

- St. Laurent Institute, Cambridge, MA, USA
The mysteries surrounding the ∼97–98% of the human genome that does not encode proteins have long captivated imagination of scientists. Does the protein-coding, 2–3% of the genome carry the 97–98% as a mere passenger and neutral “cargo” on the evolutionary path, or does the latter have biological function? On one side of the debate, many commentaries have referred to the non-coding portion of the genome as “selfish” or “junk” DNA (Orgel and Crick, 1980), while on the other side, authors have argued that it contains the real blueprint for organismal development (Penman, 1995; Mattick, 2003), and the mechanisms of developmental complexity. Thus, this question could be referred to without much exaggeration as the most important issue in genetics today.
Historically, genetic approaches have very successfully determined the function of a variety of biologically important regions of a genome (usually called “genes”), based on necessary and sufficient linkage between relatively obvious alterations in a phenotype(s) and specific changes in nucleotide sequence. The vast majority of sequences identified in the genetic screens do correspond to protein-coding portions of the genome. For example, most of the changes associated with simple Mendelian genetic diseases harbor mutations in exons of protein-coding genes or in the sequences that prevent their proper assembly into mature transcripts (near splice junctions; Cooper et al., 1995). Thus, at face value at least, the non-coding portions of the genome do not really seem to represent a reservoir of biologically or medically relevant sequences.
However, this interpretation lacks intellectual closure, primarily because of its counter-intuitive conclusion that almost all of the DNA in every cell of our body has no function. Upon closer examination, a number of reasons exist to explain why the traditional genetics methods did not uncover the genotype–phenotype relationships in the non-coding portions of the genome (Mattick, 2009). For example, in addition to the simple fact that protein-coding regions have traditionally been the primary focus of forward genetic screens, alteration of non-coding, presumably regulatory regions, may impart more subtle phenotypes than coding regions, which cause catastrophic component damage. Non-coding regions could have a higher tolerance to sequence changes compared to protein-coding regions, or a higher redundancy within cellular machineries, functioning as a major substrate for evolutionary innovation and phenotypic radiation.
Answering the basic question of the functionality of the non-coding portion of the genome has shifted more toward molecular methods, specifically toward measuring the primary output of the genome, the RNA. At its core, the central premise behind these endeavors relies on the following concept: the only functional “products” of a DNA sequence that we can identify are copies of itself, either in the form of an RNA molecule or a DNA molecule. Copying of DNA into DNA ensures replication, cell division, and DNA repair, while copying of DNA into RNA transmits information into cellular actions. Even if a regulatory DNA sequence does not directly encode RNA – its function is still measured by the eventual production of RNA from somewhere in the genome. And, while cellular processes could affect the function of a sequence of DNA in many different ways, by either covalent modification of its bases or non-covalent interaction with a plethora of DNA binding proteins, RNA output remains the only known way for a cell to use DNA-encoded information. The central posit of this concept implies that if a sequence of DNA participates in the production of some RNA or affects the quantity or type of the RNA produced, then this sequence can be functional if the RNA product has a function.
This basic hypothesis has led to several whole-genome RNA mapping experiments done during the past decade – in effect, the first attempts at genome-wide “RNA Bookkeeping.” These unbiased surveys of RNA relied on high-throughput technologies such as tiling arrays and various sequencing methods (Rinn et al., 2003; Bertone et al., 2004; Carninci et al., 2005, 2008; Kapranov et al., 2005, 2007a,b; Birney et al., 2007). In essence, the goal of all these experiments was to identify as many molecules of RNA or sites of transcription as possible in a given tissue, and catalog them into those whose localization to protein-coding regions of the genome could explain their function, and those whose localization could not. Surprisingly, the latter class grew into a pervasive and highly numerous collection (see below for more details) and became broadly dubbed as “dark matter” RNA (Johnson et al., 2005).
Originally, “dark matter” RNA referred simply to RNA produced from the regions of genome without known function, yet stable enough for detection (Johnson et al., 2005). Tiling arrays (Kapranov et al., 2003) can identify regions of genome that give rise to RNA by virtue of hybridization to probes evenly spaced throughout the non-repetitive portions of the genome. The resulting map of transcription specifies a series of RNA producing regions that could then be compared to the map of other genomic features, such as exons of protein-coding genes. The fraction of genomic sequence covered by such fragments located outside of the exons estimated the complexity of the “dark matter” RNA (Kapranov et al., 2002).
Typically, about 75% of all bases represented by all transcribed fragments detected by tiling arrays in any given human cell-line or tissue originated outside of exons of cytosolic polyA+ mRNAs, suggesting that “dark matter” transcription was prevalent in human cells (Kapranov et al., 2007a). As might be expected, this fraction was much higher for human nuclear RNA (Cheng et al., 2005). The FANTOM consortium has shown that the mouse genome could be pervasively transcribed, producing a very complex transcript architecture (Carninci et al., 2005). After combining all available microarray and sequence-based data from all biological sources, the ENCODE consortium estimated that ∼20% of all human genomic sequence might function to produce RNA (Birney et al., 2007). As a consequence of hybridization-based deconvolution of complex mixtures of nucleic acids, the signal thresholds of detection had to be set relatively high to prevent detection of spurious cross-hybridization. This resulted in one of the disadvantages of these experiments: a significant undercounting of transcribed elements. For example, using rapid amplification of cDNA ends (RACE), a more sensitive method for measurement of RNA output form specific loci, evidence of RNA production was found at 75% of randomly chosen human genomic sequences where RNA had not been detected by ENCODE consortium tiling arrays: see Supplementary Table 2 of Birney et al. (2007). This data suggested that the fraction of genome that gives to rise to RNA could far exceed the 20% figure. Indeed, when combining the regions of transcription detected by any method with the total length of all introns (always transcribed to give rise to the mature RNAs), ENCODE estimated that 93% of the human genome is transcribed (Birney et al., 2007). Thus, the matter of detecting the transcribed portion of the genome in stable RNA could depend largely on the sensitivity of the technology used.
However, these experiments have always suffered from criticism that the abundance of the “dark matter” RNAs in mammalian cells could be trivial, in part because of the sensitivity of the techniques used to detect and validate the “dark matter” transcription (van Bakel et al., 2010, 2011). Indeed, these studies were mostly aimed at giving an estimate of the fraction of genomic sequences represented in the “dark matter” RNA and thus tell us something about its complexity, but not about its relative mass (Clark et al., 2011). Perhaps “dark matter” had a very complex population of RNA, and yet represented nothing more than a trivial fraction of cellular RNA mass. Such a scenario might suggest that “dark matter” RNA resulted from non-consequential by-products of cellular processes, consistent with the overall “junk DNA” label given to the non-coding portions of the genome in general (Brosius, 2005; Struhl, 2007; van Bakel and Hughes, 2009; van Bakel et al., 2010). An opposite scenario, where the “dark matter” RNA population was indeed complex and constituted a significant mass of cellular RNA, would on the other hand, suggest that this RNA could indeed be an important and previously hidden component of the regulatory architecture controlling differentiation and development (Mattick, 2003, 2004, 2011; Kapranov et al., 2007b; St Laurent and Wahlestedt, 2007).
