


- 1 Division of Cardiology, Department of Medicine, University of Colorado Denver, Aurora, CO, USA
- 2 Department of Mathematical and Statistical Sciences, University of Colorado Denver, Denver, CO, USA
- 3 Department of Human Genetics, University of Pittsburgh, Pittsburgh, PA, USA
Endothelial lipase (LIPG) and zinc finger protein 202 (ZNF202) are two pivotal genes in high density lipoprotein (HDL metabolism). We sought to determine their genetic contribution to variation in HDL-cholesterol levels by comprehensive resequencing of both genes in 235 individuals with high or low HDL-C levels. The selected subjects were 141 Whites (High HDL Group: n = 68, Low HDL Group: n = 73, ) and 94 Hispanics (High HDL Group: n = 46, Low HDL Group: n = 48, ). We identified a total of 185 and 122 sequence variants in LIPG and ZNF202, respectively. We found only two missense variants in LIPG (T111I and N396S) and two in ZNF202 (A154V and K259E). In both genes, there were several variants unique to either the low or high HDL group. For LIPG, the proportion of unique variants differed between the high and low HDL groups in both Whites (p = 0.022) and Hispanics (p = 0.017), but for ZNF202 this difference was observed only in Hispanics (p = 0.021). We also identified a common haplotype in ZNF202 among Whites that was significantly associated with the high HDL group (p = 0.013). These findings provide insights into the genetics of LIPG and ZNF202, and suggest that sequence variants occurring with high frequency in non-exonic regions may play a prominent role in modulating HDL-C levels in the general population.
Introduction
There is an inverse relationship between the levels of high density lipoprotein cholesterol (HDL-C) and the risk of coronary heart disease (CHD; Emerging Risk Factors et al., 2009). HDL protects against the development of atherosclerotic cardiovascular disease, and thus, low HDL-C is a major CHD risk factor. About one-third of early familial CHD is attributed to low HDL-C, which is defined as below 40 mg/dl (1.0344 mmol/l) for both in men and women (ATP III, 2002). The prevalence of low HDL-C in the U.S. population was 44.6 million (16.7% of the population) in 2005 (Rosamond et al., 2008). Low HDL-C is predictive of major cardiovascular events in statin-treated patients even in the presence of low density lipoprotein cholesterol (LDL-C) levels below 70 mg/dl (Barter et al., 2007). Recent data suggest that, in addition to absolute plasma levels, the biological functions of HDL are important in assessing the role of HDL in CHD and major cardiovascular events (Vaisar et al., 2007; Besler et al., 2011). Understanding the genetic basis of the low HDL-C phenotype is, therefore, important for public health initiatives and policies.
Several studies, including twin studies, suggest that approximately half of the variation in plasma HDL-C levels is under genetic control (Prenger et al., 1992; Kronenberg et al., 2002; Lusis et al., 2004; Wang and Paigen, 2005; Qasim and Rader, 2006; Goode et al., 2007). Mutations in several genes related to lipid metabolism are associated with altered HDL-C levels, including apolipoprotein (apo) AI (Yamakawa-Kobayashi et al., 1999), apoE (Srinivasan et al., 1999), apoB (Peacock et al., 1994), lecithin:cholesterol acyltransferase (Kuivenhoven et al., 1997), lipoprotein lipase (LPL; Fisher et al., 1997), ATP-binding cassette transporter 1 (Brooks-Wilson et al., 1999), and scavenger receptor BI (Acton et al., 1999). However, in aggregate, these known mutations account for only a small proportion of the genetic contribution to HDL-C variation. The recent meta genome-wide association studies (GWAS) identified common variants in 38 loci associated with HDL-C levels, which included the aforementioned genes as well as several additional genes with as yet unknown functions (Teslovich et al., 2010). Despite the fact that this meta analysis had enormous statistical power, with a sample size of 99,900 individuals, it explained only 12% of the total HDL variation (or ∼25–30% of the genetic variance). It was suggested that additional rare or low frequency variants in these loci might explain the remaining unknown genetic contribution to the variation in HDL-C levels. Therefore, the next logical step in deciphering the genetic basis of HDL-C levels is to perform deep sequencing of these genes to identify all functional common and rare variants.
In this study, we have resequenced two important candidate genes, endothelial lipase (gene: LIPG, protein: EL), and zinc finger protein 202 (gene: ZNF202, protein: ZNF202), in selected individuals with extreme (high or low) HDL-C levels in an effort to fully explore their genetic roles in relation to HDL-C variation.
The human LIPG gene (Entrez Gene ID 9388, RefSeq ID NM_006033) is located on chromosome 18q21.1 and spans 34,945 bp with coordinates of 45,342,425 – 45,373,062. The gene consists of 10 exons with an average length of about 192 bp except for the partially coding exon 10, which is about 2.2 kb. The total transcript length of LIPGis 3,927, of which 1,755 bp is part of the open reading frame, and 2,172 bp covers the 3′ untranslated region (3′ UTR). Human EL is synthesized as a peptide of 500 amino acid residues, including a signal peptide of 20 residues. The mature protein is 480 residues with a molecular weight of 55 kDa prior to glycosylation. EL is expressed by endothelial cells in several tissues, including vessels, liver, lung, thyroid, kidney, testis, ovary, and placenta (Lindegaard et al., 2004). EL is a new member of the plasma lipase family, which also includes LPL and hepatic lipase (HL), that significantly modulates plasma HDL-C Levels (reviewed in Badellino and Rader, 2004; Das, 2005). EL is the only member of the lipase family that is synthesized by endothelial cells, and its expression is up-regulated by inflammatory cytokines, suggesting a role in endothelial homeostasis (Hirata et al., 1999; Jaye et al., 1999). Several genetic association studies have examined the role of LIPG variants in HDL metabolism or in CHD (deLemos et al., 2002; Halverstadt et al., 2003; Ma et al., 2003; Paradis et al., 2003; Yamakawa-Kobayashi et al., 2003; Mank-Seymour et al., 2004; Hutter et al., 2006; Shimizu et al., 2007; Tang et al., 2008; Brown et al., 2009; Edmondson et al., 2009; Jensen et al., 2009; Smith et al., 2009), but with conflicting results. On the other hand, several GWAS studies identified LIPG as having a significant association with HDL-C levels (Kathiresan et al., 2008; Keebler et al., 2009; Gupta et al., 2010; Ma et al., 2010; Teslovich et al., 2010).
The ZNF202 gene (Entrez Gene ID 7753, RefSeq ID NM_003455) is located on chromosome 11q23.3 and spans 36,874 bp with coordinates of 27,148,140 – 27,185,014. The ZNF202 gene encodes a 648aa protein with a molecular weight of 74.7 kDa. However, ZNF202 expression is complex, and its transcription produces 16 different mRNAs which are differentially expressed in different tissues (see AceView, Thierry-Mieg and Thierry-Mieg, 2006)1. ZNF202 is a master transcriptional repressor that affects, specifically, the expression of a number of genes involved in lipid metabolism (Wagner et al., 2000; Porsch-Özcürümez et al., 2001; Langmann et al., 2003; Schmitz et al., 2004). A linkage study of more than 100 large Utah pedigrees with early CHD found evidence of the familial low HDL-C trait linked to chromosomal region 11q23 where the ZNF202 locus is located. This region is distinct from the neighboring APOAI/CIII/AIV gene cluster (Kort et al., 2000). In contrast to the linkage and functional studies, a recent genetic association study did not find any association between the coding regions of ZNF202 variants and HDL-C levels (Stene et al., 2006a); however, the same group found a significant association between variants of this gene and the risk of atherosclerosis and ischemic heart disease (Stene et al., 2006b, 2008). None of the GWAS studies of blood lipids (Kathiresan et al., 2008; Keebler et al., 2009; Gupta et al., 2010; Ma et al., 2010; Teslovich et al., 2010) found ZNF202 to be associated with HDL-C levels suggesting that it most likely has a modest quantitative trait locus effect.
Materials and Methods
Study Design
Population
The San Luis Valley Diabetes Study (SLVDS), which was initiated in the mid-1980s, was a case-control study of Type 2 diabetes in a bi-ethnic population of non-Hispanic Whites (Whites) and Hispanics in southern Colorado. The design and objectives of the population study and methods of measuring various biometrics have been described in detail (Hamman et al., 1989). The SLVDS is ideal for association studies and for determining the role of genetic factors in the development of complex disorders, such as diabetes and cardiovascular disease. The cardiovascular risk factors of these two populations have previously been assessed (Burchfiel et al., 1990). The suitability of the SLVDS population arises from its stability based on historical demographic records, similar environmental conditions, and different genetic backgrounds with little genetic admixture between the two sub-populations (Bonilla et al., 2004). The entire SLVDS population samples (n = 1791) consisted of 659 diabetic cases and 1132 normoglycemic controls. The SI reference range for preprandial normal blood glucose level is 90–110 mg/dl (5–6 mmol/l). The control sample, which was used in this sequencing study, included 674 Whites and 458 Hispanics. The age range of study participants was 20–74 years. The quantitative traits related to lipoprotein metabolism of these control subjects have been summarized and compared between the two populations (Razzaghi et al., 2000). IRB approval for this study was obtained.
Selected subjects for resequencing
For the comprehensive DNA resequencing of the LIPG and ZNF202 genes, 235 individuals (141 Whites and 94 Hispanics) falling in the upper or lower tenth percentile ranks of the HDL-C distribution were selected from the SLVDS normoglycemic subjects: 121 subjects in the “Low HDL Group” () and 114 in the “High HDL Group” ( Figure 1A). The population normoglycemic samples (n = 1132) were first divided based on ethnicity and gender prior to ranking based on HDL-C level.
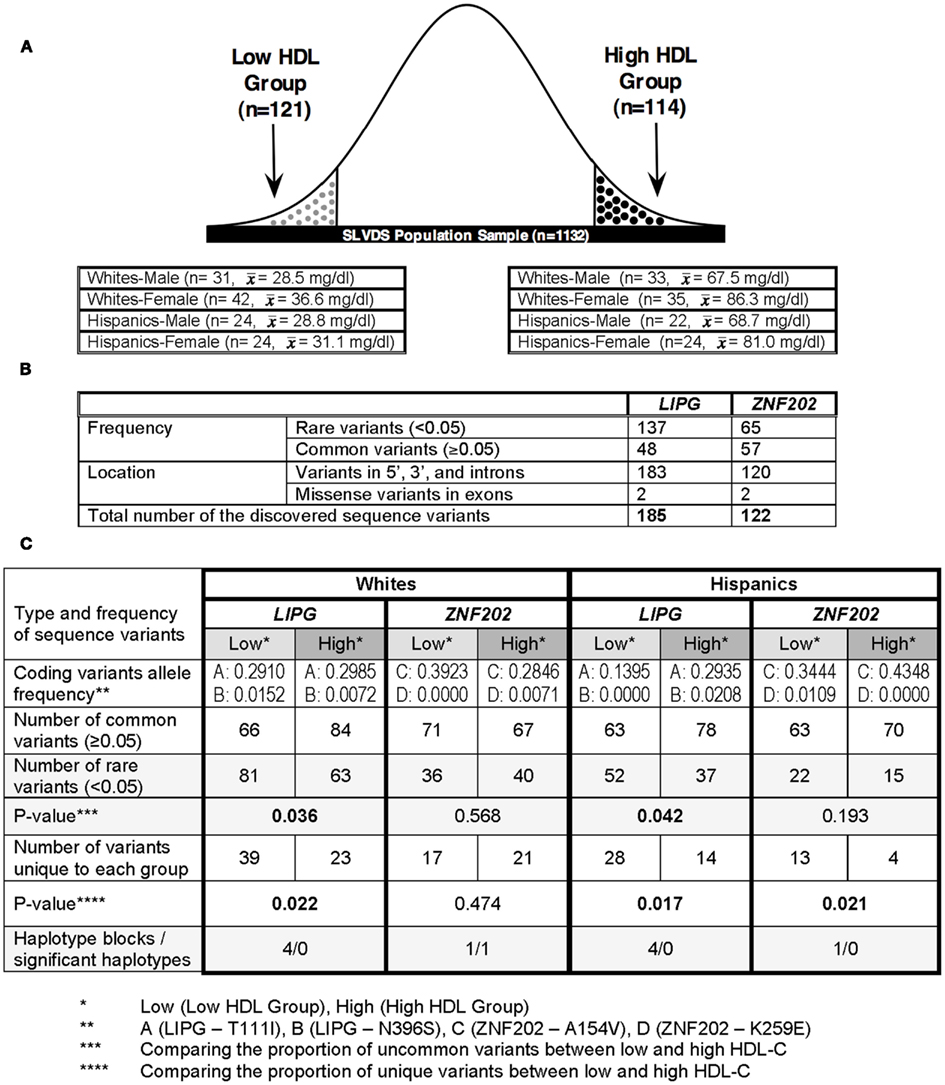
Figure 1. Study design and summary sequencing results. (A) Distribution and selection of SLVDS subjects based on their HDL-C levels. (B) Classification of the identified sequence variants in LIPG and ZNF202 loci. (C) Assignment of sequence variants to Low or High HDL Group.
