


- 1 Machine Intelligence Unit, Indian Statistical Institute, Kolkata, India
- 2 Biophysics Division, Saha Institute of Nuclear Physics, Kolkata, India
One of the important goals of most biological investigations is to classify and organize the experimental findings so that they are readily useful for deriving generalized rules. Although there is a huge amount of information on RNA structures in PDB, there are redundant files, ambiguous synthetic sequences etc. Moreover, a systematic hierarchical organization, reflecting RNA classification, is missing in PDB. In this investigation, we have classified all the available RNA structures from PDB through a programmatic approach. Hence, it would be now a simple assignment to regularly update the classification as and when new structures are released. The classification can further determine (i) a non-redundant set of RNA structures and (ii) if available, a set of structures of identical sequence and function, which can highlight structural polymorphism, ligand-induced conformational alterations etc. Presently, we have classified the available structures (2095 PDB entries having RNA chain longer than nine nucleotides solved by X-ray crystallography or NMR spectroscopy) into nine functional classes. The structures of same function and same source are mostly seen to be similar with subtle differences depending on their functional complexation. The web-server is available online at http://www.saha.ac.in/biop/www/HD-RNAS.html and is updated regularly.
Introduction
Keeping pace with advancement in the field of RNA functions, the number of RNA structures whose coordinates are available in the Protein Data Bank (PDB; Berman et al., 2000) is growing rapidly. The total number of structures of RNA with oligomeric or polymeric length, as available in July 2011, is 2095 and the number is increasing at a pace of about 100 per year. The determination of various RNA structures, such as the hammerhead ribozyme (Scott et al., 1995), SRP-RNA (Zwieb et al., 2005), and the 5S, 16S and 23S RNAs of ribosome has greatly increased our knowledge of RNA folds and the three-dimensional organization of RNA chains (Batey et al., 1999; Ferre-d’Amare and Doudna, 1999; Hermann and Patel, 1999). Collectively, these structures provide a large amount of information about RNA structural motifs (Moore, 1999). Similar exponential growth in number of crystal structures of proteins is also taking place in the PDB. Considering the need of classification of these proteins, there are a number of methods available, such as SCOP (Murzin et al., 1995; Hubbard et al., 1997), FSSP (Holm and Sander, 1997), Pisces (Wang and Dunbrack, 2005), BIPA (Lee and Blundell, 2009) etc. These methods can classify a protein structure based on its structural class, source organism, secondary structure content, resolution, etc. One can further determine a set of non-redundant structures of proteins, which are not evolutionarily related, for a statistical analysis in an unbiased method. In a similar manner, it is also necessary to organize the available RNA structures to determine different structure–function relationships. Furthermore, it is often important to compare several structures of RNA of same function and from same source, which have identical sequence, to understand effect of ligand binding, crystallization environments etc., on the three-dimensional folding. Such sets of structures could reveal significant information on structural flexibility, binding thermodynamics etc., of the biological macromolecules (Halder and Bhattacharyya, 2010; Samanta et al., 2010). They carry signatures that may indicate variations introduced in the molecular structure due to ligand binding or alteration of crystallization conditions. In our recent study, we also found that structural variability of double-helical RNA as observed in molecular dynamics simulation studies mimic that of the crystallographic ensembles (Halder and Bhattacharyya, 2010; Halder and Bhattacharyya, manuscript in preparation). Likewise, the differences in RNA structural organization among various species can be studied if a classification is available. A non-redundant set of RNA structures is also necessary to analyze the local environments at basepairing level, which provided important information in recent analyses of structure and energetics of different non-canonical basepairs (Panigrahi et al., 2011). Databases like RNABase (Murthy and Rose, 2003) and SCOR (Klosterman et al., 2002) attempted to classify the available RNA structures but failed to regularly update these only by manual curation of the RNA structures, as the number of structures is increasing quite fast. Any PDB structure released after 2004 is not classified by SCOR and RNABase database can no longer be accessed at the published address www.rnabase.org. Also, there are more activities toward classification of RNA structures on the basis of secondary structure (Tamura et al., 2004; Sarver et al., 2008), canonical as well as unusual base pairing (Lu and Olson, 2003; Das et al., 2006; Roy et al., 2008), isosteric base pairs (Leontis et al., 2002), etc., but the determination of non-redundant set of structures is only done partially by a few groups (Leontis and Westhof, 2001; Stombaugh et al., 2009).
In order to organize and classify the information of RNA structures in PDB files and make it available to the general users, we have developed a web-server, called Hierarchical Database of RNA Structures (HD-RNAS; http://www.saha.ac.in/biop/www/HD-RNAS.html, see Figure 1). Keeping in mind that some of the earlier attempts to classify RNA structures failed to keep pace with the structure determination speed, we have adapted an automated programmatic scheme with minimal or no manual intervention for the classification procedure. With the number of RNA structures increasing rapidly, there is a constant pressure of regularly updating this database. As the classification and database creation processes are done by an Octave program, our automated tool is capable of frequently classifying the newly released structures. Hence we expect that HD-RNAS can remain dynamic and would not phase out like the earlier attempts. Some manual curation is obviously involved in this automated procedure to avoid erroneous results. Whenever new structures are released in PDB, they are classified accordingly and verified manually for any inaccuracy. The programmatic scheme is flexible enough to be modified to ensure proper classification of all the RNA structures in case of discrepancies.
