormetic effect (Calabrese and Baldwin, 2001). Moreover, the exposure to PPs enhances the oxidative and thermal stress resistance. Both PPs exert no major negative impact on the reproductive output but result in a reduction of body length and fat content, two phenotypes that align well with the disposable soma patterns described by Kirkwood (1977, 1988). Utilizing various nematode mutant strains, preliminary genetic analyses identified daf-2, age-1, sek-1, and unc-43 as mediators of Q induced longevity and stress resistance (Pietsch et al., 2009) and sek-1 was shown to be important for TA mediated extension of lifespan (Saul et al., 2010). Notwithstanding these differences, some of the data obtained suggests that Q and TA induce longevity by similar genetic mechanisms, e.g., the involvement of sek-1, altering the fat metabolism, or hormesis-based dose–responses. An overview of results from previous studies can be found in Table S1 in Supplementary Material 1.
To pinpoint molecular genetic pathways, we performed genome-wide DNA microarray experiments spanning different concentrations of Q and TA, including at least two lifespan prolonging concentrations for each PP. In addition, we tested a low dose of Q (50 μM) because (i) the limitation of solubility precluded the use of doses above 200 μM Q and (ii) this concentration was shown to enhance thermal tolerance (unpublished data) possibly indicative of elevated stress resistance. For TA we included a post-effective, in single trials already toxic (Saul et al., 2010) concentration. Initially, this identified differentially expressed genes (DEGs), which were found to be significantly up- or down-regulated in response to Q and/or TA, many displayed a concentration dependent change in expression. A second layer of analysis included gene ontology (GO), Kyoto encyclopedia of genes and genomes (KEGG) pathways, as well as gene expression mountain algorithms (as introduced by Kim et al., 2001). By analyzing the overlapping transcripts identified in the respective lifespan prolonging concentrations of each PP (Q 100 and 200 μM = Qlongevity; TA 100 and 200 μM = TAlongevity), we were able to predict putative master regulators involved in PP mediated longevity. A final meta-analysis compared these core-genes with recently published transcriptional profile data linked to age-related gene expression, the genetic background of longevity mutants or infection. The alignment of common (condition-overlapping) DEGs facilitated the identification of genes and associated pathways that may act as master switches of longevity.
The primary goal of this study was to highlight that the meta-analysis of large datasets is not restricted to complement a simplistic database evaluation but has the potential to uncover compound overlapping molecular switches.
Materials and Methods
Strains and Sample Generation
N2 wild type C. elegans were maintained on nematode growth medium (NGM) plates using Escherichia coli OP50 as food source (Brenner, 1974; Sulston and Hodgkin, 1988; Lewis and Fleming, 1995). Untreated nematodes (the P0 generation) were chunked onto control and treatment plates (50, 100, and 200 μM Q; 100, 200, and 300 μM TA) and incubated at 20°C for 4 days. A synchronous culture was generated through egg preparation (Strange et al., 2007) with sodium hypochlorite (Sigma, Germany). Eggs were rotated over night (20 rpm) and the resultant hatched L1s subsequently transferred to plates containing the respective doses of Q or TA. Worms were grown to the young (pre-reproductive) adult stage, harvested by rinsing off with M9 buffer, washed at least three times with M9, shock frozen in liquid nitrogen and stored at −80°C. For each condition, samples were cultivated in triplicate.
RNA Preparation
RNA was isolated following the standard procedures as defined by the Trizol protocol (Invitrogen, Germany) but modified to include a homogenization step with 0.5 mm glass beads to maximize cell breakage. The resultant RNA was further processed by means of a RNeasy purification followed by DNase digestion (Qiagen, Germany). All samples were stored at −80°C until further use.
RNA Amplification, Biotin Labeling, and DNA Microarray Analysis
RNA was processed with the MessageAMP™ Premier RNA Amplification Kit (Ambion, Austin, TX, USA) which relies on the T7 in vitro transcription (IVT) amplification technology (Van Gelder et al., 1990). First- and second-strand cDNA synthesis, cRNA synthesis, labeling, fragmentation, GeneChip hybridization, and scanning were performed according to the manufacturer’s specifications (Affymetrix, Santa Clara, CA, USA). We utilized the C. elegans array chip (Affymetrix) which covers the whole genome (22,548 transcripts). Triplicate chips were run for each condition (designated as Q0, Q50, Q100, Q200 and TA0, TA100, TA200, TA300). Whole RNA, cDNA, and cRNA qualities and quantities were assessed at each step using capillary electrophoresis (Bioanalyzer, Agilent Technologies, UK) and micro volume spectrophotometry (NanoDrop1000, Thermo Scientific, UK).
Data Interpretation and Statistical Analysis
Processing of global transcription expression values (DNA microarray)
Pre-processing of raw microarray data included probe-specific background correction, summation of probe set values, and normalization using the GCRMA algorithm with CARMAweb 1.4, an R- and Bioconductor-based web service for microarray data analysis1 (Rainer et al., 2006). The quality of normalization was assessed by box plot and MA-graph analyses. Differences between treatments were visualized by principal component analysis (PCA) plotting with multiexperiment viewer (MeV)2 (Saeed et al., 2003). Data was initially filtered for missing values and then subjected to a CLEAR-test that combines differential expression and variability using the GEPAS web server3 (Tárraga et al., 2008). An unpaired t-test was followed by a significance analysis of microarray (SAM) test including a calculation that estimates the false discovery rate (FDR). The FDR was set to a non-stringent level of <12.5%. DEGs showing a fold change of at least 1.25 were analyzed for their molecular functions, biological processes, and cellular components using the software packages GoMiner (Zeeberg et al., 2005) and DAVID4. Due to the sound technical and experimental quality of the data, which returned strong statistically significant signal intensities, the chosen fold-cut-off values were deemed to be biologically meaningful and aligned to previous data analyses (Grigoryev et al., 2004; McCarthy and Smyth, 2009).
Comparison of datasets to screen for significant overlaps: representation factor
The overlap between differing conditions (e.g. PPs and gene expression mountains, gene classes, or datasets taken from the literature) was determined by computing the representation factors (RFs). The RFs define the fold enrichment between gene lists (Kim et al., 2001; Evans et al., 2008). The choice of N (genome) was based on the values recommended by the authors. Intersection p-values were calculated from the hypergeometric distribution. RFs were considered significant when RF >1 and *p < 0.05, **p < 0.005 or ***p < 0.001.
Results
The Relationship between Exposure to Q or TA, Lifespan Extension and the Output of Transcriptional Responses
Previous reports have stated that, whilst 50 μM of Q evoked no significant change in lifespan, 100 and 200 μM Q prolonged the mean lifespan by 11 and 10%, respectively (Pietsch et al., 2009, 2011). However, at 250 μM of Q the lifespan was reduced by −7% (see Table S1 in Supplementary Material 1) and therefore can be considered to be a toxic concentration. In addition, because concentrations above 200 μM of Q did not dissolve completely in the agar and the bacteria, we refrained from using doses above 200 μM Q for the microarray experiment. Thus, for Q, we utilized two lifespan prolonging concentrations (100 and 200 μM), and a non-effective (50 μM) pre-longevity dose for global transcriptomics.