DNA samples
After the original study, genomic DNA was extracted from blood at the University of Pittsburgh in the 1980s. Due to limited material for many of the samples, all selected DNA samples for resequencing were amplified using Whole Genomic Amplification (WGA), using Illustra GenomiPhi HY DNA Amplification Kit (GE Healthcare). Following genomic amplification, the quality of DNA was assessed by PCR and the amount was quantified using the Quant-iT PicoGreen DNA Assay Kit (Molecular Probes). All tested PCR were of adequate quality. DNA (10.0 μg) from each of the 235 samples was seeded in its appropriate coordinates on 96-well plates. The correct sequential seeding was reconfirmed by matching the order of the DNA tubes with the coordinates on the plates. DNA samples were shipped to the Seattle SNP PGA Center at the University of Washington for resequencing.
Resequencing
The entire length of the LIPG and ZNF202 genes, including 2,000 bp 5′ of exon 1, all exons and introns, and 2,000 bp 3′ of the last exon, was sequentially amplified using PCR primers tailed with universal M13 forward and reverse primer sequences. Contrary to the previously proposed 6-exon (Monaco et al., 1998) or 10-exon (Wagner et al., 2000) ZNF202 gene structure, a recent experimental-data-based capping profile for ZNF202 supports a 9-exon gene structure2. In this structure, the first three exons and part of exon 9 make up the non-coding exons of the gene. We used the latter gene structure model for this resequencing study. 5′-M13 tailed-gene-specific PCR primers were designed to cover the target region with amplicon sizes ranging from 500 to 750 bp and with a minimum of 100 bp overlap between adjacent amplicons, where applicable, and resulted in double-stranded coverage of all targeted regions. Overlapping amplicons were used to validate gene-specific primer sequences in independent experiments and to rule out the possibility of allele-specific PCR amplifications. All primer sequences were compared to the whole genome assembly to verify uniqueness against pseudogenes and gene families. Following temperature gradient optimization of small-scale reactions to determine optimal thermal cycling conditions, production level PCR amplifications were performed in 96-well plates in a volume of 7 μl comprising 0.2 μl each of 7 μM forward and reverse primers, 2.8 μl DNA (5 ng/μl), and 0.4 μl Elongase Enzyme (Invitrogen) or iProof polymerase (Bio-Rad) per well. Following evaluation by 1% agarose gel electrophoresis, reactions were diluted fourfold to sixfold in ddH2O. Dilution of the products eliminated the need for any purification of the PCR products prior to sequencing. Sequencing reactions were performed in MJ Tetrad PTC 225 thermal cyclers in 384-well format using 5% BDT v3.1 sequencing chemistry (ABI). Reaction products were precipitated in ethanol with CleanSeq magnetic beads (Agencourt). Perkin Elmer Minitrak, Multiprobe, and Evolution P3 robots were used to automate liquid handling in the setup of PCR, sequencing reactions, and precipitation reactions. Reaction products were air dried and resuspended in 30 μl with ddH2O. Chromatograms were generated from sequence reaction on an Applied Biosystems ABI 3730XL capillary sequencer. Data flow was tracked using a custom-designed laboratory information management system (LIMS). Both genes were sequenced from all 235 selected subjects.
SNP discovery and analysis
All chromatograms were base-called using Phred, assembled into contigs using Phrap, and scanned for SNPs with PolyPhred, version 6.02 (Stephens et al., 2006) to identify polymorphic sites. Data quality was monitored and assessed at multiple production checkpoints using numerous methods. For example, each chromatogram was trimmed to remove low-quality sequence (Phred score <25), resulting in analyzed reads averaging > 450 bp with an average Phred quality of 40. Following assembly of all chromatograms onto an initial reference sequence, putative polymorphic sites were selectively reviewed by sequence analysts using Consed (Gordon et al., 1998). Individual polymorphic sites in regions with lower quality data, ambiguous base calls, or those identified using laboratory quality control tools were reviewed to eliminate potential false positive positions. Outlier genotypes were scrutinized by data analysts and removed from the dataset if ambiguous. This approach generates sequence-based SNP genotypes with >99.9% accuracy. Variants were deposited into a custom postgreSQL database, formatted, and submitted to dbSNP for assignment of ss and rs identification numbers, which are provided in LIPG-section 14 and ZNF202-section 14 in Supplementary Material S1 and S2. The sequence variants were also given a descriptive ID number consisting of the gene name, followed by a six-digit number indicating the location of the variant within the sequence. For example, LIPG-007438 is the LIPG variant at a position 7438 bp from the start of sequencing (2000 bp upstream of the first exon).
Measurements
Biometric measurements
Subjects that participated in the SLVDS study had their various biometric measurements taken in the 1980s (Hamman et al., 1989). For this study, we used the biometric dataset for the following parameters: age, race, gender, body mass index (BMI), total cholesterol (TC), triglyceride (TG), LDL-C, and HDL-C. The lipid determination was described in detail in the original paper (Hamman et al., 1989). Briefly, fasting total serum cholesterol was measured by the esterase-oxidase method; HDL-C was determined enzymatically following dextran sulfate magnesium precipitation, and serum triglycerides by the enzymatic method. The LDL-C value was calculated with the Friedewald equation when the triglyceride level was less than 400 mg/dl. The means of these quantitative traits were compared using ANOVA following natural log transformation of BMI, TG, and HDL-C to achieve a more normal distribution.
Statistical Analyses
Haplotype block construction and selection of tag SNPs
Haplotype blocks were constructed based on the algorithm by Gabriel et al. (2002) as implemented in Haploview (Barrett et al., 2005). In particular, 95% confidence intervals for the standardized linkage disequilibrium (LD) coefficient (D′) were constructed for SNPs with a minor allele frequency (MAF) of 0.05 or higher. If 95% of SNPs were in strong LD, as defined by having the confidence interval for |D′| with a lower bound of at least 0.70 and an upper bound of at least 0.98, then a block was declared. For logistic regression, tag SNPs were selected to be a minimal set of markers, including non-synonymous SNPs, such that all captured alleles were correlated at r2 > 0.80. A list of the tag SNPs for each gene is provided in the LIPG-section 11 and ZNF202-section 11 in Supplementary Material S1 and S2.
Genetic association tests for extreme HDL groups
Association testing was performed by block for each haplotype with a frequency ≥0.05 using a chi-squared statistic as implemented in Haploview to compare the haplotype frequency between the high and low HDL groups. To adjust for multiple testing over haplotypes in blocks, 100,000 permutations of the data were used to build an empirical distribution for the maximum observed chi-squared statistic. Likewise, for each SNP followed up for association testing, a chi-squared statistic was used to compare allele frequencies between the high and low HDL group with multiple testing adjustment based on 100,000 permutations of the data. Permuted data sets were created by randomly shuffling the group assignment for extreme HDL. This maintains the LD structure over the entire gene. SNP association tests were performed as a follow-up to determine if a single variant could explain the haplotype results.
As a follow-up to findings from the comparison of haplotype frequencies, logistic regression was performed based on the tag SNPs. The logistic regression was adjusted for gender, age, BMI, and smoking status and utilized the most common haplotype as a reference. A global test of association (Schaid et al., 2002) was conducted to determine whether there was an association between each variant and extreme HDL grouping. The global test uses a score statistic to compare the full model containing haplotype effects to that of the null model containing no association between haplotypes and extreme HDL. p-Values were based on 1000 permutations of the data. The logistic regression analyses were conducted in R software using the haplo.stats package3.
Analysis of unique and rare variants
A variant was classified as unique if allelic variation was observed in only one of the HDL groups. The proportion of unique variants was compared between the high and low HDL groups using a two-sample test of proportions. Associations between the location (exons, introns, 5′ or 3′ flanking regions) of the unique variants and extreme HDL groups were determined by Fishers Exact Test. These analyses were repeated for rare variants, where a rare variant was defined as having a MAF < 0.05.
Results
We resequenced the entire LIPG and ZNF202 genes – including 5′ (2 kb), all exons and introns, and 3′ (2 kb) in 235 subjects (141 Whites, 94 Hispanics), divided into the high and low HDL groups. Table 1 provides a summary of variables in the subjects analyzed. By selecting these 235 subjects from both ends of the HDL-C distribution from each gender, we sought to enrich our screening for genetic variations affecting HDL-C levels (Figure 1A). A summary of the resequencing data for both genes is provided in Figure 1B and in the high and low HDL groups in Figure 1C. The details of the discovered sequence variants are provided for LIPG and ZNF202 in Supplementary Material S1 and S2. In addition, all the sequence variants have been deposited to the NCBI SNP Database4.
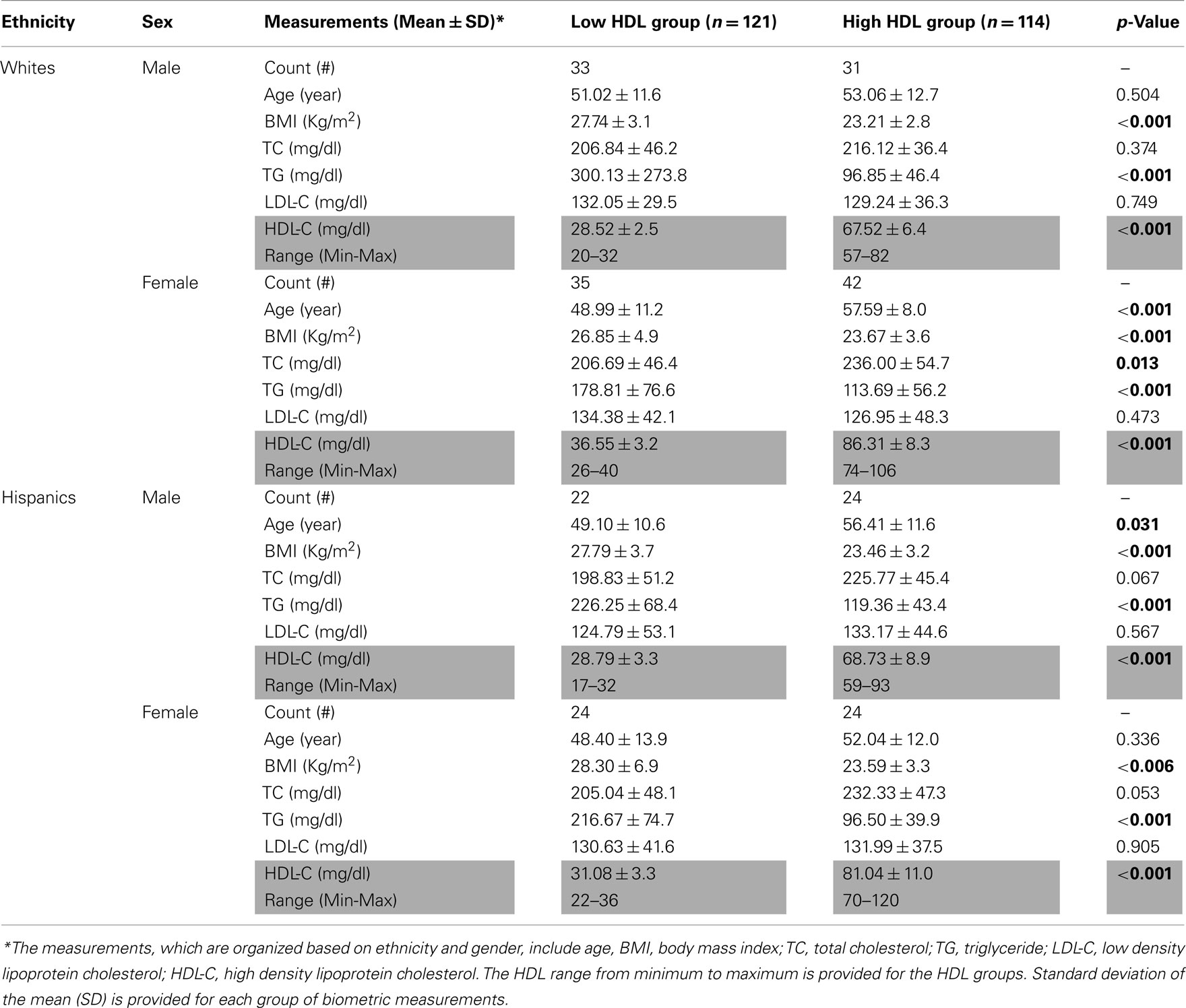
Table 1. Biometric measurements in selected subjects in high and low HDL groups.