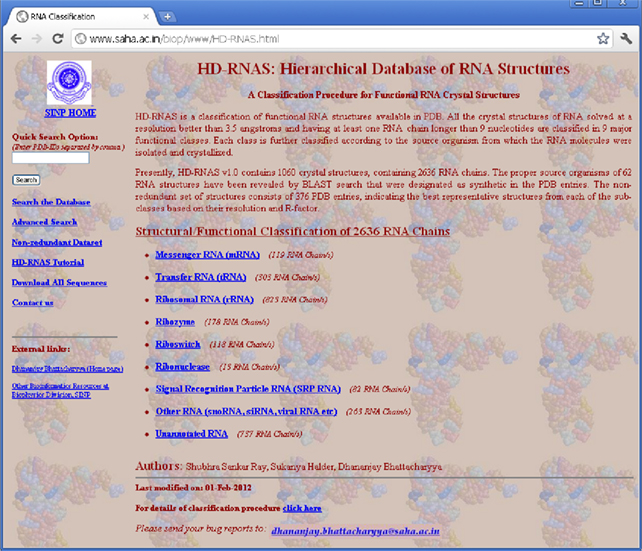
Figure 1. HD-RNAS homepage.
Classification Methodology
We focus on PDB files containing at least one RNA chain having length equal to or greater than 10 nucleotides, as shorter fragments than this length cannot be expected to fold back and form a secondary structure of biological relevance. RNA chains shorter than 10 nucleotides usually form double helix pairing with their complimentary strands, and do not form a secondary structure on their own.
A total of 2095 RNA structure entries were reported by the PDB search engine in July, 2011. We have developed a software in GNU-Octave, which is similar to MATLAB scripting language, that
i) Programmatically examines and reads the information of all the RNA structures from the PDB files and classifies them,
ii) Creates the necessary database files, and
iii) Creates the web-layout of HTML pages displayed in the server containing major information of each RNA chain.
These HTML files are published in the web. Text-based CGI-Perl codes have been created for back-end support of different search applications, which are available in HD-RNAS. The web-server is available at http://www.saha.ac.in/biop/www/HD-RNAS.html
At the first stage, the RNA structures are classified according to their functional classes, e.g., tRNA, rRNA, mRNA etc. Along with these most common ones, we have also included some other types like ribozymes, riboswitches, ribonucleases, and signal recognition particle (SRP) RNAs, keeping in mind their growing significance in maintaining cellular machinery and their specific structural patterns. A number of PDB files correspond to multi-molecular complexes of several RNA as well as protein chains and information about these individual RNA chains is given in the PDB file as MOL_ID 1, MOL_ID 2 etc. Hence, one PDB file can be classified as belonging to several different classes simultaneously. We have made no attempt to classify the structures of DNA or protein chains. The main steps of our Octave code for classifying RNA can be summarized as:
(S1) We have looked for several specific keywords in the MOL_ID field to classify each chain individually into one of the seven major classes for mRNA, SRP-RNA, tRNA, rRNA, Ribonuclease, Riboswitch, and Ribozyme. Table 1 shows the complete mapping of keywords used for the function assignment. In a few cases the functions are not clearly understood from the information in MOL_ID field alone. In those cases, we have additionally looked at HEADER and the first KEYWDS field of the PDB files also.
(S2) There are some RNA structures of various other functions. As the numbers of structures for these functional classes are very small, at present we have clustered those under the class named ‘Other-RNA’. This class also contains RNA chains of significant length (at least 10 nucleotides) and obtained from a natural source, for which no appropriate function could be assigned. There are large numbers of synthetic RNA structures for which no source organism or functional type can be determined. These sequences together comprise the “Unannotated RNA” class.
(S3) The rRNA and tRNA molecules are classified into further subclasses. The rRNA structures are classified according to 5S, 16S, 23S, and 28S (for eukaryotic organisms), depending on their sedimentation coefficients and ribosomal fragments, which group only the defined partial structures. The tRNA structures are classified according to the amino acid or stop codon names.
(S4) The structures are then classified according to the source organism from which the RNA molecules were isolated and crystallized. We have placed each chain according to the organism information as supplied by the SOURCE field of PDB files. In some of the cases, where ORGANISM_SCIENTIFIC field do not produce any source information, the source organism has been extracted from the OTHER_DETAILS field. For example, chain A of PDB entry 1YSV is annotated as “SYNTHETIC” in the “SOURCE” field, whereas OTHER_DETAILS field describes that the sequence is taken from human. Thus, we have put the structure in “Homo sapiens” mRNA class. Quite a number of PDB files contain different types of RNA molecules obtained from different source organisms. For example, PDB entry 2J00 is consisted of 16S ribosomal RNA chains from T. thermophilus, A-site, P-site, and E-site tRNAs from Escherichia coli and a synthetic mRNA. We have, therefore, placed this PDB entry into all the RNA classes along with their corresponding chain identifiers (see Figure 2).
(S5) We found that many PDB files do not contain exact source of the RNA chains, and have been indicated as synthetic. However, their size and function indicate that these sequences are from some biological organism. In order to determine the actual source of these RNA chains, we have used BLAST (Altschul et al., 1997) algorithm for sequence alignment. We have used nucleotide sequence database from NCBI as available on June, 2011 and compared our synthetic RNA sequences with all of them. We have picked up the hits having E-value less than 1.0 × 10−5, number of aligned bases greater than 99% of the complete chain length of synthetic sequence, and sequence identity 99% or greater.
(S6) Finally, we have programmatically created a master database file and all the HTML pages that describe the classification. These HTML files contain major information of each RNA chain, such as PDB-ID, chain name, functional class, source, resolution, chain length, free R-factor, and release date of the entry. Each structure is hyper-linked to the corresponding information page on PDB-site. RNA structures solved by NMR spectroscopy have poorer resolution than X-ray crystal structures and are assigned with a large resolution value and R-factor of 99.0.