For TA, the most effective concentration was 100 μM, which resulted in an increase in mean lifespan of 18%, an effect that was still significant but notably less pronounced at 200 μM (8%) and was absent at 300 μM (Saul et al., 2010; see Table S1 in Supplementary Material 1). Hence, we used two life-extending concentrations (100 and 200 μM of TA), and in addition one post-effective (borderline toxic) concentration (300 μM TA).
We chose this test design to cover a large dose-range: from pre-effective to effective in the case of Q (Q0, Q50, Q100, Q200), and from effective to post-effective/toxic for TA (TA0, TA100, TA200, TA300; Note: the numerical identifiers represent the concentration in micromolar). Pre-processing of raw microarray data included probe-specific background correction, summation of probe set values, and normalization using the GCRMA algorithm with 93 CARMAweb 1.4, an R- and Bioconductor-based web service for microarray data analysis (see text footnote 1) (Rainer et al., 2006). The CARMAweb reports can be viewed in Supplementary Material 2 and 3.
Venn diagrams (Figure 1) were compiled to summarize the level of overlap between doses (statistics for significant DEGs can be found in Supplementary Material 4: sheet “Statistics”; full genome fold change (FC-) values are listed in Supplementary Material 5). Investigating the transcriptional response of Q treatment (Figure 1A), revealed that the number of DEGs increased markedly at the lifespan modulating doses of Q100 and Q200 (3.3- and 3.8-fold compared to the non-longevity dose Q50). Interestingly, more genes were up-regulated than down-regulated at Q50, but the opposite was observed at Q100 and Q200. It is worthy of note that the majority of DEGs overlapped in Q100 and Q200 exposures, thus suggesting the presence of common response pathways.
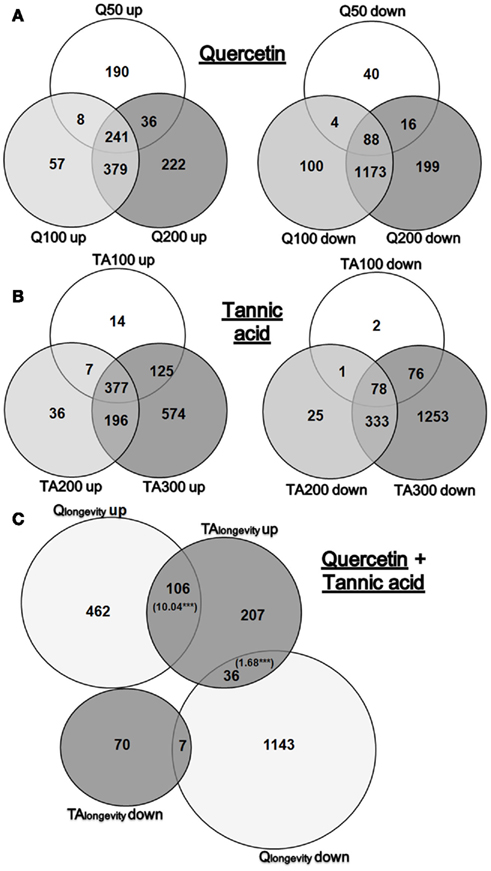
Figure 1. Venn diagrams of DEGs. Overview of significant DEGs (fold change >1.25 or <0.8) in response to Q50, Q100, Q200 or TA100, TA200, TA300. Shown is the overlap of either up- or down-regulated genes in Q (A) or TA (B) treated nematodes. (C) Displays the overlap of significantly up- and down-regulated genes in Qlongevity and TAlongevity. The bracketed numbers in the intersections display significant overrepresented RFs, ***p < 0.001.
As with Q, exposure to TA resulted in a dose dependent increase in DEGs which was most pronounced at the highest dose tested. More genes were up-regulated than down-regulated in the lifespan prolonging conditions TA100 and TA200, a ratio that was reversed at TA300 where more genes were down-regulated (Figure 1B). Striking was the proportion of DEGs that responded exclusively to TA300 (61%), a substantial proportion compared to TA100 (2%) or TA200 (6%).
By taking the intersection of DEGs shared by the lifespan prolonging concentrations of each PP, it was possible to extract gene lists for Qlongevity (Q100 and Q200 overlap), TAlongevity (T100 and T200 overlap) and Q&TAlongevity (overlap of all four groups). A Venn diagram that incorporates Qlongevity and TAlongevity identified a significant overlap (Figure 1C). As our main focus was the identification of mechanisms involved in PP triggered lifespan extension, downstream data processing focused primarily on Qlongevity and TAlongevity gene lists, however, individual GO-, KEGG-, gene expression mountains-, and gene class-analyses are provided for all concentrations in the Supplementary Material 4.
Overrepresented GO-Terms
The summary results of the GO-cluster analysis with DEGs from the Qlongevity, TAlongevity, and Q&TAlongevity gene lists are presented in Table S2 in Supplementary Material 1. The complete analyses for all single concentrations are provided in the Supplementary Material 4: sheet “GO-Analysis.” The Qlongevity analysis returned, due to the large input list of DEGs, more overrepresented GO-terms than the equivalent analysis with TAlongevity. Overrepresented GO-terms derived from the up-regulated transcripts in Qlongevity included chromatin assembly, lipid metabolic process, monooxygenase activity, and nucleosome. GO-terms from down-regulated genes included, besides others, nervous system development, regulation of multicellular organism growth, Dauer entry, regulation -of transcription, -of response to stimulus, -of cell communication, -of biological quality, -of locomotion, and -of programmed cell death. In TAlongevity, significant GO-terms were linked to muscle contraction, neurotransmitter transporter activity and cytoskeleton, embryonic development ending in birth or egg hatching, positive regulation of biological process, and chromatin. GO-terms in Q&TAlongevity were restricted to Cellular Components (pseudopodium).
Scrutinizing the GO-analysis of single concentrations (Supplementary Material 4: sheet “GO-Analysis”) revealed, for example, that the GO-terms in the pre-lifespan extending concentration Q50 (lysozyme activity and oxidoreductase activity acting on the CH–CH group of donors) may reflect an early onset of induced immunity and stress resistance. In contrast, a striking accumulation of overrepresented GO-terms were observed in TA300, which included numerous categories indicative of an unfavorable condition (DNA damage response, signal transduction, mismatch DNA binding, cell death, and others).
Overrepresented KEGGs
Whilst the assignment of GO-terms is defined by automatic/electronic annotation that is based on sequence homology, KEGG-pathways are manually curated from the literature. As with the GO-ontology, KEGG-analysis on the Q treatment lists returned a multitude of pathways (Table 1). For further details about regulated genes in the respective KEGG-terms see Table S3 in Supplementary Material 1 for Qlongevity and TAlongevity, and Supplementary Material 4: sheet “KEGG-Analysis” for single concentrations. Overrepresented KEGGs in Q50 included pathways involved in the metabolism of amino acids, glutathione, and xenobiotics as well as fatty acid elongation. At higher exposures seven (Q100) and eight (Q200) KEGG-pathways were found to be overrepresented, notably most (six) were present at both concentrations (see Qlongevity, Table 1) with an analogous mode/direction of regulation, i.e., either repressed or induced (Table S3 in Supplementary Material 1). Pathways include metabolism of amino acids, xenobiotics or drugs, transport processes (Lysosome), and signal transduction processing (Wnt and TGF-beta signaling). Whilst DEGs from amino acid metabolism and the lysosome displayed heterogeneous expression levels, transcripts belonging to the signaling pathways were predominantly repressed, whereas almost all genes associated with drug/xenobiotic metabolism were found to be induced.