Sequence Variation in the LIPG Gene
Of the 185 sequence variants observed in the LIPG gene, 137 were rare (<0.05 frequency) and 48 were common (≥0.05 frequency); 147 of them were present in Whites and 115 were present in Hispanics; 77 were found in both Whites and Hispanics, 70 were unique to Whites, and 38 were unique to Hispanics (section 8 in Supplementary Material S1). We identified two missense mutations in LIPG, one in exon 3 (LIPG-007438, T111I, MAF = 0.27) and one in exon 8 (LIPG-023529, N396S, MAF = 0.01; section 4 in Supplementary Material S1). There are at least 49 genomic repeat sequences5 within the human LIPG locus. Of the 185 sequence variants observed, 48 were located within these repeats (sections 5 and 16 in Supplementary Material S1).
Based on common variants, four haplotype blocks were identified in both Whites (Figure 2A) and Hispanics (Figure 2B). However, the structures of the haplotype blocks differed between the two ethnicities. No haplotype was associated with extreme HDL groups in Whites (p = 0.7125); but, one haplotype in block 1 (#3: TCAGCTGTTCTTC, frequency = 0.293 and 0.128 in the high and low HDL groups, respectively) was found to be associated with high HDL in Hispanics (p = 0.0073; Table 2; section 12 in Supplementary Material S1). However, after adjusting for the number of haplotypes tested, this haplotype became borderline non-significant (p = 0.0576). The individual SNPs contained in block 1 were also tested to see if their allele frequencies differed between the high and low HDL groups. Three SNPs showed significant differences, but after adjustment for multiple testing all became non-significant (Table 2). Of particular interest was the non-synonymous SNP T111I (LIPG-007438), whose frequency in the Hispanic high HDL group (MAF = 0.2935) was almost double that of the low HDL group (MAF = 0.1395; Figure 1C). Overall, no haplotype retained significance after adjustment for multiple testing in both population samples.
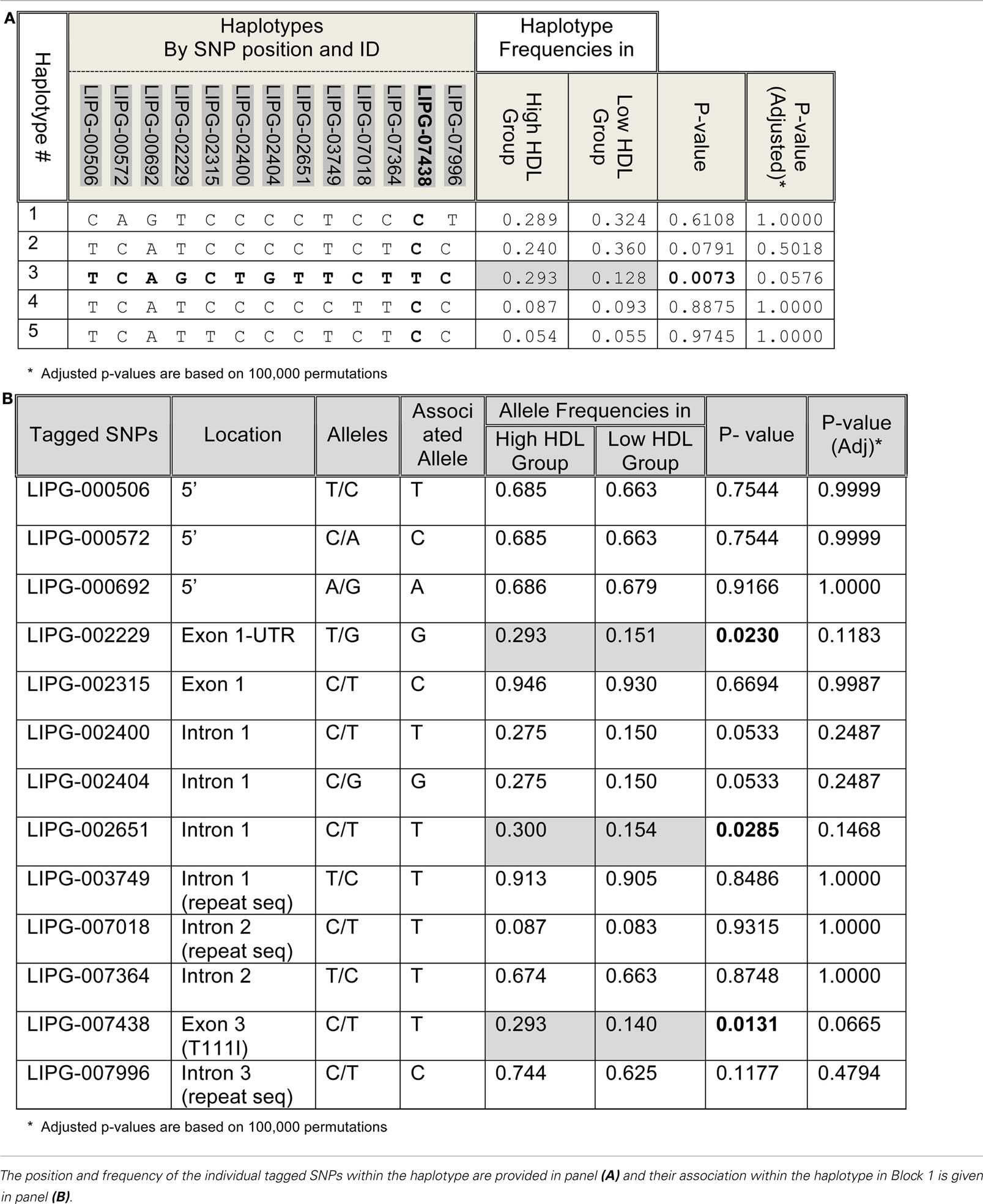
Table 2. Haplotype association of LIPG with high and low HDL groups in Hispanics.
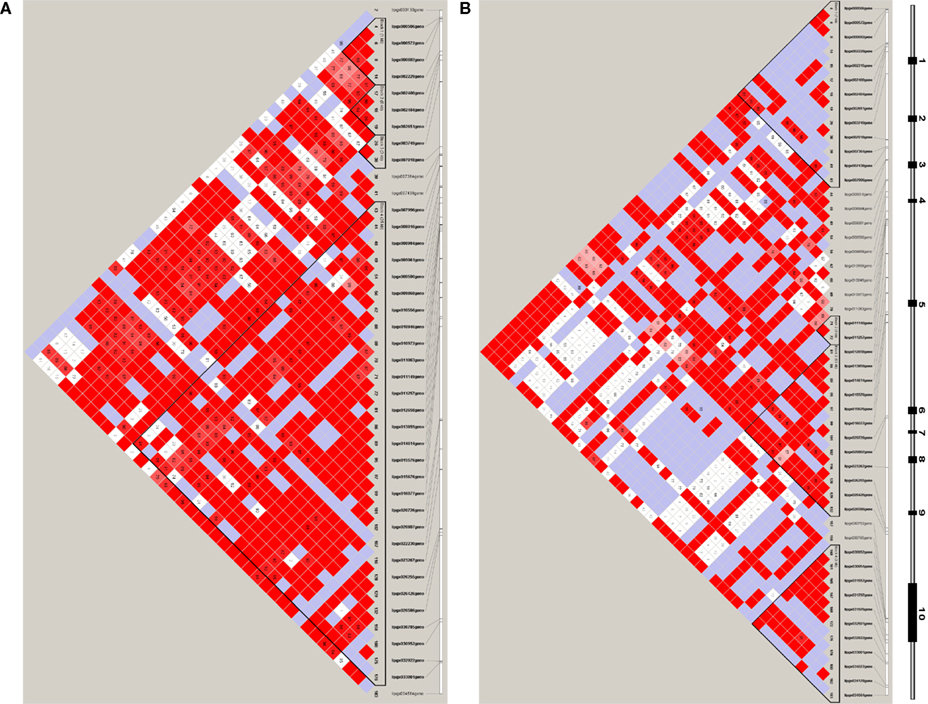
Figure 2. Haplotype block structure in LIPG. Haplotype block structure is indicated by triangular segments for LIPG in Whites (A) and in Hispanics (B); shading indicates strength of linkage disequilibrium as measured by D′, which is provided in the intersecting squares. D′ is not displayed for squares with D′ = 1. Squares with significant linkage disequilibrium (LOD ≥ 2) are shaded from light pink to red with D′ = 1 being bright red. White and blue regions lack significant linkage disequilibrium (LOD < 2) and represent D′ < 1 and D′ = 1, respectively. Spacing of SNPs along gene is given at the top of the plot. A diagram of the LIPG gene structure is provided over the plot where the first and last SNPs are on the left and right sides of the diagram, respectively. Under the SNP ID numbers are the index numbers, shown in bold, for the SNPs based on the map file. (A) LIPG, Whites, (B) LIPG, Hispanics. High resolution blocks are provided in a separate file (see Supplementary Material S3).
We observed an intriguing trend in the frequency of the rare and common variants between the high and low HDL groups in both Whites and Hispanics. The low HDL group had more rare variants but fewer common variants than the high HDL group (Figure 1C; section 10 in Supplementary Material S1). In Whites, there were 23 and 39 variants seen only in the high and low HDL groups, respectively. In Hispanics, there were 14 and 28 variants seen only in the high and low HDL groups, respectively. The proportion of rare variants differed significantly between the high and low HDL groups in both ethnicities (Whites: p-value = 0.022, Hispanics: p-value = 0.017). Interestingly, one rare variant (LIPG-022179, located in intron 6) was found in the low HDL group in both Whites and Hispanics (section 9 in Supplementary Material S1).
Next we examined whether the number of rare variants unique to the high and low HDL groups differed by location within the gene (5′, exons, introns, 3′; Table 3). We found a significant relationship between location and HDL groups in Hispanics (p = 0.0179), which was not observed in Whites (p = 0.3765). For both Whites and Hispanics, the proportion of rare variants that a person carried did not differ between the high and low HDL groups in exons, introns, 5′, 3′ regions (results not shown).
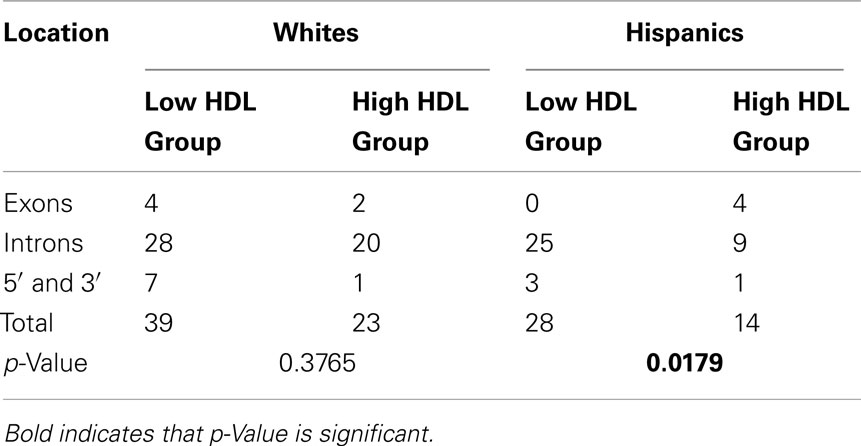
Table 3. Sequence variants unique to each HDL group based on their location within the LIPG gene.