(S7) In order to determine the non-redundant set of RNA structures at a given resolution, we pick up the structures with best resolution and R-factor (free R-value) from each subclass. Sometimes smaller fragments of a functional RNA are classified as a full-length functional RNA due to improper information in PDB and often these fragments are of better resolution than the other full-length structures. In order to avoid picking up such fragmented chains as representatives, we have put a length constraint so that the representative structure from a class should be 80% or more of the average length of that structural class. For example, the best representative structure of E. coli 23S rRNA should be chain A from PDB 1Q9A as this structure have the best resolution in its class (1.04Å). However, this structure represents only a fragmented part (27 nucleotides) of the complete RNA chain. Thus it is replaced by chain A of PDB 3R8S (resolution: 3.0Å, length: 2903 nucleotides) as the representative of its class. The non-redundant dataset is available at the web-server for various unbiased statistical analysis purposes.
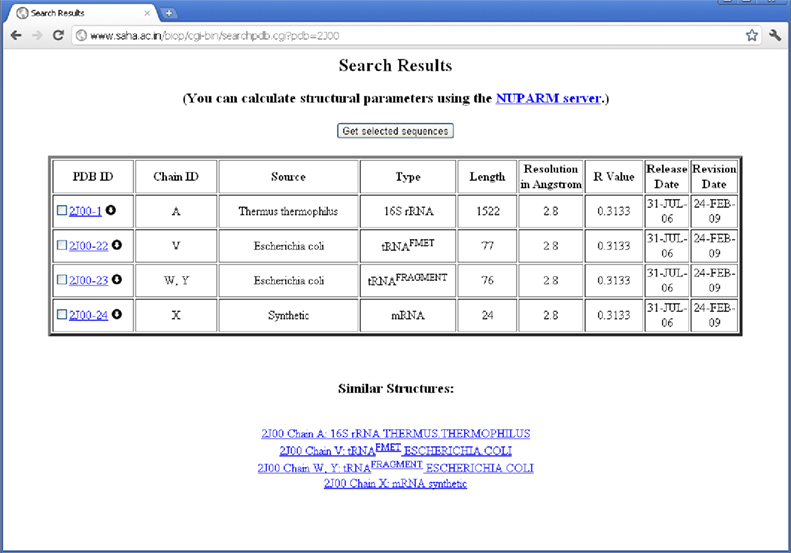
Figure 2. Proper classification of individual RNA chains present in 2J00 in HD-RNAS.

Table 1. Special keywords and their corresponding classification for a RNA chain.
Our classified database is maintained in a flat-file format, without any database management system. This has been possible since we do not keep the large PDB files at the web-server and our complete database is quite small. The web-server also provides different search options with user-specified criteria like source organism or functional types. Sequence of the RNA chains in plain text formats can be obtained from the search result pages. Similarly, one can search for PDB files in the database containing a given sequence motif.
Results and Discussion
Present Status
We found 2095 RNA structures were available in PDB in July, 2011. We further rejected 345 crystal structures as these do not contain RNA chains of significant length. Presently, the database contains 1750 PDB files having structures of 2636 RNA chains with significant length. We have 263 structures clustered in the “Other-RNA” group including IRES RNA, viral RNA, miRNA, snoRNA etc. For the “Unannotated RNA” class, it is seen that most of the unannotated RNA chains are shorter than 30 nucleotides (650 out of 737), whereas functional RNAs are generally larger. There are, however, a few entries in the unannotated class, such as 1KH6, 1P6V, 2B57 etc., which have significant length.
BLAST Search for Synthetic Sequences
There are many structures where the source organism is mentioned as “SYNTHETIC” by the depositors, whereas one expects these sequences to be derived from some natural organisms, as they correspond to quite long RNA chains. In some of the cases, however, the depositors mentioned about the source organism in the OTHER_DETAILS field of the PDB file. But the information is not provided in machine-interpretable format. The proper source organisms of 66 RNA sequences, which were imperfectly designated as synthetic, have been revealed by BLAST search. There are also some PDB entries like 1DFU, 1EHZ etc., where natural sources of the RNA sequences are mentioned in the OTHER_DETAILS field; yet, they have been designated as synthetic ones probably because they have been synthesized by in vitro transcription. We have not done BLAST search for source determination of the synthetic sequences having length smaller than 30 nucleotides, as the significance of BLAST result are poor in these cases and multiple hits with identical E-values are often observed. We could determine source of five (out of 14) E. coli tRNAGln using BLAST (see Figure 3). Wherever possible, we have manually crosschecked the validity of BLAST result from the OTHER_DETAILS field of PDB entries and the results are found to be in good agreement (see Table 2).
Table 2. Validation of BLAST search results.
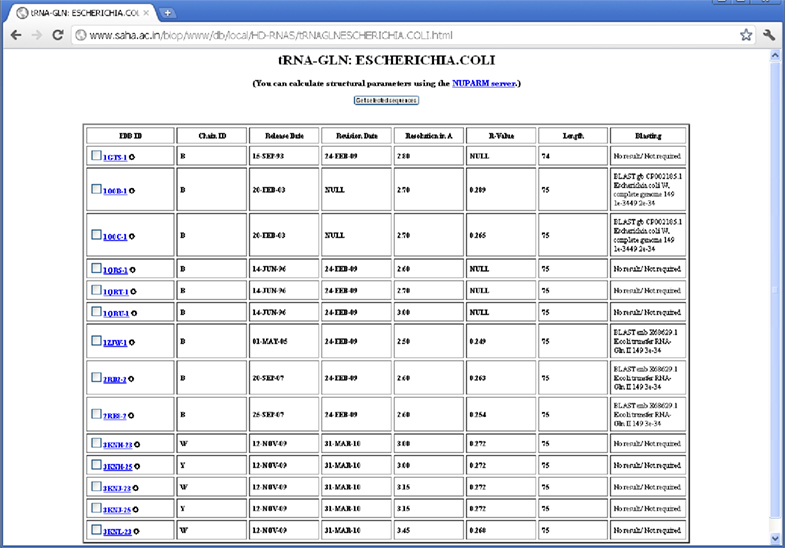
Figure 3. BLAST search results for E. coli tRNAGln.