The advent of next generation sequencing technologies has allowed for a digital output based count of reads representing short (typically on the order of 25–100 bases) stretches of RNAs from which they were derived (Cloonan and Grimmond, 2008; Wang et al., 2009). By calculating the relative fraction of such reads, one can estimate the relative mass of “dark matter” RNA as a whole, or any specific RNA or transcribed region in the total mass of the assayed RNA population. Despite the apparent simplicity of this approach, the original estimates of the fraction of non-exonic reads in human or mouse RNAseq experiments varied significantly, from as little as 7% (Mortazavi et al., 2008) to as much as 40–50% (Cloonan et al., 2008; Morin et al., 2008). A subsequent report by van Bakel et al. (2010) attempted to directly estimate the relative mass of the “dark matter” RNA and came to the conclusion that it accounts for only 12% of the polyadenylated RNA mass in human or mouse cells. In addition, this report also stated that the same conclusions could be reached by the analysis of total RNA (depleted for rRNA). One common feature of all these reports was the usage of PCR amplification as a part of the RNA preparation for sequencing, which has the potential to alter the original profile of the population (Mamanova et al., 2010; Raz et al., 2011; Sam et al., 2011). Thus, unequivocal estimation of the relative mass of the “dark matter” RNA would require RNA profiling using a sequencing approach that does not rely on amplification. Such profiling performed using single-molecule sequencing of total rRNA-depleted RNA and polyA+ RNA (Kapranov et al., 2010) found that “dark matter” RNA represents a majority of the total non-ribosomal non-mitochondrial RNA most of the human cell-lines and tissues tested (Kapranov et al., 2010). In addition, total human RNA contained a much higher complexity than the polyA+ RNA, especially in terms of “dark matter” RNAs (Kapranov et al., 2010; Raz et al., 2011). This could also explain at least in part the failure of some of the earlier reports to detect a significant fraction of the “dark matter” RNA: not only did those reports rely on PCR amplification, but also they used an RNA fraction highly enriched for polyadenylated RNAs.
Interestingly, very long (100s of kbs) stretches of intergenic space in the human genome, previously considered as “gene desert” regions produced significant levels of RNA (Kapranov et al., 2010). Hundreds of such regions (named vlincs for very long intergenic non-coding regions) spanning ∼4% of intergenic space were detected in just nine different biological sources of RNA (seven tumors and two normal tissues) used in that report. This combined with the observation that most of the vlincs tend to be highly specific to a given biological source (Kapranov et al., 2010), suggests that profiling of the pool of total cellular RNA with hundreds or thousands of different biological samples would result in detection of RNA from a large fraction of intergenic space. This assertion is supported by in-depth analysis of selected genomic regions using methods to select and enrich for all transcripts derived from such regions followed by either tiling array analysis or deep sequencing (Kapranov et al., 2005; Mercer et al., 2011a). Such studies reveal that what appears to be a low signal from either a tiling array or RNAseq experiment obtained on a complex RNA population from a single cell, can in fact represent a complex population of low abundant transcripts (Kapranov et al., 2005; Mercer et al., 2011a). Low abundance could also imply expression restricted to a sub-set of cells in a given population (from a cell-culture or especially, a tissue sample), and thus should not immediately be relegated into the realm of biological noise. These observations are important to keep in mind when interpreting the results of RNAseq or microarray experiments, especially considering that most current RNAseq experiments produce far fewer reads than the estimated minimum of ∼70 million reads required to completely cover the transcriptome from an average human cell (Kapranov et al., 2010).
These results are consistent with those of the ENCODE consortium as far as pervasive transcription is concerned. However, they differ in the estimate of how much stable RNA would remain from that pervasive transcription. The ENCODE consortium suggests that only on the order of ∼20% of human genomic sequence ever exists as stable RNA based on compilation of all available experimental data from a large number of biological sources (Birney et al., 2007). However the logical extrapolation from Kapranov et al. (2010) would suggest that most of the genomic sequence likely exists in the RNA pool when profiling a significant number of tissues using total rRNA-depleted RNA, instead of the polyA+ fraction. The discrepancy may result from the fact the ENCODE, like other similar endeavors before and after, focused on the polyadenylated fraction of RNA, that is estimated to capture only 5–25% of the total mass of the non-ribosomal non-mitochondrial RNA in a human (Kapranov et al., 2010; Raz et al., 2011). Clearly, the dominance of polyA+ RNA as the source of RNA for RNAseq experiments has significantly undercounted the complexity of RNA present in a human cell. In fact, an oligo-dT column may also not necessarily capture all the polyadenylated RNAs in a sample. For example, one can imagine that, long polyadenylated RNA molecules may not bind efficiently due to structural interference, resulting in depletion from the polyA+-selected RNA pool. In fact, depletion of longer mRNAs in polyA+ RNA pool occurs (Raz et al., 2011).
Still, the wider question of functionality of non-polyadenylated RNA as a class has received very little attention, and still remains un-answered. The absence of a polyA-tail does not mean absence of function – clearly, most short non-coding RNA species are non-polyadenylated and functional, for example tRNAs, miRNAs, snRNAs, and other classes of short RNAs. Furthermore, the presence of complex non-adenylated RNA populations in mammalian cells has been established back in 1970s (Salditt-Georgieff et al., 1981) and this type RNA occurred even in the polysomal fraction and was shown to be used for protein production (van Ness et al., 1979; Katinakis et al., 1980). More recently, a reporter mRNA engineered to contain a miRNA in its 3′ UTR served as a target for cleavage by Drosha into a polyA− RNA, and then traveled to the cytosol to function as a template for protein production (Cai et al., 2004). Thus, absence of the polyA-tail does not preclude RNAs from having a function in the cell. However, we are still at the very beginning of the exploration of the functional properties of the vast complexity of novel and apparently non-polyadenylated RNA recently discovered.
