 S. Aslibekyan1
S. Aslibekyan1 M. K. Jensen2 H. Campos2 C. D. Linkletter1 E. B. Loucks1 J. M. Ordovas3
M. K. Jensen2 H. Campos2 C. D. Linkletter1 E. B. Loucks1 J. M. Ordovas3 R. Deka4 E. B. Rimm2 and A. Baylin1,5*
R. Deka4 E. B. Rimm2 and A. Baylin1,5*- 1 Department of Community Health, Brown University, Providence, RI, USA
- 2 Department of Nutrition, Harvard School of Public Health, Boston, MA, USA
- 3 Jean Mayer USDA Human Nutrition Research Center on Aging, Tufts University, Boston, MA, USA
- 4 Department of Environmental Health, University of Cincinnati College of Medicine, Cincinnati, OH, USA
- 5 Department of Epidemiology, University of Michigan School of Public Health, Ann Arbor, MI, USA
Genetic variation in fatty acid desaturases (FADS) has previously been linked to long-chain polyunsaturated fatty acids (PUFAs) in adipose tissue and cardiovascular risk. The goal of our study was to test associations between six common FADS polymorphisms (rs174556, rs3834458, rs174570, rs2524299, rs174589, rs174627), intermediate cardiovascular risk factors, and non-fatal myocardial infarction (MI) in a matched population based case–control study of Costa Rican adults (n = 1756). Generalized linear models and multiple conditional logistic regression models were used to assess the associations of interest. Analyses involving intermediate cardiovascular risk factors and MI were also conducted in two replication cohorts, The Nurses’ Health Study (n = 1200) and The Health Professionals Follow-Up Study (n = 1295). In the Costa Rica Study, genetic variation in the FADS cluster was associated with a robust linear decrease in adipose gamma-linolenic, arachidonic, and eicosapentaenoic fatty acids, and significant or borderline significant increases in the eicosadienoic, eicosatrienoic, and dihomo-gamma-linolenic fatty acids. However, the associations with adipose tissue fatty acids did not translate into changes in inflammatory biomarkers, blood lipids, or the risk of MI in the discovery or the replication cohorts. In conclusion, fatty acid desaturase polymorphisms impact long-chain PUFA biosynthesis, but their overall effect on cardiovascular health likely involves multiple pathways and merits further investigation.
Introduction
Long-chain omega-3 polyunsaturated fatty acids (PUFAs), obtained either through dietary intake or synthesized endogenously from alpha-linolenic acid (ALA), have long been known to be protective against heart disease, diabetes, and other chronic outcomes (Harris, 2010). Previous studies showed that in populations with low intake of marine fatty acids, dietary intake of ALA also confers cardioprotective benefits, either by itself or through conversion to long-chain PUFAs (Baylin et al., 2003).
The delta5- and delta6-desaturases, encoded by FADS1 and FADS2 genes respectively, play a critical role in the conversion pathway and could have implications for chronic disease risk, especially in the context of a long-chain PUFA deficient diet (Lattka et al., 2009; Figure 1). Several studies, including a recent meta-analysis of genome-wide association (GWA) scans, linked polymorphisms in the FADS gene cluster to PUFA concentrations in serum phospholipids and erythrocyte cell membranes in several populations, including Caucasians, East Asians, and African Americans (Malerba et al., 2008; Martinelli et al., 2008; Rzehak et al., 2009; Tanaka et al., 2009; Kwak et al., 2011; Lemaitre et al., 2011; Mathias et al., 2011). Additionally, a number of studies have reported significant associations between FADS genotypes and the risk of coronary artery disease (Martinelli et al., 2008; Kwak et al., 2011). A previous analysis of our data from the Costa Rican population examined the effect of a common single nucleotide polymorphism (SNP) in the FADS2 promoter region on the risk of non-fatal myocardial infarction (MI; Baylin et al., 2007). Although a consistent decrease in adipose and plasma PUFA concentrations was observed with an increase in number of copies of the minor allele, the association with MI was not significant (Baylin et al., 2007).
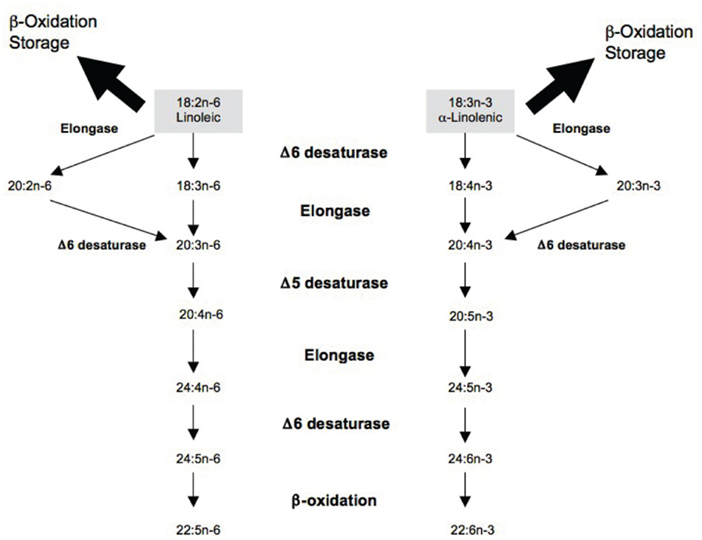
Figure 1. Biosynthetic pathway of polyunsaturated fatty acids.
The mechanisms by which genetic variation in desaturases impacts cardiovascular health are unclear and likely to involve multiple pathways. A recent study has linked the number of risk alleles in the FADS cluster to high-sensitivity C-reactive protein (hsCRP) concentrations and prevalence of coronary artery disease, suggesting inflammation as a mechanism of interest (Martinelli et al., 2008). Another potential pathway supported by extensive evidence from genome-wide studies links desaturase polymorphisms to changes in serum cholesterol and triglycerides (Aulchenko et al., 2009; Kathiresan et al., 2009; Sabatti et al., 2009; Lettre et al., 2011).
While most published studies have considered FADS gene variants and cardiovascular outcomes individually, the complex nature of biological mechanisms underlying the association between desaturases and cardiovascular risk warrants a more comprehensive approach. The objective of this study is to evaluate the association between genetic variation in the FADS cluster, adipose tissue fatty acids, serum lipids, and inflammatory biomarkers, and the risk of MI in a Costa Rican population. By considering intermediate outcomes as well as the endpoint of MI, this study aims to provide novel mechanistic insights into the relation between desaturases, dietary fatty acids, and cardiovascular disease.