Non-Redundant Dataset
To obtain an unbiased set of RNA structures, we have derived a non-redundant dataset consisting of the best representative structures from each of the classes. These representative structures are the ones with best resolution or, in case there are more than one entries having resolution values identical to the best one, the structure with smallest R-factor and larger length. As the unannotated structures include huge number of structures and most of them are unrelated, we tried to pick up more than one representatives from this clan. In order to remove redundant repeats of structures, we have calculated sequence identity among the structures of synthetic RNAs in the unannotated-RNA group. In cases where two sequences are 100% identical, we have considered that of the best resolution and R-factor as the representative one. The non-redundant dataset thus obtained contains 849 RNA chains from 702 PDB entries. Sometimes, this non-redundant set may contain more than one ribosomal RNA structures from the same group, as they contain RNA chains belonging to different classes, such as tRNA, mRNA, rRNA etc. We have not attempted to remove these, as inter-chain base pairings are also important in higher order organization of RNA structures. The web-server is also equipped with a search tool to determine a non-redundant set of structures with user-defined criteria of functional type, source organism, chain length, and resolution. Such non-redundant set of RNA structures was used recently in analyzing structure and energetics of different non-canonical basepairs (Panigrahi et al., 2011).
In our non-redundant dataset, we find that there are a large number of structures of RNA with length less than 30 nucleotides. These are mainly synthetic sequences for which functional annotation is unavailable. Among the 849 structures, 491 sequences do not carry any functional information, among which 427 sequences are of insignificant length (< 30 nucleotide). As the functional RNA molecules are generally of length larger than 30-residues, we also generate a suggested non-redundant set containing representative structures from each of the functional classes as well as representatives from the unannotated groups with larger length. The members are selected with resolution better than 3.5Å to make it a meaningful set of structures for real applications. This set has 159 structures, including only 22 functionally unannotated RNA structures of synthetic source. We have kept no structure solved by NMR spectroscopy in the non-redundant set, as there is no way one can determine quality of the data. The structures determined by cryo-electron microscopy are automatically removed because of their poor resolution.
Applications
The database can be searched for a set of RNA structural entries according to functional type or source organism. Also, a combined search can be performed using advanced options where a user can specify the chain length as well as a resolution cutoff of the crystal structures. Furthermore, one can determine if there are any structures whose sequence is identical to a given nucleotide sequence. At present, only scientific names of the organisms are accepted for search criteria in “Advanced Search Options” of our web-server.
As the classification shows, there are many RNA classes where the numbers of PDB files are 10 or greater. These subclasses have been shown in Table 3 and corresponding MOL_ID’s are shown as a suffix to the PDB-ID. They carry signatures that may indicate variations introduced in the molecular structure due to ligand binding or alteration of crystallization conditions. Eventually, they can be referred to as crystallographic ensembles in analogy with statistical ensembles obtained from molecular dynamics or Monte Carlo simulations as done recently (Halder and Bhattacharyya, 2010; Samanta et al., 2010). We have performed pair-wise secondary structure comparison for these classes to compare the structural similarity between them. For secondary structure assignment, the base pairing patterns of each RNA structure in a functional class have been obtained using BPFind software tool (Das et al., 2006). BPFind gives us the secondary structure of a nucleic acid at the basepairing level. These secondary structural information of basepairing for each chain have been converted to a one-dimensional string of characters: H, N, T, and L corresponding to Watson–Crick base pairs, non-canonical base pairs, base triplets, and unpaired bases, respectively. The secondary structures thus obtained correspond to individual RNA chains separately and do not consider inter-chain base pairing information. These secondary structural sequences are then compared with that of the best representative structure of that class using Needleman–Wunsch algorithm (Needleman and Wunsch, 1970) as implemented in EMBOSS (Rice et al., 2000). An identity scoring matrix (EDNAMAT) was used for such alignment with a gap-open penalty of −10.0 and gap-extension penalty of –0.5. We find that the average similarity is 80% or more for a set of similar structures of a given class having 30 or more RNA chains. The detailed analyses of structural variation among these crystallographic ensembles are beyond the scope of this paper and would be presented elsewhere. Here a point to note is that secondary structure comparison have not been performed for crystal structures with resolution worse than 3.5Å because of the poor quality of secondary structural information obtained from such structures. RNA structures solved by NMR spectroscopy or electron microscopy are also not included in secondary structure comparison for the same reason.