Sequence Variation in the ZNF202 Gene
A total of 122 sequence variants were observed in the ZNF202 gene; 107 were found in Whites and 85 in Hispanics; 70 were shared by both ethnic groups; 37 were present only in Whites, and 15 only in Hispanics (section 8 in Supplementary Material S2); 65 of the sequence variants were rare, and 57 were common variants (section 7 in Supplementary Material S2). The longest intron in the ZNF202 gene is intron 3 (9,132 bp), which is where 28 of 65 rare variants (43%) and 35 of 57 common variants (61%) were found. Of 122 variants, only two were missense variants, one in exon 5 (ZNF202-013891, A154V, MAF = 0.36) and the other in exon 7 (ZNF202-015468, K259E, MAF = 0.02; section 4 in Supplementary Material S2). Unlike the observed trend in LIPG, we did not find a pattern in the frequency of rare or common variants in the ZNF202 gene between the high and low HDL groups in either Whites or Hispanics (section 10 in Supplementary Material S2). Interestingly, the overall ratio of rare to common variants was significantly higher for LIPG (137/48) than for ZNF202 (65/57; p = 0.000219). There are 19 genomic repeat sequences (see text footnote 5) within the human ZNF202 locus, and 19 of the 122 total sequence variants were located within these repeats (sections 5 and 16 in Supplementary Material S2).
There was one haplotype block in the ZNF202 gene in both ethnic groups (Figures 3A,B), and haplotypes in this block revealed significant associations between the two extreme groups of HDL (section 12 in Supplementary Material S2). In Whites there were five haplotypes with frequency greater than 0.05, and two of them (#1-W and #2-W) were associated with HDL-C differences, but only one of them (#2-W) retained significance after adjustment for testing of multiple haplotypes (p = 0.0133; Table 4). The frequency of this haplotype in the low HDL group (0.187) was almost half the frequency in the high HDL group (0.321). In Hispanics, four haplotypes had frequencies greater than 0.05, and one of them (#1-H) with the frequency of 0.317 showed significant association (p = 0.0246) with high HDL group, but after adjustment for multiple testing became non-significant (p = 0.0608; Table 4). Interestingly, this 50 bp haplotype in Hispanics (#1-H) differed in only seven bases from the haplotype in Whites (#1-W), but each had association with different HDL groups (Table 4; section 12 in Supplementary Material S2). Similar observation can be extended to the next set of the haplotypes comparison (#2-H vs. #2-W) as well.
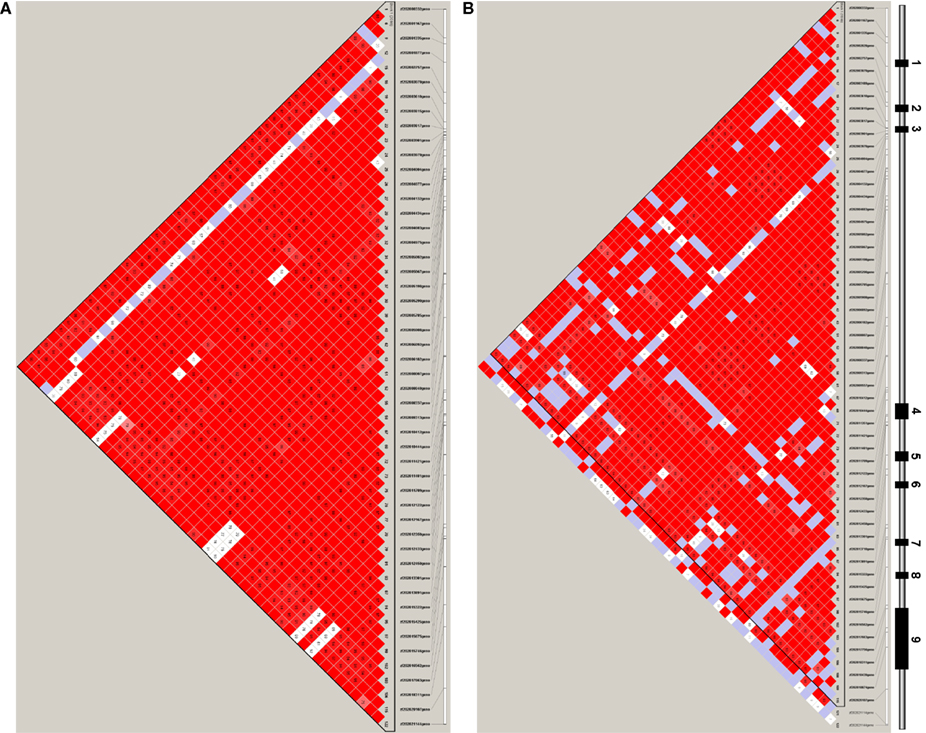
Figure 3. Haplotype block structure in ZNF202. Haplotype block structure is indicated by triangular segments for ZNF202 in Whites (A) and in Hispanics (B); shading indicates strength of linkage disequilibrium as measured by D′, which is provided in the intersecting squares. D′ is not displayed for squares with D′ = 1. Squares with significant linkage disequilibrium (LOD ≥ 2) are shaded from light pink to red with D′ = 1 being bright red. White and blue regions lack significant linkage disequilibrium (LOD < 2) and represent D′ < 1 and D′ = 1, respectively. Spacing of SNPs along gene is given at the top of the plot. A diagram of the ZNF202 gene structure is provided over the plot where the first and last SNPs are on the left and right sides of the diagram, respectively. Under the SNP ID numbers are the index numbers, shown in bold, for the SNPs based on the map file. (A) ZNF202, Whites, (B) ZNF202, Hispanics. High resolution blocks are provided in a separate file (Supplementary Material S3).
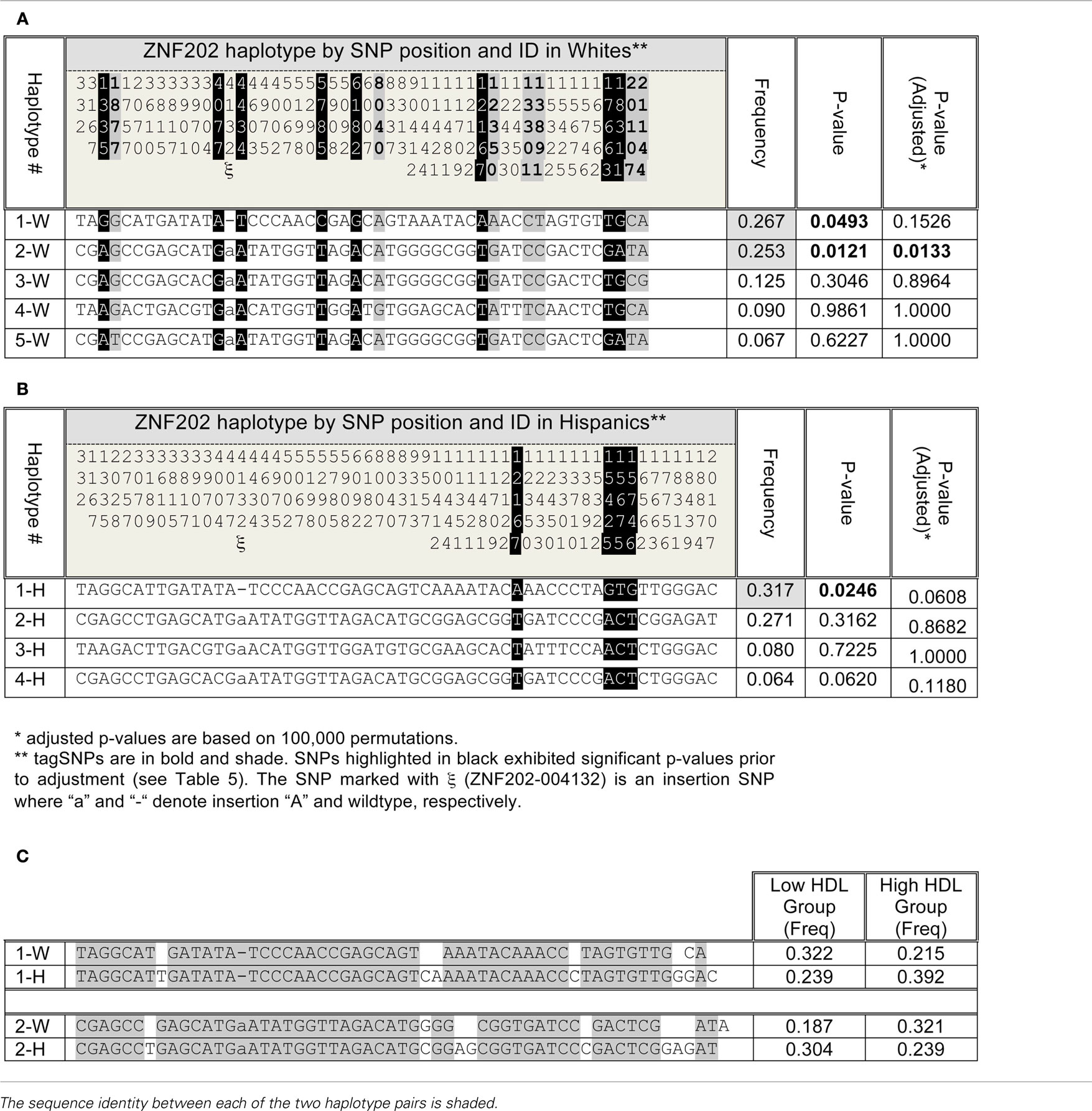
Table 4. Haplotype association of ZNF202 between high and low HDL groups in Whites (A) and in Hispanics (B). The sequence identity between the two significant haplotypes associated with low and high HDL groups across Whites and Hispanics (C).
To follow-up the haplotype results, 13 SNPs selected from the haplotypes were tested for allele frequency differences between the high and low HDL groups (Table 5). No SNPs remained significant after correction for multiple testing. These SNPs were in strong LD with one another.