Materials and Methods
Study Population
The population of the Costa Rica Study, described in detail in prior publications, included 4,548 unrelated Hispanics who resided in the Central Valley of Costa Rica between 1994 and 2004 (Baylin and Campos, 2004; Kabagambe et al., 2007). Cases of first non-fatal acute MI were ascertained by two independent cardiologists in the participating hospitals and deemed eligible if they met the World Health Organization criteria, survived hospitalization, were under 75 years of age on the day of their first MI, and able to answer the questionnaire (Tunstallpedoe et al., 1994). Eligible cases were matched by 5-year age group, sex, and area of residence to population controls. Participation was 98% for cases and 88% for controls. The study population is appropriate for investigating genetic markers of disease due to its origin in a small number of founders and low rates of migration (Baylin et al., 2007).
The original sample size was 2,274 cases and 2,274 controls. Participants missing information on outcomes, exposure, or covariates were excluded from the analysis. Excluded participants did not significantly differ from those who remained in the analyses on key demographic, genetic, and dietary variables. Data were missing due to lost laboratory samples as well as non-differential response patterns with respect to the exposure, outcome, or covariates. All participants provided written informed consent. The study was approved by the Human Subjects Committee of the Harvard School of Public Health and the University of Costa Rica.
The replication study populations consisted of the Nurses Health Study (NHS) and Health Professionals Follow-Up Study (HPFS) participants. The NHS enrolled 121,701 female nurses aged 30–55 who returned a mailed questionnaire in 1976 regarding lifestyle and medical history. The HPFS enrolled 51,529 males aged 40–75 who returned a similar questionnaire in 1986. Participants of both cohorts have received follow-up questionnaires biennially to record newly diagnosed illnesses. Detailed descriptions of the study cohorts have been published previously (Colditz et al., 1997).
In both replication cohorts, nested case–control studies were designed using incident coronary heart disease (CHD), with non-fatal MI and fatal CHD as the outcome. Diagnosis of MI was confirmed on the basis of the criteria of the World Health Organization (Tunstallpedoe et al., 1994). Fatal CHD was confirmed by an examination of hospital or autopsy records. Among participants who provided blood samples and who were free of diagnosed cardiovascular disease or cancer at blood draw, 474 women and 454 men with incident CHD between blood draw and June of 2004 were identified. Using risk-set sampling (Prentice and Breslow, 1978), controls were selected randomly and matched in a 1:2 ratio on age, smoking, and month of blood return, yielding final sample sizes of 1200 for NHS and 1295 for HPFS.
Biochemical Measurements
In the Costa Rica Study, exposures were ascertained via adipose tissue biomarkers for the following fatty acids: 18:3n−3 (ALA), 18:2n−6 (LA), 18:3n−6 (gamma-linolenic acid, GLA), 20:3n−3 (eicosatrienoic acid, ETA), 20:2n−6 (eicosadienoic acid, EDA), 20:3n−6 (dihomo-gamma-linolenic acid, DGA), 20:4n−6 (arachidonic acid, AA), 20:5n−3 (eicosapentaenoic acid, EPA), and 22:6n−3 (docosahexaenoic acid, DHA). Advantages of using adipose tissue biomarkers to characterize long-term nutritional intake include slow turnover, absence of recall bias, and lack of response to conditions of acute disease (Baylin et al., 2002). However, adipose tissue concentrations of AA are poorly correlated with dietary intake due to high metabolic regulation (Baylin et al., 2002). Therefore, we used adipose tissue AA as a metabolic marker representing the endogenous component explained by genetic variation, adjusted for dietary intake as measured by the previously validated food frequency questionnaire (Kabagambe et al., 2001). Subcutaneous adipose biopsies, collected following an overnight fast, were performed using a modification of the Beynen and Katan method with a plastic syringe instead of a vacutainer (Beynen and Katan, 1985). Fatty acids from adipose tissue were quantified by gas–liquid chromatography (Baylin et al., 2002). Peak retention times and area percentages of total fatty acids were analyzed with the ChemStation A.08.03 software (Agilent Technologies; Truong et al., 2009). Average coefficients of variation for 12 blind duplicates were 3.6% for LA, 6.4% for ALA, 21.7% for GLA, 7.3% for EDA, 7.6% for DGA, 43.6% for ETA, 11.0% for AA, 20.3% for EPA, and 14.8% for DHA. Samples were stored at −80°C and run within a year of collection. Control samples were run from 2001 to 2003 and no evidence of analyte instability was found.
Biomarker analyses of inflammation and serum lipids were restricted to control subjects to preclude reverse causation. Blood samples were collected during the home visits; the tubes were centrifuged within 6 h at 2,500 rpm for 20 min to separate plasma. Plasma triglycerides, low density lipoprotein (LDL), high density lipoprotein (HDL), and total cholesterol were assayed with enzymatic reagents (Boehringer-Mannheim) and standardized according to the program specified by the Centers for Disease Control and the National Heart, Lung, and Blood Institute (Williams et al., 2007). To measure inflammation, the study used hsCRP and vascular cellular adhesion molecule-1 (VCAM-1), two plasma biomarkers that have been extensively validated and linked to cardiovascular risk in large-scale prospective studies (Ridker et al., 2000; Blankenberg et al., 2001). Concentrations of hsCRP were measured using immunoturbidimetry on Roche Modular P chemistry autoanalyzer (Hoffman La Roche, average coefficient of variation = 1.42%). The Quantikine Human sVCAM-1 assay (R&D Systems, kit lot # 258820) was used to measure sVCAM-1 (average coefficient of variation = 9.58%).
Between 1989 and 1990, a blood sample was requested from all active participants in NHS and collected from 32,826 women. Similarly, blood samples were requested between 1993 and 1995 and obtained from 18,225 HPFS participants. Plasma lipids and inflammatory markers were assessed using standard methods with reagents from Roche Diagnostics (Indianapolis, IN, USA) and Bayer Diagnostics (New York, NY, USA; Pai et al., 2002).