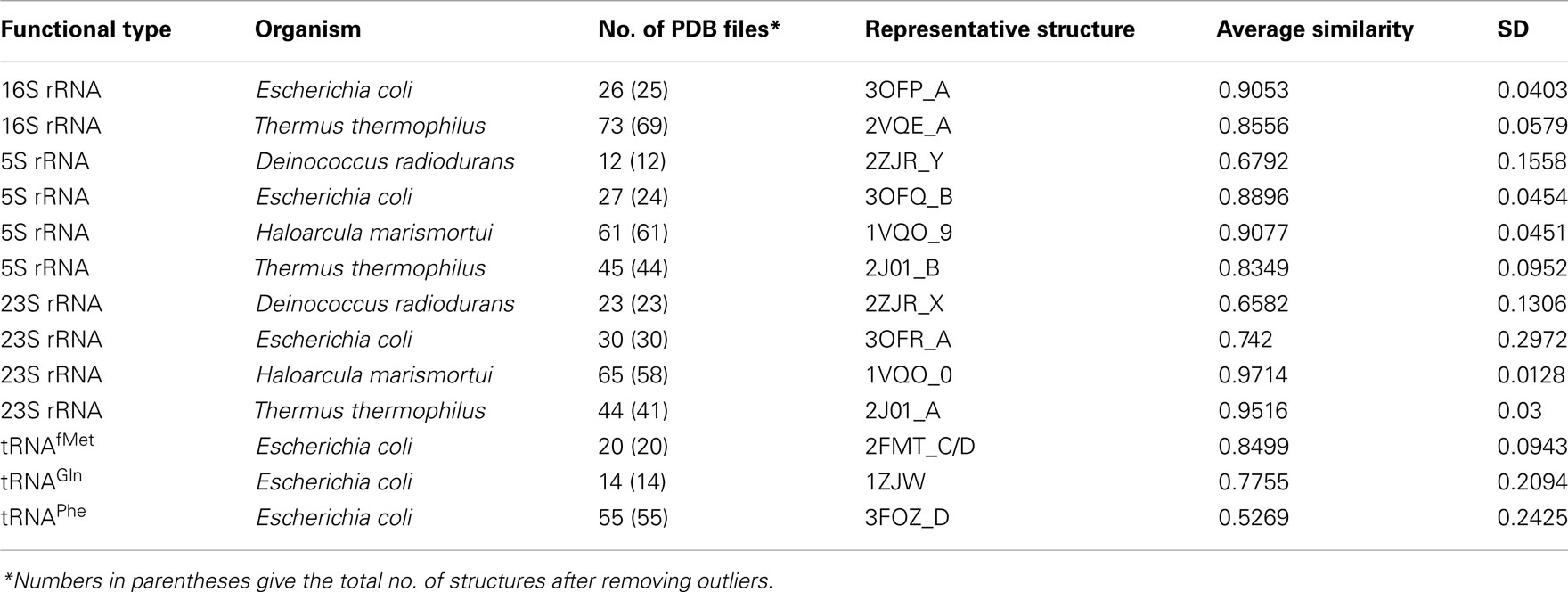
Table 3. Subclasses where the number of PDB files are 10 or greater and their structural variation.
Comparison of secondary structure can also be useful to predict the functions of unannotated RNAs. As mentioned in the earlier section, the functional types of many RNA chains that have been placed under the other-RNA and unannotated-RNA classes could not be obtained from the PDB files unambiguously. However, we find that there are a number of structures in these two types, which are of significant length (30 nucleotide or more). We have performed structural comparison of these RNA chains with known functional RNAs available in the suggested non-redundant dataset, consisting of only X-ray crystal structures with resolution better than 3.5Å. For the secondary structure alignment, the basepairing pattern for each RNA structure in the non-redundant dataset, obtained using BPFind software, comprises a database of known structural forms of well-classified functional RNAs. In a similar way, the secondary structural data for each of the unannotated RNA chains were generated and compared against the database of known structural forms. Thus, the secondary structural information of an unannotated RNA chain has been aligned with secondary structural information of each RNA chain present in the non-redundant dataset using Needleman–Wunsch algorithm for global sequence alignment. We have assigned probable function of an unannotated RNA as identical to some known structural form with which the best similarity score is obtained. For example, chain A of 1FIR shows highest score of 362 against chain A of 1EVV (Table 4). To validate the results of our function prediction method and to remove any false positive, we have used the bootstrapping method. For each pair of unannotated query and its best match in the non-redundant dataset, the best match secondary structure has been shuffled to generate a set of 100 random secondary structures having the same composition. Thus, the secondary structure sequence for chain A of 1EVV has been shuffled to generate 100 random sequence files and aligned secondary structure of 1FIR (chain: A) with all these 100 random sequences. Average and SD of these alignment scores have been calculated (Tables 4 and 5). We find that in many cases the original best match score and the average score against random sequences are quite similar (e.g., 3NVK, 1VFG, etc.). Figure 4 shows distributions of the random scores along with the actual predictive scores for two representative systems. Obviously, prediction of function of w-chain of 3KIS is questionable (Figure 4b) while that of v-chain of 3KIS is a good prediction. This quality depends on (original score for best match – average score from random sequences)/SD of random scores, as given in the last column of Tables 4 and 5. It is found that when these values are larger than 6 or 7, the predictions are generally correct. We have tried to manually examine our results against the original functions as obtained from literature study and found them to be in very good agreement.
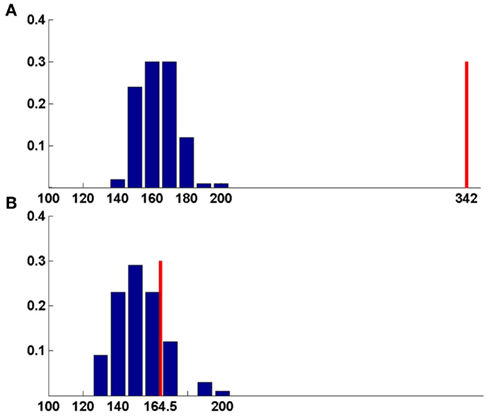
Figure 4. Bootstrapping method for function prediction. Blue-random scores, red-best match score. (A) Chain v of 3KIS, (B) chain w of 3KIS. For chain v, the best score is significantly larger than random average score giving correct prediction, whereas for chain w, best match score lies within the range of random scores, leading to false prediction (Table 4).