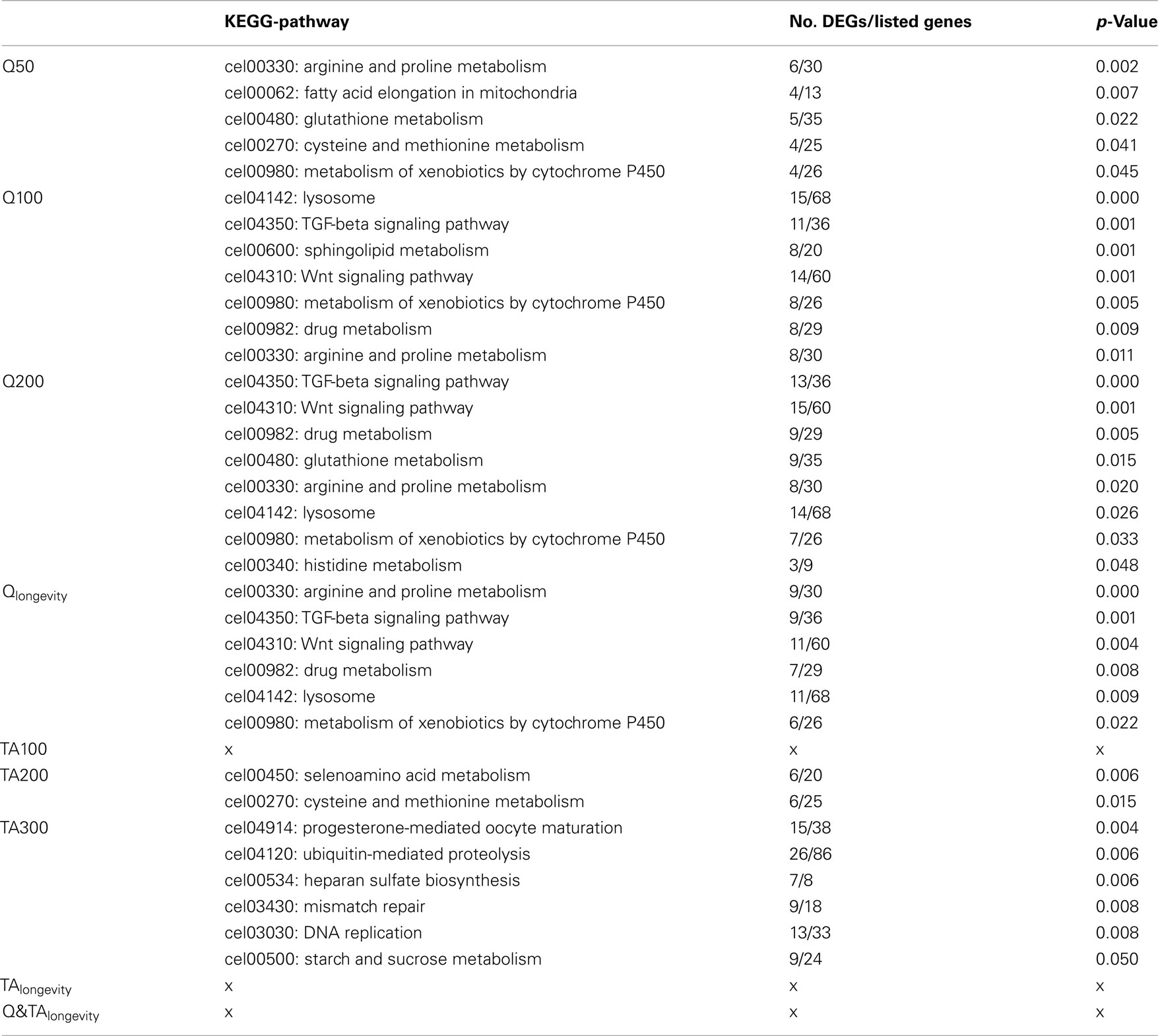
Table 1. Overview of significant KEGG-pathways (see Table S3 in Supplementary Material 1 for pooled groups and Supplementary Material 4: sheet “KEGG-Analysis” for genes with FC-values).
The dynamic response patterns of Q exposure were not mirrored by the TA treatments. No KEGG-pathways could be linked to the most potent lifespan extending concentration, TA100; hence, none were assigned to TAlongevity. For TA200 only two KEGG-pathways were found to be overrepresented, both derived from amino acid metabolism. Analysis of the TA300 gene list returned six KEGG-pathways, notably DNA replication, mismatch repair, and Ubiquitin-mediated proteolysis.
Meta-Analysis: Comparison of Global Transcriptional Patterns in Qlongevity and TAlongevity to Selected Datasets in the Literature
To date, several microarray studies have identified age-related transcriptional changes in the nematode C. elegans (summarized in Golden and Melov, 2007). To investigate if a systematic comparison would reveal common age-related pathways, we correlated our datasets with the published expression profiles obtained from long-lived daf-12(rh273) (Fisher and Lithgow, 2006), daf-2 mutants (Evans et al., 2008), TGF-beta mutants (Shaw et al., 2007), and immune-responsive Pseudomonas aeruginosa infected nematodes (Evans et al., 2008). The dataset from Evans et al. (2008) includes a meta-analysis of different daf-2 alleles (Murphy et al., 2003; McElwee et al., 2004) and immune-challenged nematodes (Shapira et al., 2006; Troemel et al., 2006).
The meta-analysis aimed to indentify similar molecular mechanisms of longevity in Qlongevity, as well as TAlongevity and define (if possible) the correlation to long-living mutants. Datasets were analyzed by focusing on (i) overrepresented gene expression mountains (according to Kim et al., 2001), which characterize the global patterns of a transcriptional response and (ii) DEGs, to identify new target genes. The significance of overlap between two gene lists were assigned by calculating the RF, and significances were determined by hypergeometric probability scoring. Supplementary Material 5 provides Gene Expression Mountains, Gene Classes and Groups, and further studies in a table. By using the filter function in Excel, all results that are described in the following sections can be reconstructed.
Overrepresented expression mountains
Kim et al. (2001) assembled data from several hundred C. elegans DNA microarray experiments. This allowed a three-dimensional gene expression map to be computed consisting of 44 co-regulated gene-groups (mountains), 30 of which could be assigned to specific gene classes and therefore to a potential physiological importance.