SNP Selection
Seven SNPs located on the FADS gene cluster were selected for analysis based on previously published evidence of their role in fatty acid metabolism, linkage disequilibrium patterns, and availability in the Costa Rican population: rs174556 (C/T), rs3834458 (T/deletion), rs174570 (C/T), rs2524299 (A/T), rs174589 (C/G), rs174611 (T/C), rs174627 (C/T; Baylin et al., 2002; Malerba et al., 2008; Tanaka et al., 2009).
Genotyping
A DNA stock sample of ∼200 mg was collected from all study participants and stored at −80°C in Costa Rica. This sample was extracted from frozen buffy coats using the Qiagen QIAamp DNA Blood Kit. Purity was determined by the ratio of absorbance at 260–280 (A260/A280) to be between 1.7 and 1.9 for all samples. For high-throughput genotyping 15 ng/ml “working solutions” were prepared and aliquoted into 96-well plates. Genotyping was performed at the University of Cincinnati using the SNPlex Genotyping System (Applied Biosystems). Fragmented genomic DNA (50 ng) was dried into each well of a 384-well plate (∼1 ng DNA per genotype). After phosphorylation of oligonucleotide ligation assay (OLA) probes and universal linkers, allele-specific ligation, and enzymatic purification were performed. Polymerase chain reaction (PCR) utilized universal biotinylated primers, so amplicons could be captured on streptavidin-coated plates. Single-strand PCR products were hybridized with a universal set of fluorescently dye-labeled mobility modifiers, the ZipChute probes that have a unique sequence corresponding to each SNP. ZipChute probes were eluted and separated for detection by capillary electrophoresis on ABI PRISM 3130XL DNA Analyzer (Applied Biosystems). Data were collected, formatted, processed, and analyzed using the GeneMapper Analysis Software (Version 4.0), which assigned individual genotypes. Genotyping was attempted on 4,082 individuals (90% of the total study population); of those, call rates ranged from 74% (for rs174570) to 93% (for rs3834458).
Ancestry was estimated using a set of 39 informative markers selected from published lists of validated SNPs with allele frequencies from Amerindian, European, and West African samples (Ruiz-Narváez et al., 2010). Based on the set of selected markers, the expected variance of estimated individual ancestral proportions for any particular set of loci was calculated using a maximum likelihood approach with a high degree of precision (SE ≈0.15; Ruiz-Narváez et al., 2010).
For the NHS and the HPFS, genotyping was performed at Merck Research Laboratories (North Wales, PA, USA). Genotyping was done using the Affymetrix Genome-Wide Human 6.0 array and the Birdseed calling algorithm (Korn et al., 2008). Ninety-six percentage of the NHS samples and 98% of the HFPS samples were successfully genotyped. SNPs that were monomorphic, had a missing call rate ≥2%, a Hardy–Weinberg equilibrium p value < 1 × 10−4, or a minor allele frequency <0.02 were excluded, leaving a total of 724,881 in HPFS and 721,316 in NHS for analysis of called genotypes. Imputation of ∼2.5 million SNPs was performed using MACH software (Version 1.0.16) with HapMap CEU phased II data (Release 22) as the reference panel. Of SNPs used in the analyses, three (rs174556, rs147570, rs2524299) were on the Affymetrix chip, while two (rs174589 and rs174627) were imputed with r2 values > 0.85.
Statistical Analysis
Data were analyzed using the SAS software package (Version 9.2; SAS Institute Inc, Cary, NC, USA). To assess the significance of differences in general characteristics and potential confounders, we used paired t-tests for continuous variables, McNemar’s tests for categorical variables, and Fisher’s exact test for minor allele frequencies. The ALLELE procedure was used to test for deviations from Hardy–Weinberg equilibrium among controls. Of all SNPs, only rs174611 was found to be in violation of the Hardy–Weinberg equilibrium and removed from all subsequent analyses.
Linear regression models were fit among controls to evaluate the association between each FADS cluster SNP, adipose tissue long-chain PUFAs, and plasma hsCRP, VCAM-1, and serum lipids. Least square means and 95% confidence intervals were used to report the relation between the outcomes and FADS genetic variants. Normal probability plots were constructed, log-transformations were carried out for non-normally distributed variables (GLA, hsCRP, and triglycerides), and geometric means were reported. The intermediate risk factors models were adjusted for age, sex, and residence area, while the PUFA models were additionally adjusted for dietary intake of all nine fatty acids as assessed by the food frequency questionnaire. Adipose tissue PUFAs and intermediate risk factors were modeled as continuous variables. The relation between the SNPs and the dichotomous MI outcome was modeled using conditional logistic regression, adjusted for age, sex, and residence area. Additionally, models fit to the Costa Rica Study data were adjusted for ancestry due to evidence of population stratification (Ruiz-Narváez et al., 2010); models fit to the NHS and the HPFS data were not adjusted for ancestry as the cohorts were found to be genetically homogeneous. Least square means of adipose fatty acids, blood lipids, and inflammatory markers were estimated using PROC GLM, adjusted for potential confounders listed above. P values from single SNP analyses were adjusted for multiple comparisons using the false discovery rate controlling procedure (Benjamini and Hochberg, 1995).
Linear and conditional logistic regression analyses with individual SNPs as predictors and inflammation markers, blood lipids, and MI as outcomes as described above were replicated in the NHS and HPFS. Genotype information at the rs3834458 locus was not available in the replication cohorts. Because minor allele frequencies varied considerably between the discovery and replication cohorts, a meta-analysis was not attempted. All analyses were adjusted for sex and age.
Results
The general characteristics of the three populations are summarized by case/control status in Table 1. None of the selected SNPs differed significantly in minor allele frequency by disease status. Cases were more likely to report MI risk factors, specifically smoking and history of chronic disease. Additionally, cases in the Costa Rica Study had significantly lower adipose tissue concentrations of ALA and LA. Ancestral admixture proportions did not vary by case–control status in the discovery cohort and were estimated at 58% European, 38% Amerindian, and 4% West African.