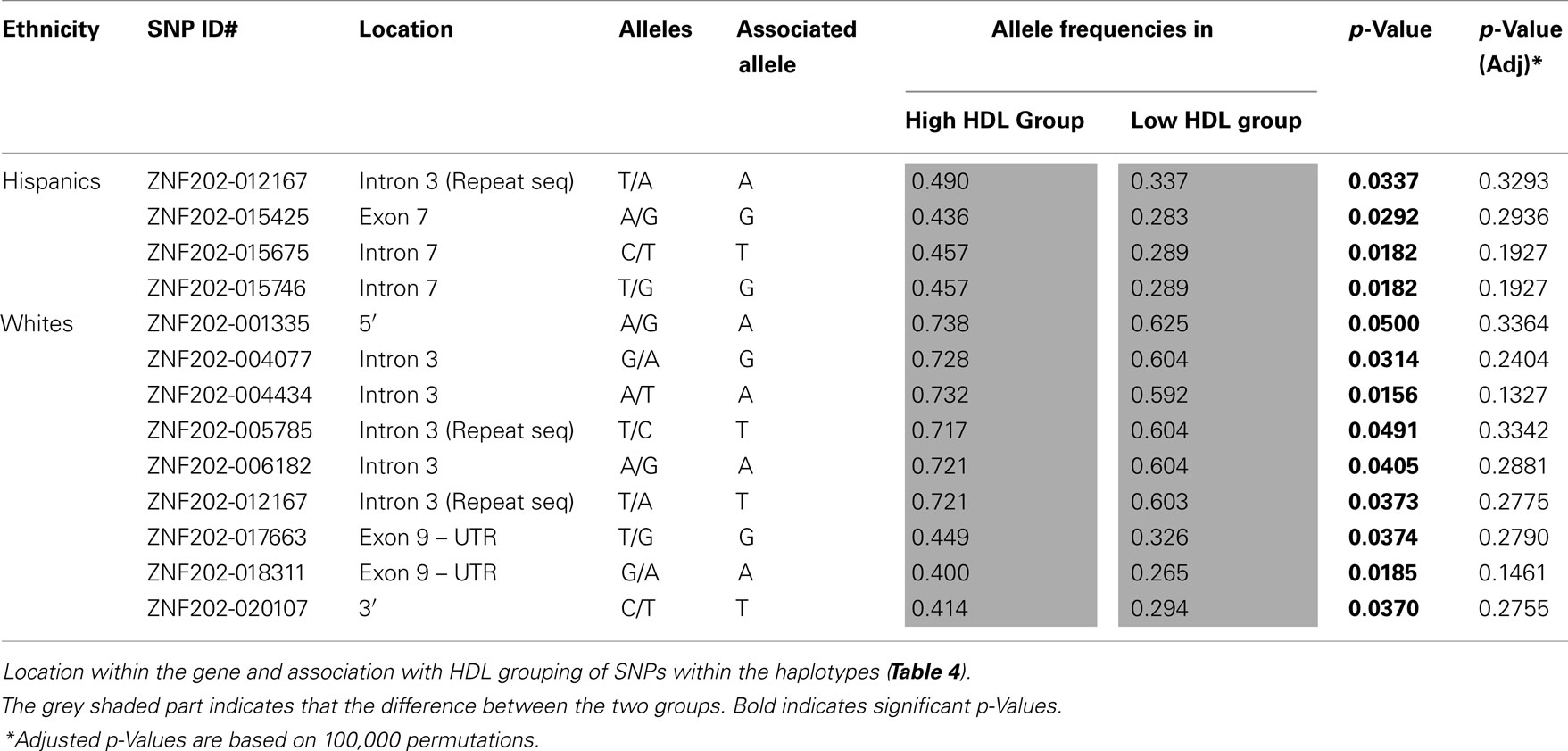
Table 5. Association results for SNPs in ZNF202.
Also of interest was the number of rare variants unique to the low HDL group or to the high HDL group. While the observed number of rare variants was similar between the high and low HDL groups in Whites (21 vs. 17; p = 0.474), the number of rare variants was significantly different in Hispanics (4 vs. 13; p = 0.0213; Figure 1C; section 9 in Supplementary Material S2). One rare variant located in intron 3 (ZNF202-006898, MAF = 0.01) was present in the high HDL group of both Whites and Hispanics. One of the two non-synonymous variants identified in our samples was K259E (ZNF202-015468, MAF = 0.02). This rare missense variant was predicted by the bioinformatic program SIFT (Ng and Henikoff, 2001) to be intolerant and by PolyPhen (Ramensky et al., 2002) to possibly be damaging.
The distribution of rare ZNF202 variants over exons, introns, and the 5′ and 3′ regions was not significantly different between high and low HDL groups. Additionally, there was no significant relationship between the proportions of rare variants carried by an individual and their HDL group (results not shown).
To better classify the association between ZNF202 tagSNPs and the HDL groups, a logistic regression was performed (Table 6), and based on the global test of association, only Whites demonstrated a significant association between low and high HDL groups (p = 0.00596).
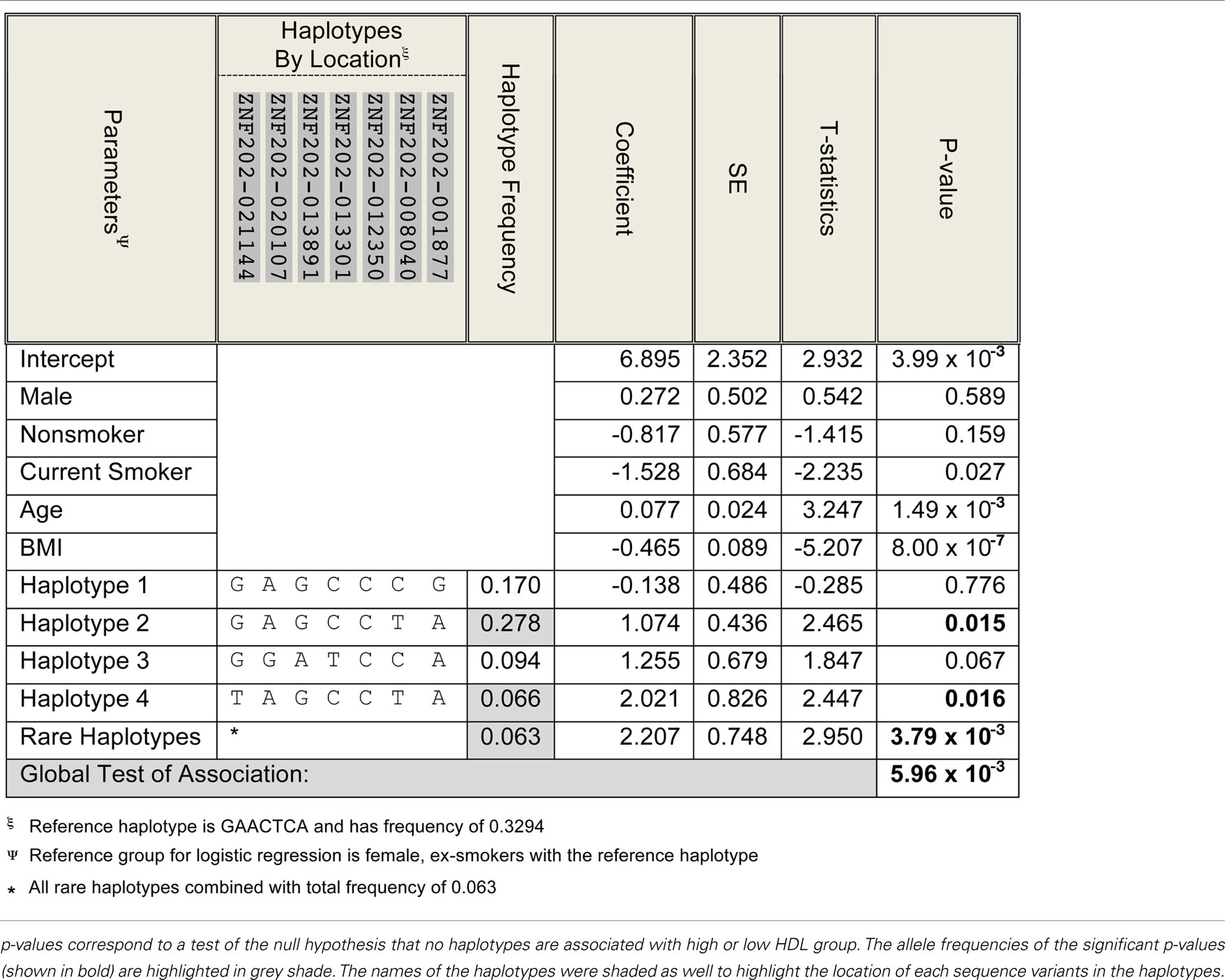
Table 6. Logistic regression of ZNF202 haplotypes between high and low HDL groups in Whites.
In light of the significance of microRNAs (miR) in gene regulation, we looked at the 3′UTR regions of LIPG and ZNF202 for potential miR sites, and determined if any of the identified variants in our study were contained in those sites (section 13 in Supplementary Material S1 and S2). We found one rare variant (ZNF202-018021) in one individual within the putative binding site of miR-127, about 360 bases from the stop codon. Because of the sporadic presence of this variant it was not possible to examine its impact on HDL-C.
Discussion
The genetic basis of HDL-C levels is complex (Weissglas-Volkov and Pajukanta, 2010), and inconsistency between genetic associations and functional studies of its candidate genes can add further confusion to this complexity. Two genes, LIPG and ZNF202, exemplify the complexity. Several functional studies have firmly established the role of these two genes in HDL metabolism. Yet, genetic association studies have produced contradictory results regarding their roles in modulating HDL-C levels. The goal of this study was, therefore, to resequence the entire length of these two genes in 235 subjects with extreme HDL-C levels in order to identify all variants associated with HDL-C. The subjects were selected from a bi-ethnic population of Whites and Hispanics and classified into high HDL (n = 114) or low HDL (n = 121) groups.
We identified 185 and 122 sequence variants in LIPG and ZNF202 genes, respectively; both common and rare variants were identified. We constructed detailed haplotype (multi-site) maps for both genes in Whites and Hispanics and identified haplotype association of ZNF202 and marginal association of LIPG with HDL-C. The associated sequence variants were common variants occurring in non-coding regions of the gene. Rare and missense variants were not associated with HDL-C levels. This pattern suggests that the mechanism of association with HDL-C levels is most likely mediated through modulatory effects on the expression of these two genes.
LIPG
At least 18 different missense mutations in the human LIPG gene have been reported in scientific databases to-date. All but one (T111I) are rare variants. Our sequence analysis of the LIPG gene identified only two coding variants: one common, T111I (LIPG-007438), and the other rare, N396S (LIPG-023529), with minor allele frequencies of 0.27 and 0.01, respectively.
Several GWAS studies have identified LIPG as having significant association with HDL-C levels (Kathiresan et al., 2008; Keebler et al., 2009; Gupta et al., 2010; Ma et al., 2010; Teslovich et al., 2010). However, the results pertaining to the association of T111I with HDL-C levels have been inconsistent, as some studies found an association (Ma et al., 2003; Mank-Seymour et al., 2004; Hutter et al., 2006; Tang et al., 2008; Smith et al., 2009) while others did not (Halverstadt et al., 2003; Paradis et al., 2003; Shimizu et al., 2007; Jensen et al., 2009). Although the impact of the T111I variant on the bridging function of EL has yet to be evaluated, recent functional studies indicate T111I has normal lipase activity (Edmondson et al., 2009; Smith et al., 2009), and thus it may not have a major impact on HDL-C levels. In our dataset, the occurrence of T111I in Hispanics was twice as common in the high HDL group as in the low HDL group; in Whites there was no difference, confirming its elusive association pattern (Figure 1C). However the elusive association of T111I with HDL-C levels can be explained by its location within a haplotype block. The T111I variant (SNP #41, LIPG-007438) resides on different haplotype blocks, depending on ethnicity (Figure 2). We found in Hispanics, it is part of block 1 and hence carrying the influence of the other SNPs in the block on HDL; whereas, in Whites, it is located in a small but an independent region between Blocks 3 and 4. Therefore, it has no influence on HDL by itself.
The rare missense variant, N396S, has been shown to be a causal variant. Edmondson et al. (2009) showed that N396S is a loss-of-function variant in EL, and it is associated with an increase in HDL-C levels of about 8 mg/dl. Furthermore, this same group found a significant excess of rare LIPG missense mutations with loss-of-function unique to subjects with elevated HDL-C. Thus, the N396S variant may impact HDL-C levels at the population level only in combination with other rare causal variants. A similar mechanism, where multiple rare alleles contribute to low HDL-C, was demonstrated by Cohen et al. (2004) and for hypertriglyceridemia by Johansen et al. (2010). However, our data show this is not the underlying mechanism in our population sample, as N396S was the only rare and causal missense variant we found.
Our data suggest that structure/function changes to EL as a result of these two missense mutations are unlikely to have a major impact on HDL-C levels, at least in our population samples. The remaining sequence variants (n = 183) identified in our study do not affect structure or function of EL, but some may affect the regulation of LIPG expression. This obvious deducible conclusion is in line with the hypothesis that most weak-effect causal variants are non-coding (Cooper and Shendure, 2011), which is supported by the collective GWAS evidence that shows about 88% of trait-associated variants are non-coding (Hindorff et al., 2009), and functional studies have defined a regulatory role for some of these variants (Musunuru et al., 2010).