Figure 2 displays all gene expression mountains, in which DEGs of Qlongevity and/or TAlongevity (Figures 2A,B) are overrepresented (additional RF values for single concentrations in all 44 gene expression mountains can be found in Supplementary Material 4: sheet “Gene Expression Mountains” and “Gene Classes and Groups”). DEGs from Qlongevity and TAlongevity could both be assigned to mounts 4 (sperm-enriched genes, protein kinases and phosphatases, MSPs), 8 (enriched for intestine-, protease-, carboxylesterase-, lipase-genes, antibacterial proteins and UGTs), 19 (enriched for genes from amino acid and lipid metabolism and CYPs), 22 (collagen enriched), and 24 (also enriched for amino acid and lipid metabolism genes, as well as fatty acid oxidation). In addition, Qlongevity was significantly linked to mounts 1 (enriched for muscle, neuronal, and PDZ genes), 14 and 35 (enriched for collagen), 15 (no specific gene-groups), 21 (enriched for lipid metabolism genes), and 32 (enriched for nucleosomal histones). Comparing the direction of regulation, it was noticeable that in TAlongevity the majority of DEGs in the respective mounts were up-regulated, whereas no trend was observed in the Qlongevity gene list (with the exception of mountain 4).
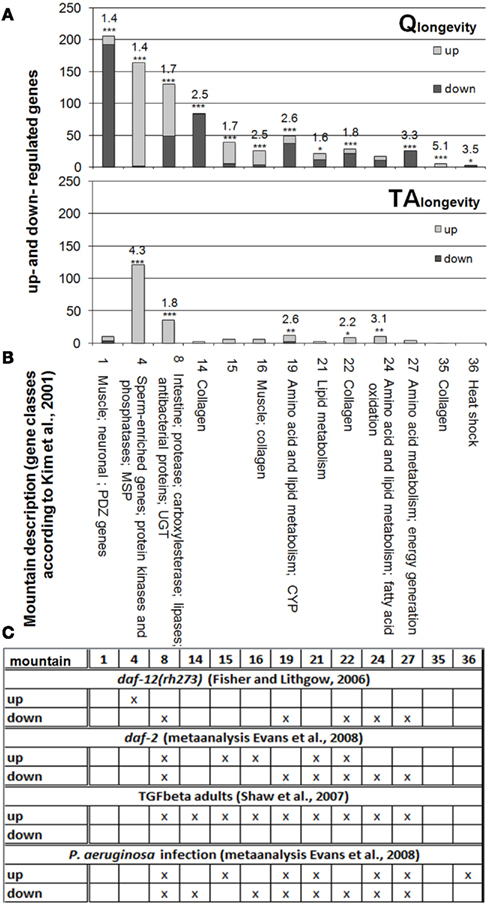
Figure 2. Overrepresented gene expression mountains. Identification of overrepresented gene expression mountains (based on Kim et al., 2001) from Qlongevity(A) and TAlongevity (B), as well as from datasets of longevity mutants and P. aeruginosa infected C. elegans (Fisher and Lithgow, 2006; Shaw et al., 2007; Evans et al., 2008) (C). Depicted are only RFs from mounts which were significantly affected by at least either Qlongevity or TAlongevity. *p < 0.05, **p < 0.005, ***p < 0.001. (The complete dataset and RFs for all 44 gene expression mountains in each and pooled PP concentrations, as well as RFs for previously published datasets can be found in Supplementary Material 4: sheet “Gene Expression Mountains”; results can be reconstructed using the filter function in Supplementary Material 5.)
The comparison of PP treatments to overrepresented mounts from experiments with long-lived mutants and P. aeruginosa infected nematodes (Figure 2C; Supplementary Material 4: sheet “Gene Expression Mountains”) revealed large overlaps. For example, Fisher and Lithgow (2006) reported for daf-12(rh273) an enrichment of induced DEGs in mount 4, a finding that matches well with both PPs tested. Further differentially regulated mounts in daf-12(rh273) are also overrepresented in Qlongevity and TAlongevity, suggesting parallels in molecular mechanisms to daf-12(rh273) and possibly DAF-12’s involvement in Q and TA mediated longevity.
Similarly, distinct mounts of Qlongevity are highly similar to daf-2 (Evans et al., 2008) and TGF-beta mutants (Shaw et al., 2007), as well as P. aeruginosa infected nematodes (Evans et al., 2008). The pattern of overrepresented mounts in TAlongevity best resembles the results from adult TGF-beta mutants (Shaw et al., 2007), but also overlaps to some extent with the other conditions. These marked similarities will be probed in more detail in the next section.
Overlapping DEGs from selected microarray studies in C. elegans
Figure 3 demonstrates the relationship between Qlongevity (left section), TAlongevity (right section), and Q&TAlongevity (middle section) to daf-12(rh273) mutants (Figure 3A), daf-2 mutants (Figure 3B), adult TGF-beta mutants (Figure 3C), and P. aeruginosa infected nematodes, respectively. [A comparison to further microarray studies (Hill et al., 2000; Wang and Kim, 2003; Viswanathan et al., 2005) and single concentration results can be found in Supplementary Material 4: sheet “Meta-Analysis.”] To gain a deep functional insight into aging-related genes, significantly overlapping DEGs were examined further with a GO- and InterPro-cluster analysis (Table S4 in Supplementary Material 1).