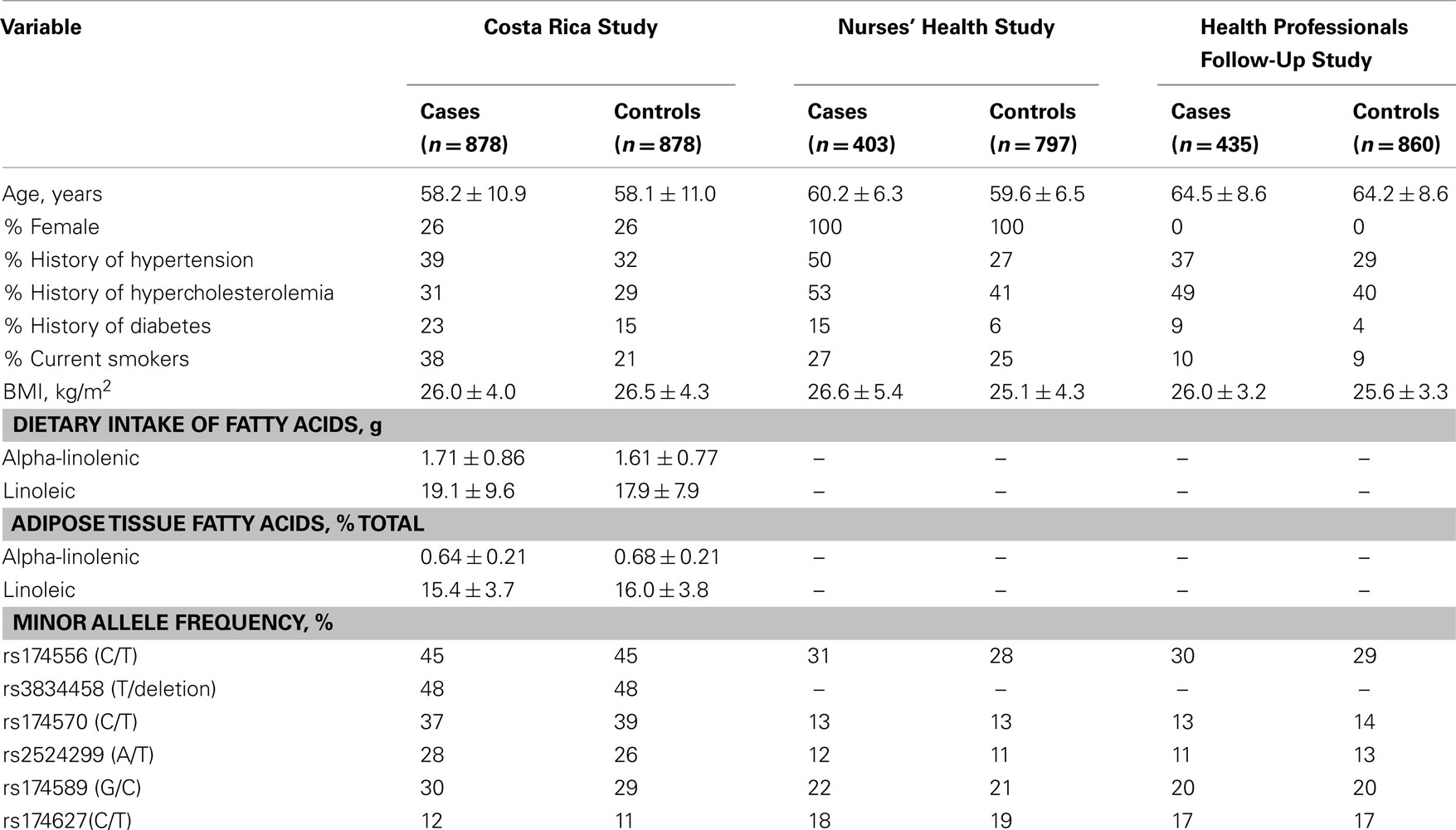
Table 1. General characteristics of the study populations.
As shown in Table 2, least square means of adipose tissue ALA and LA did not vary significantly by FADS genotype in the Costa Rica Study. Upon adjustment for multiple testing, variation at the rs174627 locus was not associated with any of the fatty acid outcomes. For all the remaining SNPs, the number of variant allele copies was significantly associated with a decrease in adipose AA. Additionally, variation at all loci except rs174589 and rs174627 was associated with decreased adipose GLA and EPA, while for most SNPs, adipose EDA, DGA, and ETA increased with the number of variant allele copies. The direction of association was consistent across SNPs.
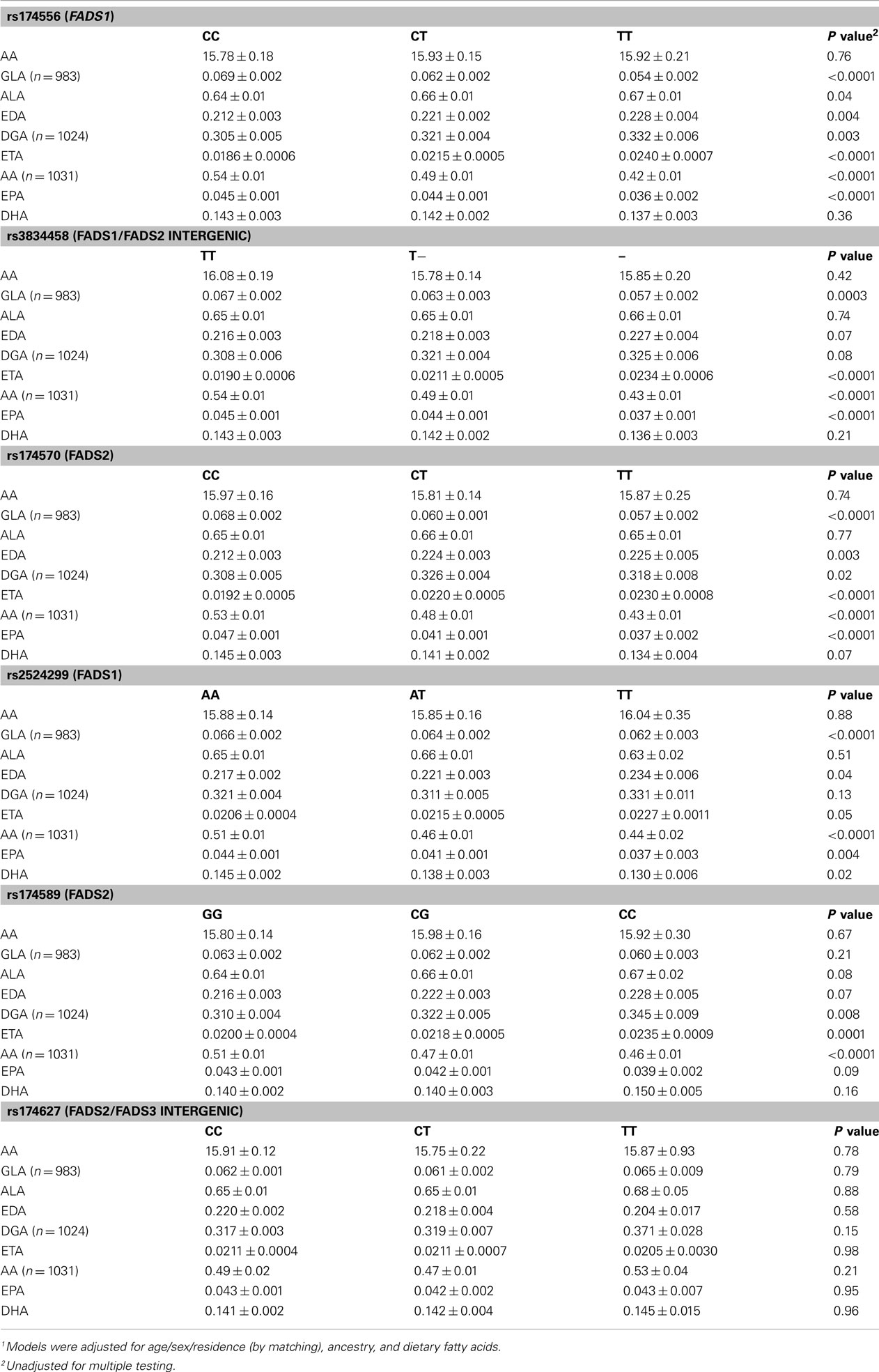
Table 2. Least square means1 (±SE) of selected adipose fatty acids (% of total adipose fatty acids) by genotype among controls in the Costa Rica Study (n = 1032 unless indicated otherwise).