Perhaps one of the greatest hurdles in accepting the biological relevance of “dark matter” transcription is the fact that a large proportion of it comes from intronic regions of already annotated genes. Based on single-molecule RNAseq data, it is estimated that the intronic “dark matter” RNA constitutes 70–80% of all mass of the human “dark matter” RNA (Kapranov et al., 2010). The report van Bakel et al. (2010) obtained a similarly high estimate of the fraction of the intronic RNA, but proposed that it simply represents un-processed pre-mRNA. This conclusion was further supported by the data presented in that report where the fraction of intronic RNA amounted only 5.8% of the mass of the human cell’s total RNA not including the ribosomal and mitochondrial RNA (van Bakel et al., 2010). However, as mentioned above, this estimate could result from the choice of polyadenylated RNA used in that study, combined with the effect of PCR amplification. Single-molecule sequencing of total RNA revealed a much higher fraction of intronic RNAs in a human cell, on the order of 30–50% of non-ribosomal, non-mitochondrial RNA (Kapranov et al., 2010). The latter estimate should at least cause us to pause before any unambiguous acceptance of the trivial explanation above – as much as half of nuclear-encoded non-ribosomal RNA in the cell is probably not something one should dismiss outright as noise. In addition, different genes vary in terms of how much intronic RNA they produce, as do different introns of the same gene, and even different regions of the same intron (Kapranov et al., 2010). These observations are not consistent with noise expected from pre-mRNA en-route to splicing or excised introns en-route to degradation. Furthermore, intronic signal does not necessarily mean that it arises from excised introns or pre-mRNA. Since RNAseq does not provide information on the complete structure of an RNA molecule, we do not know what kind of transcripts make up the intronic signal observed in RNAseq experiments. In fact, it could represent different types of elements: alternative exons, exon isoforms of known transcripts, independent stand-alone transcripts, or excised introns (Figure 1). Moreover, one can imagine that any given gene could have a collection of such different types of novel transcripts buried in its introns (Mattick, 1994; Kapranov et al., 2005). Overall, it is fair to say that we are at the beginning of our understanding of role of intronic RNAs in a cell and we should maintain an open mind as to its functional importance (Clark et al., 2011).
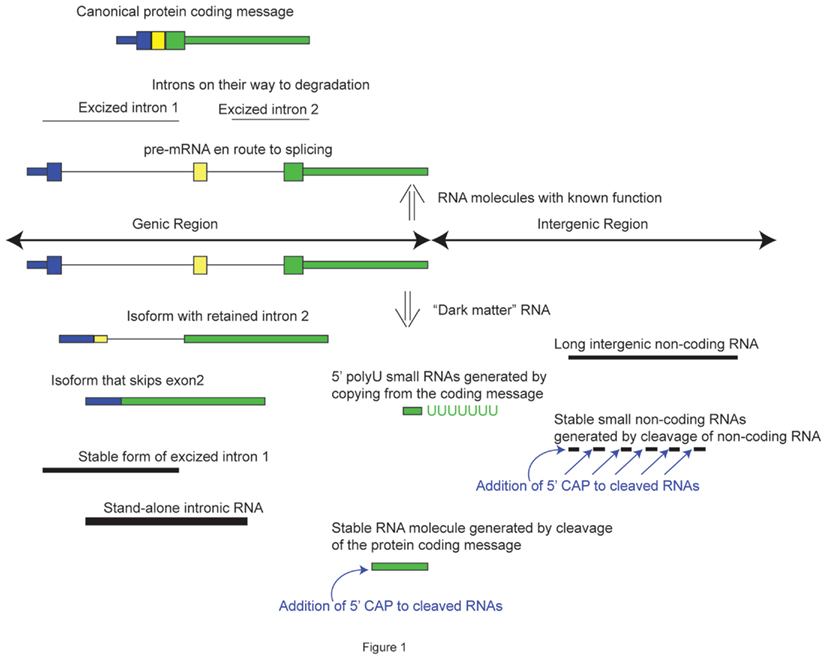
Figure 1. Types of RNA molecules derived from annotated and unannotated loci that constitute “dark matter” RNAs.
Another class of sequences deserves special mention in the context of the transcriptional activity of genomic “dark matter” – the repetitive regions of a genome, which until very recently had been largely avoided by genome-wide RNA profiling studies for technical reasons. For example, tiling microarray designs typically exclude these regions (Kapranov et al., 2007a) because the signal from the probes cannot resolve into an attribute for a specific repeat element. However, the nucleotide-level precision of the next generation sequencing technologies allows mapping of reads with a relatively high specificity, even to repeat regions of the genome. This in turn allows for interrogation of RNAs produced from repeats. For example, one such study relied on mapping of CAGE tags that mark the 5′ ends of capped RNAs (Kodzius et al., 2006) to profile expression of different types of repeats in mammalian cells (Faulkner et al., 2009). Interestingly, a significant fraction of transcription in that study coincided with repeats. Different tissues express different levels and types of repetitive elements, with embryonic tissues having the highest levels of CAGE tags (Faulkner et al., 2009). Interestingly, that study also found that a certain class of repetitive elements, retrotransposons, might provide alternative or tissue-specific promoters for protein-coding genes (Faulkner et al., 2009), and a recent paper has shown that these sequences mobilize to effect somatic transposition events in the human brain (Baillie et al., 2011).
However, the last decade of transcriptome exploration also revealed additional dimensions of its complexity. The first added level of complexity arises from the fact that any given locus can be criss-crossed by different transcripts on both strands, described as “transcriptional forests” by the RIKEN researchers after a large scale effort aimed at sequencing full-length cDNAs from mammalian samples (Carninci et al., 2005). The transcriptional forests are common in the protein-coding loci, where the transcripts that form the complex lattices of overlapping transcription often borrow sequences from known exons and non-exonic regions; however, the function of most of the additional RNA isoforms, which are presumably context-specific, is not understood. For example, based on EST evidence, the GENCODE consortium has shown that a human protein-coding locus specifies on average 5.4 isoforms (Harrow et al., 2006). However, only 2.4 of those could encode a protein, while the function of the rest remains an enigma (Harrow et al., 2006).
Other studies have reached similar conclusions using RACE in combination with tiling arrays to profile the complexity of transcripts sharing exons of ∼400 human protein-coding genes (Birney et al., 2007; Denoeud et al., 2007). More than 80% of all transcripts had alternative 5′ ends or novel exons (Denoeud et al., 2007). In-depth analysis of one human locus encoding the MeCP2 proteins using the RACE/array method revealed 15 new isoforms that have exons derived from intronic and intergenic sequences with often perfectly correct splice sites (Djebali et al., 2008). In most cases, however, additional isoforms identified either do not appear to change the open reading frame or do not encode proteins (Denoeud et al., 2007; Djebali et al., 2008), consistent with previous GENCODE results (Harrow et al., 2006).
The recent realization that RNA could be cleaved and capped at the newly formed 5′ end to produce a separate stable RNA species provides an additional conceptual dimension of the complexity of the mammalian cell’s RNA population (Affymetrix/CSHL ENCODE Project, 2009; Otsuka et al., 2009; Mercer et al., 2010, 2011b). This opens a whole new realm of possibilities where the final, apparently mature and spliced RNA species may not represent the final and/or the only functional product. Conversely, shorter RNAs that would otherwise be considered as simple degradation products, may have function. One tantalizing possibility suggests that they might function in a manner similar to that observed in RNA-mediated inheritance, carried out by apparent RNA degradation products loaded into germ line cells to mediate regulation of gene expression in the subsequent generation of mice (Rassoulzadegan et al., 2006).