In both Whites and Hispanics, the high HDL groups had more common variants but fewer rare variants than did the low HDL groups (Figure 1C; sections 9 and 10 in Supplementary Material S1). When we determined the locations (5′, exons, introns, 3′) of variants unique to either low or high HDL group, we observed a significant difference between the two groups. In Hispanics, the low HDL group had a significantly higher number of variants located in intronic regions of the gene than the high HDL group (Table 3). This suggests the presence of functional motif(s) in the intronic regions of the gene that modulate LIPG expression, which strengthens our hypothesis that causal variants in putative regulatory motifs in the non-coding regions of the gene may explain the wide variation in HDL-C levels observed in the general population. Several LIPG functional studies have shown that even a modest increase in EL expression has profound effects on HDL metabolism (Jaye et al., 1999; Ishida et al., 2003; Jin et al., 2003; Ma et al., 2003; Maugeais et al., 2003; Khetarpal et al., 2011). Thus, these functional studies support the notion that in a population where causal missense mutations are rare and few, common SNPs in regulatory regions of a gene may play a major role in modulating HDL-C levels.
Our data also suggest that the genetic architecture (haplotype blocks and SNP patterns) of LIPG is different between Whites and Hispanics. Although both ethnic groups had four haplotype blocks, the size of each block was substantially different between them (Figure 2). One of the interesting findings in the haplotype analyses was the borderline association of one haplotype (#3: TCAGCTGTTCTTC) with the high HDL group in Hispanics (p = 0.0576). This haplotype was present at a relatively high frequency (0.293 and 0.128 in the high and low HDL groups, respectively; Table 2). Within this haplotype, there were three significant SNPs prior to adjustment for multiple testing (Table 2), which are located in Exon 1-UTR (LIPG-002229, MAF = 0.29), Intron 1 (LIPG-002651, MAF = 0.28), and Exon 3 (LIPG-007438, T111I, MAF = 0.27). The first two SNPs may be involved in the regulation of LIPG expression and are excellent candidates for functional studies. Indeed, using luciferase reporter assay, Khetarpal et al. (2011) recently showed the common variant LIPG-002229(rs34474737) decreased promoter activity and confirmed our finding that this variant was in strong LD with non-functional T111I, which was enriched in high HDL group. We suspect that the borderline adjusted p-value is due to our moderate sample size and hence moderate statistical power; therefore, we report both the adjusted and unadjusted p-values in the tables. None of the haplotypes in Whites showed significant associations with either HDL group. Another mechanism that is emerging in the genetics of complex disorders is the possible involvement of many common variants, with each having a small effect below a significance threshold, but which collectively may explain most of the observed variance. This phenomenon has been demonstrated, for example, in the genetic risk of schizophrenia (International Schizophrenia Consortium et al., 2009). The SNPs discussed in the above haplotype may well represent another example of this mechanism.
ZNF202
In contrast to LIPG, there has been only one genetic study that examined the association of ZNF202 with the plasma lipid profile. Stene et al. (2006b) screened the promoter and protein-coding exons of the ZNF202 in 190 individuals with extreme high or low levels of HDL-C selected from 9259 Danish adults. They identified a total of 17 sequence variants, of which four (A154V, K259E, V274L, R605W) were in the protein-coding exons. Except A154V, which had high frequency among the Danish cohort (MAF = 0.30), the other three were rare variants. None of the missense variants identified had a major contribution to HDL-C levels. The group also acknowledged limitations of their study resulting from focusing only on promoter and the protein-coding exons.
In this study we identified only two coding variants, A154A (ZNF202-013891, MAF = 0.36) and K259E (ZNF202-015468, MAF = 0.02). The latter is a rare missense variant, which by itself cannot explain the extreme HDL-C levels in our population sample. The other variant, A154V, is common but little is known regarding its impact on ZNF202 structure or function. Stene et al. (2006a) found a significant association between this SNP and the risk of ischemic heart disease (IHD) and myocardial infarction (MI) in women but not in men. In another follow-up study by the same group (Stene et al., 2008), it was shown that SNP −660A/G (MAF = 30%) in the ZNF202 promoter predicted severe atherosclerosis and increased risk of IHD. Furthermore, homozygosity of the variant (GG) reduced the expression of ZNF202 by 60%, indicating the G allele is associated with reduced transcriptional activity. Interestingly, they found that the A154V SNP was in tight LD and strongly correlated with the −660A/G SNP. Our study did not find a significant association between A154V and HDL-C. In our sample, the 660A/G SNP (ZNF202-001310) was present at only 0.03 MAF and did not show significant association with HDL-C. Nevertheless, the significance of the A154V SNP on the structure and function of ZNF202 should be assessed independently of its LD with the 660A/G SNP, as by itself, it could be a causal factor for alteration in ZNF202 function.
Unlike LIPG, the haplotype block structure for ZNF202 was similar between Hispanics and Whites and consisted of one block (Figure 3). Also, the ratio of rare to common variants was significantly lower for ZNF202 than for LIPG. This implies that evolutionary forces have limited leverage on this highly conserved transcriptional factor gene. We identified two high frequency haplotypes in Whites, and both showed significant association with HDL grouping (Table 4; section 12 in Supplementary Material S2). One haplotype (#1-W) had a frequency of 0.267 and was associated with low HDL, while the other (#2-W) had a frequency of 0.253 and was associated with high HDL. However, only the latter remained significant after adjustment for multiple testing. In Hispanics, there was a haplotype (#1-H) with frequency of 0.317 that was significantly associated with high HDL group prior to adjusting for multiple testing (Table 4; section 12 in Supplementary Material S2). It is noteworthy that of the 50 nucleotides defining the haplotypes, the difference between the low HDL haplotype in Whites (#1-W) and the high HDL haplotype in Hispanics (#1-H) is only seven nucleotides (Table 4). These seven may be causal variants with functional significance.
SNPs from these haplotypes were tested for HDL group association, but none retained significance after adjustment for multiple testing (Table 5). Two of these SNPs (ZNF202-012167 and ZNF202-005785) were within the genomic dispersed repeat sequences. The role these repeats may play in modulating HDL-C levels is currently unclear. Another common SNP (2T ⇒ G), ZNF202-176663, with MAF of 0.449 and 0.326 in high and low HDL groups, respectively, is located in the 3′UTR, which is 2 bp downstream of the translation stop codon. Stene et al. (2006b) also found this SNP in their Danish population sample, where it exhibited an allele frequency of 0.39, and was marginally associated with low HDL-C levels in men but not women. It will be of interest to assess this SNP in future functional studies to determine if it impacts the expression of the ZNF202 gene.
In order to assess differences in findings between our study and Stene et al. (2006b), we used logistic regression between extreme HDL groups and haplotypes carrying tagged SNPs. We found significant association, again, only in Whites (Table 6). Furthermore, this association is not with missense variants but is with variants in non-coding regions of the gene.
In conclusion, comprehensive resequencing may reveal a more complete picture of genetic architecture underlying complex traits than exome-resequencing approach. The finding in this study that a few non-coding SNPs in LIPG and ZNF202 associated with HDL-C levels should provide impetus for pursuing future studies aimed at elucidating the mechanisms by which non-coding SNPs affect expression of these two genes.
Limitation of this study
The resequencing of LIPG in this study failed to screen 5773 bp within intronic segments of the gene (section 3 in Supplementary Material S1). As a result, there may be additional SNPs in the missed regions that may be relevant to HDL-C, which could not be analyzed.
Author Contributions
Study concept and design: Razzaghi, Kamboh. Acquisition of data: Razzaghi. Analysis and interpretation of data: Razzaghi, Santorico, Kamboh. Administrative, technical, and material support: Razzaghi. Study supervision: Razzaghi. Drafting of the manuscript: Razzaghi. Critical revision of the manuscript for important intellectual content: Razzaghi, Santorico, Kamboh. For the authorship and acknowledgment assignment and order, the criteria set by the International Committee of Medical Journal Editors (http://www.icmje.org) were followed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute (NHLBI; #R82 and #HL080630) to Dr. Hamid Razzaghi and in part by NHLBI grant (HL084613) to Dr. Ilyas Kamboh. Resequencing services were provided by the University of Washington, Department of Genome Sciences, under U.S. Federal Government contract number N01-HV-48194 from NHLBI. In addition, we wish to acknowledge Dr. Mark Rieder, Dr. Debbie Nickerson, and Mr. Eric Torskey of the University of Washington for the outstanding resequencing service. The SLVDS DNA samples and the associated biometric data were kindly provided by Dr. Richard Hamman of the University of Colorado Denver. We appreciate Dr. Jonathan Cohen from the University of Texas Southwestern Medical Center for his review and comments on the manuscript.
Supplementary Material
The Supplementary Material S1, S2 and S3 for this article can be found online at http://www.frontiersin.org/Applied_Genetic_Epidemiology/10.3389/fgene.2012.00089/abstract
Abbreviations
apoAI, apolipoprotein AI; apoE, apolipoprotein E; apoB, apolipoprotein B; BMI, body mass index; EL, endothelial lipase; HDL-C, high density lipoproteins-cholesterol; LDL-C, low density lipoproteins-cholesterol; LOD score logarithm of odds; LPL, lipoprotein lipase; MAF, minor allele frequency; SD, standard deviation; SLVDS, San Luis Valley diabetes study; TC, total cholesterol; TG, triglycerides; WGA, whole genome amplification; ZNF202, zinc finger protein 202.
Footnotes
References
Acton, S., Osgood, D., Donoghue, M., Corella, D., Pocovi, M., Cenarro, A., Mozas, P., Keilty, J., Squazzo, S., Woolf, E. A., and Ordovas, J. M. (1999). Association of polymorphisms at the SR-BI gene locus with plasma lipid levels and body mass index in a white population. Arterioscler. Thromb. Vasc. Biol. 19, 1734–1743.
Badellino, K. O., and Rader, D. J. (2004). The role of endothelial lipase in high-density lipoprotein metabolism. Curr. Opin. Cardiol. 19, 392–395.
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265.
Barter, P., Gotto, A. M., LaRosa, J. C., Maroni, J., Szarek, M., Grundy, S. M., Kastelein, J. J., Bittner, V., Fruchart, J. C., and Treating to New Targets Investigators. (2007). HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 357, 1301–1310.
Besler, C., Heinrich, K., Rohrer, L., Doerries, C., Riwanto, M., Shih, D. M., Chroni, A., Yonekawa, K., Stein, S., Schaefer, N., Mueller, M., Akhmedov, A., Daniil, G., Manes, C., Templin, C., Wyss, C., Maier, W., Tanner, F. C., Matter, C. M., Corti, R., Furlong, C., Lusis, A. J., von Eckardstein, A., Fogelman, A. M., Lüscher, T. F., and Landmesser, U. (2011). Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Invest. 121, 2693–2708.
Bonilla, C., Parra, E. J., Pfaff, C. L., Dios, S., Marshall, J. A., Hamman, R. F., Ferrell, R. E., Hoggart, C. L., McKeigue, P. M., and Shriver, M. D. (2004). Admixture in the Hispanics of the San Luis Valley, Colorado, and its implications for complex trait gene mapping. Ann. Hum. Genet. 68, 139–153.
Brooks-Wilson, A., Marcil, M., Clee, S. M., Zhang, L. H., Roomp, K., van Dam, M., Yu, L., Brewer, C., Collins, J. A., Molhuizen, H. O., Loubser, O., Ouelette, B. F., Fichter, K., Ashbourne-Excoffon, K. J., Sensen, C. W., Scherer, S., Mott, S., Denis, M., Martindale, D., Frohlich, J., Morgan, K., Koop, B., Pimstone, S., Kastelein, J. J., Genest, J. Jr., and Hayden, M. R. (1999). Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 22, 336–345.
Brown, R. J., Edmondson, A. C., Griffon, N., Hill, T. B., Fuki, I. V., Badellino, K. O., Li, M., Wolfe, M. L., Reilly, M. P., and Rader, D. J. (2009). A naturally occurring variant of endothelial lipase associated with elevated HDL exhibits impaired synthesis. J. Lipid Res. 50, 1910–1916.
Burchfiel, C. M., Hamman, R. F., Marshall, J. A., Baxter, J., Kahn, L. B., and Amirani, J. J. (1990). Cardiovascular risk factors and impaired glucose tolerance: the San Luis Valley Diabetes Study. Am. J. Epidemiol. 131, 57–70.