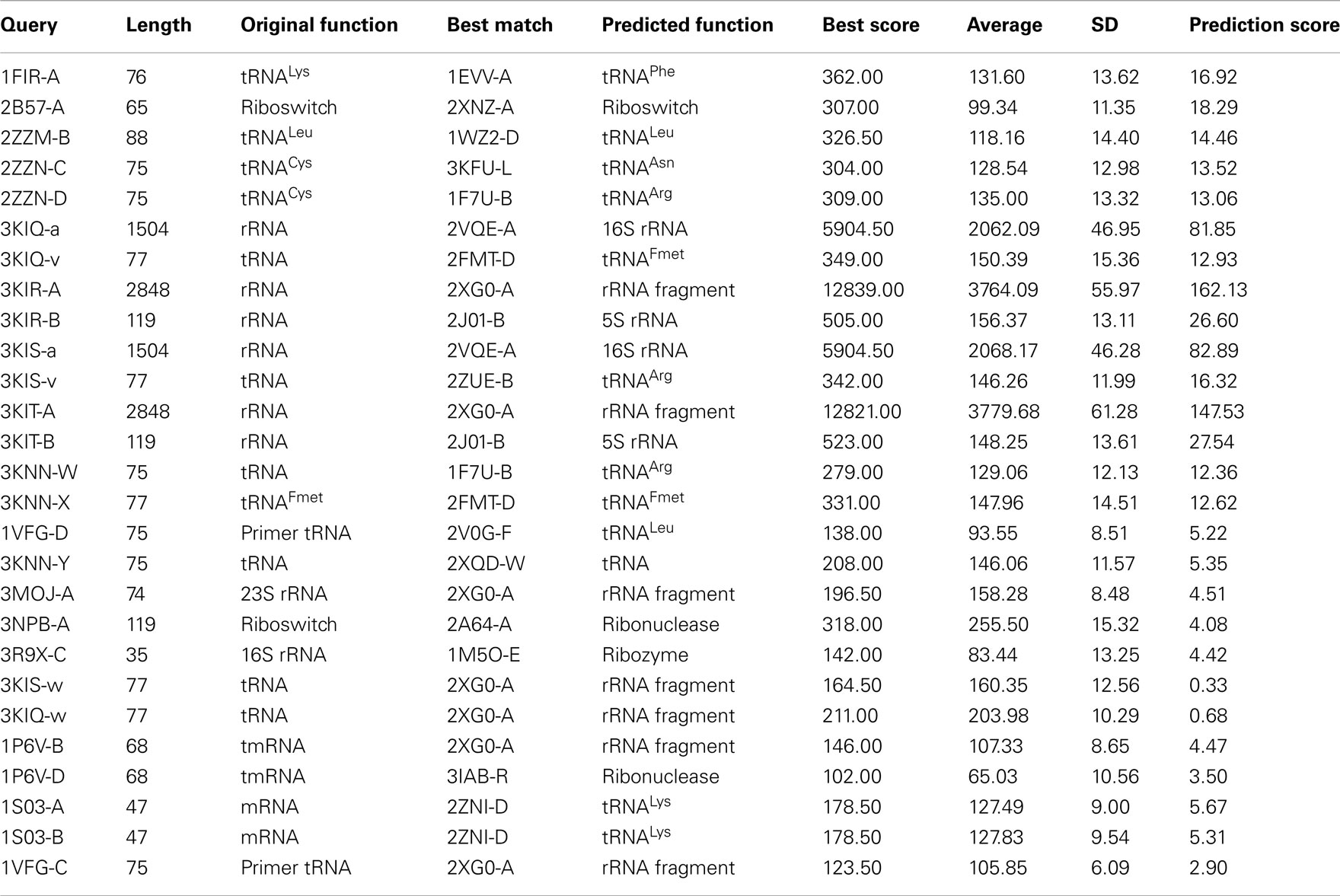
Table 4. Function assignment to RNAs of the other-RNA class.