Taken together, all of these added complexities suggest a complete reconsideration of the definition of RNA “dark matter.” We would like to posit that it includes not just the RNAs that are made from the “dark matter” regions of the genome, but any RNA molecule whose function we do not understand (Figure 1). For example, an RNA molecule consisting solely of exons of an otherwise protein-coding transcript, but spliced into an RNA with no open reading frame, can be considered as an RNA molecule whose function we do not understand, even though it is assembled from individual sequences with known function. Likewise, an RNA molecule processed from a protein-coding gene or pseudogene and having lost its protein-coding capacity can be considered a “dark matter” transcript as long as we do not understand its function. The “dark matter” RNAs can therefore comprise both coding and non-coding RNAs, as long as their function currently remains unclear. While, for the most part, “dark matter” RNAs have features of non-coding RNAs (Carninci et al., 2005; Cheng et al., 2005; Djebali et al., 2008), it remains possible that some of the RNAs previously considered as non-coding do encode short peptides (Kondo et al., 2010). In fact, recent results based on profiling of sites in RNA molecules bound by ribosomes suggest that many mouse “dark matter” RNAs indeed encode short peptides (Ingolia et al., 2011). Undoubtedly, the prevalence and biological relevance of these peptides will remain a very interesting and important question for years to come.
If the entire genome is transcribed and represented as stable RNAs at least in some biological samples, then we should re-evaluate as a community our strategies in terms of annotating the “dark matter” RNAs. Despite the ongoing efforts to annotate the lncRNAs (Amaral et al., 2011; Cabili et al., 2011; Chen et al., 2011; Wang and Chang, 2011), the lists obtained from different experiments do not overlap significantly. For example, only ∼19% of base pairs covered by the human vlinc regions in the intergenic space overlap those found by lncRNAs (Kapranov et al., 2010; also see Figure 2A). This suggests that current databases only scratch the surface of the immense complexity of the RNA population of human cells.
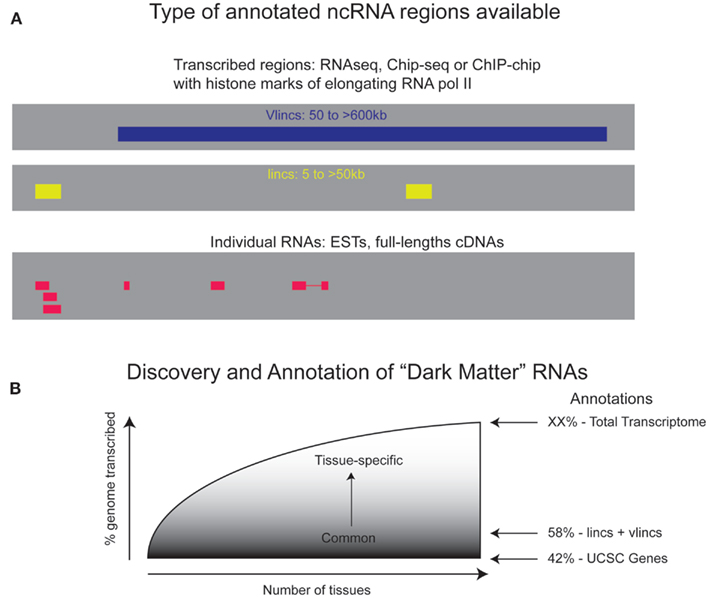
Figure 2. Coverage of the genome by “dark matter” RNAs. (A) Information currently available about the regions of dark matter transcription and the actual RNA molecules made from these region comes from various types of experiments and databases. There is relatively little overlap between these different databases suggesting that the actual extend of dark matter transcription is far greater than any one database suggests. (B) A theoretical curve showing expected results of the fraction of the genome that is transcribed as a function of the number of biological sources whose RNA is profiled. The coverage of transcribed genome by protein coding genes including their introns is 42% and lincRNAs bring it up to 58%. However, the full extent of the transcribed genome is expected to be much greater than that.
In retrospect, this is not surprising when one considers that current genomic annotations, such as the human GenBank mRNA track on the UCSC browser (Kent et al., 2002), depend primarily on sequenced full-length cDNAs, each one representing only a single-molecule of RNA. GenBank currently contains ∼300K such entries, which closely approximates estimates of the total number of polyadenylated RNA molecules contained in a single cell (∼300K; Hastie and Bishop, 1976). Thus, based on these numbers, it is fair to say that all we know in terms of the complete sequences of RNAs from the human transcriptome represents just one cell’s worth of polyadenylated RNA! Of the 300K human GenBank mRNA entries, ∼88% are represented by unique cDNAs, pointing to the fact that many of the current gene models and annotations are based on a single (!!!) fully sequenced RNA molecule. This is reinforced by the recent application of targeted RNA sequencing, which revealed a plethora of new coding and non-coding transcripts, even from intensively studied human loci such as p53, HOX, and sonic hedgehog (SHH) that are either only expressed in a very limited number of cells in what was previously considered a homogenous culture, or where otherwise missed in the cDNA libraries (Kapranov et al., 2005; Mercer et al., 2011a). In addition, most of the annotated cDNAs have been characterized from the polyadenylated transcriptome, thus the non-polyadenylated fraction remains virtually un-uncharted from the point of view of full-length cDNA sequencing. Considering how many molecules of RNA a given human locus must make during the lifetime of an individual, evidently, this depth of knowledge only scratches the surface of RNA complexity.
Finally, we believe that understanding of the true extent and function of human transcription remains one of the most important philosophical and scientific questions of our time. Considering this, we suggest that the community should undertake a directed approach aimed at answering this question. We envision the profiling of a reasonably large number of carefully chosen samples based on total RNA depleted of rRNA, rather than polyA+, using amplification free RNAseq approaches. Given the high-tissue specificity of dark matter RNAs, samples would include at least 100–200 key tissues or cell-lines, rich in intergenic RNAs, such as Ewing Sarcomas. We expect the curve of detected dark matter transcripts to reach a plateau steeply – the big un-answered question so far is where this plateau will be and how much further the curve will rise as more samples are added (Figure 2B). RNAseq will yield regions of transcription, while additional methods will unravel the complexities of individual transcripts in each region of transcription. This could be accomplished by a site-directed methods similar to the one described by Djebali et al. (2008).
As our understanding of the function of the novel RNA expands, the domain of “dark matter” transcripts will shrink. Unfortunately, for the most part we cannot yet predict in silico which of these “non-canonical” RNA molecules are functional and what function they might fulfill, like we usually can for protein-coding mRNAs. This is probably the greatest challenge to our understanding and acceptance of this type of RNA – our general inability to predict what an RNA species might do when it does not have an obvious open reading frame. However, this should not stop us from exploring the function of these RNAs in biological or medical context. Even if a function of a given RNA molecule or transcribed region in a genome may not be known, its association with a disease should provide novel mechanistic insights, and novel diagnostic tools for the disease. The fact that “dark matter” RNAs tend to be highly specific to their biological source emphasizes the promise of this approach (Cheng et al., 2005; Kapranov et al., 2010). Surprisingly perhaps, it seems remarkably easy to detect phenotypes associated with siRNA-mediated knockdown or over-expression of non-coding RNAs, even in cell-culture, and to correlate these phenotypes with aberrant expression of the non-coding RNAs in disease states like cancer and neurological diseases (see, e.g., Mattick, 2009; Gupta et al., 2010; Askarian-Amiri et al., 2011; Gibb et al., 2011; Khaitan et al., 2011; Ulitsky et al., 2011). Moreover, one of the first examples of association of “dark matter” transcripts emanating from a family of repetitive regions with a particular type of cancer has been recently provided by Ting et al., 2011. Overall, it would not be surprising if “dark matter” transcripts would eventually occupy a central place in our conceptual understanding of the molecular events underlying human development and disease, and thereby enter the arsenal of therapeutic targets as prominently as those gene products whose function we currently understand.