Table 3 shows the least square means of blood lipids and inflammatory biomarkers by FADS genotype. After adjustment for multiple comparisons, no significant associations were observed between the FADS cluster SNPs and intermediate cardiovascular risk factors, with the sole exception of a statistically significant increase in triglycerides across the number of variant copies in rs174570 in the Costa Rica Study; however, the association was not replicated in the NHS or the HFPS. Variation in the FADS genes was not associated with the risk of MI in any of the three study populations (Table 4).
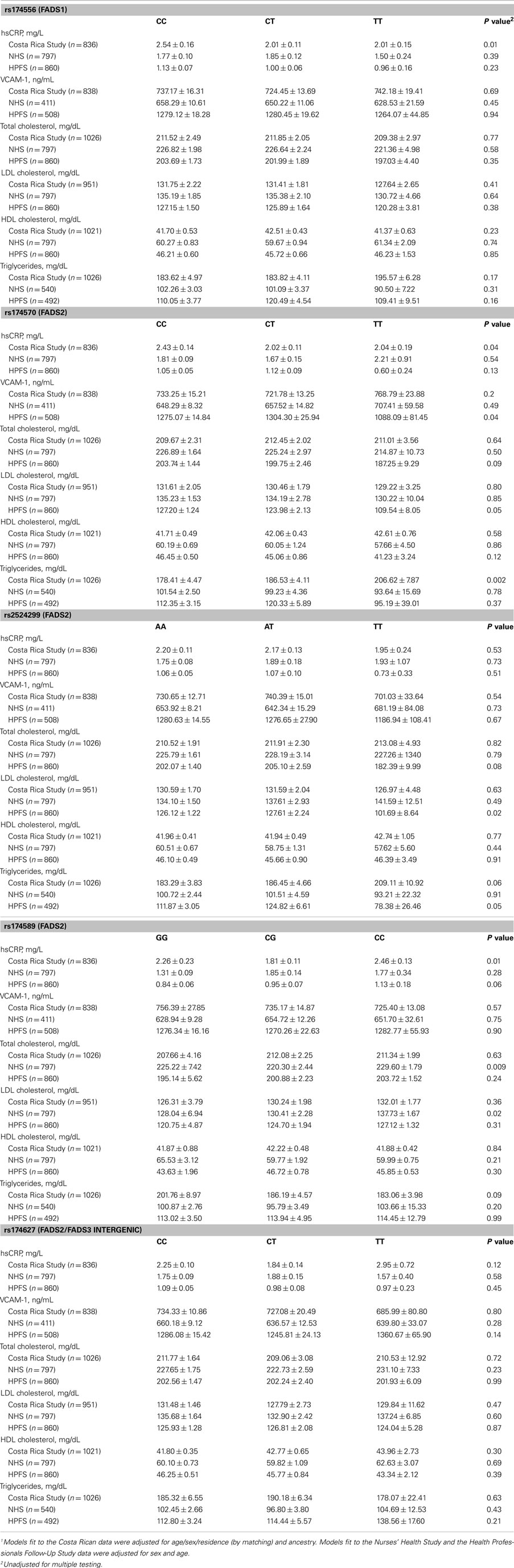
Table 3. Least square means1 (±SE) of blood lipids and inflammatory markers by FADS genotype among controls.