Cohen, J. C., Kiss, R. S., Pertsemlidis, A., Marcel, Y. L., McPherson, R., and Hobbs, H. H. (2004). Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305, 869–872.
Cooper, G. M., and Shendure, J. (2011). Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 12, 628–640.
Das, U. N. (2005). Long-chain polyunsaturated fatty acids, endothelial lipase and atherosclerosis. Prostaglandins Leukot. Essent. Fatty Acids 72, 173–179.
deLemos, A. S., Wolfe, M. L., Long, C. J., Sivapackianathan, R., and Rader, D. J. (2002). Identification of genetic variants in endothelial lipase in persons with elevated high-density lipoprotein cholesterol. Circulation 106, 1321–1326.
Edmondson, A. C., Brown, R. J., Kathiresan, S., Cupples, L. A., Demissie, S., Manning, A. K., Jensen, M. K., Rimm, E. B., Wang, J., Rodrigues, A., Bamba, V., Khetarpal, S. A., Wolfe, M. L., Derohannessian, S., Li, M., Reilly, M. P., Aberle, J., Evans, D., Hegele, R. A., and Rader, D. J. (2009). Loss-of-function variants in endothelial lipase are a cause of elevated HDL cholesterol in humans. J. Clin. Invest. 119, 1042–1050.
Emerging Risk Factors, Collaboration Di Angelantonio, E., Sarwar, N., Perry, P., Kaptoge, S., Ray, K. K., Thompson, A., Wood, A. M., Lewington, S., Sattar, N., Packard, C. J., Collins, R., Thompson, S. G., and Danesh, J. (2009). Major lipids, apolipoproteins, and risk of vascular disease. JAMA 302, 1993–2000.
Fisher, R. M., Humphries, S. E., and Talmud, P. J. (1997). Common variation in the lipoprotein lipase gene: effects on plasma lipids and risk of atherosclerosis. Atherosclerosis 135, 145–159.
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., Higgins, J., DeFelice, M., Lochner, A., Faggart, M., Liu-Cordero, S. N., Rotimi, C., Adeyemo, A., Cooper, R., Ward, R., Lander, E. S., Daly, M. J., and Altshuler, D. (2002). The structure of haplotype blocks in the human genome. Science 296, 2225–2229.
Goode, E. L., Cherny, S. S., Christian, J. C., Jarvik, G. P., and de Andrade, M. (2007). Heritability of longitudinal measures of body mass index and lipid and lipoprotein levels in aging twins. Twin Res. Hum. Genet. 10, 703–711.
Gordon, D., Abajian, C., and Green, P. (1998). Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202.
Gupta, R., Ejebe, K., Butler, J., Lettre, G., Lyon, H., Guiducci, C., Wilks, R., Bennett, F., Forrester, T., Tayo, B., Musunuru, K., Hirschhorn, J., Kathiresan, S., Cooper, R. S., and McKenzie, C. A. (2010). Association of common DNA sequence variants at 33 genetic loci with blood lipids in individuals of African ancestry from Jamaica. Hum. Genet. 128, 557–561.
Halverstadt, A., Phares, D. A., Ferrell, R. E., Wilund, K. R., Goldberg, A. P., and Hagberg, J. M. (2003). High-density lipoprotein-cholesterol, its subfractions, and responses to exercise training are dependent on endothelial lipase genotype. Metab. Clin. Exp. 52, 1505–1511.
Hamman, R. F., Marshall, J. A., Baxter, J., Kahn, L. B., Mayer, E. J., Orleans, M., Murphy, J. R., and Lezotte, D. C. (1989). Methods and prevalence of non-insulin-dependent diabetes mellitus in a biethnic Colorado population. The San Luis Valley Diabetes Study. Am. J. Epidemiol. 129, 295–311.
Hindorff, L. A., Sethupathy, P., Junkins, H. A., Ramos, E. M., Mehta, J. P., Collins, F. S., and Manolio, T. A. (2009). Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 106, 9362–9367.
Hirata, K., Dichek, H. L., Cioffi, J. A., Choi, S. Y., Leeper, N. J., Quintana, L., Kronmal, G. S., Cooper, A. D., and Quertermous, T. (1999). Cloning of a unique lipase from endothelial cells extends the lipase gene family. J. Biol. Chem. 274, 14170–14175.
Hutter, C. M., Austin, M. A., Farin, F. M., Viernes, H. M., Edwards, K. L., Leonetti, D. L., McNeely, M. J., and Fujimoto, W. Y. (2006). Association of endothelial lipase gene (LIPG) haplotypes with high-density lipoprotein cholesterol subfractions and apolipoprotein AI plasma levels in Japanese Americans. Atherosclerosis 185, 78–86.
International Schizophrenia Consortium, Purcell, S. M., Wray, N. R., Stone, J. L., Visscher, P. M., O’Donovan, M. C., Sullivan, P. F., and Sklar, P. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752.
Ishida, T., Choi, S., Kundu, R. K., Hirata, K., Rubin, E. M., Cooper, A. D., and Quertermous, T. (2003). Endothelial lipase is a major determinant of HDL level. J. Clin. Invest. 111, 347–355.
Jaye, M., Lynch, K. J., Krawiec, J., Marchadier, D., Maugeais, C., Doan, K., South, V., Amin, D., Perrone, M., and Rader, D. J. (1999). A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 21, 424–428.
Jensen, M. K., Rimm, E. B., Mukamal, K. J., Edmondson, A. C., Rader, D. J., Vogel, U., Tjønneland, A., Sørensen, T. I., Schmidt, E. B., and Overvad, K. (2009). The T111I variant in the endothelial lipase gene and risk of coronary heart disease in three independent populations. Eur. Heart J. 30, 1584–1589.
Jin, W., Millar, J. S., Broedl, U., Glick, J. M., and Rader, D. J. (2003). Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J. Clin. Invest. 111, 357–362.
Johansen, C. T., Wang, J., Lanktree, M. B., Cao, H., McIntyre, A. D., Ban, M. R., Martins, R. A., Kennedy, B. A., Hassell, R. G., Visser, M. E., Schwartz, S. M., Voight, B. F., Elosua, R., Salomaa, V., O’Donnell, C. J., Dallinga-Thie, G. M., Anand, S. S., Yusuf, S., Huff, M. W., Kathiresan, S., and Hegele, R. A. (2010). Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 42, 684–687.
Kathiresan, S., Melander, O., Guiducci, C., Surti, A., Burtt, N. P., Rieder, M. J., Cooper, G. M., Roos, C., Voight, B. F., Havulinna, A. S., Wahlstrand, B., Hedner, T., Corella, D., Tai, E. S., Ordovas, J. M., Berglund, G., Vartiainen, E., Jousilahti, P., Hedblad, B., Taskinen, M. R., Newton-Cheh, C., Salomaa, V., Peltonen, L., Groop, L., Altshuler, D. M., and Orho-Melander, M. (2008). Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 40, 189–197.
Keebler, M. E., Sanders, C. L., Surti, A., Guiducci, C., Burtt, N. P., and Kathiresan, S. (2009). Association of blood lipids with common DNA sequence variants at 19 genetic loci in the multiethnic United States National Health and Nutrition Examination Survey III. Circ. Cardiovasc. Genet. 2, 238–243.
Khetarpal, S. A., Edmondson, A. C., Raghavan, A., Neeli, H., Jin, W., Badellino, K. O., Demissie, S., Manning, A. K., DerOhannessian, S. L., Wolfe, M. L., Cupples, L. A., Li, M., Kathiresan, S., and Rader, D. J. (2011). Mining the LIPG allelic spectrum reveals the contribution of rare and common regulatory variants to HDL cholesterol. PLoS Genet. 7, e1002393. doi:10.1371/journal.pgen.1002393
Kort, E. N., Ballinger, D. G., Ding, W., Hunt, S. C., Bowen, B. R., Abkevich, V., Bulka, K., Campbell, B., Capener, C., Gutin, A., Harshman, K., McDermott, M., Thorne, T., Wang, H., Wardell, B., Wong, J., Hopkins, P. N., Skolnick, M., and Samuels, M. (2000). Evidence of linkage of familial hypoalphalipoproteinemia to a novel locus on chromosome 11q23. Am. J. Hum. Genet. 66, 1845–1856.
Kronenberg, F., Coon, H., Ellison, R. C., Borecki, I., Arnett, D. K., Province, M. A., Eckfeldt, J. H., Hopkins, P. N., and Hunt, S. C. (2002). Segregation analysis of HDL cholesterol in the NHLBI Family Heart Study and in Utah pedigrees. Eur. J. Hum. Genet. 10, 367–374.
Kuivenhoven, J. A., Pritchard, H., Hill, J., Frohlich, J., Assmann, G., and Kastelein, J. (1997). The molecular pathology of lecithin: cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 38, 191–205.
Langmann, T., Schumacher, C., Morham, S. G., Honer, C., Heimerl, S., Moehle, C., and Schmitz, G. (2003). ZNF202 is inversely regulated with its target genes ABCA1 and apoE during macrophage differentiation and foam cell formation. J. Lipid Res. 44, 968–977.
Lindegaard, M. L., Nielsen, J. E., Hannibal, J., and Nielsen, L. B. (2004). Expression of the endothelial lipase gene in murine embryos and reproductive organs. J. Lipid Res. 46, 439–444.
Lusis, A. J., Mar, R., and Pajukanta, P. (2004). Genetics of atherosclerosis. Annu. Rev. Genomics Hum. Genet. 5, 189–218.
Ma, K., Cilingiroglu, M., Otvos, J. D., Ballantyne, C. M., Marian, A. J., and Chan, L. (2003). Endothelial lipase is a major genetic determinant for high-density lipoprotein concentration, structure, and metabolism. Proc. Natl. Acad. Sci. U.S.A. 100, 2748–2753.
Ma, L., Yang, J., Runesha, H. B., Tanaka, T., Ferrucci, L., Bandinelli, S., and Da, Y. (2010). Genome-wide association analysis of total cholesterol and high-density lipoprotein cholesterol levels using the Framingham heart study data. BMC Med. Genet. 11, 55. doi:10.1186/1471-2350-11-55
Mank-Seymour, A. R., Durham, K. L., Thompson, J. F., Seymour, A. B., and Milos, P. M. (2004). Association between single-nucleotide polymorphisms in the endothelial lipase (LIPG) gene and high-density lipoprotein cholesterol levels. Biochem. Biophys. Acta 1636, 40–46.
Maugeais, C., Tietge, U. J., Broedl, U. C., Marchadier, D., Cain, W., McCoy, M. G., Lund-Katz, S., Glick, J. M., and Rader, D. J. (2003). Dose-dependent acceleration of high-density lipoprotein catabolism by endothelial lipase. Circulation 108, 2121–2126.
Monaco, C., Helmer Citterich, M., Caprini, E., Vorechovsky, I., Russo, G., Croce, C. M., Barbanti-Brodano, G., and Negrini, M. (1998). Molecular cloning and characterization of ZNF202: a new gene at 11q23.3 encoding testis-specific zinc finger proteins. Genomics 52, 358–362.
Musunuru, K., Strong, A., Frank-Kamenetsky, M., Lee, N. E., Ahfeldt, T., Sachs, K. V., Li, X., Li, H., Kuperwasser, N., Ruda, V. M., Pirruccello, J. P., Muchmore, B., Prokunina-Olsson, L., Hall, J. L., Schadt, E. E., Morales, C. R., Lund-Katz, S., Phillips, M. C., Wong, J., Cantley, W., Racie, T., Ejebe, K. G., Orho-Melander, M., Melander, O., Koteliansky, V., Fitzgerald, K., Krauss, R. M., Cowan, C. A., Kathiresan, S., and Rader, D. J. (2010). From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466, 714–719.
Ng, P. C., and Henikoff, S. (2001). Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874.
Paradis, M. E., Couture, P., Bosse, Y., Despres, J. P., Perusse, L., Bouchard, C., Vohl, M. C., and Lamarche, B. (2003). The T111I mutation in the EL gene modulates the impact of dietary fat on the HDL profile in women. J. Lipid Res. 44, 1902–1908.