Glossary
CAGE: cap analysis of gene expression, a method based on selection of RNAs containing the 5′ CAP modification and obtaining short sequences or tags near the 5′ end of these RNAs. Typically, millions of tags are obtained in each experiment.
ENCODE: encyclopedia of DNA elements, an NHGRI-sponsored project aimed at empirically identifying functionally important element in the human genome sequence, http://www.genome.gov/10005107.
Genomic dark matter: usually refers to the portion of a genome that does not correspond to exons (coding or non-coding) of annotated mRNAs.
RACE: rapid amplification of cDNA Ends, a PCR-approach with “outward” positioned primers to amplify toward the 5′ and 3′ end of an RNA molecule from a point inside the molecule.
RNAseq: a method to quantify and profile RNA population in a cell based on massive sequencing of short (typically less than 100 bases) regions of a large number of RNA molecules. Typically, sequencing is conducted on cDNA, rather than RNA, thus cDNAseq would have been more appropriate. However, RNAseq is used for historical reasons. A note, since direct RNA sequencing is now possible, a bona fide RNAseq analysis should somehow be distinguished from cDNAseq.
Single-molecule sequencing: a method where a single-molecule of a nucleic acid is sequenced directly as opposed methods that obtain sequence signal from a population of molecules.
Tiling array: a microarray platform designed to interrogate genomic sequence with a certain resolution set by the distance between the probes. Opposite in concept to exon arrays where probes are designed only to the annotated regions of interest.
Vlinc: very long intergenic non-coding RNA region, identified based on continuous RNAseq signal that spans genomic regions of 50 kb or longer (often much longer) in the area of genome where no annotated gene has been found.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thanks Dr. John Mattick for very helpful suggestions on the manuscript, and Drs. Robert Arceci, Tim Triche, Jason Farrar, Patrice Milos, John Thompson, and Claes Wahlestedt for helpful and encouraging discussions.
References
Affymetrix/CSHL ENCODE Project. (2009). Post-transcriptional processing generates a diversity of 5’-modified long and short RNAs. Nature 457, 1028–1032.
Amaral, P. P., Clark, M. B., Gascoigne, D. K., Dinger, M. E., and Mattick, J. S. (2011). lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 39, D146–D151.
Askarian-Amiri, M. E., Crawford, J., French, J. D., Smart, C. E., Smith, M. A., Clark, M. B., Ru, K., Mercer, T. R., Thompson, E. R., Lakhani, S. R., Vargas, A. C., Campbell, I. G., Brown, M. A., Dinger, M. E., and Mattick, J. S. (2011). SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA 17, 878–891.
Baillie, J. K., Barnett, M. W., Upton, K. R., Gerhardt, D. J., Richmond, T. A., De Sapio, F., Brennan, P. M., Rizzu, P., Smith, S., Fell, M., Talbot, R. T., Gustincich, S., Freeman, T. C., Mattick, J. S., Hume, D. A., Heutink, P., Carninci, P., Jeddeloh, J. A., and Faulkner, G. J. (2011). Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479, 534–537.
Bertone, P., Stolc, V., Royce, T. E., Rozowsky, J. S., Urban, A. E., Zhu, X., Rinn, J. L., Tongprasit, W., Samanta, M., Weissman, S., Gerstein, M., and Snyder, M. (2004). Global identification of human transcribed sequences with genome tiling arrays. Science 306, 2242–2246.
Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigo, R., Gingeras, T. R., Margulies, E. H., Weng, Z., Snyder, M., Dermitzakis, E. T., Thurman, R. E., Kuehn, M. S., Taylor, C. M., Neph, S., Koch, C. M., Asthana, S., Malhotra, A., Adzhubei, I., Greenbaum, J. A., Andrews, R. M., Flicek, P., Boyle, P. J., Cao, H., Carter, N. P., Clelland, G. K., Davis, S., Day, N., Dhami, P., Dillon, S. C., Dorschner, M. O., Fiegler, H., Giresi, P. G., Goldy, J., Hawrylycz, M., Haydock, A., Humbert, R., James, K. D., Johnson, B. E., Johnson, E. M., Frum, T. T., Rosenzweig, E. R., Karnani, N., Lee, K., Lefebvre, G. C., Navas, P. A., Neri, F., Parker, S. C., Sabo, P. J., Sandstrom, R., Shafer, A., Vetrie, D., Weaver, M., Wilcox, S., Yu, M., Collins, F. S., Dekker, J., Lieb, J. D., Tullius, T. D., Crawford, G. E., Sunyaev, S., Noble, W. S., Dunham, I., Denoeud, F., Reymond, A., Kapranov, P., Rozowsky, J., Zheng, D., Castelo, R., Frankish, A., Harrow, J., Ghosh, S., Sandelin, A., Hofacker, I. L., Baertsch, R., Keefe, D., Dike, S., Cheng, J., Hirsch, H. A., Sekinger, E. A., Lagarde, J., Abril, J. F., Shahab, A., Flamm, C., Fried, C., Hackermuller, J., Hertel, J., Lindemeyer, M., Missal, K., Tanzer, A., Washietl, S., Korbel, J., Emanuelsson, O., Pedersen, J. S., Holroyd, N., Taylor, R., Swarbreck, D., Matthews, N., Dickson, M. C., Thomas, D. J., Weirauch, M. T., Gilbert, J., Drenkow, J., Bell, I., Zhao, X., Srinivasan, K. G., Sung, W. K., Ooi, H. S., Chiu, K. P., Foissac, S., Alioto, T., Brent, M., Pachter, L., Tress, M. L., Valencia, A., Choo, S. W., Choo, C. Y., Ucla, C., Manzano, C., Wyss, C., Cheung, E., Clark, T. G., Brown, J. B., Ganesh, M., Patel, S., Tammana, H., Chrast, J., Henrichsen, C. N., Kai, C., Kawai, J., Nagalakshmi, U., Wu, J., Lian, Z., Lian, J., Newburger, P., Zhang, X., Bickel, P., Mattick, J. S., Carninci, P., Hayashizaki, Y., Weissman, S., Hubbard, T., Myers, R. M., Rogers, J., Stadler, P. F., Lowe, T. M., Wei, C. L., Ruan, Y., Struhl, K., Gerstein, M., Antonarakis, S. E., Fu, Y., Green, E. D., Karaöz, U., Siepel, A., Taylor, J., Liefer, L. A., Wetterstrand, K. A., Good, P. J., Feingold, E. A., Guyer, M. S., Cooper, G. M., Asimenos, G., Dewey, C. N., Hou, M., Nikolaev, S., Montoya-Burgos, J. I., Löytynoja, A., Whelan, S., Pardi, F., Massingham, T., Huang, H., Zhang, N. R., Holmes, I., Mullikin, J. C., Ureta-Vidal, A., Paten, B., Seringhaus, M., Church, D., Rosenbloom, K., Kent, W. J., Stone, E. A., NISC