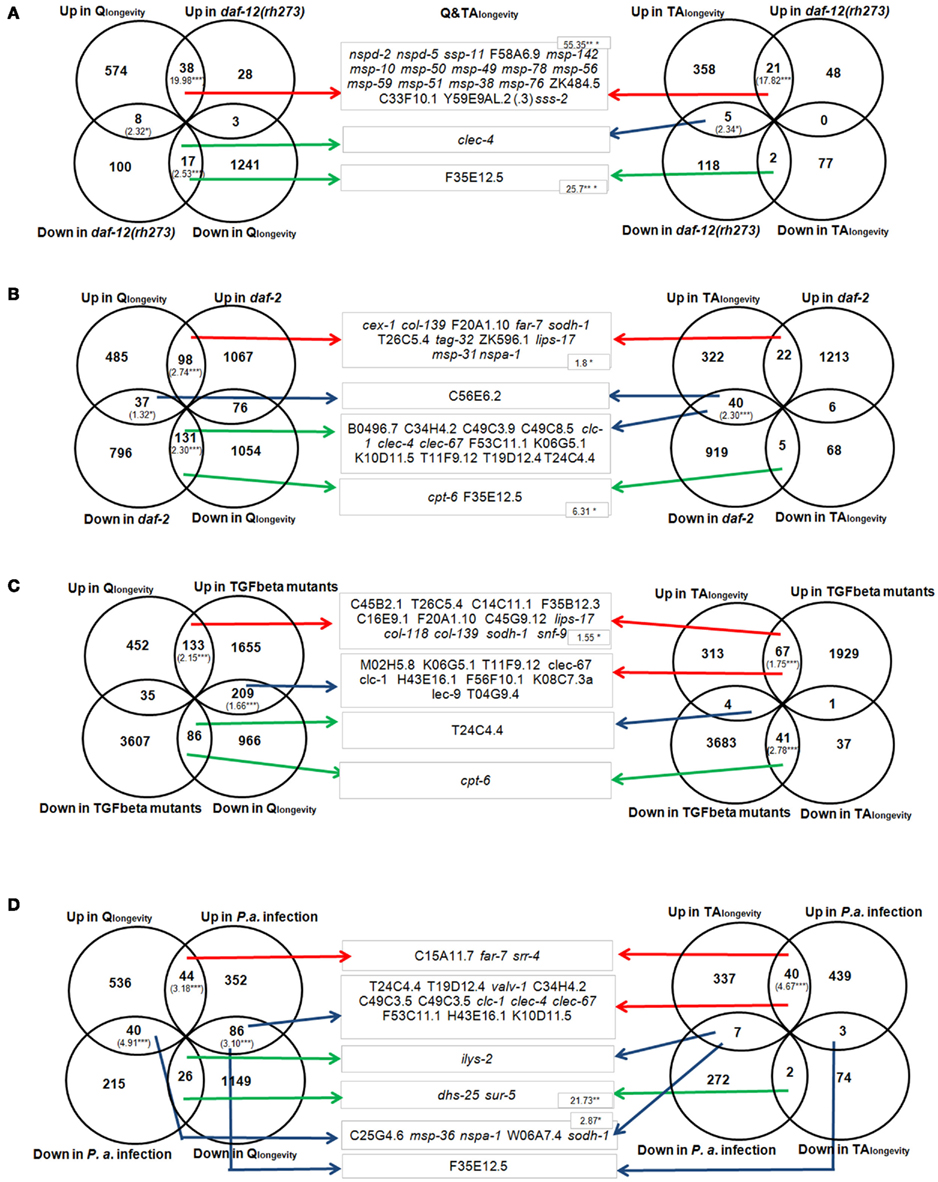
Figure 3. Comparison of DEGs derived from Qlongevity, TAlongevity, and daf-12(rh273) mutants (Fisher and Lithgow, 2006) (A); daf-2 mutants (Evans et al., 2008) (B); TGF-beta adults (Shaw et al., 2007) (C); and P. aeruginosa challenged C. elegans (Evans et al., 2008) (D). Significant overlap is indicated by RF values in brackets. The middle section represents the overlap of pooled DEGs in Q&TAlongevity and respective conditions. Arrow colors define the mode of regulation, where red defines intersections containing jointly up-regulated DEGs, green down-regulated DEGs and blue DEGs that are regulated in opposite directions. The overlap is significant where RF values (numbers in brackets) are shown. *p < 0.05, **p < 0.005, ***p < 0.001. (Additional information about all RF values in single PP concentrations and published data can be found in Supplementary Material 4: sheet “Meta-Analysis.” A GO- and InterPro-cluster analysis of overlapping DEGs in respective intersections can be found in Table S4 in Supplementary Material 1. All results can be reconstructed using the filter function in Supplementary Material 5.)
A comparison to daf-12(rh273). The RFs for overlapping genes (both up- and down-regulated) of Qlongevity and daf-12(rh273) were significantly enriched (Figure 3A), suggesting that they share similar genetic mechanisms. Interestingly, eight DEGs (bre-1, clec-4, clec-66, dod-22, F35E12.5, F55G11.5, M02F4.7, pho-1) were down-regulated in both conditions, transcripts which were also repressed in daf-2 and previously thought to be down-regulated in several longevity backgrounds (Fisher and Lithgow, 2006). The downstream analysis revealed one prominent cluster in each intersection (major sperm protein for commonly up-regulated genes and C-type lectin-like for commonly down-regulated genes). It is however noteworthy that a group of eight genes of the CUB-like region proteins were up-regulated in Qlongevity but down-regulated in daf-12(rh273) mutants.
In TAlongevity, the overlap with daf-12(rh273) was significant for common up-regulated DEGs and also the category “up in TAlongevity/down in daf-12” (Figure 3A). Cluster analyses for these intersections returned transcripts involved in major sperm protein and hydrolase activity, respectively. Comparing DEGs of Q&TAlongevity and daf-12(rh273) revealed a strong overrepresentation of common up- and down-regulated transcripts. All up-regulated genes, 18 in total, assign to the gene-groups msp, cell structure, and/or sperm- and male enriched (notably 17 are members of gene expression mountain 4).
A comparison to daf-2. Qlongevity and daf-2 mutants share a significant proportion of DEGs (Figure 3B). The GO- and InterPro-cluster analyses of the intersection of up-regulated genes revealed five statistically significant clusters, including determination of adult lifespan, lipid metabolic process, and oxidoreductase activity (Table S4 in Supplementary Material 1). The intersection of down-regulated transcripts returned four clusters, again determination of adult lifespan and UDP-glycosyltransferase activity. In contrast, TAlongevity differs from Qlongevity when compared to daf-2 mutants; only the section “up in TAlongevity/down in daf-2” revealed an overrepresentation (Figure 3B).
Since a substantial part of the category “up in TAlongevity/down in daf-2” are also up-regulated in TGF-beta mutants (17 genes) and in P. aeruginosa infected nematodes (18 genes), suggests that a network of DEGs are present in all three conditions. Four clusters emerge from this intersection, notably as in Qlongevity, the term lipid metabolic process like stands out. The fact, that no gene of this GO-term was present in the intersection Qlongevity/daf-2 confirms that both PPs modulate lipid metabolism, but via differing routes. Overall, Q&TAlongevity and daf-2 share 11 up-regulated genes and two down-regulated genes (Figure 3B, middle) but 13 genes are expressed in opposite direction (two CUB-like genes (F53C11.1, K10D11.5), two belonging to the defense response category (C49C3.9, T19D12.4) and two coding for proteins with a von Willebrand factor type A (vWA) domain).
A comparison to adult TGF-beta mutants. The overlap between the gene lists for Qlongevity and TGF-beta mutants is significant both in the up-regulated section and the section “Qlongevity down/TGF-beta mutants up” (Figure 3C). The cluster analysis revealed, e.g., the terms catalytic activity, active transmembrane transporter activity, and peptidase 28, carbohydrate metabolic process, respectively (Table S4 in Supplementary Material 1). Indeed, 45 genes of the “Qlongevity down/TGF-beta mutants up” section are also down-regulated in daf-2, suggesting that some overlap of TGF-beta and ILS signaling prevails, albeit with contrasting regulation patterns.
The comparison of TAlongevity and TGF-beta mutants (Figure 3C) revealed that approximately 25% of the DEGs overlapped. The cluster analysis highlighted two clusters for each intersection: peptidase 28 and lipid metabolic process in the section comprising induced DEGs and chromatin organization and embryonic development ending in birth or egg hatching in the group of repressed DEGs. Comparing DEGs from Q&TAlongevity and TGF-beta mutants (Figure 3C, middle) returned only 12 genes that were marginally overrepresented in the up-regulated intersection which were also up-regulated in daf-2 mutants.