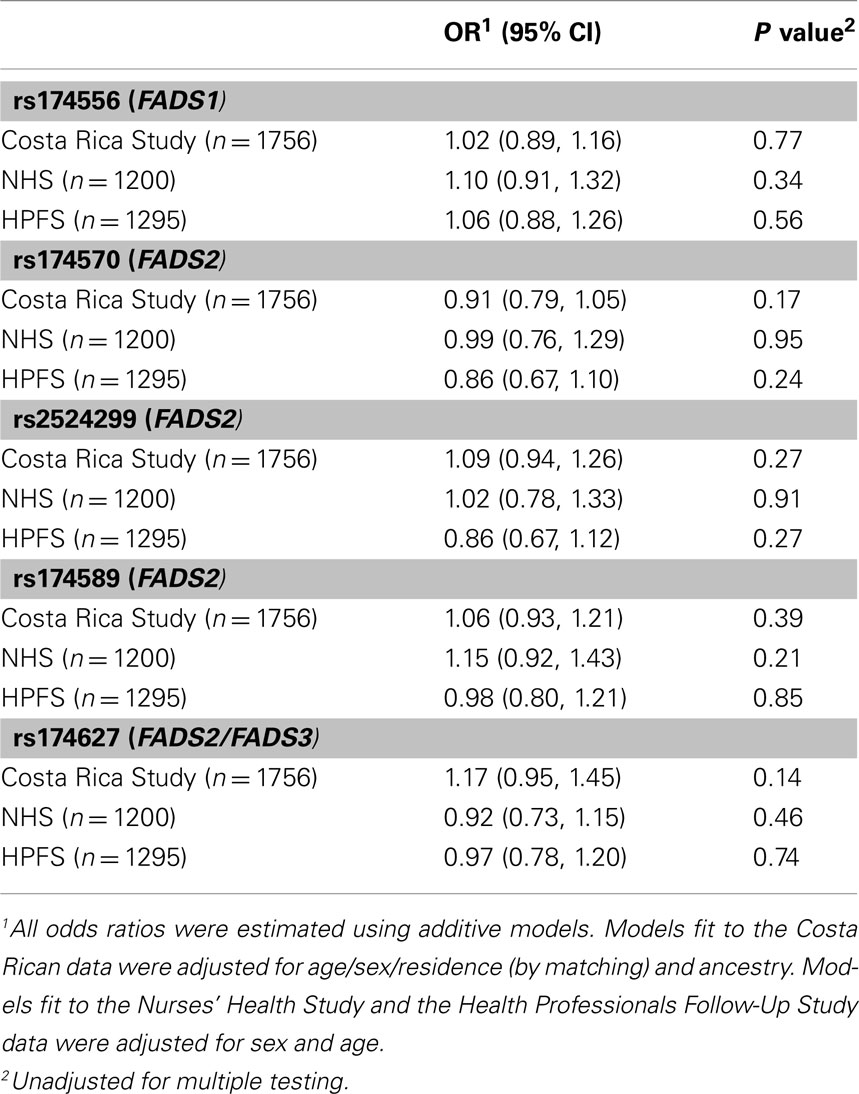
Table 4. The risk of non-fatal myocardial infarction associated with the number of copies of the variant allele.
Discussion
Our findings show that genetic variation in the FADS cluster is associated with adipose tissue fatty acids in the Costa Rica Study. In particular, we demonstrated associations of multiple desaturase SNPs with decreases in adipose AA, GLA, and EPA as well as with increases in adipose EDA, ETA, and DGA. Our results contribute to the body of evidence in support of robust associations between long-chain PUFAs and FADS cluster polymorphisms, including a recent meta-analysis of GWA studies from the CHARGE consortium (Lemaitre et al., 2011). Specifically, in concordance with our findings, the strongest signal in the FADS cluster reported by Tanaka et al. (2009, rs174537) was similarly associated with a decrease in long-chain PUFA (namely AA and EPA) concentrations.
The observed associations of desaturase polymorphisms with adipose fatty acids, however, do not translate into significant changes in serum lipids, inflammatory biomarkers, or the risk of MI in any of the three study populations. Given the evidence of manifold physiological effects of PUFAs, is likely that the overall effect of FADS polymorphisms on cardiovascular health is determined by a combination of mechanisms such as inflammation, hyperlipidemia, endothelial function, cardiac rhythm, thrombosis, and other pathways that were outside the scope of this investigation. Additionally, little is known about the putative cardiovascular effects of the fatty acids in the side branches of the n−3 and the n−6 pathways, i.e. ETA and EDA, which were significantly increased among carriers of the variant FADS genotypes in the Costa Rica Study.
The strengths of our study include its large size, high response rates, the representativeness of the sample of the Costa Rican population, and extensive information on genetic and dietary covariates that included biomarker measures. The observed associations between some of the SNPs and the adipose fatty acids in the Costa Rica Study were robust and remained statistically significant even after applying the more conservative Bonferroni correction. However, our findings should be interpreted in light of several important limitations. First, the observational nature of the Costa Rica Study, the NHS, and the HFPS precludes from establishing any causal relations between the genetic and dietary exposures and the outcomes. On a related note, the selected SNPs may merely be in linkage disequilibrium with the true causal variant and not have any physiologic effects of their own. Second, the fatty acid findings were not replicated in independent populations due to current unavailability of another large-scale cohort with adipose tissue data; future such studies may consider conducting similar analyses to establish the validity of our results. Third, the expression of fatty acid concentrations as percentages instead of absolute measurements, which is inherent to gas–liquid chromatography, implies that the fatty acid outcomes are not independent and thus the results need to be interpreted in the context of the entire pathway. Fourth, adipose tissue concentrations of fatty acids are dependent on a combination of genetic and environmental predictors including but not limited to genetic variation in desaturases. Specifically, we acknowledge that other relevant SNPs in the desaturase cluster, particularly those that were identified subsequently to our genotyping efforts, may mediate the relationship between PUFA intake and cardiovascular health. Finally, there were important differences in design between the discovery and the replication studies, including but not limited to different matching criteria for cases and controls.
In conclusion, we established associations between genetic variation in the desaturase cluster and adipose tissue fatty acids in the Costa Rica Study, while demonstrating null associations of the selected FADS variants with cardiovascular risk across three cohorts. Future studies investigating mechanisms of PUFA action will further our understanding of the biological interplay between genetic variation in desaturases and cardiovascular health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All authors read and approved the final manuscript, and report no conflicts of interest. This study was supported by grants HL081549, HL34594, CA87969, CA55075, and HL35464 from the National Institutes of Health.
References
Aulchenko, Y. S., Ripatti, S., Lindqvist, I., Boomsma, D., Heid, I. M., Pramstaller, P. P., Penninx, B. W., Janssens, A. C., Wilson, J. F., Spector, T., Martin, N. G., Pedersen, N. L., Kyvik, K. O., Kaprio, J., Hofman, A., Freimer, N. B., Jarvelin, M. R., Gyllensten, U., Campbell, H., Rudan, I., Johansson, A., Marroni, F., Hayward, C., Vitart, V., Jonasson, I., Pattaro, C., Wright, A., Hastie, N., Pichler, I., Hicks, A. A., Falchi, M., Willemsen, G., Hottenga, J. J., de Geus, E. J., Montgomery, G. W., Whitfield, J., Magnusson, P., Saharinen, J., Perola, M., Silander, K., Isaacs, A., Sijbrands, E. J., Uitterlinden, A. G., Witteman, J. C., Oostra, B. A., Elliott, P., Ruokonen, A., Sabatti, C., Gieger, C., Meitinger, T., Kronenberg, F., Döring, A., Wichmann, H. E., Smit, J. H., McCarthy, M. I., van Duijn, C. M., and Peltonen L; ENGAGE, Consortium. (2009). Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 41, 47–55.