Comparative Sequencing Program, Baylor College of Medicine Human Genome Sequencing Center, Washington University Genome Sequencing Center, Broad Institute, Children’s Hospital Oakland Research Institute, Batzoglou, S., Goldman, N., Hardison, R. C., Haussler, D., Miller, W., Sidow, A., Trinklein, N. D., Zhang, Z. D., Barrera, L., Stuart, R., King, D. C., Ameur, A., Enroth, S., Bieda, M. C., Kim, J., Bhinge, A. A., Jiang, N., Liu, J., Yao, F., Vega, V. B., Lee, C. W., Ng, P., Shahab, A., Yang, A., Moqtaderi, Z., Zhu, Z., Xu, X., Squazzo, S., Oberley, M. J., Inman, D., Singer, M. A., Richmond, T. A., Munn, K. J., Rada-Iglesias, A., Wallerman, O., Komorowski, J., Fowler, J. C., Couttet, P., Bruce, A. W., Dovey, O. M., Ellis, P. D., Langford, C. F., Nix, D. A., Euskirchen, G., Hartman, S., Urban, A. E., Kraus, P., Van Calcar, S., Heintzman, N., Kim, T. H., Wang, K., Qu, C., Hon, G., Luna, R., Glass, C. K., Rosenfeld, M. G., Aldred, S. F., Cooper, S. J., Halees, A., Lin, J. M., Shulha, H. P., Zhang, X., Xu, M., Haidar, J. N., Yu, Y., Ruan, Y., Iyer, V. R., Green, R. D., Wadelius, C., Farnham, P. J., Ren, B., Harte, R. A., Hinrichs, A. S., Trumbower, H., Clawson, H., Hillman-Jackson, J., Zweig, A. S., Smith, K., Thakkapallayil, A., Barber, G., Kuhn, R. M., Karolchik, D., Armengol, L., Bird, C. P., de Bakker, P. I., Kern, A. D., Lopez-Bigas, N., Martin, J. D., Stranger, B. E., Woodroffe, A., Davydov, E., Dimas, A., Eyras, E., Hallgrímsdóttir, I. B., Huppert, J., Zody, M. C., Abecasis, G. R., Estivill, X., Bouffard, G. G., Guan, X., Hansen, N. F., Idol, J. R., Maduro, V. V., Maskeri, B., McDowell, J. C., Park, M., Thomas, P. J., Young, A. C., Blakesley, R. W., Muzny, D. M., Sodergren, E., Wheeler, D. A., Worley, K. C., Jiang, H., Weinstock, G. M., Gibbs, R. A., Graves, T., Fulton, R., Mardis, E. R., Wilson, R. K., Clamp, M., Cuff, J., Gnerre, S., Jaffe, D. B., Chang, J. L., Lindblad-Toh, K., Lander, E. S., Koriabine, M., Nefedov, M., Osoegawa, K., Yoshinaga, Y., Zhu, B., and de Jong, P. J. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816.
Brosius, J. (2005). Waste not, want not – transcript excess in multicellular eukaryotes. Trends Genet. 21, 287–288.
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., and Rinn, J. L. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927.
Cai, X., Hagedorn, C. H., and Cullen, B. R. (2004). Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10, 1957–1966.
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., Oyama, R., Ravasi, T., Lenhard, B., Wells, C., Kodzius, R., Shimokawa, K., Bajic, V. B., Brenner, S. E., Batalov, S., Forrest, A. R., Zavolan, M., Davis, M. J., Wilming, L. G., Aidinis, V., Allen, J. E., Ambesi-Impiombato, A., Apweiler, R., Aturaliya, R. N., Bailey, T. L., Bansal, M., Baxter, L., Beisel, K. W., Bersano, T., Bono, H., Chalk, A. M., Chiu, K. P., Choudhary, V., Christoffels, A., Clutterbuck, D. R., Crowe, M. L., Dalla, E., Dalrymple, B. P., De Bono, B., Della Gatta, G., Di Bernardo, D., Down, T., Engstrom, P., Fagiolini, M., Faulkner, G., Fletcher, C. F., Fukushima, T., Furuno, M., Futaki, S., Gariboldi, M., Georgii-Hemming, P., Gingeras, T. R., Gojobori, T., Green, R. E., Gustincich, S., Harbers, M., Hayashi, Y., Hensch, T. K., Hirokawa, N., Hill, D., Huminiecki, L., Iacono, M., Ikeo, K., Iwama, A., Ishikawa, T., Jakt, M., Kanapin, A., Katoh, M., Kawasawa, Y., Kelso, J., Kitamura, H., Kitano, H., Kollias, G., Krishnan, S. P., Kruger, A., Kummerfeld, S. K., Kurochkin, I. V., Lareau, L. F., Lazarevic, D., Lipovich, L., Liu, J., Liuni, S., Mcwilliam, S., Madan Babu, M., Madera, M., Marchionni, L., Matsuda, H., Matsuzawa, S., Miki, H., Mignone, F., Miyake, S., Morris, K., Mottagui-Tabar, S., Mulder, N., Nakano, N., Nakauchi, H., Ng, P., Nilsson, R., Nishiguchi, S., Nishikawa, S., Nori, F., Ohara, O., Okazaki, Y., Orlando, V., Pang, K. C., Pavan, W. J., Pavesi, G., Pesole, G., Petrovsky, N., Piazza, S., Reed, J., Reid, J. F., Ring, B. Z., Ringwald, M., Rost, B., Ruan, Y., Salzberg, S. L., Sandelin, A., Schneider, C., Schönbach, C., Sekiguchi, K., Semple, C. A., Seno, S., Sessa, L., Sheng, Y., Shibata, Y., Shimada, H., Shimada, K., Silva, D., Sinclair, B., Sperling, S., Stupka, E., Sugiura, K., Sultana, R., Takenaka, Y., Taki, K., Tammoja, K., Tan, S. L., Tang, S., Taylor, M. S., Tegner, J., Teichmann, S. A., Ueda, H. R., van Nimwegen, E., Verardo, R., Wei, C. L., Yagi, K., Yamanishi, H., Zabarovsky, E., Zhu, S., Zimmer, A., Hide, W., Bult, C., Grimmond, S. M., Teasdale, R. D., Liu, E. T., Brusic, V., Quackenbush, J., Wahlestedt, C., Mattick, J. S., Hume, D. A., Kai, C., Sasaki, D., Tomaru, Y., Fukuda, S., Kanamori-Katayama, M., Suzuki, M., Aoki, J., Arakawa, T., Iida, J., Imamura, K., Itoh, M., Kato, T., Kawaji, H., Kawagashira, N., Kawashima, T., Kojima, M., Kondo, S., Konno, H., Nakano, K., Ninomiya, N., Nishio, T., Okada, M., Plessy, C., Shibata, K., Shiraki, T., Suzuki, S., Tagami, M., Waki, K., Watahiki, A., Okamura-Oho, Y., Suzuki, H., Kawai, J., Hayashizaki, Y., FANTOM Consortium, and RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group). (2005). The transcriptional landscape of the mammalian genome. Science 309, 1559–1563.