Peacock, R. E., Hamsten, A., Johansson, J., Nilsson-Ehle, P., and Humphries, S. E. (1994). Associations of genotypes at the apolipoprotein AI-CIII-AIV, apolipoprotein B and lipoprotein lipase gene loci with coronary atherosclerosis and high density lipoprotein subclasses. Clin. Genet. 46, 273–282.
Porsch-Özcürümez, M., Langmann, T., Heimerl, S., Borsukova, H., Kaminski, W. E., Drobnik, W., Honer, C., Schumacher, C., and Schmitz, G. (2001). The zinc finger protein 202 (ZNF202) is a transcriptional repressor of ATP binding cassette transporter A1 (ABCA1) and ABCG1 gene expression and a modulator of cellular lipid efflux. J. Biol. Chem. 276, 12427–12433.
Prenger, V. L., Beaty, T. H., and Kwiterovich, P. O. (1992). Genetic determination of high-density lipoprotein-cholesterol and apolipoprotein A-1 plasma levels in a family study of cardiac catheterization patients. Am. J. Hum. Genet. 51, 1047–1057.
Qasim, A., and Rader, D. J. (2006). Human genetics of variation in high-density lipoprotein cholesterol. Curr. Atheroscler. Rep. 8, 198–205.
Ramensky, V., Bork, P., and Sunyaev, S. (2002). Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–3900.
Razzaghi, H., Aston, C. E., Hamman, R. F., and Kamboh, M. I. (2000). Genetic screening of the lipoprotein lipase gene for mutations associated with high triglyceride/low HDL-cholesterol levels. Hum. Genet. 107, 257–267.
Rosamond, W., Flegal, K., Furie, K., Go, A., Greenlund, K., Haase, N., Hailpern, S. M., Ho, M., Howard, V., Kissela, B., Kittner, S., Lloyd-Jones, D., McDermott, M., Meigs, J., Moy, C., Nichol, G., O’Donnell, C., Roger, V., Sorlie, P., Steinberger, J., Thom, T., Wilson, M., Hong, Y., and American Heart Association Statistics Committee and Stroke Statistics Subcommittee. (2008). Heart disease and stroke statistics – 2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 117, e25–e146.
Schaid, D. J., Rowland, C. M., Tines, D. E., Jacobson, R. M., and Poland, G. A. (2002). Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 70, 425–434.
Schmitz, G., Heimerl, S., and Langmann, T. (2004). Zinc finger protein ZNF202 structure and function in transcriptional control of HDL metabolism. Curr. Opin. Lipidol. 15, 199–208.
Shimizu, M., Kanazawa, K., Hirata, K., Ishida, T., Hiraoka, E., Matsuda, Y., Iwai, C., Miyamoto, Y., Hashimoto, M., Kajiya, T., Akita, H., and Yokoyama, M. (2007). Endothelial lipase gene polymorphism is associated with acute myocardial infarction, independently of high-density lipoprotein-cholesterol levels. Circ. J. 71, 842–846.
Smith, C. E., Arnett, D. K., Tsai, M. Y., Lai, C. Q., Parnell, L. D., Shen, J., Laclaustra, M., Junyent, M., and Ordovás, J. M. (2009). Physical inactivity interacts with an endothelial lipase polymorphism to modulate high density lipoprotein cholesterol in the GOLDN study. Atherosclerosis 206, 500–504.
Srinivasan, S. R., Ehnholm, C., Elkasabany, A., and Berenson, G. (1999). Influence of apolipoprotein E polymorphism on serum lipids and lipoprotein changes from childhood to adulthood: the Bogalusa Heart Study. Atherosclerosis 143, 435–443.
Stene, M. C., Frikke-Schmidt, R., Nordestgaard, B. G., Grande, P., Schnohr, P., and Tybjaerg-Hansen, A. (2008). Functional promoter variant in zinc finger protein 202 predicts severe atherosclerosis and ischemic heart disease. J. Am. Coll. Cardiol. 52, 369–377.
Stene, M. C., Frikke-Schmidt, R., Nordestgaard, B. G., Steffensen, R., Schnohr, P., and Tybjaerg-Hansen, A. (2006a). Zinc Finger Protein 202: a new candidate gene for ischemic heart disease: the Copenhagen City Heart Study. Atherosclerosis 2, 43–50.
Stene, M. C., Frikke-Schmidt, R., Nordestgaard, B. G., and Tybjaerg-Hansen, A. (2006b). Zinc Finger Protein 202, genetic variation, and HDL cholesterol in the general population. J. Lipid Res. 47, 944–952.
Stephens, M., Sloan, J. S., Robertson, P. D., Scheet, P., and Nickerson, D. A. (2006). Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat. Genet. 38, 375–381.
Tang, N. P., Wang, L. S., Yang, L., Zhou, B., Gu, H. J., Sun, Q. M., Cong, R. H., Zhu, H. J., and Wang, B. (2008). Protective effect of an endothelial lipase gene variant on coronary artery disease in a Chinese population. J. Lipid Res. 49, 369–375.
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M., Pirruccello, J. P., Ripatti, S., Chasman, D. I., Willer, C. J., Johansen, C. T., Fouchier, S. W., Isaacs, A., Peloso, G. M., Barbalic, M., Ricketts, S. L., Bis, J. C., Aulchenko, Y. S., Thorleifsson, G., Feitosa, M. F., Chambers, J., Orho-Melander, M., Melander, O., Johnson, T., Li, X., Guo, X., Li, M., Shin Cho, Y., Jin Go, M., Jin Kim, Y., Lee, J. Y., Park, T., Kim, K., Sim, X., Twee-Hee Ong, R., Croteau-Chonka, D. C., Lange, L. A., Smith, J. D., Song, K., Hua Zhao, J., Yuan, X., Luan, J., Lamina, C., Ziegler, A., Zhang, W., Zee, R. Y., Wright, A. F., Witteman, J. C., Wilson, J. F., Willemsen, G., Wichmann, H. E., Whitfield, J. B., Waterworth, D. M., Wareham, N. J., Waeber, G., Vollenweider, P., Voight, B. F., Vitart, V., Uitterlinden, A. G., Uda, M., Tuomilehto, J., Thompson, J. R., Tanaka, T., Surakka, I., Stringham, H. M., Spector, T. D., Soranzo, N., Smit, J. H., Sinisalo, J., Silander, K., Sijbrands, E. J., Scuteri, A., Scott, J., Schlessinger, D., Sanna, S., Salomaa, V., Saharinen, J., Sabatti, C., Ruokonen, A., Rudan, I., Rose, L. M., Roberts, R., Rieder, M., Psaty, B. M., Pramstaller, P. P., Pichler, I., Perola, M., Penninx, B. W., Pedersen, N. L., Pattaro, C., Parker, A. N., Pare, G., Oostra, B. A., O’Donnell, C. J., Nieminen, M. S., Nickerson, D. A., Montgomery, G. W., Meitinger, T., McPherson, R., McCarthy, M. I., McArdle, W., Masson, D., Martin, N. G., Marroni, F., Mangino, M., Magnusson, P. K., Lucas, G., Luben, R., Loos, R. J., Lokki, M. L., Lettre, G., Langenberg, C., Launer, L. J., Lakatta, E. G., Laaksonen, R., Kyvik, K. O., Kronenberg, F., König, I. R., Khaw, K. T., Kaprio, J., Kaplan, L. M., Johansson, A., Jarvelin, M. R., Janssens, A. C., Ingelsson, E., Igl, W., Kees Hovingh, G., Hottenga, J. J., Hofman, A., Hicks, A. A., Hengstenberg, C., Heid, I. M., Hayward, C., Havulinna, A. S., Hastie, N. D., Harris, T. B., Haritunians, T., Hall, A. S., Gyllensten, U., Guiducci, C., Groop, L. C., Gonzalez, E., Gieger, C., Freimer, N. B., Ferrucci, L., Erdmann, J., Elliott, P., Ejebe, K. G., Döring, A., Dominiczak, A. F., Demissie, S., Deloukas, P., de Geus, E. J., de Faire, U., Crawford, G., Collins, F. S., Chen, Y. D., Caulfield, M. J., Campbell, H., Burtt, N. P., Bonnycastle, L. L., Boomsma, D. I., Boekholdt, S. M., Bergman, R. N., Barroso, I., Bandinelli, S., Ballantyne, C. M., Assimes, T. L., Quertermous, T., Altshuler, D., Seielstad, M., Wong, T. Y., Tai, E. S., Feranil, A. B., Kuzawa, C. W., Adair, L. S., Taylor, H. A. Jr., Borecki, I. B., Gabriel, S. B., Wilson, J. G., Holm, H., Thorsteinsdottir, U., Gudnason, V., Krauss, R. M., Mohlke, K. L., Ordovas, J. M., Munroe, P. B., Kooner, J. S., Tall, A. R., Hegele, R. A., Kastelein, J. J., Schadt, E. E., Rotter, J. I., Boerwinkle, E., Strachan, D. P., Mooser, V., Stefansson, K., Reilly, M. P., Samani, N. J., Schunkert, H., Cupples, L. A., Sandhu, M. S., Ridker, P. M., Rader, D. J., van Duijn, C. M., Peltonen, L., Abecasis, G. R., Boehnke, M., and Kathiresan, S. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713.
Thierry-Mieg, D., and Thierry-Mieg, J. (2006). AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 7(Suppl. 1), S12.1–S12.14.
ATP III. (2002). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 106, 3143–3421.
Vaisar, T., Pennathur, S., Green, P. S., Gharib, S. A., Hoofnagle, A. N., Cheung, M. C., Byun, J., Vuletic, S., Kassim, S., Singh, P., Chea, H., Knopp, R. H., Brunzell, J., Geary, R., Chait, A., Zhao, X. Q., Elkon, K., Marcovina, S., Ridker, P., Oram, J. F., and Heinecke, J. W. (2007). Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Invest. 117, 746–756.
Wagner, S., Hess, M. A., Ormonde-Hanson, P., Malandro, J., Hu, H., Chen, M., Kehrer, R., Frodsham, M., Schumacher, C., Beluch, M., Honer, C., Skolnick, M., Ballinger, D., and Bowen, B. R. (2000). A broad role for the zinc finger protein ZNF202 in human lipid metabolism. J. Biol. Chem. 275, 15685–15690.
Wang, X., and Paigen, B. (2005). Genetics of variation in HDL cholesterol in humans and mice. Circ. Res. 96, 27–42.
Weissglas-Volkov, D., and Pajukanta, P. (2010). Genetic causes of high and low serum HDL-cholesterol. J. Lipid Res. 51, 2032–2057.
Yamakawa-Kobayashi, K., Yanagi, H., Endo, K., Arinami, T., and Hamaguchi, H. (2003). Relationship between serum HDL-C levels and common genetic variants of the endothelial lipase gene in Japanese school-aged children. Hum. Genet. 113, 311–315.
Yamakawa-Kobayashi, K., Yanagi, H., Fukayama, H., Hirano, C., Shimakura, Y., Yamamoto, N., Arinami, T., Tsuchiya, S., and Hamaguchi, H. (1999). Frequent occurrence of hypoalphalipoproteinemia due to mutant apolipoprotein A-I gene in the population: a population-based survey. Hum. Mol. Genet. 8, 331–336.
Keywords: LIPG, ZNF202, endothelial lipase, zinc finger protein 202, HDL, genetic association, resequencing
Citation: Razzaghi H, Santorico SA and Kamboh MI (2012) Population-based resequencing of LIPG and ZNF202 genes in subjects with extreme HDL levels. Front. Gene. 3:89. doi: 10.3389/fgene.2012.00089
Received: 17 February 2012; Accepted: 03 May 2012;
Published online: 14 June 2012.
Edited by:
Ali Samadikuchaksaraei, Imperial College London, UKReviewed by:
Sarah Buxbaum, Jackson State University, USAYing Wu, University of North Carolina, USA
Copyright: © 2012 Razzaghi, Santorico and Kamboh. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hamid Razzaghi, PhD, Division of Cardiology, Department of Medicine, University of Colorado Denver, Aurora, CO 80045, USA. e-mail:cmF6emFnaGlfaGFtaWRAeWFob28uY29t