Baylin, A., and Campos, H. (2004). Arachidonic acid in adipose tissue is associated with nonfatal acute myocardial infarction in the Central Valley of Costa Rica. J. Nutr. 134, 3095–3099.
Baylin, A., Kabagambe, E. K., Ascherio, A., Spiegelman, D., and Campos, H. (2003). Adipose tissue α-linolenic acid and nonfatal acute myocardial infarction in Costa Rica. Circulation 107, 1586–1591.
Baylin, A., Kabagambe, E. K., Siles, X., and Campos, H. (2002). Adipose tissue biomarkers of fatty acid intake. Am. J. Clin. Nutr. 76, 750–757.
Baylin, A., Ruiz-Narvaez, E., Kraft, P., and Campos, H. (2007). alpha-linolenic acid, Delta(6)-desaturase gene polymorphism, and the risk of nonfatal myocardial infarction. Am. J. Clin. Nutr. 85, 554–560.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate – a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Beynen, A., and Katan, M. (1985). Rapid sampling and long-term storage of subcutaneous adipose-tissue biopsies for determination of fatty acid composition. Am. J. Clin. Nutr. 42, 317–322.
Blankenberg, S., Rupprecht, H. J., Bickel, C., Peetz, D., Hafner, G., Tiret, L., and Meyer, J. (2001). Circulating cell adhesion molecules and death in patients with coronary artery disease. Circulation 104, 1336–1342.
Colditz, G. A., Manson, J. E., and Hankinson, S. E. (1997). The Nurses’ Health Study: 20-year contribution o the understanding of health among women. J. Womens Health 6, 49–62.
Harris, W. (2010). Omega-6 and omega-3 fatty acids: partners in prevention. Curr. Opin. Clin. Nutr. 13, 125–129.
Kabagambe, E. K., Baylin, A., Allan, D. A., Siles, X., Spiegelman, D., and Campos, H. (2001). Application of the method of triads to evaluate the performance of food frequency questionnaires and biomarkers as indicators of long-term dietary intake. Am. J. Epidemiol. 154, 1126–1135.
Kabagambe, E. K., Baylin, A., and Campos, H. (2007). Nonfatal acute myocardial infarction in Costa Rica – modifiable risk factors, population-attributable risks, and adherence to dietary guidelines. Circulation 115, 1075–1081.
Kathiresan, S., Willer, C. J., Peloso, G. M., Demissie, S., Musunuru, K., Schadt, E. E., Kaplan, L., Bennett, D., Li, Y., Tanaka, T., Voight, B. F., Bonnycastle, L. L., Jackson, A. U., Crawford, G., Surti, A., Guiducci, C., Burtt, N. P., Parish, S., Clarke, R., Zelenika, D., Kubalanza, K. A., Morken, M. A., Scott, L. J., Stringham, H. M., Galan, P., Swift, A. J., Kuusisto, J., Bergman, R. N., Sundvall, J., Laakso, M., Ferrucci, L., Scheet, P., Sanna, S., Uda, M., Yang, Q., Lunetta, K. L., Dupuis, J., de Bakker, P. I., O’Donnell, C. J., Chambers, J. C., Kooner, J. S., Hercberg, S., Meneton, P., Lakatta, E. G., Scuteri, A., Schlessinger, D., Tuomilehto, J., Collins, F. S., Groop, L., Altshuler, D., Collins, R., Lathrop, G. M., Melander, O., Salomaa, V., Peltonen, L., Orho-Melander, M., Ordovas, J. M., Boehnke, M., Abecasis, G. R., Mohlke, K. L., and Cupples, L. A. (2009). Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 41, 56–65.
Korn, J. M., Kuruvilla, F. G., McCarroll, S. A., Wysoker, A., Nemesh, J., Cawley, S., Hubbell, E., Veitch, J., Collins, P. J., Darvishi, K., Lee, C., Nizzari, M. M., Gabriel, S. B., Purcell, S., Daly, M. J., and Altschuler, D. (2008). Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 40, 1253–1260.
Kwak, J. H., Paik, J. K., Kim, O. Y., Jang, Y. S., Lee, S. H., Ordovas, J. M., and Lee, J. H. (2011). FADS gene polymorphisms in Koreans: association with omega-6 polyunsaturated fatty acids in serum phospholipids, lipid peroxides, and coronary artery disease. Atherosclerosis 214, 94–100.
Lattka, E., Illig, T., Heinrich, J., and Koletzko, B. (2009). FADS gene cluster polymorphisms: important modulators of fatty acid levels and their impact on atopic diseases. J. Nutrigenet. Nutrigenomics 2, 119–128.
Lemaitre, R. N., Tanaka, T., Tang, W., Manichaikul, A., Foy, M., Kabagambe, E. K., Nettleton, J. A., King, I. B., Weng, L. C., Bhattacharya, S., Bandinelli, S., Bis, J. C., Rich, S. S., Jacobs, D. R., Cherubini, A., McKnight, B., Liang, S., Gu, X., Rice, K., Laurie, C. C., Lumley, T., Browning, B. L., Psaty, B. M., Chen, Y. D. I., Friedlander, Y., Djousse, L., Wu, J. H. Y., Siscovick, D. S., Uitterlinden, A. G., Arnett, D. K., Ferrucci, L., Fornage, M., Tsai, M. Y., Mozaffarian, D., and Steffen, L. M. (2011). Genetic loci associated with plasma phospholipid n-3 fatty acids: a meta-analysis of genome-wide association studies from the CHARGE consortium. PLoS Genet. 7, e1002193. doi: 10.1371/journal.pgen.1002193
Lettre, G., Palmer, C. D., Young, T., Ejebe, K. G., Allayee, H., Benjamin, E. J., Bennett, F., Bowden, D. W., Chakravarti, A., Dreisbach, A., Farlow, D. N., Folsom, A. R., Fornage, M., Forrester, T., Fox, E., Haiman, C. A., Hartiala, J., Harris, T. B., Hazen, S. L., Heckbert, S. R., Henderson, B. E., Hirschhorn, J. N., Keating, B. J., Kritchevsky, S. B., Larkin, E., Li, M., Rudock, M. E., McKenzie, C. A., Meigs, J. B., Meng, Y. A., Mosley, T. H., Newman, A. B., Newton-Cheh, C. H., Paltoo, D. N., Papanicolaou, G. J., Patterson, N., Post, W. S., Psaty, B. M., Qasim, A. N., Qu, L., Rader, D. J., Redline, S., Reilly, M. P., Reiner, A. P., Rich, S. S., Rotter, J. I., Liu, Y., Shrader, P., Siscovick, D. S., Tang, W. H., Taylor, H. A., Tracy, R. P., Vasan, R. S., Waters, K. M., Wilks, R., Wilson, J. G., Fabsitz, R. R., Gabriel, S. B., Kathiresan, S., and Boerwinkle, E. (2011). Genome-Wide Association Study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet. 7, e1001300. doi: 10.1371/journal.pgen.1001300
Malerba, G., Schaeffer, L., Xumerle, L., Klopp, N., Trabetti, E., Biscuola, M., Cavallari, U., Galavotti, R., Martinelli, N., Guarini, P., Girelli, D., Olivieri, O., Corrocher, R., Heinrich, J., Pignatti, P. F., and Illig, T. (2008). SNPs of the FADS gene cluster are associated with polyunsaturated fatty acids in a cohort of patients with cardiovascular disease. Lipids 43, 289–299.