Carninci, P., Yasuda, J., and Hayashizaki, Y. (2008). Multifaceted mammalian transcriptome. Curr. Opin. Cell Biol. 20, 274–280.
Chen, F., Evans, A., Gaskell, E., Pham, J., and Tsai, M. C. (2011). Regulatory RNA: the new age. Mol. Cell 43, 851–852.
Cheng, J., Kapranov, P., Drenkow, J., Dike, S., Brubaker, S., Patel, S., Long, J., Stern, D., Tammana, H., Helt, G., Sementchenko, V., Piccolboni, A., Bekiranov, S., Bailey, D. K., Ganesh, M., Ghosh, S., Bell, I., Gerhard, D. S., and Gingeras, T. R. (2005). Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308, 1149–1154.
Clark, M. B., Amaral, P. P., Schlesinger, F. J., Dinger, M. E., Taft, R. J., Rinn, J. L., Ponting, C. P., Stadler, P. F., Morris, K. V., Morillon, A., Rozowsky, J. S., Gerstein, M. B., Wahlestedt, C., Hayashizaki, Y., Carninci, P., Gingeras, T. R., and Mattick, J. S. (2011). The reality of pervasive transcription. PLoS Biol. 9, e1000625; discussion e1001102. doi:10.1371/journal.pbio.1000625
Cloonan, N., Forrest, A. R., Kolle, G., Gardiner, B. B., Faulkner, G. J., Brown, M. K., Taylor, D. F., Steptoe, A. L., Wani, S., Bethel, G., Robertson, A. J., Perkins, A. C., Bruce, S. J., Lee, C. C., Ranade, S. S., Peckham, H. E., Manning, J. M., Mckernan, K. J., and Grimmond, S. M. (2008). Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 5, 613–619.
Cloonan, N., and Grimmond, S. M. (2008). Transcriptome content and dynamics at single-nucleotide resolution. Genome Biol. 9, 234.
Cooper, D. N., Krawczak, M., and Antonarakis, S. E. (1995). “The nature and mechanisms of human gene mutation,” in The Metabolic and Molecular Bases of Inherited Disease, 7th Edn, eds C. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (New York: McGraw-Hill), 259–291.
Denoeud, F., Kapranov, P., Ucla, C., Frankish, A., Castelo, R., Drenkow, J., Lagarde, J., Alioto, T., Manzano, C., Chrast, J., Dike, S., Wyss, C., Henrichsen, C. N., Holroyd, N., Dickson, M. C., Taylor, R., Hance, Z., Foissac, S., Myers, R. M., Rogers, J., Hubbard, T., Harrow, J., Guigo, R., Gingeras, T. R., Antonarakis, S. E., and Reymond, A. (2007). Prominent use of distal 5’ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res. 17, 746–759.
Djebali, S., Kapranov, P., Foissac, S., Lagarde, J., Reymond, A., Ucla, C., Wyss, C., Drenkow, J., Dumais, E., Murray, R. R., Lin, C., Szeto, D., Denoeud, F., Calvo, M., Frankish, A., Harrow, J., Makrythanasis, P., Vidal, M., Salehi-Ashtiani, K., Antonarakis, S. E., Gingeras, T. R., and Guigo, R. (2008). Efficient targeted transcript discovery via array-based normalization of RACE libraries. Nat. Methods 5, 629–635.
Faulkner, G. J., Kimura, Y., Daub, C. O., Wani, S., Plessy, C., Irvine, K. M., Schroder, K., Cloonan, N., Steptoe, A. L., Lassmann, T., Waki, K., Hornig, N., Arakawa, T., Takahashi, H., Kawai, J., Forrest, A. R., Suzuki, H., Hayashizaki, Y., Hume, D. A., Orlando, V., Grimmond, S. M., and Carninci, P. (2009). The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 41, 563–571.
Gibb, E. A., Brown, C. J., and Lam, W. L. (2011). The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 10, 38.
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., Tsai, M. C., Hung, T., Argani, P., Rinn, J. L., Wang, Y., Brzoska, P., Kong, B., Li, R., West, R. B., Van De Vijver, M. J., Sukumar, S., and Chang, H. Y. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076.
Harrow, J., Denoeud, F., Frankish, A., Reymond, A., Chen, C. K., Chrast, J., Lagarde, J., Gilbert, J. G., Storey, R., Swarbreck, D., Rossier, C., Ucla, C., Hubbard, T., Antonarakis, S. E., and Guigo, R. (2006). GENCODE: producing a reference annotation for ENCODE. Genome Biol. 7(Suppl. 1), S41–S49.
Hastie, N. D., and Bishop, J. O. (1976). The expression of three abundance classes of messenger RNA in mouse tissues. Cell 9, 761–774.
Ingolia, N. T., Lareau, L. F., and Weissman, J. S. (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802.
Johnson, J. M., Edwards, S., Shoemaker, D., and Schadt, E. E. (2005). Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 21, 93–102.
Kapranov, P., Cawley, S. E., Drenkow, J., Bekiranov, S., Strausberg, R. L., Fodor, S. P., and Gingeras, T. R. (2002). Large-scale transcriptional activity in chromosomes 21 and 22. Science 296, 916–919.
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., Stadler, P. F., Hertel, J., Hackermuller, J., Hofacker, I. L., Bell, I., Cheung, E., Drenkow, J., Dumais, E., Patel, S., Helt, G., Ganesh, M., Ghosh, S., Piccolboni, A., Sementchenko, V., Tammana, H., and Gingeras, T. R. (2007a). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488.
Kapranov, P., Willingham, A. T., and Gingeras, T. R. (2007b). Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 8, 413–423.
Kapranov, P., Drenkow, J., Cheng, J., Long, J., Helt, G., Dike, S., and Gingeras, T. R. (2005). Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 15, 987–997.
Kapranov, P., Sementchenko, V. I., and Gingeras, T. R. (2003). Beyond expression profiling: next generation uses of high density oligonucleotide arrays. Brief. Funct. Genomic. Proteomic. 2, 47–56.
Kapranov, P., St Laurent, G., Raz, T., Ozsolak, F., Reynolds, C. P., Sorensen, P. H., Reaman, G., Milos, P., Arceci, R. J., Thompson, J. F., and Triche, T. J. (2010). The majority of total nuclear-encoded non-ribosomal RNA in a human cell is “dark matter” un-annotated RNA. BMC Biol. 8, 149. doi:10.1186/1741-7007-8-149
Katinakis, P. K., Slater, A., and Burdon, R. H. (1980). Non-polyadenylated mRNAs from eukaryotes. FEBS Lett. 116, 1–7.