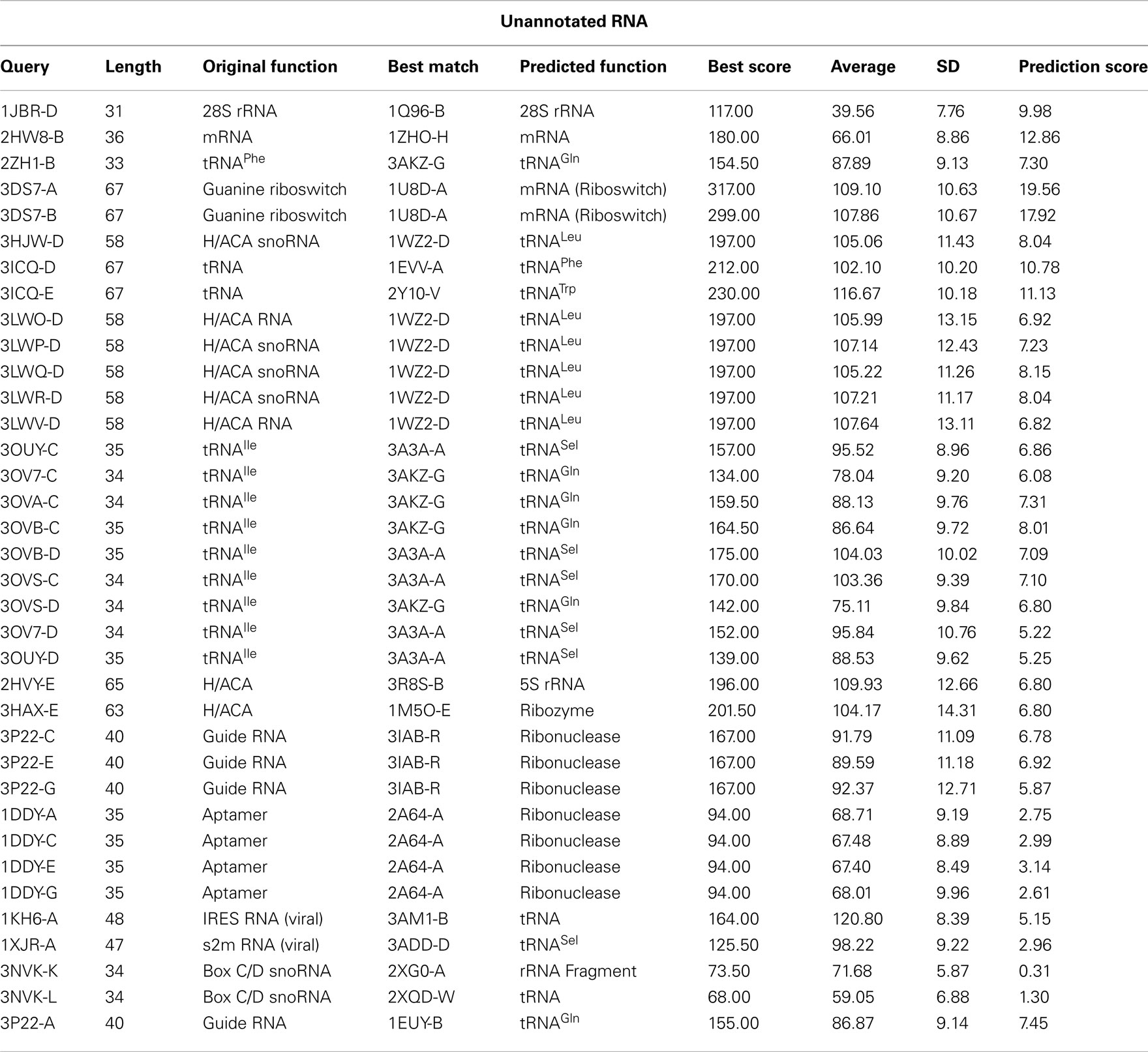
Table 5. Function annotation to unannotated RNAs.
Using the above procedure, we have been able to predict the functions of 17 RNA structures from the “Unannotated-RNA” class and 18 more from the “Other-RNA” class. The results of the function assignments are shown in Tables 4 and 5. Structures of H/ACA box snoRNAs, like 3HJW (chain D), 3LWP (chain D), 3LWQ (chain D), 3LWR (chain D), and 3P22 (chain A), have been predicted as tRNAs. In these cases, the snoRNAs are bound with tRNA-processing proteins and thus have tRNA-like structural motifs for proper recognition. Also, the sub-divisions of different tRNAs can be predicted with limited accuracy only, as the secondary structure of tRNAs is nearly universal among different sub-types.
Conclusion
Hierarchical Database of RNA Structures is an evolving resource that is expected to grow and incorporate more and more RNA structures as and when they are solved and made available from PDB. We have used the PDB files in plain text format, instead of XML files, as these do not contain any extra information but require significantly more storage due to their huge size. The classification of RNA structures are done by an automated tool, a code written in high-level GNU-Octave language, which takes roughly 3 h to classify 2095 PDB files in a 3.0 GHz “Pentium 4” processor with 1GB RAM. Most importantly, out of these 3 h, the classification job takes only a part and nearly 2 h are used by the BLAST search program. However, this task is carried out once in a month during creation of the database and does not affect users. There is also an inbuilt function called “pdbread” in the recent versions of Matlab which can read a PDB file into a structure and can also store the relevant information. Although, a small part of our program and the function “pdbread” are similar, the function “pdbread” demands more time to gather the complete information including the coordinate data from a big PDB file. However, our routine skips the coordinate data in the PDB file to reduce CPU time. The program code is a flexible one and certainly there would be necessity for modifications when structures of altogether new RNA with function as yet unknown or when structures of more siRNA, miRNA, virus etc., would be available. However, we can easily modify the code in future to characterize these structures into new functional classes. Similarly, when several structures of a sub–sub-class would be available, it is expected to open up new directions of research toward understanding ligand or environment-induced structural alterations. For instance, it would be interesting to understand conformational variations between ligand-bound and ligand-free states of riboswitches, between ribosome structures with or without tRNA bound to it, free tRNA and tRNA complexed with synthatase etc. Such structural comparisons can also be used to detect the functional class of unannotated RNA structures. We have used a simple mechanism for prediction of function of unknown RNA structures but it can be improved by betterment of the scoring matrix or by using graph-theoretic approach. We are planning to include the predicted functions in later versions of the database.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to Dr. Devapriya Chowdhury, JNU, New Delhi, India, and Dr. Rahul Banerjee, SINP, India, for suggestions. We are thankful to Ms. Shreya Pakrashi, Ms. Tania Sen, and Ms. Rishika Sengupta for technical support. Funding: This work has been partially supported by Dept. of Biotechnology, India and CBAUNP project of Dept. of Atomic Energy, Govt. of India.
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J. H., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST a new generation of protein database search programs. Nucl. Acids Res. 25, 3389–3402.