A comparison to P. aeruginosa infected nematodes. Qlongevity or TAlongevity were compared to the transcriptional response induced by the infection with the pathogenic bacteria P. aeruginosa (Shapira et al., 2006; Troemel et al., 2006; summarized in Evans et al., 2008). Venn diagrams (Figure 3D) illustrate the presence of significant intersections of DEGs in Qlongevity and immune-challenged nematodes (both up; up in Qlongevity/down in infection; both down; up in infection/down in Qlongevity). The cluster analysis revealed a common up-regulation in, for example, lipid metabolic processes, monooxygenase activity, and genes coding for UDP-glucuronosyl/UDP-glucosyltransferase as well as a concurrent down-regulation in particular parts of catalytic activity. The intersections of oppositely responding DEGs contain genes coding for oxidoreductases and serine-type peptidase activity (Qlongevity up/infection down) and for structural constitutes of cuticle (Qlongevity down/infection up), respectively. A large proportion of DEGs from the intersection “up in infection/down in Qlongevity” is down-regulated in TGF-beta, daf-2 and daf-12(rh273) (for exact numbers and genes see Supplementary Material 5 in respective filter condition), and 21 genes of “up in Qlongevity/down in infection” are up-regulated in TGF-beta mutants.
TAlongevity shares highly significant patterns with the list of up-regulated DEGs from infected nematodes. The cluster analysis returned only one term, genes with a CUB-like region, however, interestingly, CUB-like genes were previously found to be activated in the induced immune response pathway modulated by PMK-1 (Troemel et al., 2006).
Only five genes are regulated in Q&TAlongevity as well as in nematodes infected with P. aeruginosa (up-regulated: C15A11.7, far-7 and srr-4; down-regulated: dhs-25, sur-5). Of the 15 common up-regulated DEGs in “TAlongevity and P. aeruginosa infection and down-regulated in Qlongevity,” 11 are also down-regulated in daf-2 mutants, including two C-type lectins (clec-4, clec-67), two genes coding for proteins with a CUB-like region (F53C11.1, K10D11.5) and two defense response genes (C49C3.9, T19D12.4).
Finally, we evaluated the overlap of DEGs in Q and TA treated nematodes with the DEGs regulated by PMK-1 (Troemel et al., 2006; Supplementary Material 4: sheet “Meta-Analysis”) and found a significant overlap for Qlongevity and TAlongevity in the up- regulated intersection and the “down-regulated by PP/up due to PMK-1.” These results underline the involvement of the p38 MAP kinase pathway in both PP actions.
Discussion
Polyphenols transform the transcriptional output and thereby alter the physiological status of an animal. By performing DNA microarray experiments and comparing the resulting data with previously published gene expression screens, we revealed an interlaced interplay of genetic pathways affected by Q and TA. Moreover, our findings support results from previous studies (Pietsch et al., 2009, 2011; Saul et al., 2010).
Concentration Dependent Variations in Global Transcriptional Responses
Low concentrations of Quercetin (Q50) modulate processes which may contribute to the wellbeing of the nematode (e.g. the gene classes gsts, peroxidases, lysozymes; GO-terms: oxidoreductase activity; KEGG-analysis: glutathione metabolism and metabolism of xenobiotics by cytochrome P450), but not sufficiently to significantly extend lifespan (Pietsch et al., 2011). In contrast, Q100 and Q200 impact significantly on the global transcriptome which manifests itself in a strong longevity phenotype. This effect is, at least in part, driven by transcriptional repression mechanisms as indicated by the high number of down-regulated genes. As shown recently, increasing the concentration to Q250 reverts the beneficial effects on longevity (Pietsch et al., 2011). High dose toxicity is particularly apparent at, for example, TA300 (GO-terms: DNA repair, DNA damage response, response to stress, and cell death and KEGG-analysis: Ubiquitin-mediated proteolysis, mismatch repair, DNA replication).
Genetic basis of Qlongevity and TAlongevity
Qlongevity shares transcriptional patterns with long-lived daf-12(rh273), daf-2 mutants, TGF-beta mutants, as well as P. aeruginosa challenged nematodes (as identified by the meta-analysis and overlapping gene expression mountains). These results suggest that Q operates through a complex network of interlinked pathways. Combining the microarray analysis with lifespan data from Q exposed nematode mutants (Pietsch et al., 2009) allowed the construction of a hypothetical model that describes the mode of action of Q induced longevity (Figure 4A).
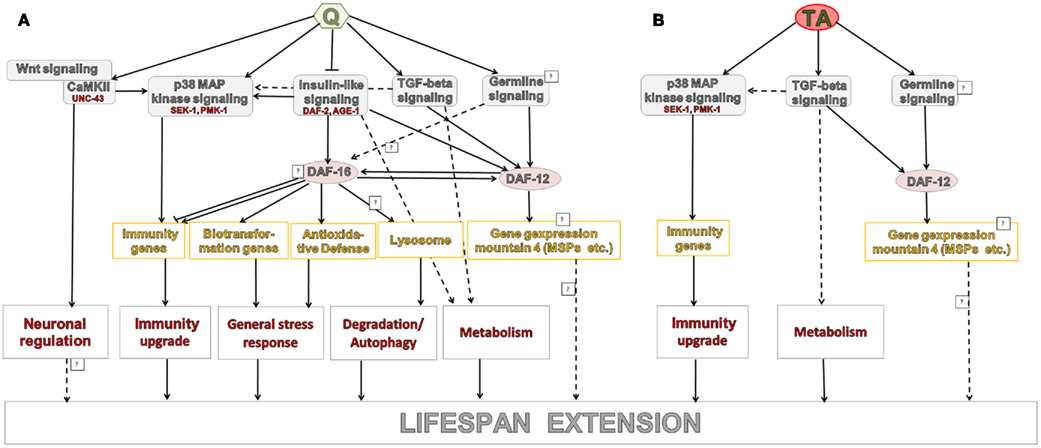
Figure 4. Hypothetical model of Q’s (A) and TA’s (B) mode of action.
The overlap between TAlongevity and the conditions assessed as part of the meta-analysis revealed a less pronounced, yet still significant correlation. Beside activation of amino acid metabolism pathways, the strongest overlap was with data from the TGF-beta mutants. This suggests that the TGF-beta pathway plays a prominent role in the TA mediated life extension which is summarized in Figure 4B.
It is intriguing to note that TA treatment has, compared to Q treatment, a lesser effect on the transcriptome (as defined by the number of significant DEGs) but a more marked life-extending property. It is currently not known if this is an indirect effect or whether transcriptional changes diminish/suppress the positive output of Q action. Notwithstanding the observed differences in genetic action modes, TGF-beta and p38 MAPK pathways, as well as the nuclear hormone receptor DAF-12 seem to be involved in both PPs. DAF-12, which is downstream of the TGF-beta- and ILS pathways, is a member of the steroid hormone receptor superfamily. It is linked to Dauer formation and, together with DAF-16, also influences gonad-dependent adult longevity. Given that neither Blueberry polyphenols (Wilson et al., 2006), Caffeic acid nor Rosmarinic acid (Pietsch et al., 2011) could extend the lifespan of sek-1 mutants (sek-1 is a genetic player in the p38 MAPK pathway) suggests that the innate immunity may act as a prominent downstream effector of PPs.