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., and Haussler, D. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006.
Khaitan, D., Dinger, M. E., Mazar, J., Crawford, J., Smith, M. A., Mattick, J. S., and Perera, R. J. (2011). The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 71, 3852–3862.
Kodzius, R., Kojima, M., Nishiyori, H., Nakamura, M., Fukuda, S., Tagami, M., Sasaki, D., Imamura, K., Kai, C., Harbers, M., Hayashizaki, Y., and Carninci, P. (2006). CAGE: cap analysis of gene expression. Nat. Methods 3, 211–222.
Kondo, T., Plaza, S., Zanet, J., Benrabah, E., Valenti, P., Hashimoto, Y., Kobayashi, S., Payre, F., and Kageyama, Y. (2010). Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science 329, 336–339.
Mamanova, L., Andrews, R. M., James, K. D., Sheridan, E. M., Ellis, P. D., Langford, C. F., Ost, T. W., Collins, J. E., and Turner, D. J. (2010). FRT-seq: amplification-free, strand-specific transcriptome sequencing. Nat. Methods 7, 130–132.
Mattick, J. S. (2003). Challenging the dogma: the hidden layer of non-protein-coding RNAs in complex organisms. Bioessays 25, 930–939.
Mattick, J. S. (2009). The genetic signatures of noncoding RNAs. PLoS Genet. 5, e1000459. doi:10.1371/journal.pgen.1000459
Mattick, J. S. (2011). The central role of RNA in human development and cognition. FEBS Lett. 585, 1600–1616.
Mercer, T. R., Dinger, M. E., Bracken, C. P., Kolle, G., Szubert, J. M., Korbie, D. J., Askarian-Amiri, M. E., Gardiner, B. B., Goodall, G. J., Grimmond, S. M., and Mattick, J. S. (2010). Regulated post-transcriptional RNA cleavage diversifies the eukaryotic transcriptome. Genome Res. 20, 1639–1650.
Mercer, T. R., Gerhardt, D. J., Dinger, M. E., Crawford, J., Trapnell, C., Jeddeloh, J. A., Mattick, J. S., and Rinn, J. L. (2011a). Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 30, 99–104.
Mercer, T. R., Wilhelm, D., Dinger, M. E., Solda, G., Korbie, D. J., Glazov, E. A., Truong, V., Schwenke, M., Simons, C., Matthaei, K. I., Saint, R., Koopman, P., and Mattick, J. S. (2011b). Expression of distinct RNAs from 3’ untranslated regions. Nucleic Acids Res. 39, 2393–2403.
Morin, R., Bainbridge, M., Fejes, A., Hirst, M., Krzywinski, M., Pugh, T., Mcdonald, H., Varhol, R., Jones, S., and Marra, M. (2008). Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. BioTechniques 45, 81–94.
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 5, 621–628.
Otsuka, Y., Kedersha, N. L., and Schoenberg, D. R. (2009). Identification of a cytoplasmic complex that adds a cap onto 5’-monophosphate RNA. Mol. Cell. Biol. 29, 2155–2167.
Rassoulzadegan, M., Grandjean, V., Gounon, P., Vincent, S., Gillot, I., and Cuzin, F. (2006). RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature 441, 469–474.
Raz, T., Kapranov, P., Lipson, D., Letovsky, S., Milos, P. M., and Thompson, J. F. (2011). Protocol dependence of sequencing-based gene expression measurements. PLoS ONE 6, e19287. doi:10.1371/journal.pone.0019287
Rinn, J. L., Euskirchen, G., Bertone, P., Martone, R., Luscombe, N. M., Hartman, S., Harrison, P. M., Nelson, F. K., Miller, P., Gerstein, M., Weissman, S., and Snyder, M. (2003). The transcriptional activity of human chromosome 22. Genes Dev. 17, 529–540.
Salditt-Georgieff, M., Harpold, M. M., Wilson, M. C., and Darnell, J. E. Jr. (1981). Large heterogeneous nuclear ribonucleic acid has three times as many 5’ caps as polyadenylic acid segments, and most caps do not enter polyribosomes. Mol. Cell. Biol. 1, 179–187.
Sam, L. T., Lipson, D., Raz, T., Cao, X., Thompson, J., Milos, P. M., Robinson, D., Chinnaiyan, A. M., Kumar-Sinha, C., and Maher, C. A. (2011). A comparison of single molecule and amplification based sequencing of cancer transcriptomes. PLoS ONE 6, e17305. doi:10.1371/journal.pone.0017305
St Laurent, G. III, and Wahlestedt, C. (2007). Noncoding RNAs: couplers of analog and digital information in nervous system function? Trends Neurosci. 30, 612–621.
Struhl, K. (2007). Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 14, 103–105.
Ting, D. T., Lipson, D., Paul, S., Brannigan, B. W., Akhavanfard, S., Coffman, E. J., Contino, G., Deshpande, V., Iafrate, A. J., Letovsky, S., Rivera, M. N., Bardeesy, N., Maheswaran, S., and Haber, D. A. (2011). Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 331, 593–596.
Ulitsky, I., Shkumatava, A., Jan, C. H., Sive, H., and Bartel, D. P. (2011). Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550.
van Bakel, H., and Hughes, T. R. (2009). Establishing legitimacy and function in the new transcriptome. Brief. Funct. Genomic. Proteomic. 8, 424–436.
van Bakel, H., Nislow, C., Blencowe, B. J., and Hughes, T. R. (2010). Most “dark matter” transcripts are associated with known genes. PLoS Biol. 8, e1000371. doi:10.1371/journal.pbio.1000371
van Bakel, H., Nislow, C., Blencowe, B. J., and Hughes, T. R. (2011). Response to “the reality of pervasive transcription.” PLoS Biol. 9, e1001102. doi:10.1371/journal.pbio.1001102
van Ness, J., Maxwell, I. H., and Hahn, W. E. (1979). Complex population of nonpolyadenylated messenger RNA in mouse brain. Cell 18, 1341–1349.
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914.
Keywords: dark matter RNA, genomics, transcriptome, non-coding, intronic RNA, vlinc, linc, gene
Citation: Kapranov P and St. Laurent G (2012) Dark matter RNA: existence, function, and controversy. Front. Gene. 3:60. doi: 10.3389/fgene.2012.00060
Received: 27 October 2011; Accepted: 30 March 2012;
Published online: 23 April 2012.
Edited by:
Cheng-Han Huang, New York Blood Center, USAReviewed by:
Cheng-Han Huang, New York Blood Center, USAVladimir Benes, European Molecular Biology Laboratory, Germany
Eduardo M. Reis, Universidade de Sao Paulo, Brazil
Copyright: © 2012 Kapranov and St. Laurent. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Philipp Kapranov and Georges St. Laurent, St. Laurent Institute, One Kendall Square Suite 200LL, Cambridge, MA 02139, USA. e-mail:cGhpbGlwcGswOEBnbWFpbC5jb20=;Z2Vvcmdlc3Q5OEB5YWhvby5jb20=