Martinelli, N., Girelli, D., Malerba, G., Guarini, P., Illig, T., Trabetti, E., Sandri, M., Friso, S., Pizzolo, F., Schaeffer, L., Heinrich, J., Pignatti, P. F., Corrocher, R., and Olivieri, O. (2008). FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am. J. Clin. Nutr. 88, 941–949.
Mathias, R. A., Sergeant, S., Ruczinski, I., Torgerson, D. G., Hugenschmidt, C. E., Kubala, M., Vaidya, D., Suktitipat, B., Ziegler, J. T., Ivester, P., Case, D., Yanek, L. R., Freedman, B. I., Rudock, M. E., Barnes, K. C., Langefeld, C. D., Becker, L. C., Bowden, D. W., Becker, D. M., and Chilton, F. H. (2011). The impact of FADS genetic variants on omega-6 polyunsaturated fatty acids in African Americans. BMC Genet. 12, 50. doi: 10.1186/1471-2156-12-50
Pai, J. K., Curhan, G. C., Cannuscio, C. C., Rifai, N., Ridker, P. M., and Rimm, E. B. (2002). Stability of novel plasma markers associated with cardiovascular disease: processing within 36 hours of specimen collection. Clin. Chem. 48, 1781–1784.
Prentice, R. L., and Breslow, N. E. (1978). Retrospective studies and failure time models. Biometrika 65, 153–158.
Ridker, P. M., Hennekens, C. H., Buring, J. E., and Rifai, N. (2000) C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 342, 836–843.
Ruiz-Narváez, E., Bare, L., Arellano, A., Catanese, J., and Campos, H. (2010). West African and Amerindian ancestry and risk of myocardial infarction and metabolic syndrome in the Central Valley population of Costa Rica. Hum. Genet. 127, 629–638.
Rzehak, P., Heinrich, J., Klopp, N., Schaeffer, L., Hoff, S., Wolfram, G., Illig, T., and Linseisen, J. (2009). Evidence for an association between genetic variants of the fatty acid desaturase 1 fatty acid desaturase 2 (FADS1 FADS2) gene cluster and the fatty acid composition of erythrocyte membranes. Br. J. Nutr. 101, 20–26.
Sabatti, C., Service, S. K., Hartikainen, A.-L., Pouta, A., Ripatti, S., Brodsky, J., Jones, C. G., Zaitlen, N. A., Varilo, T., Kaakinen, M., Sovio, U., Ruokonen, A., Laitinen, J., Jakkula, E., Coin, L., Hoggart, C., Collins, A., Turunen, H., Gabriel, S., Elliot, P., McCarthy, M. I., Daly, M. J., Järvelin, M. R., Freimer, N. B., and Peltonen, L. (2009). Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 41, 35–46.
Tanaka, T., Shen, J., Abecasis, G. R., Kisialiou, A., Ordovas, J. M., Guralnik, J. M., Singleton, A., Bandinelli, S., Cherubini, A., Arnett, D., Tsai, M. Y., and Ferrucci, L. (2009). Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI study. PLoS Genet. 5, e1000338. doi: 10.1371/journal.pgen.1000338
Truong, H., DiBello, J. R., Ruiz-Narvaez, E., Kraft, P., Campos, H., and Baylin, A. (2009). Does genetic variation in the Δ6-desaturase promoter modify the association between α-linolenic acid and the prevalence of metabolic syndrome? Am. J. Clin. Nutr. 89, 920–925.
Tunstallpedoe, H., Kuulasmaa, K., Amouyel, P., Arveiler, D., Rajakangas, A. M., and Pajak, A. (1994). Myocardial infarction and coronary deaths in the World Health Organization MONICA Project – registration procedures, event rates, and case fatality rates in 38 populations from 21 countries in 4 continents. Circulation 90, 583–612.
Keywords: desaturases, polyunsaturated fatty acids, inflammation, lipids, myocardial infarction
Citation: Aslibekyan S, Jensen MK, Campos H, Linkletter CD, Loucks EB, Ordovas JM, Deka R, Rimm EB and Baylin A (2012) Fatty acid desaturase gene variants, cardiovascular risk factors, and myocardial infarction in the Costa Rica Study. Front. Gene. 3:72. doi: 10.3389/fgene.2012.00072
Received: 08 March 2012; Accepted: 13 April 2012;
Published online: 03 May 2012.
Edited by:
Jill Barnholtz-Sloan, Case Western Reserve University School of Medicine, USAReviewed by:
Cheryl L. Thompson, Case Western Reserve University, USAMichael Scheurer, Baylor College of Medicine, USA
Copyright: © 2012 Aslibekyan, Jensen, Campos, Linkletter, Loucks, Ordovas, Deka, Rimm and Baylin. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: A. Baylin, Department of Epidemiology, University of Michigan School of Public Health, 1420 Washington Heights, Ann Arbor, MI 48109-2029, USA. e-mail:YWJheWxpbkB1bWljaC5lZHU=