Based on the meta-analysis with Q&TAlongevity and relevant microarray studies (Hill et al., 2000; Wang and Kim, 2003; Viswanathan et al., 2005; Fisher and Lithgow, 2006; Shaw et al., 2007; Evans et al., 2008), we were able to identify a subset of transcripts that are possibly relevant for aging (see Table S5 in Supplementary Material 1). Clearly further investigations into their aging-modulating activities are warranted. Likewise, we call for further experimentation to establish the regulatory interlink between TGF-beta signaling and DAF-12, as well as p38 MAPK.
Conclusion
The meta-analysis displayed an extensive overlap between PP treatment and numerous mutants as well as immunity challenging conditions, however, frequently the mode of regulation was in opposite direction. This strongly suggests the presence of a complex regulatory interplay between the input and multiple downstream targets. Clearly, PPs action cannot be reduced to the activation or inhibition of single genes and pathways; nevertheless it is apparent that TGF-beta, ILS, and p38 MAPK play a prominent role in PPs’ mode of action. Furthermore, we were able to demonstrate that an extensive comparison with data from the literature can provide a deep insight into the transcriptome to a level that goes beyond a simple GO- and KEGG-analysis. Given the convolution observed with single gene knockout alleles and exposures to pure compounds, one can envisage the complexity that will arise with multidimensional mixture toxicity experiments. The development of comparative databases and most importantly powerful, yet intuitive, bioinformatic tools will undoubtedly aid in the streamlining of large datasets. Overall, our results strengthen the notion that both PPs act in conserved genetic pathways that overlap, or at least correlate, with the longevity phenotypes and transcriptional fingerprints of certain mutant strains. Clearly, further future tests are needed to confirm single genetic players and specify the interplay of conserved pathways.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank members of the King’s College Genomics Center (Dr. Matthew Arno, Dr. Estibaliz Aldecoa-Otalora Astarloa) for assistance in the microarray experimentation. Furthermore, we thank the Caenorhabditis Genetics Centre, which is funded by the National Institutes of Health National Centre for Research Resources, for the supply of the Caenorhabditis elegans strains. This work was supported, in part, by a grant awarded by the Biotechnology and Biological Sciences Research Council (BBSRC grant BB/E025099 and a BBSRC Underwood Fellowship). Ralph Menzel was supported by the DFG grant STE673/16-1.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Toxicogenomics_/10.3389/fgene.2012.00048/abstract
Footnotes
References
Boots, A. W., Haenen, G. R. M. M., and Bast, A. (2008). Health effects of quercetin: from antioxidant to nutraceutical. Eur. J. Pharmacol. 585, 325–337.
Calabrese, E. J., and Baldwin, L. A. (2001). Hormesis: U-shaped dose responses and their centrality in toxicology. Trends Pharmacol. Sci. 22, 285–291.
Evans, E. A., Kawli, T., and Tan, M. W. (2008). Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog. 4, e1000175. doi:10.1371/journal.ppat.1000175
Fisher, A. L., and Lithgow, G. J. (2006). The nuclear hormone receptor DAF-12 has opposing effects on Caenorhabditis elegans lifespan and regulates genes repressed in multiple long-lived worms. Aging Cell 5, 127–138.
Gami, M. S., and Wolkow, C. A. (2006). Studies of Caenorhabditis elegans DAF-2/insulin signaling reveal targets for pharmacological manipulation of lifespan. Aging Cell 5, 31–37.
Gill, M. S. (2006). Endocrine targets for pharmacological intervention in ageing in Caenorhabditis elegans. Aging Cell 5, 23–30.
Golden, T. R., and Melov, S. (2007). “Gene expression changes associated with aging in C. elegans,” in WormBook, ed. . Available at: http://www.wormbook.org
Grigoryev, D. N., Ma, S.-F., Irizarry, R. A., Ye, S. Q., Quackenbush, J., and Garcia, J. G. N. (2004). Orthologous gene-expression profiling in multi-species models: search for candidate genes. Genome Biol. 5, R34.
Grünz, G., Haas, K., Soukup, S., Klingenspor, M., Kulling, S. E., Daniel, H., and Spanier, B. (2012). Structural features and bioavailability of four flavonoids and their implications for lifespan-extending and antioxidant actions in C. elegans. Mech. Ageing Dev. 133, 1–10.
Hill, A. A., Hunter, C. P., Tsung, B. T., Tucker-Kellogg, G., and Brown, E. L. (2000). Genomic analysis of gene expression in C. elegans. Science 290, 809–812.
Kaletta, T., and Hengartner, M. O. (2006). Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 5, 387–399.
Kampkötter, A., Nkwonkam, C. G., Zurawski, R. F., Timpel, C., Chovolou, Y., Wätjen, W., and Kahl, R. (2007a). Effects of the flavonoids kaempferol and fisetin on thermotolerance, oxidative stress and FoxO transcription factor DAF-16 in the model organism Caenorhabditis elegans. Arch. Toxicol. 81, 849–858.
Kampkötter, A., Nkwonkam, C. G., Zurawski, R. F., Timpel, C., Chovolou, Y., Wätjen, W., and Kahl, R. (2007b). Investigations of protective effects of the flavonoids quercetin and rutin on stress resistance in the model organism Caenorhabditis elegans. Toxicology 234, 113–123.
Kampkötter, A., Timpel, C., Zurawski, R. F., Ruhl, S., Chovolou, Y., Proksch, P., and Wätjen, W. (2008). Increase of stress resistance and lifespan of Caenorhabditis elegans by quercetin. Comp. Biochem. Physiol. B 149, 314–323.
Kim, S. K., Lund, J., Kiraly, M., Duke, K., Jiang, M., Stuart, J. M., Eizinger, A., Wylie, B. N., and Davidson, G. S. (2001). A gene expression map for Caenorhabditis elegans. Science 293, 2087–2092.
Koleckar, V., Kubikova, K., Rehakova, Z., Kuca, K., Jun, D., Jahodar, L., and Opletal, L. (2008). Condensed and hydrolysable tannins as antioxidants influencing the health. Mini Rev. Med. Chem. 8, 436–447.
Lewis, J. A., and Fleming, J. T. (1995). “Basic culture methods,” in Caenorhabditis elegans, Modern Biological Analysis of an Organism, eds H. F. Epstein, and D. C. Shakes (San Diego, CA: Academic Press), 3–29.
Lublin, A., Isoda, F., Patel, H., Yen, K., Nguyen, L., Hajje, D., Schwartz, M., and Mobbs, C. (2011). FDA-approved drugs that protect mammalian neurons from glucose toxicity slow aging dependent on cbp and protect against proteotoxicity. PLoS ONE 6, e27762. doi:10.1371/journal.pone.0027762
McCarthy, D. J., and Smyth, G. (2009). Testing significance relative to a fold-change threshold. Bioinformatics 25, 765–771.
McElwee, J. J., Schuster, E., Blanc, E., Thomas, J. H., and Gems, D. (2004). Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 279, 44533–44543.
Murphy, C. T., McCarroll, S. A., Bargmann, C. I., Fraser, A., Kamath, R. S., Ahringer, J., Li, H., and Kenyon, C. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283.