Batey, R. T., Rambo, R. P., and Doudna, J. A. (1999). Tertiary motifs in RNA structure and folding. Angew. Chem. Int. Ed. Engl. 38, 2327–2343.
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., and Bourne, P. E. (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242.
Das, J., Mukherjee, S., Mitra, A., and Bhattacharyya, D. (2006). Non-canonical base pairs and higher order structures in nucleic acids crystal structure database analysis. J. Biomol. Struct. Dynam. 24, 149–161.
Ferre-d’Amare, A. R., and Doudna, J. A. (1999). RNA FOLDS insights from recent crystal structures. Ann. Rev. Biophys. Biomol. Struct. 28, 57–73.
Halder, S., and Bhattacharyya, D. (2010). Structural stability of tandemly occurring noncanonical basepairs within double helical fragments: molecular dynamics studies of functional RNA. J. Phys. Chem. B 114, 14028–14040.
Hermann, T., and Patel, D. J. (1999). Stitching together RNA tertiary architectures. J. Mol. Biol., 294, 829–849.
Holm, L., and Sander, C. (1997). Dali/FSSP: classification of three-dimensional protein folds. Nucl. Acids Res. 25, 231–234.
Hubbard, T. J. P., Murzin, A. G., Brenner, S. E., and Chothia, C. (1997). SCOP: A structural classification of proteins database. Nucl. Acids Res. 25, 236–239.
Klosterman, P. S., Tamura, M., Holbrook, S. R., and Brenner, S. E. (2002). SCOR: a structural classification of RNA database. Nucl. Acids Res. 30, 392–394.
Lee, S., and Blundell, T. L. (2009) , BIPA: a database for protein-nucleic acid interaction in 3D structures. Bioinformatics 15, 1559–1560.
Leontis, N. B., Stombaugh, J., and Westhof, E. (2002). The non-Watson-Crick base pairs and their associated isostericity matrices. Nucl. Acids Res. 30, 3497–3531.
Leontis, N. B., and Westhof, E. (2001). Geometric nomenclature and classification of RNA base pairs. RNA 7, 499–512.
Lu, X. J., and Olson, W. K. (2003). 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucl. Acids Res. 31, 5108–5121.
Murthy, V. L., and Rose, G. D. (2003). RNABase: an annotated database of RNA structures. Nucl. Acids Res. 31, 502–504.
Murzin, A. G., Brenner, S. E., Hubbard, T., and Chothia, C. (1995). SCOP – a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol., 247, 536–540.
Needleman, S. B., and Wunsch, C. D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol., 48, 443–453.
Panigrahi, S., Pal, R., and Bhattacharyya, D. (2011). Structure and energy of non-canonical basepairs: comparison of various computational chemistry methods with crystallographic ensembles. J. Biomol. Struct. Dynam. 29, 541–556.
Rice, P., Longden, I., and Bleasby, A. (2000). EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277.
Roy, A., Panigrahi, S., Bhattacharyya, M., and Bhattacharyya, D. (2008). Structure, stability and dynamics of canonical and noncanonical base pairs: quantum chemical studies. J. Phys. Chem. B 112, 3786–3796.
Samanta, S., Chakrabarti, J., and Bhattacharyya, D. (2010). Changes in thermodynamic properties of DNA base pairs in protein-DNA recognition. J. Biomol. Struct. Dynam. 27, 429–442.
Sarver, M., Zirbel, C. L., Stombaugh, J., Mokdad, A., and Leontis, N. B. (2008). FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol., 56, 215–252.
Scott, W. G., Finch, J. T., and Klug, A. (1995). The crystal-structure of an all-RNA hammerhead ribozyme – a proposed mechanism for RNA catalytic cleavage. Cell 81, 991–1002.
Stombaugh, J., Zirbel, C. L., Westhof, E., and Leontis, N. B. (2009). Frequency and isostericity of RNA base pairs. Nucl. Acids Res. 37, 2294–2312.
Tamura, M., Hendrix, D. K., Klosterman, P. S., Schimmelman, N. R. B., Brenner, S. E., and Holbrook, S. R. (2004). SCOR: structural classification of RNA, version 2.0 . Nucl. Acids Res. 32, D182–D184.
Wang, G. L., and Dunbrack, R. L. (2005). PISCES: recent improvements to a PDB sequence culling server. Nucl. Acids Res. 33, W94–W98.
Keywords: RNA classification, RNA crystal structures, RNA database, functional RNA, structure prediction, functional annotation
Citation: Ray SS, Halder S, Kaypee S and Bhattacharyya D (2012) HD-RNAS: an automated hierarchical database of RNA structures. Front. Gene. 3:59. doi: 10.3389/fgene.2012.00059
Received: 26 December 2011; Accepted: 29 March 2012;
Published online: 18 April 2012.
Edited by:
Fengfeng Zhou, Shenzhen Institutes of Advanced Technology, ChinaReviewed by:
Peng Jiang, University of Iowa, USAGaurav Sablok, Huazhong Agricultural University, China
Ao Li, University of Science and Technology of China, China
Yi Zhao, Institute of Computing Technology,C.A.S., China
Bin Song, Oracle, USA
Copyright: © 2012 Ray, Halder, Kaypee and Bhattacharyya. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Dhananjay Bhattacharyya, Biophysics Division, Saha Institute of Nuclear Physics, 1/AF Bidhannagar, Kolkata 700 064, India. e-mail:ZGhhbmFuamF5LmJoYXR0YWNoYXJ5eWFAc2FoYS5hYy5pbg==
†They contributed equally and can be considered as joint first authors.