Pietsch, K., Saul, N., Chakrabarti, S., Stürzenbaum, S. R., Menzel, R., and Steinberg, C. E. W. (2011). Hormetins, antioxidants and prooxidants: defining quercetin-, caffeic acid- and rosmarinic acid- mediated life extension in C. elegans. Biogerontology 12, 329–347.
Pietsch, K., Saul, N., Menzel, R., Stürzenbaum, S. R., and Steinberg, C. E. W. (2009). Quercetin mediated lifespan extension in Caenorhabditis elegans is modulated by age-1, daf-2, sek-1, and unc-43. Biogerontology 10, 565–578.
Rainer, J., Sanchez-Cabo, F., Stocker, G., Sturn, A., and Trajanoski, Z. (2006). CARMAweb: comprehensive R- and bioconductor-based web service for microarray data analysis. Nucleic Acids Res. 34, W498–W503.
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., Braisted, J., Klapa, M., Currier, T., Thiagarajan, M., Sturn, A., Snuffin, M., Rezantsev, A., Popov, D., Ryltsov, A., Kostukovich, E., Borisovsky, I., Liu, Z., Vinsavich, A., Trush, V., and Quackenbush, J. (2003). MeV/TM4:a free, open-source system for microarray data management and analysis. BioTechniques 34, 374–378.
Saul, N., Pietsch, K., Menzel, R., and Steinberg, C. E. W. (2008). Quercetin-mediated longevity in Caenorhabditis elegans: is DAF-16 involved? Mech. Ageing Dev. 129, 611–613.
Saul, N., Pietsch, K., Menzel, R., Stürzenbaum, S. R., and Steinberg, C. E. W. (2010). The longevity effect of tannic acid in Caenorhabditis elegans: disposable soma meets hormesis. J. Gerontol. A Biol. Sci. Med. Sci. 65, 626–635.
Saul, N., Pietsch, K., Stürzenbaum, S. R., Menzel, R., and Steinberg, C. E. W. (2011). Diversity of polyphenol action in Caenorhabditis elegans: between toxicity and longevity. J. Nat. Prod. 74, 1713–1720.
Shapira, M., Hamlin, B. J., Rong, J., Chen, K., Ronen, M., and Tan, M. W. (2006). A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 14086–14091.
Shaw, W. M., Luo, S., Landis, J., Ashraf, J., and Murphy, C. T. (2007). The C. elegans TGF-beta Dauer pathway regulates longevity via insulin signaling. Curr. Biol. 17, 1635–1645.
Strange, K., Christensen, M., and Morrison, R. (2007). Primary culture of Caenorhabditis elegans developing embryo cells for electrophysiological, cell biological and molecular studies. Nat. Protoc. 2, 1003–1012.
Sulston, J. E., and Hodgkin, J. (1988). “Methods,” in The Nematode Caenorhabditis elegans, ed. W. B. Wood (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), 587–606.
Surco-Laos, F., Cabello, J., Gómez-Orte, E., González-Manzano, S., González-Paramás, A. M., Santos-Buelga, C., and Dueñas, M. (2011). Effects of O-methylated metabolites of quercetin on oxidative stress, thermotolerance, lifespan and bioavailability on Caenorhabditis elegans. Food Funct. 2, 445–456.
Tárraga, J., Medina, I., Carbonell, J., Huerta-Cepas, J., Minguez, P., Alloza, E., Al-Shahrour, F., Vegas-Azcárate, S., Goetz, S., Escobar, P., Garcia-Garcia, F., Conesa, A., Montaner, D., and Dopazo, J. (2008). GEPAS, a web-based tool for microarray data analysis and interpretation. Nucleic Acids Res. 36, W308–W314.
The C. elegans Sequencing Consortium. (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018.
Troemel, E. R., Chu, S. W., Reinke, V., Lee, S. S., Ausubel, F. M., and Kim, D. H. (2006). p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2, e183. doi:10.1371/journal.pgen.0020183
Van Gelder, R. N., von Zastrow, M. E., Yool, A., Dement, W. C., Barchas, J. D., and Eberwine, J. H. (1990). Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. U.S.A. 87, 1663–1667.
Viswanathan, M., Kim, S. K., Berdichevsky, A., and Guarente, L. (2005). A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans lifespan. Dev. Cell 5, 605–615.
Wang, J., and Kim, S. K. (2003). Global analysis of Dauer gene expression in Caenorhabditis elegans. Development 130, 1621–1634.
Wilson, M. A., Shukitt-Hale, B., Kalt, W., Ingram, D. K., Joseph, J. A., and Wolkow, C. A. (2006). Blueberry polyphenols increase lifespan and thermotolerance in Caenorhabditis elegans. Aging Cell 5, 59–68.
Zeeberg, B. R., Qin, H., Narasimhan, S., Sunshine, M., Cao, H., Kane, D. W., Reimers, M., Stephens, R. M., Bryant, D., Burt, S. K., Elnekave, E., Hari, D. M., Wynn, T. A., Cunningham-Rundles, C., Stewart, D. M., Nelson, D., and Weinstein, J. N. (2005). High-throughput gominer, an “industrial-strength” integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of common variable immune deficiency (CVID). BMC Bioinformatics 6, 168. doi:10.1186/1471-2105-6-168
Keywords: Quercetin, Tannic acid, TGF-beta, ILS, DAF-12, p38 MAPK, C. elegans
Citation: Pietsch K, Saul N, Swain SC, Menzel R, Steinberg CEW and Stürzenbaum SR (2012) Meta-analysis of global transcriptomics suggests that conserved genetic pathways are responsible for Quercetin and Tannic acid mediated longevity in C. elegans. Front. Gene. 3:48. doi: 10.3389/fgene.2012.00048
Received: 27 January 2012; Paper pending published: 09 February 2012;
Accepted: 16 March 2012; Published online: 05 April 2012.
Edited by:
Michael Aschner, Vanderbilt University Medical Center, USAReviewed by:
Kim A. Caldwell, The University of Alabama, USAChris Vulpe, University of California, USA
Pan Chen, Vanderbilt University, USA
Copyright: © 2012 Pietsch, Saul, Swain, Menzel, Steinberg and Stürzenbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kerstin Pietsch, Laboratory of Freshwater and Stress Ecology, Department of Biology, Humboldt-Universität zu Berlin, Spaethstr. 80/81, 12437 Berlin, Germany. e-mail:a3BpZXRzY2hAZ214LmRl; Stephen R. Stürzenbaum, Analytical and Environmental Science Division, School of Biomedical Sciences, King’s College London, 150 Stamford Street, London SE1 9NH, UK. e-mail:c3RlcGhlbi5zdHVyemVuYmF1bUBrY2wuYWMudWs=
†Present address: Suresh C. Swain, Institute of Biological, Environmental and Rural Sciences, Aberystwyth University, Penglais, Aberystwyth, Ceredigion SY23 3DA, UK.
The microarray data is MIAME compliant and has been deposited in the NCBI Gene Expression Omnibus, GEO (http://www.ncbi.nlm.nih.gov/geo) accession number GSE35354.